
Ursolic Acid Suppresses Colorectal Cancer Through Autophagy–Lysosomal Degradation of β-Catenin

Abstract

1. Introduction
2. Results
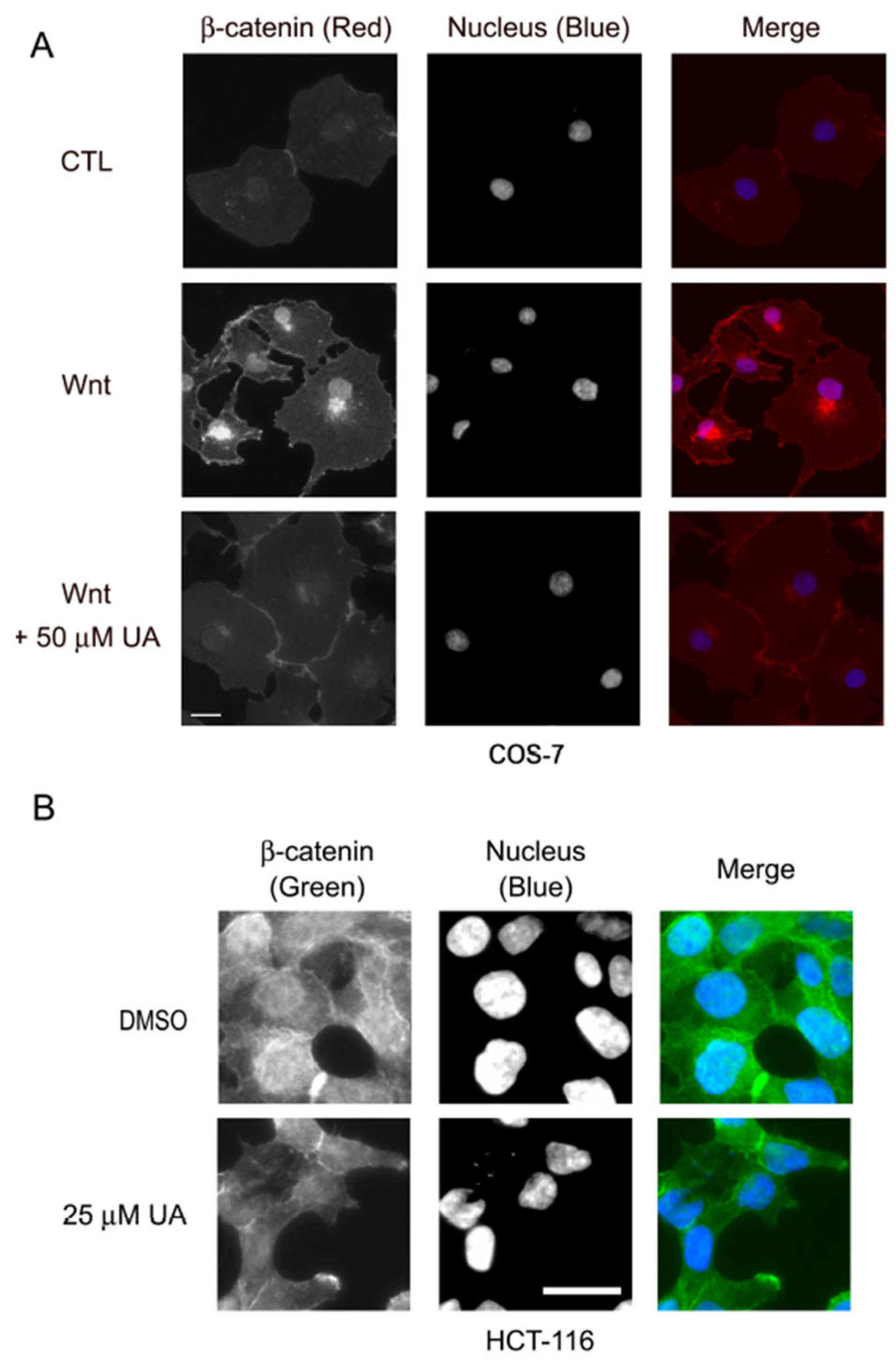
2.1. UA Reduces Wnt/β-Catenin Signaling in Wnt-Stimulated P19 Cells and COS-7 Cells
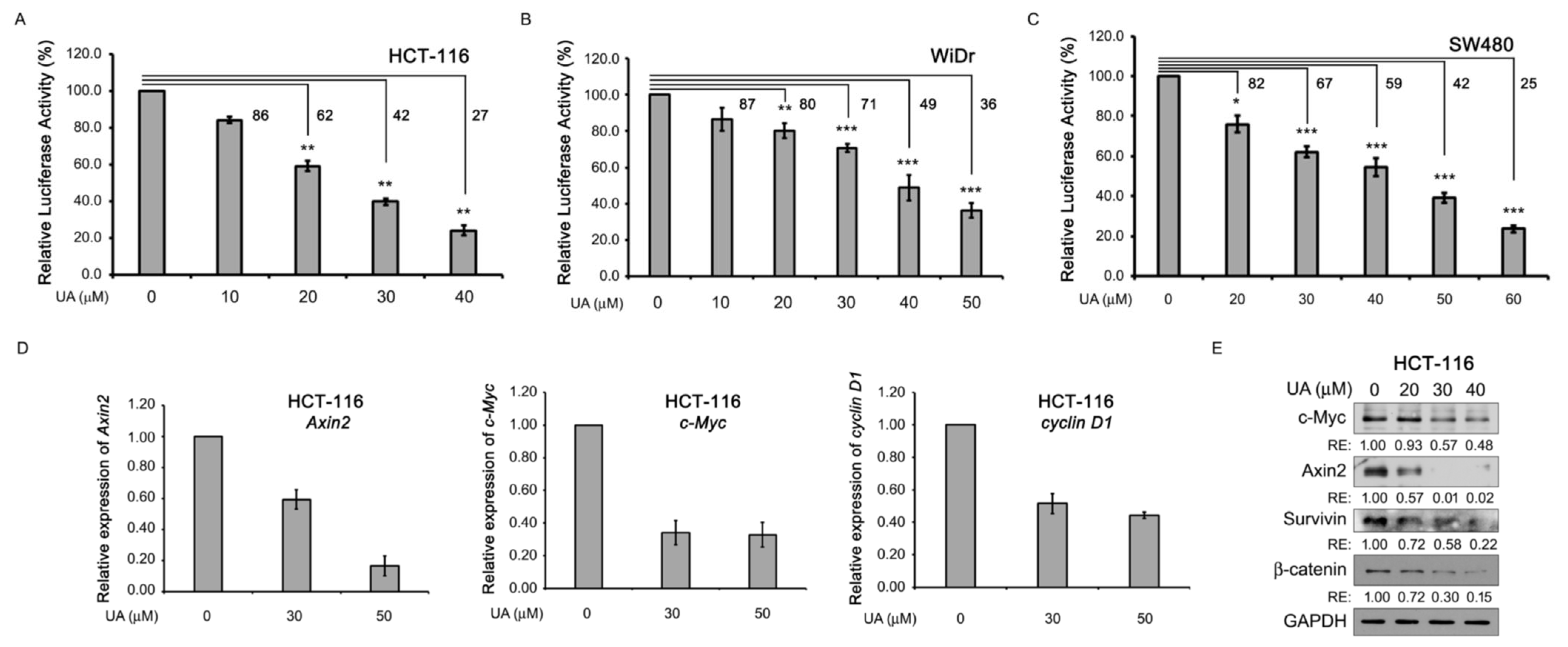
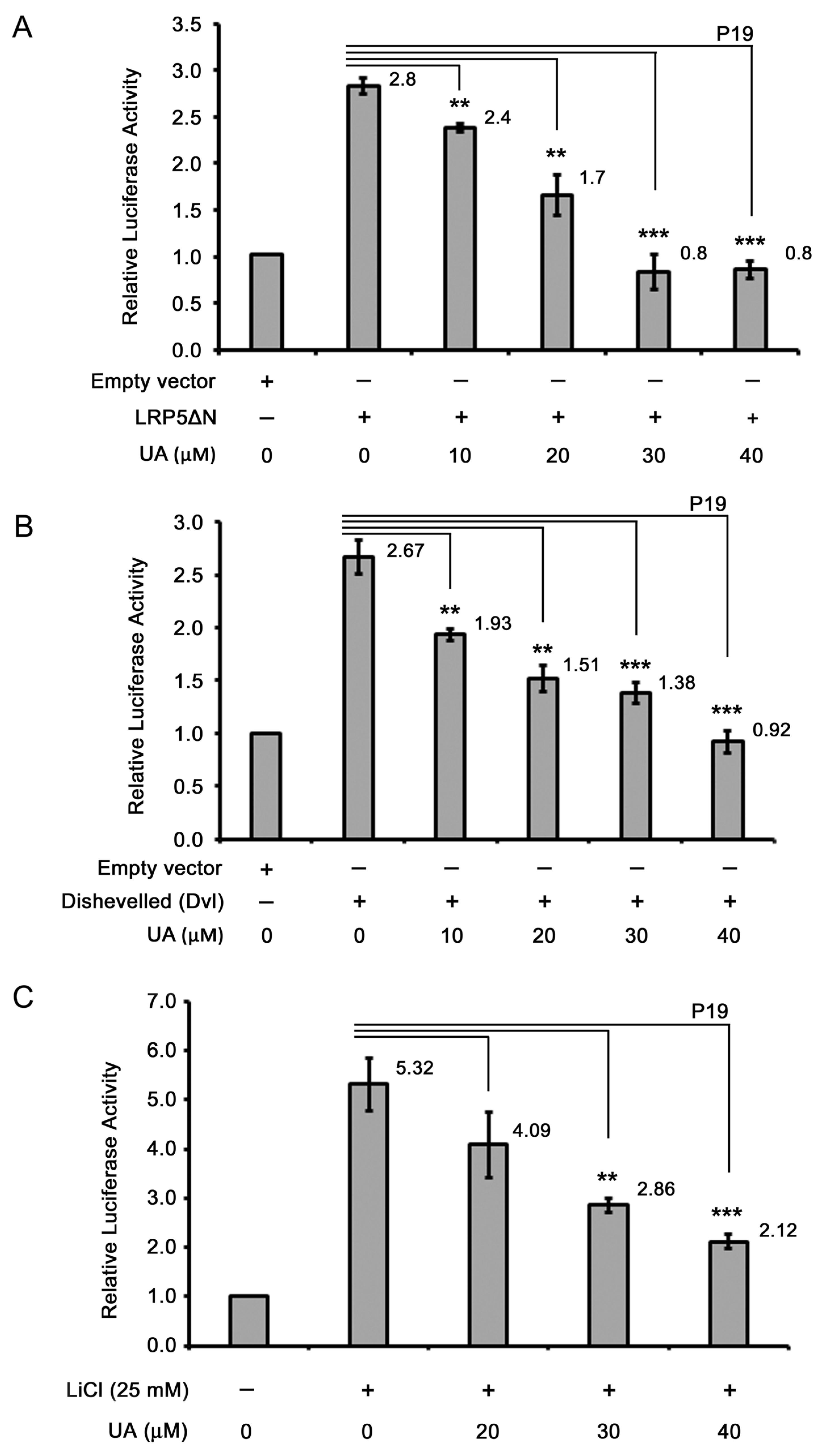
2.2. UA Reduces Wnt/β-Catenin Signaling in Colorectal Cancer Cells
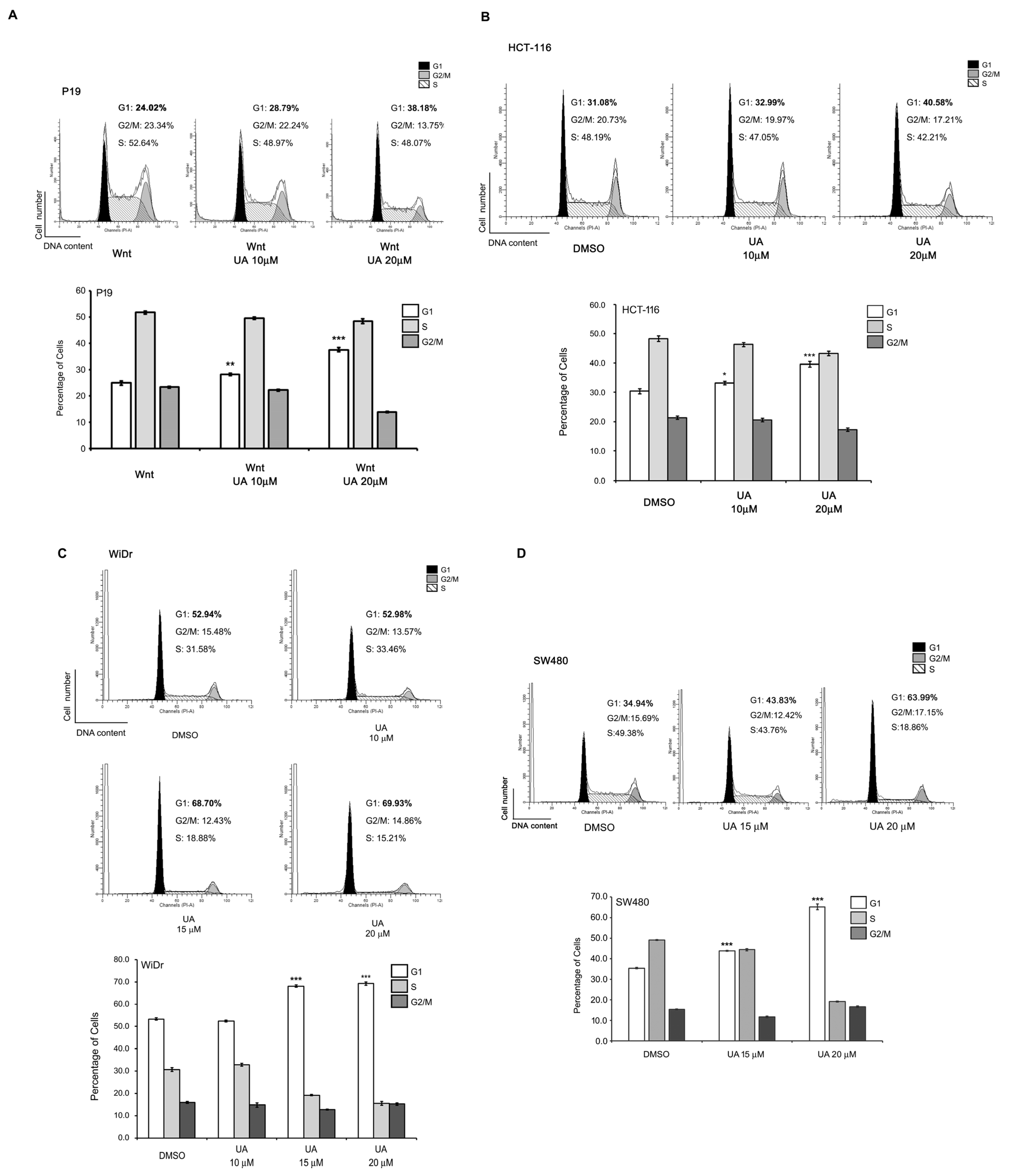
2.3. UA Suppresses the Proliferation of Wnt-Stimulated Cells and Colorectal Cancer Cells
2.4. UA Downregulates β-Catenin in Wnt Signaling
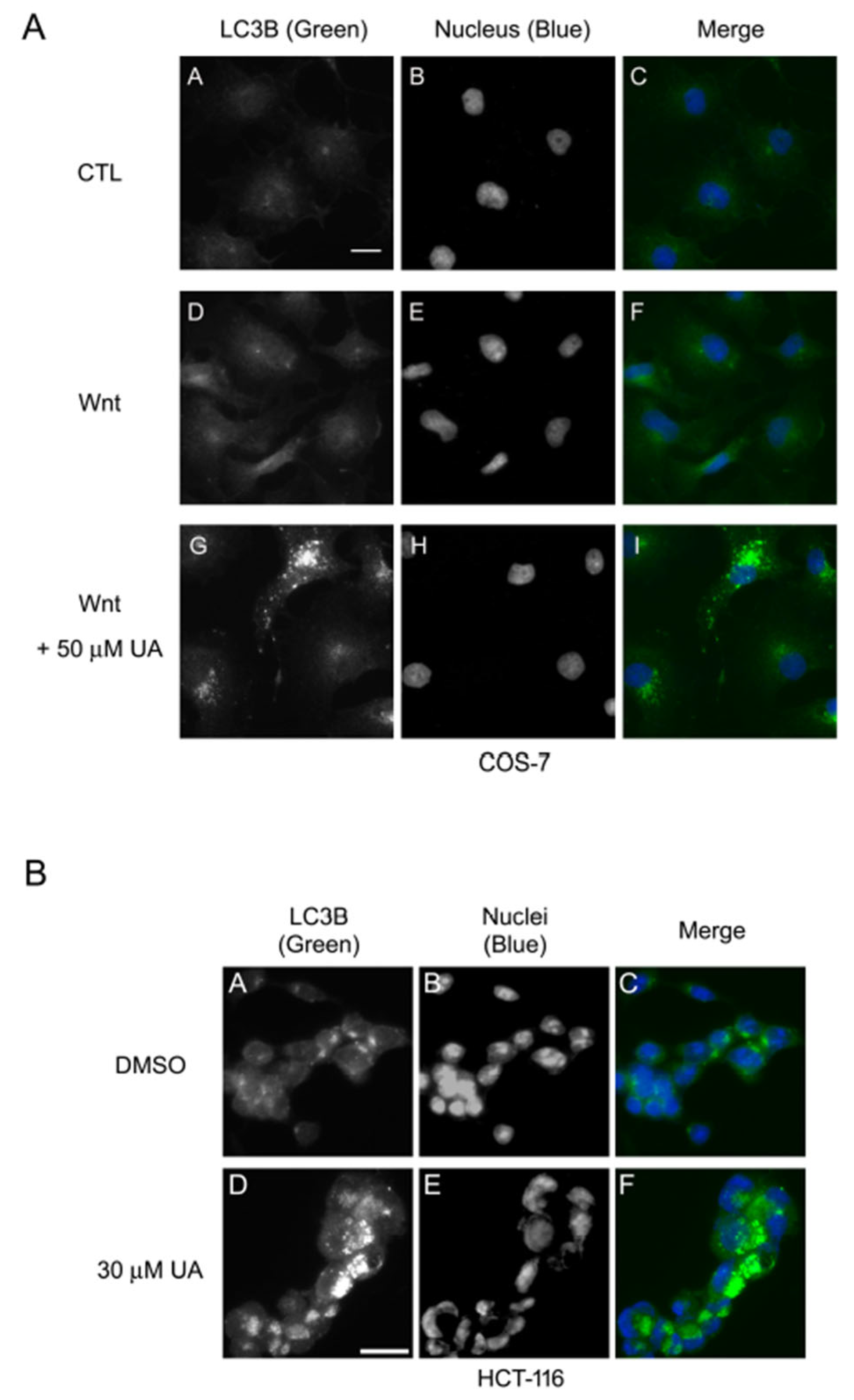
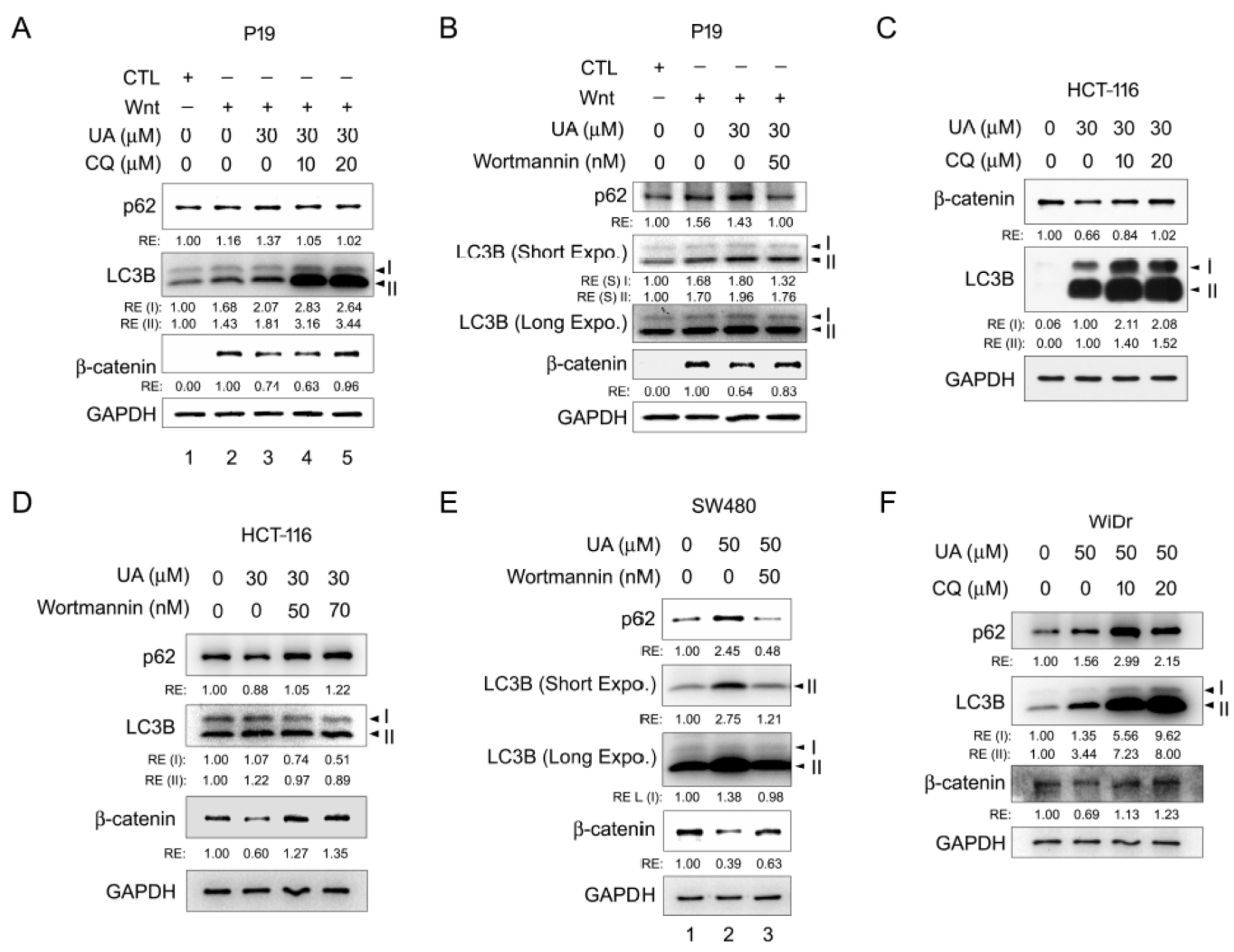
2.5. UA Induces Autophagy–Lysosomal Degradation of β-Catenin in Wnt Signaling
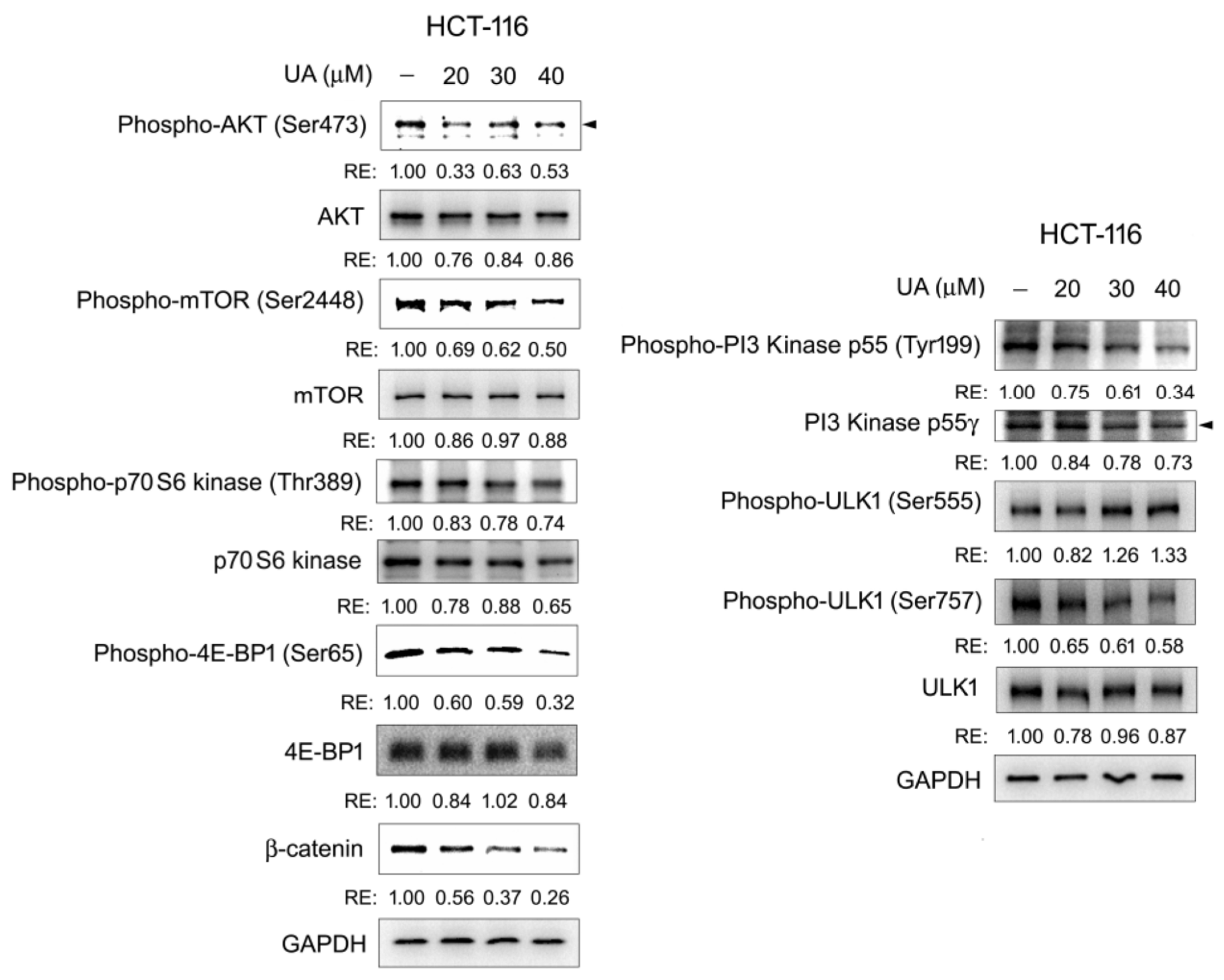
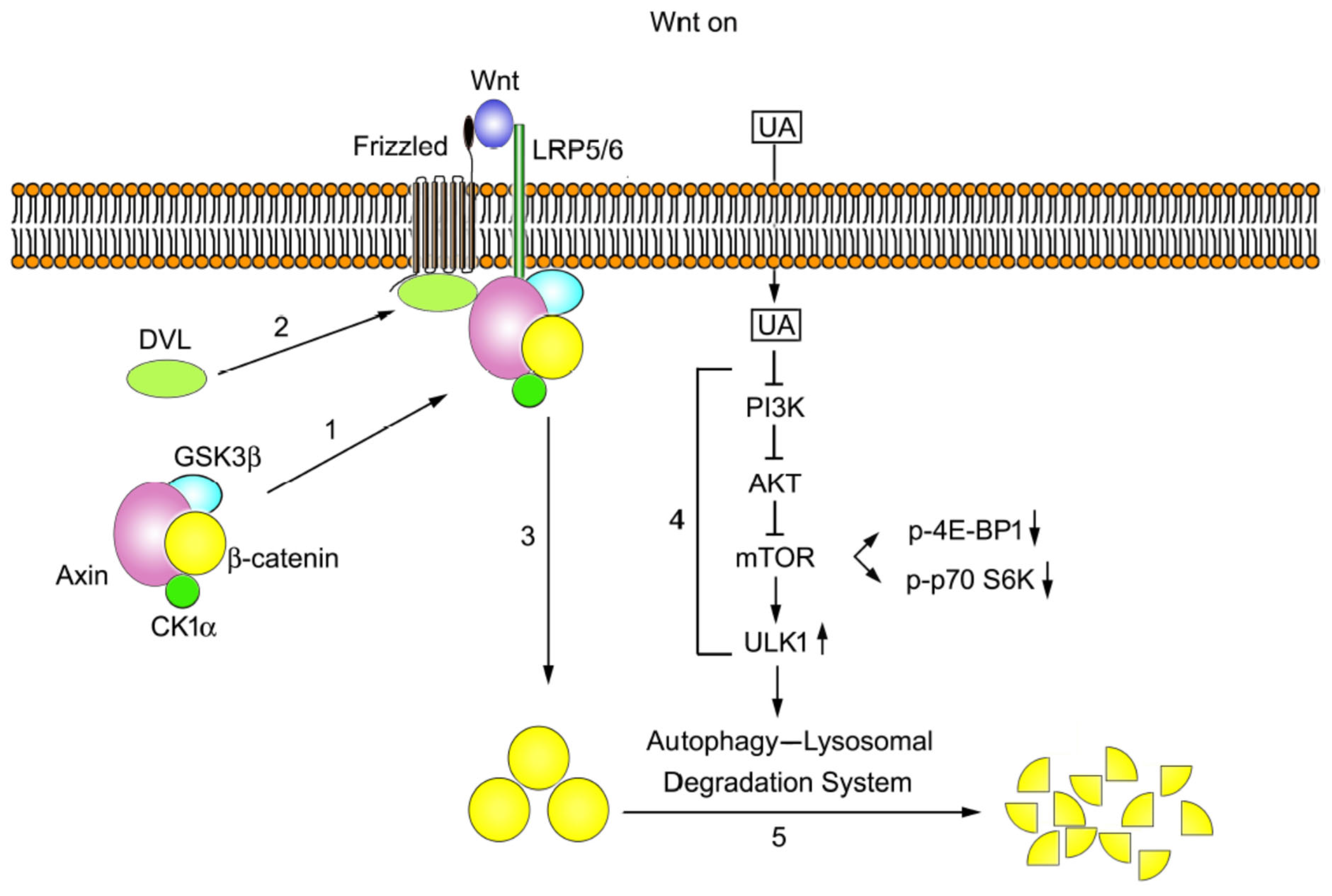
2.6. The PI3K/AKT/mTOR/ULK Pathway Is Involved in UA-Induced Autophagy–Lysosomal Degradation of β-Catenin in Wnt Signaling
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Western Blotting
4.4. Reverse Transcription Quantitative Polymerase Chain Reaction
4.5. Immunofluorescence Staining
4.6. Dual Luciferase Activity Assay
4.7. Cell Viability Assay
4.8. Cell Cycle Analysis Through Flow Cytometry
4.9. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Steinhart, Z.; Angers, S. Wnt signaling in development and tissue homeostasis. Development 2018, 145, dev146589. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, L.; Zhang, L. Therapeutic Potential of Natural Compounds from Herbs and Nutraceuticals in Alleviating Neurological Disorders: Targeting the Wnt Signaling Pathway. J. Agric. Food Chem. 2024, 72, 2411–2433. [Google Scholar] [CrossRef] [PubMed]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, T.; Chen, G.Y. Flavonoids and Colorectal Cancer Prevention. Antioxidants 2018, 7, 187. [Google Scholar] [CrossRef]
- Keum, N.; Giovannucci, E. Global burden of colorectal cancer: Emerging trends, risk factors and prevention strategies. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 713–732. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Tomasetti, C.; Marchionni, L.; Nowak, M.A.; Parmigiani, G.; Vogelstein, B. Only three driver gene mutations are required for the development of lung and colorectal cancers. Proc. Natl. Acad. Sci. USA 2015, 112, 118–123. [Google Scholar] [CrossRef]
- Morin, P.J.; Sparks, A.B.; Korinek, V.; Barker, N.; Clevers, H.; Vogelstein, B.; Kinzler, K.W. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 1997, 275, 1787–1790. [Google Scholar] [CrossRef]
- Vacante, M.; Borzi, A.M.; Basile, F.; Biondi, A. Biomarkers in colorectal cancer: Current clinical utility and future perspectives. World J. Clin. Cases 2018, 6, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Tariq, K.; Ghias, K. Colorectal cancer carcinogenesis: A review of mechanisms. Cancer Biol. Med. 2016, 13, 120–135. [Google Scholar] [CrossRef] [PubMed]
- Biondi, A.; Vacante, M.; Ambrosino, I.; Cristaldi, E.; Pietrapertosa, G.; Basile, F. Role of surgery for colorectal cancer in the elderly. World J. Gastrointest. Surg. 2016, 8, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Biondi, A.; Grosso, G.; Mistretta, A.; Marventano, S.; Toscano, C.; Drago, F.; Gangi, S.; Basile, F. Laparoscopic vs. open approach for colorectal cancer: Evolution over time of minimal invasive surgery. BMC Surg. 2013, 13 Suppl. S2, S12. [Google Scholar] [CrossRef]
- Varghese, A.M.; Saltz, L.B. BRAF mutation as a biomarker in colorectal cancer. Adv. Genom. Genet. 2015, 5, 347–353. [Google Scholar] [CrossRef]
- Bahrami, A.; Hassanian, S.M.; ShahidSales, S.; Farjami, Z.; Hasanzadeh, M.; Anvari, K.; Aledavood, A.; Maftouh, M.; Ferns, G.A.; Khazaei, M.; et al. Targeting RAS signaling pathway as a potential therapeutic target in the treatment of colorectal cancer. J. Cell Physiol. 2018, 233, 2058–2066. [Google Scholar] [CrossRef]
- Watanabe, T.; Itabashi, M.; Shimada, Y.; Tanaka, S.; Ito, Y.; Ajioka, Y.; Hamaguchi, T.; Hyodo, I.; Igarashi, M.; Ishida, H.; et al. Japanese Society for Cancer of the Colon and Rectum (JSCCR) Guidelines 2014 for treatment of colorectal cancer. Int. J. Clin. Oncol. 2015, 20, 207–239. [Google Scholar] [CrossRef]
- Jager, S.; Trojan, H.; Kopp, T.; Laszczyk, M.N.; Scheffler, A. Pentacyclic triterpene distribution in various plants—Rich sources for a new group of multi-potent plant extracts. Molecules 2009, 14, 2016–2031. [Google Scholar] [CrossRef]
- Lopez-Hortas, L.; Perez-Larran, P.; Gonzalez-Munoz, M.J.; Falque, E.; Dominguez, H. Recent developments on the extraction and application of ursolic acid. A review. Food Res. Int. 2018, 103, 130–149. [Google Scholar] [CrossRef]
- Kunkel, S.D.; Elmore, C.J.; Bongers, K.S.; Ebert, S.M.; Fox, D.K.; Dyle, M.C.; Bullard, S.A.; Adams, C.M. Ursolic acid increases skeletal muscle and brown fat and decreases diet-induced obesity, glucose intolerance and fatty liver disease. PLoS ONE 2012, 7, e39332. [Google Scholar] [CrossRef]
- Sun, Q.; He, M.; Zhang, M.; Zeng, S.; Chen, L.; Zhou, L.; Xu, H. Ursolic acid: A systematic review of its pharmacology, toxicity and rethink on its pharmacokinetics based on PK-PD model. Fitoterapia 2020, 147, 104735. [Google Scholar] [CrossRef]
- Abdulhussin, A.J.; HattabMutlag, S.; Salih, M.K.; Abdul-Wahab, A.H. Autophagy and Cancer Treatment Review. Eur. J. Mol. Clin. Med. 2021, 8, 2494–2503. [Google Scholar]
- Lu, G.; Wang, Y.; Shi, Y.; Zhang, Z.; Huang, C.; He, W.; Wang, C.; Shen, H.M. Autophagy in health and disease: From molecular mechanisms to therapeutic target. MedComm 2022, 3, e150. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X. Targeting the Wnt/beta-catenin signaling pathway in cancer. J. Hematol. Oncol. 2020, 13, 165. [Google Scholar] [CrossRef]
- McBurney, M.W. P19 embryonal carcinoma cells. Int. J. Dev. Biol. 1993, 37, 135–140. [Google Scholar]
- Chen, H.J.; Hsu, L.S.; Shia, Y.T.; Lin, M.W.; Lin, C.M. The beta-catenin/TCF complex as a novel target of resveratrol in the Wnt/beta-catenin signaling pathway. Biochem. Pharmacol. 2012, 84, 1143–1153. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.J.; Lin, C.M.; Lin, C.S.; Perez-Olle, R.; Leung, C.L.; Liem, R.K. The role of microtubule actin cross-linking factor 1 (MACF1) in the Wnt signaling pathway. Genes Dev. 2006, 20, 1933–1945. [Google Scholar] [CrossRef]
- Rowan, A.J.; Lamlum, H.; Ilyas, M.; Wheeler, J.; Straub, J.; Papadopoulou, A.; Bicknell, D.; Bodmer, W.F.; Tomlinson, I.P. APC mutations in sporadic colorectal tumors: A mutational “hotspot” and interdependence of the “two hits”. Proc. Natl. Acad. Sci. USA 2000, 97, 3352–3357. [Google Scholar] [CrossRef]
- El-Bahrawy, M.; Poulsom, R.; Rowan, A.J.; Tomlinson, I.T.; Alison, M.R. Characterization of the E-cadherin/catenin complex in colorectal carcinoma cell lines. Int. J. Exp. Pathol. 2004, 85, 65–74. [Google Scholar] [CrossRef]
- Mu, F.; Huang, J.; Xing, T.; Jing, Y.; Cui, T.; Guo, Y.; Yan, X.; Li, H.; Wang, N. The Wnt/beta-Catenin/LEF1 Pathway Promotes Cell Proliferation at Least in Part Through Direct Upregulation of miR-17-92 Cluster. Front. Genet. 2019, 10, 525. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.M.; Chen, H.H.; Lin, C.A.; Wu, H.C.; Sheu, J.J.; Chen, H.J. Apigenin-induced lysosomal degradation of beta-catenin in Wnt/beta-catenin signaling. Sci. Rep. 2017, 7, 372. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Chen, Y.G. Dishevelled: The hub of Wnt signaling. Cell Signal 2010, 22, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Smalley, M.J.; Signoret, N.; Robertson, D.; Tilley, A.; Hann, A.; Ewan, K.; Ding, Y.; Paterson, H.; Dale, T.C. Dishevelled (Dvl-2) activates canonical Wnt signalling in the absence of cytoplasmic puncta. J. Cell Sci. 2005, 118 Pt 22, 5279–5289. [Google Scholar] [CrossRef]
- Garcia de Herreros, A.; Dunach, M. Intracellular Signals Activated by Canonical Wnt Ligands Independent of GSK3 Inhibition and beta-Catenin Stabilization. Cells 2019, 8, 1148. [Google Scholar] [CrossRef]
- Zhao, L.; Zhao, J.; Zhong, K.; Tong, A.; Jia, D. Targeted protein degradation: Mechanisms, strategies and application. Signal Transduct. Target. Ther. 2022, 7, 113. [Google Scholar] [CrossRef]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Niklaus, M.; Adams, O.; Berezowska, S.; Zlobec, I.; Graber, F.; Slotta-Huspenina, J.; Nitsche, U.; Rosenberg, R.; Tschan, M.P.; Langer, R. Expression analysis of LC3B and p62 indicates intact activated autophagy is associated with an unfavorable prognosis in colon cancer. Oncotarget 2017, 8, 54604–54615. [Google Scholar] [CrossRef]
- Blommaart, E.F.; Krause, U.; Schellens, J.P.; Vreeling-Sindelarova, H.; Meijer, A.J. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur. J. Biochem. 1997, 243, 240–246. [Google Scholar] [CrossRef]
- Lorzadeh, S.; Kohan, L.; Ghavami, S.; Azarpira, N. Autophagy and the Wnt signaling pathway: A focus on Wnt/beta-catenin signaling. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118926. [Google Scholar] [CrossRef]
- Chen, C.; Gao, H.; Su, X. Autophagy-related signaling pathways are involved in cancer (Review). Exp. Ther. Med. 2021, 22, 710. [Google Scholar] [CrossRef]
- Deng, S.; Shanmugam, M.K.; Kumar, A.P.; Yap, C.T.; Sethi, G.; Bishayee, A. Targeting autophagy using natural compounds for cancer prevention and therapy. Cancer 2019, 125, 1228–1246. [Google Scholar] [CrossRef]
- Seo, D.Y.; Lee, S.R.; Heo, J.W.; No, M.H.; Rhee, B.D.; Ko, K.S.; Kwak, H.B.; Han, J. Ursolic acid in health and disease. Korean J. Physiol. Pharmacol. 2018, 22, 235–248. [Google Scholar] [CrossRef]
- Lewinska, A.; Adamczyk-Grochala, J.; Kwasniewicz, E.; Deregowska, A.; Wnuk, M. Ursolic acid-mediated changes in glycolytic pathway promote cytotoxic autophagy and apoptosis in phenotypically different breast cancer cells. Apoptosis 2017, 22, 800–815. [Google Scholar] [CrossRef]
- Huang, M.T.; Ho, C.T.; Wang, Z.Y.; Ferraro, T.; Lou, Y.R.; Stauber, K.; Ma, W.; Georgiadis, C.; Laskin, J.D.; Conney, A.H. Inhibition of skin tumorigenesis by rosemary and its constituents carnosol and ursolic acid. Cancer Res. 1994, 54, 701–708. [Google Scholar]
- Yan, S.L.; Huang, C.Y.; Wu, S.T.; Yin, M.C. Oleanolic acid and ursolic acid induce apoptosis in four human liver cancer cell lines. Toxicol. In Vitro 2010, 24, 842–848. [Google Scholar] [CrossRef]
- Li, J.; Liang, X.; Yang, X. Ursolic acid inhibits growth and induces apoptosis in gemcitabine-resistant human pancreatic cancer via the JNK and PI3K/Akt/NF-kappaB pathways. Oncol. Rep. 2012, 28, 501–510. [Google Scholar] [CrossRef]
- Leng, S.; Hao, Y.; Du, D.; Xie, S.; Hong, L.; Gu, H.; Zhu, X.; Zhang, J.; Fan, D.; Kung, H.F. Ursolic acid promotes cancer cell death by inducing Atg5-dependent autophagy. Int. J. Cancer 2013, 133, 2781–2790. [Google Scholar] [CrossRef]
- Song, Y.H.; Jeong, S.J.; Kwon, H.Y.; Kim, B.; Kim, S.H.; Yoo, D.Y. Ursolic acid from Oldenlandia diffusa induces apoptosis via activation of caspases and phosphorylation of glycogen synthase kinase 3 beta in SK-OV-3 ovarian cancer cells. Biol. Pharm. Bull. 2012, 35, 1022–1028. [Google Scholar] [CrossRef]
- Zheng, Q.Y.; Jin, F.S.; Yao, C.; Zhang, T.; Zhang, G.H.; Ai, X. Ursolic acid-induced AMP-activated protein kinase (AMPK) activation contributes to growth inhibition and apoptosis in human bladder cancer T24 cells. Biochem. Biophys. Res. Commun. 2012, 419, 741–747. [Google Scholar] [CrossRef]
- Prasad, S.; Yadav, V.R.; Sung, B.; Reuter, S.; Kannappan, R.; Deorukhkar, A.; Diagaradjane, P.; Wei, C.; Baladandayuthapani, V.; Krishnan, S.; et al. Ursolic acid inhibits growth and metastasis of human colorectal cancer in an orthotopic nude mouse model by targeting multiple cell signaling pathways: Chemosensitization with capecitabine. Clin. Cancer Res. 2012, 18, 4942–4953. [Google Scholar] [CrossRef]
- Li, W.; Zhang, H.; Nie, M.; Tian, Y.; Chen, X.; Chen, C.; Chen, H.; Liu, R. Ursolic acid derivative FZU-03,010 inhibits STAT3 and induces cell cycle arrest and apoptosis in renal and breast cancer cells. Acta Biochim. Biophys. Sin. 2017, 49, 367–373. [Google Scholar] [CrossRef]
- Pohl, C.; Dikic, I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 2019, 366, 818–822. [Google Scholar] [CrossRef]
- Hosseini, M.; Najmabadi, H.; Kahrizi, K. Calpains: Diverse Functions but Enigmatic. Arch. Iran. Med. 2018, 21, 170–179. [Google Scholar]
- Ravanan, P.; Srikumar, I.F.; Talwar, P. Autophagy: The spotlight for cellular stress responses. Life Sci. 2017, 188, 53–67. [Google Scholar] [CrossRef]
- Heras-Sandoval, D.; Perez-Rojas, J.M.; Hernandez-Damian, J.; Pedraza-Chaverri, J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell Signal 2014, 26, 2694–2701. [Google Scholar] [CrossRef]
- Liu, J.; Xiao, Q.; Xiao, J.; Niu, C.; Li, Y.; Zhang, X.; Zhou, Z.; Shu, G.; Yin, G. Wnt/beta-catenin signalling: Function, biological mechanisms, and therapeutic opportunities. Signal Transduct. Target. Ther. 2022, 7, 3. [Google Scholar] [CrossRef]
- Cui, C.; Zhou, X.; Zhang, W.; Qu, Y.; Ke, X. Is beta-Catenin a Druggable Target for Cancer Therapy? Trends Biochem. Sci. 2018, 43, 623–634. [Google Scholar] [CrossRef]
- Li, X.; Pu, W.; Zheng, Q.; Ai, M.; Chen, S.; Peng, Y. Proteolysis-targeting chimeras (PROTACs) in cancer therapy. Mol. Cancer 2022, 21, 99. [Google Scholar] [CrossRef]
- Liao, H.; Li, X.; Zhao, L.; Wang, Y.; Wang, X.; Wu, Y.; Zhou, X.; Fu, W.; Liu, L.; Hu, H.G.; et al. A PROTAC peptide induces durable beta-catenin degradation and suppresses Wnt-dependent intestinal cancer. Cell Discov. 2020, 6, 35. [Google Scholar] [CrossRef]
- Lashuel, H.A. Rethinking protein aggregation and drug discovery in neurodegenerative diseases: Why we need to embrace complexity? Curr. Opin. Chem. Biol. 2021, 64, 67–75. [Google Scholar] [CrossRef]
- Kusoglu, A.; Biray Avci, C. Cancer stem cells: A brief review of the current status. Gene 2019, 681, 80–85. [Google Scholar] [CrossRef]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.G.; Lee, S.H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and their Therapeutic Implications in Cancer Treatment. Stem. Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef]
- Liao, W.; Zhang, L.; Chen, X.; Xiang, J.; Zheng, Q.; Chen, N.; Zhao, M.; Zhang, G.; Xiao, X.; Zhou, G.; et al. Targeting cancer stem cells and signalling pathways through phytochemicals: A promising approach against colorectal cancer. Phytomedicine 2023, 108, 154524. [Google Scholar] [CrossRef]
- Katoh, M. Canonical and non-canonical WNT signaling in cancer stem cells and their niches: Cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity (Review). Int. J. Oncol. 2017, 51, 1357–1369. [Google Scholar] [CrossRef]
- Leszczynski, P.; Smiech, M.; Teeli, A.S.; Zolocinska, A.; Slysz, A.; Pojda, Z.; Pierzchala, M.; Taniguchi, H. Neurogenesis Using P19 Embryonal Carcinoma Cells. J. Vis. Exp. 2019, 146, e58225. [Google Scholar]
- Wang, Z.; Zhao, T.; Zhang, S.; Wang, J.; Chen, Y.; Zhao, H.; Yang, Y.; Shi, S.; Chen, Q.; Liu, K. The Wnt signaling pathway in tumorigenesis, pharmacological targets, and drug development for cancer therapy. Biomark. Res. 2021, 9, 68. [Google Scholar] [CrossRef]
- Hsieh, C.Y.; Chang, W.C.; Lin, C.C.; Chen, J.H.; Lin, C.Y.; Liu, C.H.; Lin, C.; Hung, M.C. Combination treatment of arsenic trioxide and osimertinib in recurrent and metastatic head and neck squamous cell carcinoma. Am. J. Cancer Res. 2022, 12, 5049–5061. [Google Scholar]
- Lin, L.C.; Yeh, C.T.; Kuo, C.C.; Lee, C.M.; Yen, G.C.; Wang, L.S.; Wu, C.H.; Yang, W.C.; Wu, A.T. Sulforaphane potentiates the efficacy of imatinib against chronic leukemia cancer stem cells through enhanced abrogation of Wnt/beta-catenin function. J. Agric. Food Chem. 2012, 60, 7031–7039. [Google Scholar] [CrossRef]
- Hseu, Y.C.; Tsai, T.J.; Korivi, M.; Liu, J.Y.; Chen, H.J.; Lin, C.M.; Shen, Y.C.; Yang, H.L. Antitumor properties of Coenzyme Q(0) against human ovarian carcinoma cells via induction of ROS-mediated apoptosis and cytoprotective autophagy. Sci. Rep. 2017, 7, 8062. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Species | Dilution | Application | Catalogue Number | Brand Name |
|---|---|---|---|---|---|
| AKT (pan) (C67E7) | Rabbit mab a | 1:1000 | WB b | #4691 | Cell Signaling Technology |
| Phospho-AKT (Ser473) (D9E) XP | Rabbit mab | 1:2000 | WB | #4060 | Cell Signaling Technology |
| Axin2 (76G6) | Rabbit mab | 1:1000 | WB | #2151 | Cell Signaling Technology |
| LC3B | Rabbit | 1:1000 | WB; immune c | #2775 | Cell Signaling Technology |
| mTOR | Rabbit mab | 1:1000 | WB | #2972 | Cell Signaling Technology |
| Phospho-mTOR (Ser2448) | Rabbit | 1:1000 | WB | #2971 | Cell Signaling Technology |
| p70 S6 kinase | Rabbit mab | 1:1000 | WB | #9202 | Cell Signaling Technology |
| Phospho-p70 S6 kinase (Thr389) | Rabbit mab | 1:1000 | WB | #9205 | Cell Signaling Technology |
| 4E-BP1 (53H11) | Rabbit mab | 1:1000 | WB | #9644 | Cell Signaling Technology |
| Phospho-4E-BP1 (Ser65) | Rabbit | 1:1000 | WB | #9451 | Cell Signaling Technology |
| ULK1 (D8H5) | Rabbit mab | 1:1000 | WB | #8054 | Cell Signaling Technology |
| Phospho-ULK1 (Ser555) (D1H4) | Rabbit mab | 1:1000 | WB | #5869 | Cell Signaling Technology |
| Phospho-ULK1 (Ser757) (D1H4) | Rabbit | 1:1000 | WB | #6888 | Cell Signaling Technology |
| Phospho-PI3 Kinase p85 (Tyr458/p55Tyr199) | Rabbit | 1:1000 | WB | #4228 | Cell Signaling Technology |
| GAPDH (6C5) | Mouse mab | 1:3000 | WB | sc-32233 | Santa Cruz Biotechnology |
| SQSTM1 (D-3) | Mouse mab | 1:500 | WB | sc-28359 | Santa Cruz Biotechnology |
| PI3-Kinase p55γ (E-9) | Mouse mab | 1:500 | WB | sc-376615 | Santa Cruz Biotechnology |
| Survivin (D-8) | Mouse mab | 1:500 | WB | sc-17779 | Santa Cruz Biotechnology |
| c-Myc (9E10) | Mouse mab | 1:500 | WB | sc-40 | Santa Cruz Biotechnology |
| β-catenin | Mouse mab | 1:1000 | WB; immune | 610154 | BD Transduction Laboratories |
| MMP2 | rabbit | 1:1000 | WB | GTX104577 | GeneTex |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, C.-M.; Chao, M.-C.; Chen, H.-H.; Chen, H.-J. Ursolic Acid Suppresses Colorectal Cancer Through Autophagy–Lysosomal Degradation of β-Catenin. Int. J. Mol. Sci. 2025, 26, 6210. https://doi.org/10.3390/ijms26136210
Lin C-M, Chao M-C, Chen H-H, Chen H-J. Ursolic Acid Suppresses Colorectal Cancer Through Autophagy–Lysosomal Degradation of β-Catenin. International Journal of Molecular Sciences. 2025; 26(13):6210. https://doi.org/10.3390/ijms26136210
Chicago/Turabian StyleLin, Chung-Ming, Min-Chih Chao, Hsin-Han Chen, and Hui-Jye Chen. 2025. "Ursolic Acid Suppresses Colorectal Cancer Through Autophagy–Lysosomal Degradation of β-Catenin" International Journal of Molecular Sciences 26, no. 13: 6210. https://doi.org/10.3390/ijms26136210
APA StyleLin, C.-M., Chao, M.-C., Chen, H.-H., & Chen, H.-J. (2025). Ursolic Acid Suppresses Colorectal Cancer Through Autophagy–Lysosomal Degradation of β-Catenin. International Journal of Molecular Sciences, 26(13), 6210. https://doi.org/10.3390/ijms26136210