ML integration with QM data accelerates predictions, as in SmartCADD’s billion-compound screening [
15]. The QMugs dataset supports ML models for 665,000 molecules [
40]. Linear-scaling DFT and sparse qubitization enable the analysis of larger systems (~50 orbitals) [
32]. Quantum computing advancements, such as VQE and QAOA, enhance quantum mechanical (QM) calculations [
30]. Open-source tools, like ORCA and Qiskit, democratize access [
20,
34]. Industry partnerships, like Pfizer-XtalPi and Aqemia-Janssen, drive adoption [
17,
33]. A review highlights QM’s role in streamlining drug discovery [
41].
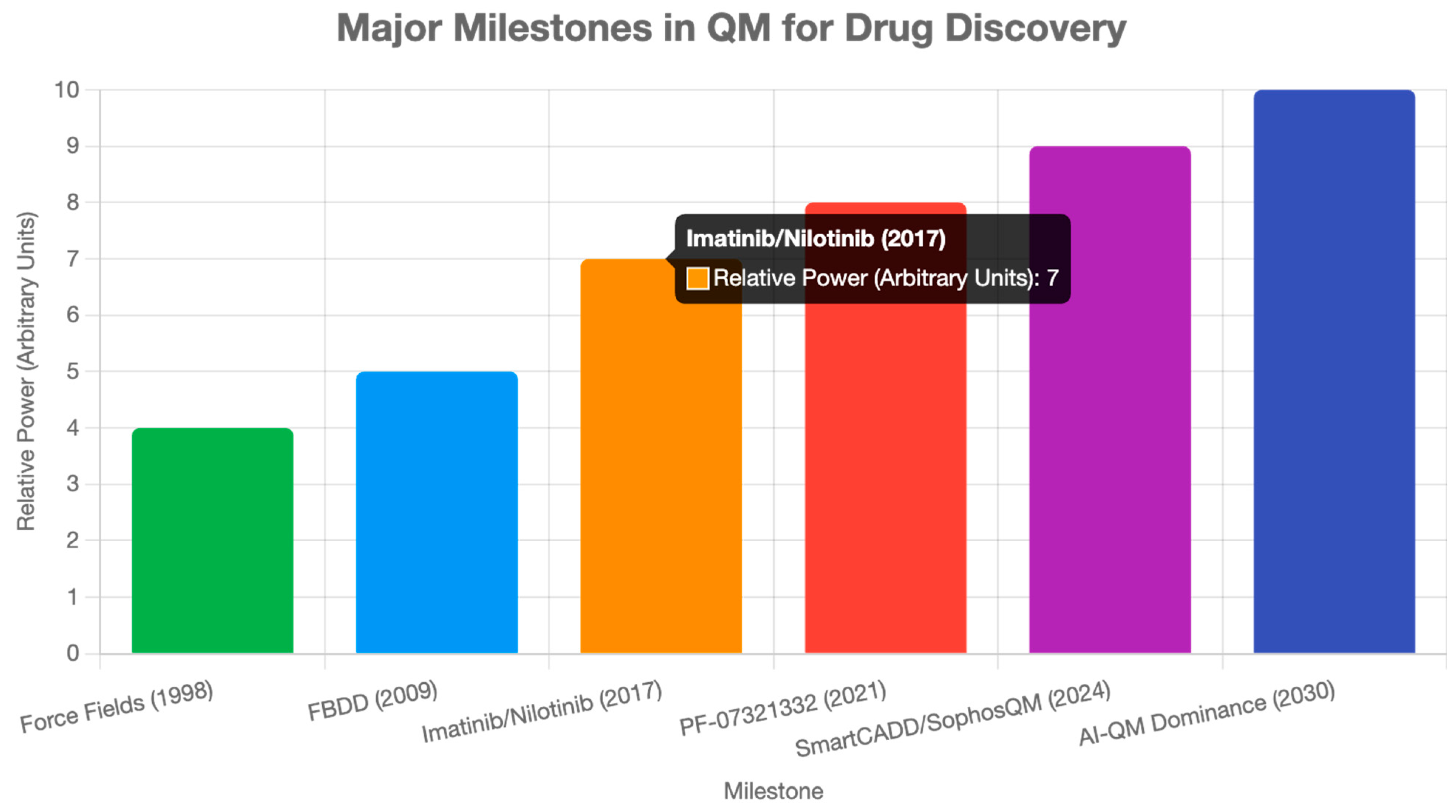
5.2. Future Projections for QM in Drug Discovery
Quantum mechanics is expected to be central to computer-aided drug design (CADD) by 2030, driven by advancements in quantum mechanics methods, quantum computing, and artificial intelligence (AI). AI-QM platforms, such as SmartCADD and Aqemia’s pipeline, will reduce drug discovery timelines by integrating QM precision with ML efficiency [
15,
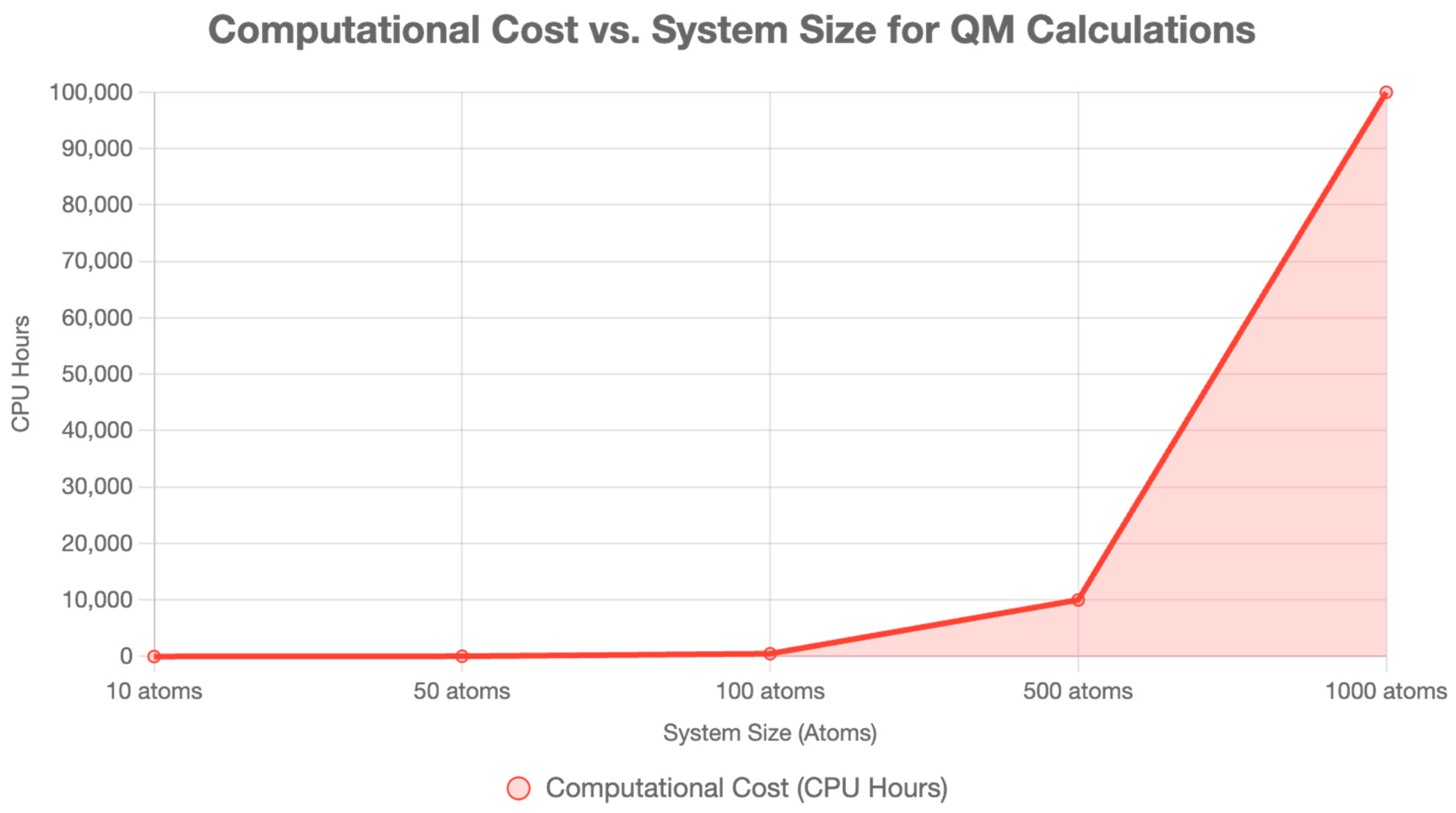
33]. Quantum computing will enable large-scale quantum mechanical (QM) calculations, with fault-tolerant systems projected by the late 2020s, allowing the analysis of complex biomolecular systems (~500–1000 atoms) with density functional theory (DFT)-level accuracy [
30,
32]. Efficient QM methods, such as linear-scaling DFT and FMO, will be extended to larger systems, thereby enhancing SBDD and FBDD [
12,
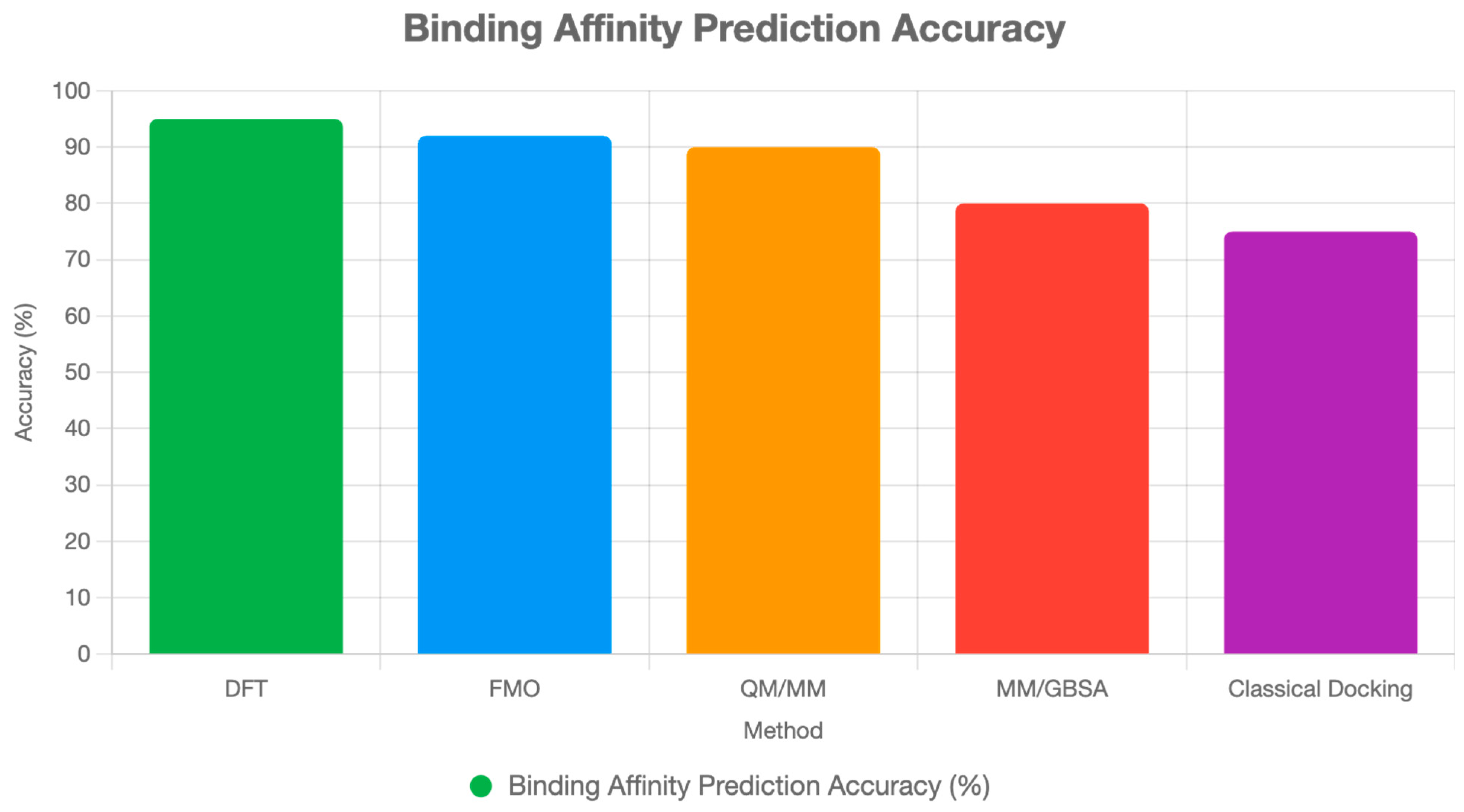
32]. QM scoring functions are expected to outperform classical methods in predicting binding affinities, particularly for SMKIs and covalent inhibitors [
11]. By the mid-2030s, QM and quantum computing are expected to influence clinical trial design and personalized medicine, optimizing drug–target interactions at the patient level [
35]. Industry adoption is expected to grow through collaborations, supported by cloud-based quantum platforms and open-source tools such as Qiskit and ORCA [
20,
33,
34].
5.3. Projections of Novel Applications for Biological Drugs
The application of quantum mechanics (QM) and quantum computing in drug discovery is poised to extend beyond small molecules to biological drugs, including gene therapies, monoclonal antibodies, and biosimilars, by 2030–2035. Biological drugs, which include large proteins, nucleic acids, and cell-based therapies, present unique challenges due to their size, conformational flexibility, and complex interactions. However, advancements in quantum mechanics (QM) methods and quantum computing offer transformative opportunities.
Gene therapies, which deliver genetic material (e.g., DNA, RNA) to treat diseases, such as cystic fibrosis or hemophilia, rely on delivery vehicles, including viral vectors (e.g., adeno-associated viruses, AAVs) or lipid nanoparticles (LNPs). QM can enhance the design of these delivery systems by modeling critical interactions at the atomic level. For instance, DFT and QM/MM can optimize the electronic properties of LNP components, such as ionizable lipids, to enhance nucleic acid encapsulation and endosomal escape, as observed in mRNA vaccines like those for COVID-19 [
26,
27]. Quantum computing could accelerate these calculations, enabling high-throughput screening of LNP formulations by 2030, using algorithms like VQE to predict lipid–nucleic acid binding energies [
30,
34]. For viral vectors, QM/MM can model capsid–protein interactions with cell receptors, thereby enhancing targeting specificity—a current limitation in AAV therapies [
43]. However, the large size of viral capsids (~10
5 atoms) exceeds current quantum mechanical (QM) capabilities, requiring hybrid quantum mechanical/molecular mechanical (QM/MM) approaches or quantum-enhanced coarse-grained models [
16,
32]. By 2035, fault-tolerant quantum computers could simulate entire capsid–receptor complexes, streamlining gene therapy development [
35].
Monoclonal antibodies (mAbs), such as trastuzumab for breast cancer, are large biologics (~150 kDa, comprising around 20,000 atoms) that bind to specific antigens with high affinity. QM applications in mAb design are limited by computational cost; however, QM/MM and FMO can model critical regions, such as complementarity-determining regions (CDRs), which govern antigen binding [
8,
9,
11]. DFT could predict electronic interactions (e.g., hydrogen bonds, π-π stacking) in CDRs, improving binding affinity and specificity, as demonstrated in small-molecule SBDD [
11]. Quantum computing could enhance these calculations by 2030, using QAOA to optimize CDR sequences across vast combinatorial spaces, potentially reducing off-target effects [
30]. QM could also inform glycosylation patterns, which affect mAb stability and immunogenicity, by modeling sugar–protein interactions [
13]. Compared to small molecules, mAbs are less amenable to complete QM analysis because of their size; however, hybrid approaches can focus on key binding interfaces, offering precision that is unattainable with classical MD [
5,
16]. By 2035, quantum computers could simulate entire antigen-binding fragments (~500 atoms), enabling the design of de novo monoclonal antibodies (mAbs) [
35].
Biosimilars, which are highly similar to approved biologics, such as monoclonal antibodies (mAbs), aim to reduce costs while maintaining efficacy and safety. Quality Management (QM) plays a critical role in biosimilar development by ensuring structural and functional equivalence to reference products. For example, QM/MM could compare the electronic properties of CDRs in a biosimilar versus its reference mAb, verifying identical binding interactions with antigens [
8]. DFT could predict post-translational modifications (e.g., oxidation, deamidation), which impact biosimilar stability, as seen in comparability studies for adalimumab biosimilars [
44]. Quantum computing could accelerate these analyses by 2030, utilizing VQE to compute energy landscapes of modified residues, thereby ensuring that biosimilars match reference mAbs in critical quality attributes [
34]. Unlike originator mAbs, biosimilars require extensive analytical characterization to demonstrate similarity, making QM’s precision valuable for regulatory approval [
44]. However, biosimilars face the same computational challenges as mAbs, with large molecular sizes limiting complete QM analysis, necessitating focused QM/MM studies [
16]. By 2035, quantum-enhanced workflows could streamline biosimilar characterization, reducing development costs and timelines [
35].
5.4. Protein Binding and Structure Prediction in Dynamic Environments
By 2030–2035, QM and quantum computing are projected to revolutionize protein binding to receptors and protein structure prediction from sequence data, accounting for dynamic environments such as solvent effects, conformational flexibility, and biological context. Unlike static Protein Data Bank (PDB) structures, which represent frozen snapshots of proteins under specific experimental conditions, these approaches aim to capture the ensemble of conformations and interactions in physiological settings, critical for accurate drug discovery.
Protein–receptor binding involves complex interactions, including hydrogen bonding, van der Waals forces, and electrostatic interactions, which are modulated by the protein’s dynamic conformational landscape. QM methods, such as DFT and QM/MM, can model these interactions with high precision, particularly for critical binding interfaces. For instance, QM/MM can treat the receptor’s active site and ligand-binding region with DFT while modeling the surrounding protein and solvent with MM force fields, thereby capturing electronic polarization and solvent effects that are absent in classical MD [
8,
11,
16]. By 2030, advances in linear-scaling DFT and FMO will enable the analysis of larger binding interfaces (~500 atoms), thereby improving binding affinity predictions for receptors such as G-protein-coupled receptors (GPCRs) or tyrosine kinases [
9,
12]. Quantum computing will enhance this by accelerating quantum mechanical (QM) calculations, utilizing the variational quantum eigensolver (VQE) to compute energy landscapes of binding poses in real time, potentially reducing errors in docking studies for small-molecule kinase inhibitors (SMKIs) [
30,
34]. Enhanced sampling techniques, such as metadynamics integrated with QM/MM, will explore the free-energy landscape of binding, accounting for conformational changes and entropic contributions in dynamic environments [
45].
5.5. New Drug Indications Enabled by Quantum Mechanics
By 2030–2035, QM and quantum computing are projected to enable the discovery of new drug indications by leveraging their precision in modeling molecular interactions, facilitating drug repurposing, targeting undruggable proteins, addressing rare diseases, and developing precision medicine therapies. These advancements will expand the therapeutic landscape, identifying novel uses for existing drugs and designing treatments for previously intractable conditions.
Drug repurposing, the identification of new indications for approved drugs, benefits from QM’s ability to predict off-target interactions with high accuracy. DFT and QM/MM can model the binding affinities of existing drugs to novel protein targets, revealing unexpected therapeutic potential [
11,
16]. For example, QM calculations could identify kinase inhibitors, such as imatinib, as candidates for non-oncological diseases, such as pulmonary fibrosis, by predicting interactions with fibrotic pathways [
25,
29]. Quantum computing is expected to accelerate this process by 2030, utilizing QAOA to screen vast protein–ligand interaction spaces, thereby identifying repurposing opportunities across thousands of targets in days rather than months [
30]. By 2035, quantum-enhanced AI platforms could integrate QM-derived binding data with clinical datasets, predicting repurposing candidates for complex diseases such as Alzheimer’s, where polypharmacology is crucial [
15,
29,
35] (
Figure 3).





{kind=link}
{kind=link}
{kind=link}
{kind=link}