Abstract
The title compound has been fully characterised using 1H and 13C NMR spectroscopy, which reveals (E) to (Z) isomerisation upon dissolution. In the solid state, X-ray diffraction shows exclusively the (E)-isomer with two geometrically near-identical independent molecules each with the outer rings tilted with respect to the central ring.
1. Introduction
The title compound 1 was first described in the literature in 1956 [], when it was prepared by oxidation of the corresponding diamine using mercuric oxide and subsequently reacted with a large excess of different anilines to give substituted azophenines (Scheme 1). This route provides perhaps the most efficient route to unsymmetrically substituted azophenines 2 which have been of recent interest as potential molecular switches when adsorbed onto a metal surface []. Because of the early date of the previous work, the degree of characterisation of 1 is limited with only the melting point, elemental analysis, and UV and IR spectra being reported [].
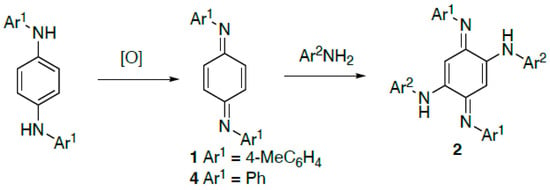
Scheme 1.
General formation of N,N′-diaryl-1,4-benzoquinonediimines and their reaction to give azophenines.
We recently prepared compound 1 on a large scale for new studies on the molecular switching behaviour of derived azophenines, and in this paper we describe a detailed 1H and 13C NMR analysis of 1 as well as its X-ray structure determination.
2. Results
The required starting diamine N,N′-di-p-tolyl-p-phenylenediamine 3 is reported to be readily accessible from the inexpensive starting materials hydroquinone and p-toluidine. However, the earliest procedures for this reaction used rather harsh conditions involving the prolonged heating of a melt of the neat materials with zinc chloride at over 200 °C [,]. A somewhat later procedure replaced the zinc chloride with iodine []. In our hands, none of these were satisfactory and so we adapted a patent procedure employing phosphoric acid as a dehydration catalyst [] (Scheme 2). Using this method, the diamine 3 was reliably obtained in low yield and had a melting point and NMR data in accordance with expectations.
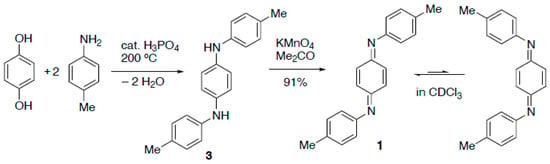
Scheme 2.
Synthetic route used to prepare compound 1.
The original oxidation of 3 to give the target diimine 1 used mercuric oxide in boiling benzene [], but we found that potassium permanganate in acetone, reported for the synthesis of the corresponding diphenyl compound 4 [], was less hazardous and just as effective. In this way compound 1 was obtained in high yield as dark red crystals with a melting point in good agreement with expectation.
A study of the compound using 1H and 13C NMR spectroscopy revealed interesting behaviour. Separate signals were observed for (E)- and (Z)-isomers and, in addition, their ratio varied with time. Specifically, a spectrum run immediately (5 min) after dissolution in CDCl3 showed an (E)/(Z) ratio of 5:1 but after 15 min this had changed to 3:1 and eventually after >12 h came to an equilibrium value of 1.25:1. When the equilibrated solution was allowed to evaporate, however, the deposited crystals were again the (E)-isomer. We conclude that the compound exists in the solid state exclusively as the (E)-isomer but this equilibrates in solution to a 55:45 mixture. A similar phenomenon has previously been observed for the diphenyl analogue 4 which was observed to equilibrate from the all-(E) solid to a mixture of (E)- and (Z)-isomers on dissolving in CDCl3 [], with variable temperature kinetic studies allowing an estimation of the energy barrier to isomerisation. In that case, the full assignment of 1H and 13C signals for both isomers was reported which was in excellent agreement with our independent assignments for 1 (Figure 1). A more recent study also reports 1H and 13C shifts for (E)- and (Z)-isomers of 4 in CD3SOCD3 with the 1H signals fully assigned [].
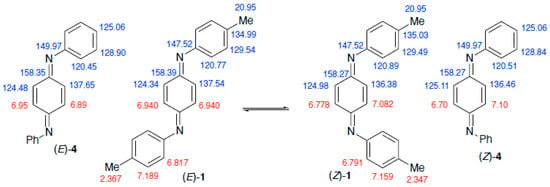
Figure 1.
Assignment of 1H (red) and 13C (blue) NMR signals for (E)- and (Z)-isomers of 1 and comparison with values for 4 [].
By comparing the spectra at different times after dissolution we were able to unambiguously assign all signals for the (E)- and (Z)-isomers (see Supplementary Materials). As shown in Figure 1, the data show some unexpected features. First, most chemical shift values are very similar between the two isomers, so we present them with one additional decimal place compared with the normal convention. While the 1H signals for the p-tolyl groups are all distinct, the 13C signals for N–C and CH3 are coincident between the two isomers. Perhaps more surprising is the observation that while the two 1H and 13C signals for the central quinonoid ring are both well separated for the (Z)-isomer, distinctly different 13C signals for the (E)-isomer correspond to a single 1H environment. A similar phenomenon is also observed for 4 (Figure 1).
For compound 1 the recrystallised material proved to be suitable for X-ray diffraction and the resulting structure features two independent molecules, each symmetrical about an inversion centre located at the centre of the quinonoid ring (Figure 2). We were aware that the crystal initially selected might not have been representative of the bulk material so, to exclude this possibility, six additional crystals were subjected to preliminary crystallographic examination but all gave the same unit cell parameters.
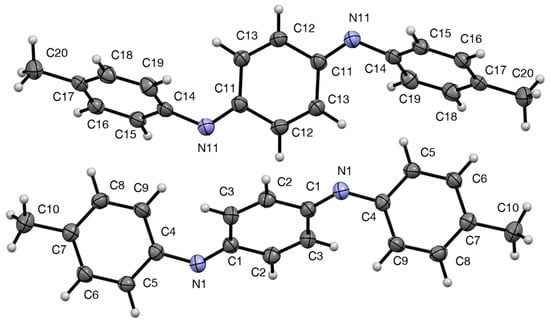
Figure 2.
Structures of the two independent molecules of 1 with numbering system used and 50% probability ellipsoids.
The dimensions of the two molecules are very similar (the fit of non-hydrogen atoms of the two independent molecules gives a root-mean-squared deviation of 0.057 Å) but there is a slight difference in the angles between the rings. In each case the two outer tolyl rings are exactly parallel and are tilted with respect to the central quinonediimine plane by 51.47(4)° for the C-1 molecule but 55.34(4)° for the C-11 molecule. In fact this structural feature is also common to most of the analogous N,N′-diaryl-1,4-benoquinonediimines whose structures have been determined (Figure 3).
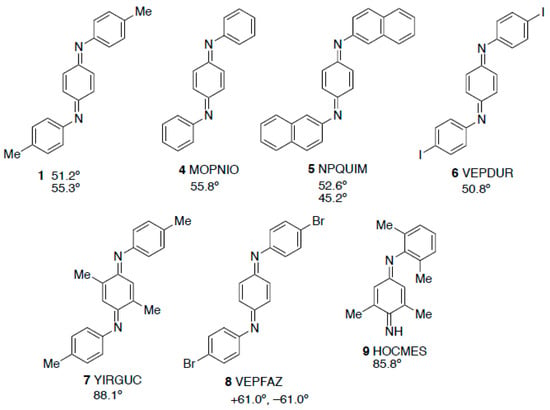
Figure 3.
Structure of 1 compared with diarylbenzoquinonediimines that have previously been crystallographically characterised with CSD Refcodes and dihedral angles between the N-aryl rings and the central ring.
Specifically, the diphenyl compound 4 [], the two independent molecules in the structure of the dinaphthyl compound 5 [], and the diiodo compound 6 [] all have angles in a similar range, while the extra methyl groups in 7 [] lead to a value in that case that is much larger. The exception to this trend is the dibromo compound 8 [] where the two outer rings are tilted to the same extent with respect to the central ring but in opposite directions, a surprising feature not commented upon by the original authors. In the more hindered monoaryldiimine 9 the angle between planes is again higher in a similar way to 7 [].
In the crystal, the molecules of 1 are arranged in rows with alternation of the two forms when viewed along the a-axis (Figure 4).
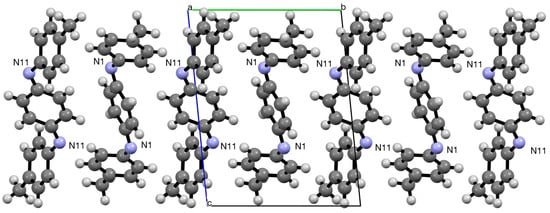
Figure 4.
Alternating arrangement of molecules in the crystal viewed along the a-axis.
In summary, we have found that compound 1 gives very similar NMR data to the diphenyl analogue 4 and exhibits the same (E) to (Z) isomerisation upon dissolution in CDCl3. In the solid state the X-ray structure shows purely the (E)-isomer with the coplanar outer rings tilted with respect to the central one to a similar degree to comparable structures.
3. Experimental
3.1. General Experimental Details
Melting points were recorded on a Reichert hot-stage microscope (Reichert, Vienna, Austria) and are uncorrected. NMR spectra were obtained using Bruker AVII 400 or AVIII 500 instruments (Bruker, Billerica, MA, USA). Spectra were run in CDCl3 and chemical shifts are reported in ppm to high frequency of the reference and are referenced to internal Me4Si (δ 0.00) for 1H and to the solvent signal (δ 77.00) for 13C, and coupling constants are in Hz. NMR spectra were processed using iNMR reader, version 6.3.3 (Mestrelab Research, Santiago de Compostela, Spain).
3.2. N,N′-Di-p-tolyl-p-phenylenediamine 3
Following a literature procedure [], a mixture of hydroquinone (27.5 g, 0.25 mol), p-toluidine (20.8 g, 0.19 mol), and orthophosphoric acid (1.33 mL, 0.025 mol) was heated in a sand bath at 200 °C until it melted. With continued heating, additional p-toluidine (41.8 g, 0.39 mol) was added through a funnel over 50 min. The mixture was heated with removal of the water produced by distillation for 4.5 h. The molten material was carefully poured into rapidly stirred ethanol (250 mL) and the resulting crystals were filtered off, washed with ethanol and dried to give the product (3.65 g, 5%) as brown crystals, m.p. 182–184 °C (lit. [,], 182 °C). 1H and 13C NMR data were in agreement with reported values [].
3.3. N,N′-Di-(p-tolyl)-1,4-benzoquinonediimine 1
A solution of diamine 1 (2.01 g, 6.97 mmol) in AR acetone (50 mL) was stirred while a solution of potassium permanganate (0.93 g, 6.0 mmol) in AR acetone (100 mL) was added over a period of 30 min. After a further 30 min, the mixture was filtered to remove MnO2 and the filtrate evaporated. Recrystallisation of the residue from acetone followed by toluene gave the product (1.82 g, 91%) as dark red crystals, m.p. 121–123 °C (lit. [], 123–123.5 °C).
- E-isomer
- 1H NMR (500 MHz): δ 7.189 (4 H, d, J 8.0), 6.940 (4H, s), 6.817 (4 H, d, J 8.0) and 2.367 (6 H, s); 13C NMR (125 MHz): δ 158.39 (2C, C=N), 147.52 (2C, C–N), 137.54 (2CH), 134.99 (2C), 129.54 (4CH), 124.34 (2CH), 120.77 (4CH), and 20.95 (2CH3).
- Z-isomer
- 1H NMR (500 MHz): δ 7.159 (4 H, d, J 8.0), 7.082 (2H, d, J 1.0), 6.791 (4H, d, J 8.0), 6.778 (2 H, d, J 1.0) and 2.347 (6 H, s); 13C NMR (125 MHz): δ 158.27 (2C, C=N), 147.52 (2C, C–N), 136.38 (2CH), 135.03 (2C), 129.49 (4CH), 124.98 (2CH), 120.89 (4CH), and 20.95 (2CH3).
3.4. X-Ray Structure Determination of 1
X-ray diffraction data for compound 1 were collected at 125 K using the St Andrews Automated Robotic Diffractometer (STANDARD) [], consisting of a Rigaku sealed-tube generator equipped with a SHINE monochromator [Mo Kα radiation (λ = 0.71075 Å)], and a Saturn 724 CCD area detector, coupled with a Microglide goniometer head and an ACTOR SM robotic sample changer. Data for the compound were collected and processed (including correction for Lorentz, polarisation, and absorption) using CrystalClear []. The structure was solved by direct methods (SIR2011) [] and refined by full-matrix least-squares against F2 (SHELXL-2014/7) []. Methyl hydrogens were modelled as disordered across two idealised orientations. All calculations were performed using the CrystalStructure (version 4.1) [] interface.
Crystal data for C20H18N2: M = 286.38 g mol−1, red prism, crystal dimensions 1.00 × 0.20 × 0.03 mm, triclinic, space group P (No. 2), a = 6.8083(10), b = 9.7827(15), c = 12.2576(19) Å, a = 82.1608(10), b = 80.782(10), g = 74.958(8)°, V = 774.4(2) Å3, Z = 2, Dcalc = 1.228 g cm−3, T = 125 K, goodness of fit on F2 0.947, 11611 reflections measured, 3502 unique (Rint = 0.0542), which were used in all calculations. The final R1 [I > 2σ(I)] was 0.0412 and wR2 (all data) was 0.1118. Data have been deposited at the Cambridge Crystallographic Data Centre as CCDC 2482921. The data can be obtained free of charge from the Cambridge Crystallographic Data Centre via http://www.ccdc.cam.ac.uk/structures.
Supplementary Materials
The following are available online: Figure S1. 500 MHz 1H NMR spectrum of 1 (spectrum after 5 min); Figure S2. 500 MHz 1H NMR spectrum of 1 (spectrum after 5 min, expansion); Figure S3. 500 MHz 1H NMR spectrum of 1 (spectrum after 15 min); Figure S4. 500 MHz 1H NMR spectrum of 1 (spectrum after 15 min, expansion); Figure S5. 400 MHz 1H NMR spectrum of 1 (spectrum after 13 h); Figure S6. 125 MHz DEPTQ 13C NMR spectrum of 1 (after 13 h); Figure S7. 125 MHz DEPTQ 13C NMR spectrum of 1 (after 13 h, expansion); Figure S8. HSQC NMR spectrum of 1; Figure S9. HMBC NMR spectrum of 1.
Author Contributions
R.B. prepared the compounds and recorded the spectroscopic data; A.M.Z.S. collected the X-ray diffraction data and solved the structure; R.A.A. designed the study, analysed the data, and wrote the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The research data supporting this publication (original NMR files) can be accessed at https://doi.org/10.17360/86d6ec80-2aff-433b-8000-d5d9a77b293d.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Hughes, G.K.M.; Saunders, B.C. Studies in peroxidase action. Part XI. The oxidation of a mixture of amines. J. Chem. Soc. 1956, 3814–3820. [Google Scholar] [CrossRef]
- Simpson, G.J.; Hogan, S.W.L.; Caffio, M.; Adams, C.J.; Früchtl, H.; van Mourik, T.; Schaub, R. New class of metal bound molecular switches involving H-tautomerism. Nano Lett. 2014, 14, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Calm, A. Ueber die Einwerkung des Anilins auf Resorcin und Hydrochinon. Ber. Dtsch. Chem. Ges. 1883, 16, 2786–2814. [Google Scholar] [CrossRef]
- Hatschek, A.; Zega, A. Ueber die Einwirkung von Paratoluidin auf Resorcin und Hydrochinon. J. Prakt. Chem. 1886, 33, 209–241. [Google Scholar] [CrossRef]
- Buu-Hoi, N.P. The scope of the Knoevenagel synthesis of aromatic secondary amines. J. Chem. Soc. 1952, 4346–4349. [Google Scholar] [CrossRef]
- Hardman, A.F. Process for Preparing Secondary Amines. U.S. Patent 2238320, 15 April 1941. [Google Scholar]
- Gadomska, A.V.; Varlamov, V.T. New method of the synthesis of N,N′-diphenyl-1,4-benzoquinone diimine. Russ. J. Gen. Chem. 2014, 84, 1299–1301. [Google Scholar] [CrossRef]
- Sandberg, M.; Hjertberg, T. E/Z isomerism in polyaniline. A model study. Synth. Met. 1989, 29, E257–E264. [Google Scholar] [CrossRef]
- Han, C.-C.; Balakumar, R.; Thirumalai, D.; Chung, M.-T. The different electronic natures displayed by the alkylthio groups in simple and higher conjugated aniline systems. Org. Biomol. Chem. 2006, 4, 3511–3516. [Google Scholar] [CrossRef] [PubMed]
- Gawlicka-Chruszcz, A.; Stadnicka, K. A comparative study of intermolecular interactions in the crystal structures of phenyl/phenyl end-capped oligoanilines. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2002, 58, o416–o420. [Google Scholar] [CrossRef] [PubMed]
- Povet’eva, Z.P.; Chetkina, L.A.; Kopylov, V.V. X-Ray structure analysis of N,N′-di-b-naphthyl-p-benzoquinone diimine. J. Struct. Chem. 1980, 21, 216–219. [Google Scholar] [CrossRef]
- Kenny, T.; Lamare, S.; Aly, S.M.; Fortin, D.; Brisard, G.; Harvey, P.D. Reduced and oxidized forms of the Pt-organometallic version of polyaniline. Inorg. Chem. 2012, 51, 13081–13095. [Google Scholar] [CrossRef] [PubMed]
- Jian, F.-F.; Zhuang, R.-R.; Wang, K.-F.; Wang, J. (6E)-N-[(4Z)-2,5-Dimethyl-4-(p-tolylimino)cyclohexa-2,5-dienylidene]-4-methylaniline. Acta Crystallogr. Sect. E Struct. Rep. Online 2008, 64, o78. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Xi, C. CuCl-catalyzed aerobic oxidative reaction of primary aromatic amines. Tetrahedron Lett. 2008, 49, 4011–4015. [Google Scholar] [CrossRef]
- Kampmann, S.S.; Sobolev, A.N.; Koutsantonis, G.A.; Stewart, S.G. Stable nickel(0) phosphites as catalysts for C–N cross-coupling reactions. Adv. Synth. Catal. 2014, 356, 1967–1973. [Google Scholar] [CrossRef]
- Fuller, A.L.; Scott-Hayward, L.A.S.; Li, Y.; Bühl, M.; Slawin, A.M.Z.; Woollins, J.D. Automated Chemical Crystallography. J. Am. Chem. Soc. 2010, 132, 5799–5802. [Google Scholar] [CrossRef] [PubMed]
- CrystalClear-SM Expert, version 2.1; Rigaku Americas: The Woodlands, TX, USA; Rigaku Corporation: Tokyo, Japan, 2015.
- Burla, M.C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G.; Spagna, R. SIR2011: A new package for crystal structure determination and refinement. J. Appl. Crystallogr. 2012, 45, 357–361. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- CrystalStructure, version 4.1; Rigaku Americas: The Woodlands, TX, USA; Rigaku Corporation: Tokyo, Japan, 2014.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).