
Enhancement of 3-MA in Paclitaxel Treatment of MDA-MB-231 Tumor-Bearing Nude Mice and Its Mechanisms

 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Results
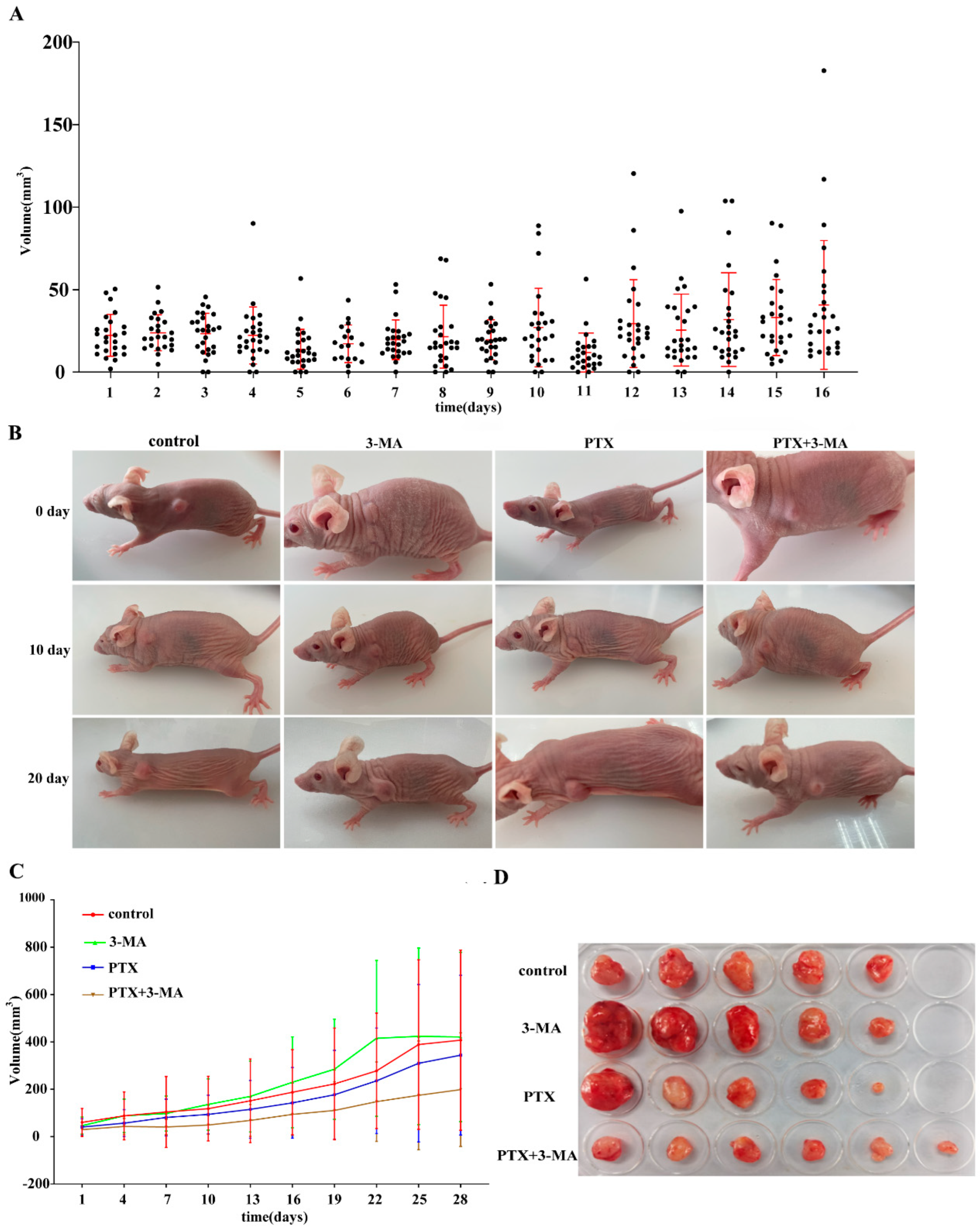
2.1. Establishment of TNBC Nude Mouse Model
2.2. Effects of Combined Use of PTX and 3-MA on Tumor Treatment in Tumor-Bearing Nude Mice
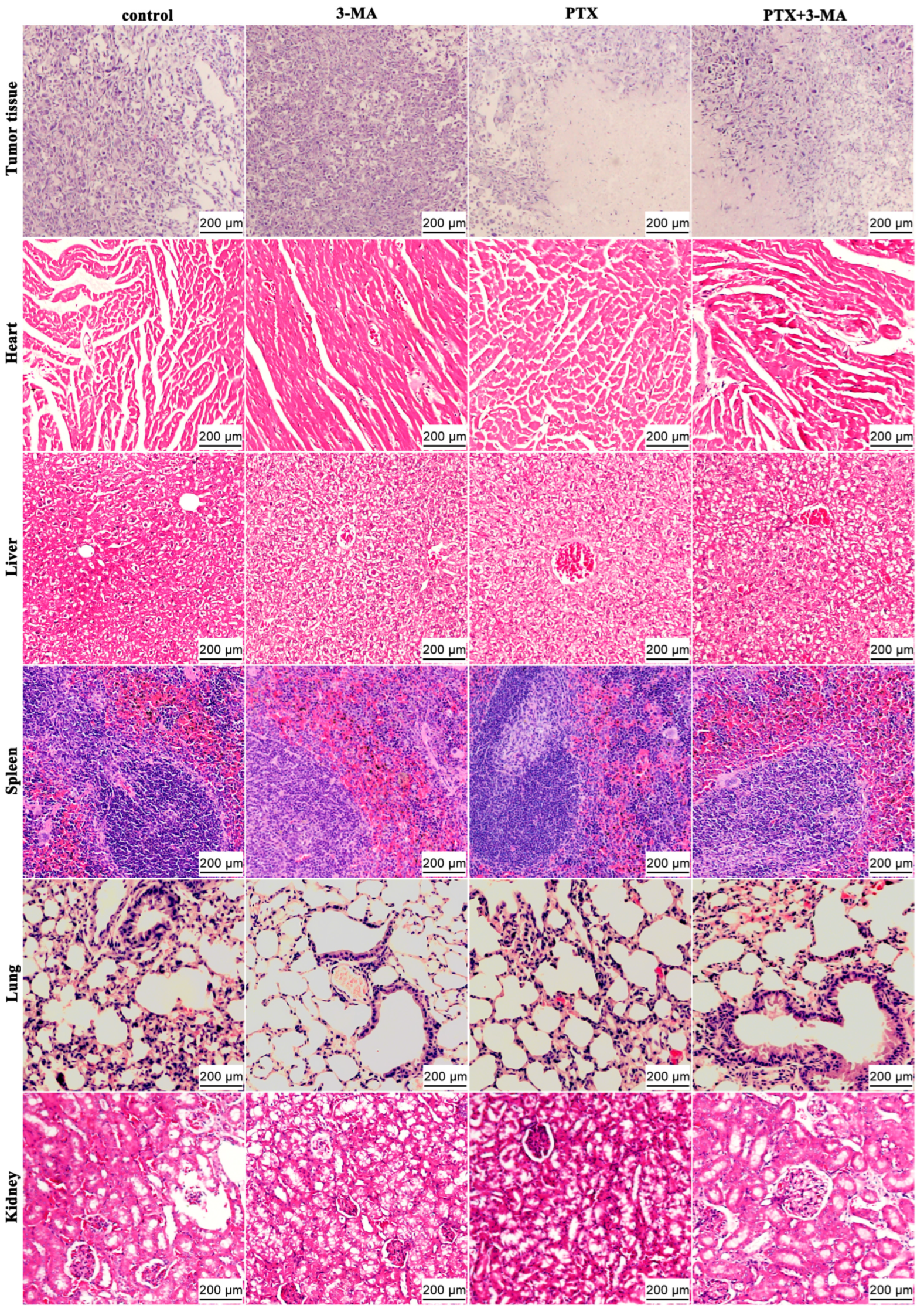
2.3. HE Staining of Tumor and Organ Tissues in MDA-MB-231 Tumor-Bearing Nude Mice After Combined Treatment of PTX and 3-MA
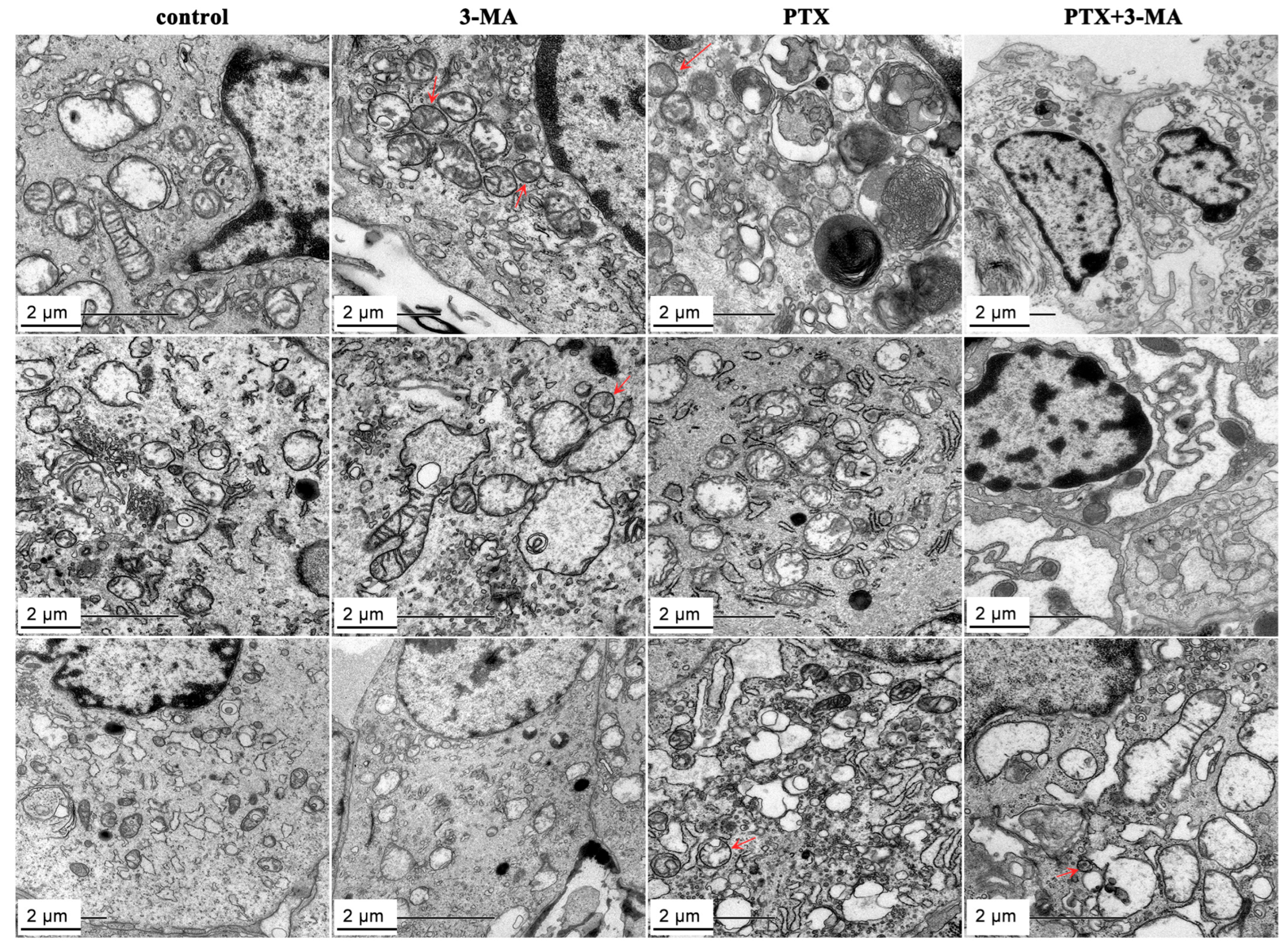
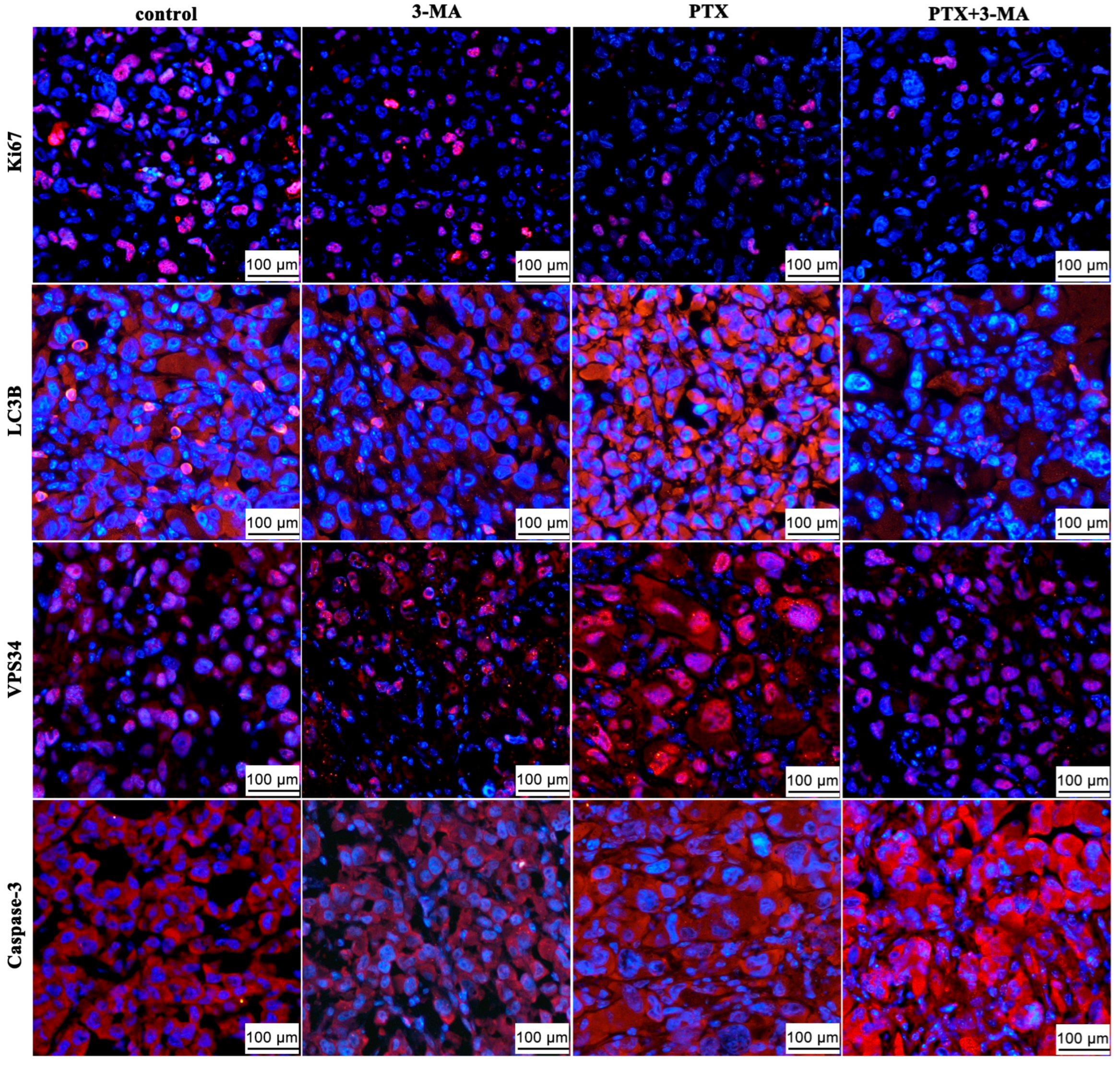
2.4. Effects of 3-MA on the PTX Induced Autophagy in MDA-MB-231 Tumor-Bearing Nude Mouse
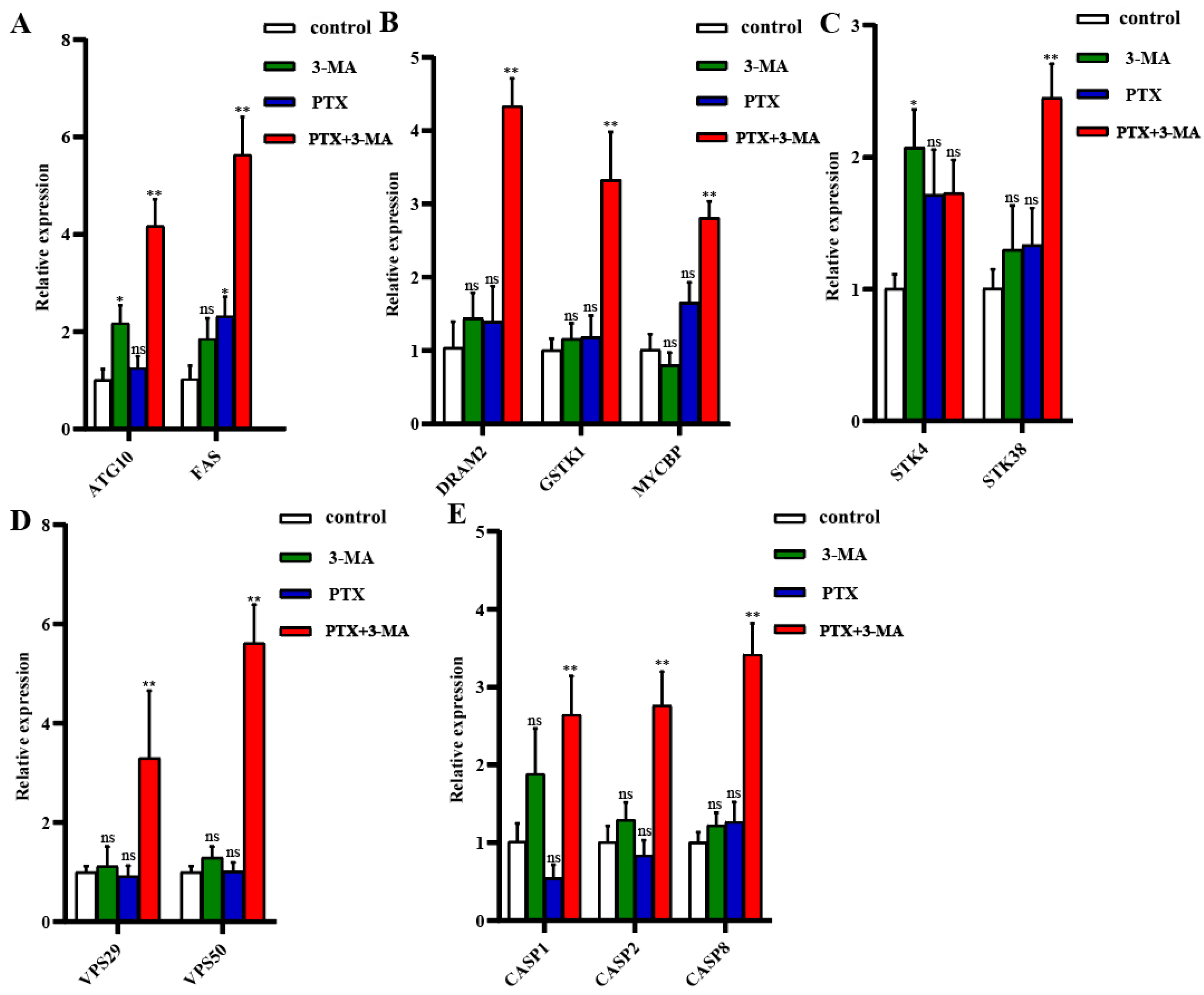
2.5. Gene Expression in MDA-MB-231 Tumor-Bearing Nude Mice After Combined Treatment of PTX with 3-MA
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Animals
4.3. Establishment of TNBC Mouse Model
4.4. PTX and 3-MA Administration in TNBC Mouse Model
4.5. Histopathological Analysis
4.6. Transmission Electron Microscopy Analysis
4.7. Immunofluorescence
4.8. q-PCR
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Howard, F.M.; Olopade, O.I. Epidemiology of Triple-Negative Breast Cancer: A Review. Cancer J. 2021, 27, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Bilani, N.; Zabor, E.C.; Elson, L.; Elimimian, E.B.; Nahleh, Z. Breast Cancer in the United States: A Cross-Sectional Overview. J. Cancer Epidemiol. 2020, 2020, 6387378. [Google Scholar] [CrossRef]
- Borri, F.; Granaglia, A. Pathology of triple negative breast cancer. Semin. Cancer Biol. 2021, 72, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A. Breast Cancer Statistics: Recent Trends. Adv. Exp. Med. Biol. 2019, 1152, 1–7. [Google Scholar]
- Bai, J.; Gao, Y.; Zhang, G. The treatment of breast cancer in the era of precision medicine. Cancer Biol. Med. 2025, 22, 322–347. [Google Scholar] [CrossRef]
- Nagini, S. Breast Cancer: Current Molecular Therapeutic Targets and New Players. Anticancer Agents Med. Chem. 2017, 17, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, E.; Zhang, X.; Zhang, Z.; Huang, G.; Tang, B.; Chen, C.; Lai, L.; Pei, Z.; Zhang, Y.; et al. Autophagy-related biological targets and network mechanisms of juglone against bladder cancer. J. Cancer Res. Clin. Oncol. 2025, 151, 194. [Google Scholar] [CrossRef]
- Yang, A.; Rajeshkumar, N.V.; Wang, X.; Yabuuchi, S.; Alexander, B.M.; Chu, G.C.; Von Hoff, D.D.; Maitra, A.; Kimmelman, A.C. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov. 2014, 4, 905–913. [Google Scholar] [CrossRef]
- Yang, A.; Herter-Sprie, G.; Zhang, H.; Lin, E.Y.; Biancur, D.; Wang, X.; Deng, J.; Hai, J.; Yang, S.; Wong, K.K.; et al. Autophagy Sustains Pancreatic Cancer Growth through Both Cell-Autonomous and Nonautonomous Mechanisms. Cancer Discov. 2018, 8, 276–287. [Google Scholar] [CrossRef]
- Pérez, E.; Das, G.; Bergmann, A.; Baehrecke, E.H. Autophagy regulates tissue overgrowth in a context-dependent manner. Oncogene 2015, 34, 3369–3376. [Google Scholar] [CrossRef]
- Kim, M.J.; Woo, S.J.; Yoon, C.H.; Lee, J.S.; An, S.; Choi, Y.H.; Hwang, S.G.; Yoon, G.; Lee, S.J. Involvement of autophagy in oncogenic K-Ras-induced malignant cell transformation. J. Biol. Chem. 2011, 286, 12924–12932. [Google Scholar] [CrossRef] [PubMed]
- Lock, R.; Roy, S.; Kenific, C.M.; Su, J.S.; Salas, E.; Ronen, S.M.; Debnath, J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Biol. Cell 2011, 22, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Qian, W.; Qin, T.; Xiao, Y.; Cheng, L.; Cao, J.; Chen, X.; Ma, Q.; Wu, Z. Mouse-Derived Allografts: A Complementary Model to the KPC Mice on Researching Pancreatic Cancer In Vivo. Comput. Struct. Biotechnol. J. 2019, 17, 498–506. [Google Scholar] [CrossRef] [PubMed]
- Ishaq, M.; Ojha, R.; Sharma, A.P.; Singh, S.K. Autophagy in cancer: Recent advances and future directions. Semin. Cancer Biol. 2020, 66, 171–181. [Google Scholar] [CrossRef]
- Blommaart, E.F.; Krause, U.; Schellens, J.P.; Vreeling-Sindelárová, H.; Meijer, A.J. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur. J. Biochem. 1997, 243, 240–246. [Google Scholar] [CrossRef]
- Dong, Y.; Wu, Y.; Zhao, G.L.; Ye, Z.Y.; Xing, C.G.; Yang, X.D. Inhibition of autophagy by 3-MA promotes hypoxia-induced apoptosis in human colorectal cancer cells. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 1047–1054. [Google Scholar] [CrossRef]
- Zheng, N.; Liu, L.; Liu, W.W.; Li, F.; Hayashi, T.; Tashiro, S.I.; Onodera, S.; Ikejima, T. Crosstalk of ROS/RNS and autophagy in silibinin-induced apoptosis of MCF-7 human breast cancer cells in vitro. Acta Pharmacol. Sin. 2017, 38, 277–289. [Google Scholar] [CrossRef]
- Chen, X.; Liu, H.; Chen, K.; Guo, Y.; Mo, T.; Gao, T. Inhibition of autophagy impairs free fatty acid-induced excessive lipid accumulation in hepatocellular carcinoma and hepatic cells. J. Biosci. 2022, 47, 27. [Google Scholar] [CrossRef]
- Abu Samaan, T.M.; Samec, M.; Liskova, A.; Kubatka, P.; Büsselberg, D. Paclitaxel’s Mechanistic and Clinical Effects on Breast Cancer. Biomolecules 2019, 9, 789. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Kimmelman, A.C.; Debnath, J. Targeting Autophagy in Cancer: Recent Advances and Future Directions. Cancer Discov. 2019, 9, 1167–1181. [Google Scholar] [CrossRef]
- Tyutyunyk-Massey, L.; Gewirtz, D.A. Roles of autophagy in breast cancer treatment: Target, bystander or benefactor. Semin. Cancer Biol. 2020, 66, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Abouzeid, H.A.; Kassem, L.; Liu, X.; Abuelhana, A. Paclitaxel resistance in breast cancer: Current challenges and recent advanced therapeutic strategies. Cancer Treat. Res. Commun. 2025, 43, 100918. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.J.; Ha, H.; Lee, B.S.; Kim, B.H.; Song, H.K.; Kim, Y.K. LC3B is an RNA-binding protein to trigger rapid mRNA degradation during autophagy. Nat. Commun. 2022, 13, 1436. [Google Scholar] [CrossRef]
- Xu, K.; Zhu, W.; Xu, A.; Xiong, Z.; Zou, D.; Zhao, H.; Jiao, D.; Qing, Y.; Jamal, M.A.; Wei, H.J.; et al. Inhibition of FOXO1-mediated autophagy promotes paclitaxel-induced apoptosis of MDA-MB-231 cells. Mol. Med. Rep. 2022, 25, 72. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, J.; Lin, X. Manual Delineation of the Region of Interest Combined With Clinical Image Analysis to Predict the Ki-67 Expression Level in Non-small Cell Lung Cancer. Sage Open Pathol. 2025, 18, 30502098251336608. [Google Scholar] [CrossRef]
- Chang, Y.; Jia, H.Q.; Xu, B.; Yang, L.; Xu, Y.T.; Zhang, J.Y.; Wang, M.Q.; Yang, L.X.; Song, Z.C. Metadherin inhibits chemosensitivity of triple-negative breast cancer to paclitaxel via activation of AKT/GSK-3β signaling pathway. Chem. Biol. Drug Des. 2024, 103, e14416. [Google Scholar] [CrossRef]
- Kendall, A.K.; Chandra, M.; Xie, B.; Wan, W.; Jackson, L.P. Improved mammalian retromer cryo-EM structures reveal a new assembly interface. J. Biol. Chem. 2022, 298, 102523. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Her, S.; Kim, M.; Jang, I.S.; Park, J. The expression of damage-regulated autophagy modulator 2 (DRAM2) contributes to autophagy induction. Mol. Biol. Rep. 2012, 39, 1087–1093. [Google Scholar] [CrossRef]
- Vasconcelos, C.S.; Salgado, M.R.T.; Martins, M.R.; Anunciação, C.E.C.; Sobral, D.V.; Torres, L.C. Soluble sFas, sFasL, sPD1, and sPDL1 Analyses in the Peripheral Blood of Locally Advanced Breast Cancer Patients Before and After Neoadjuvant Chemotherapy. J. Surg. Oncol. 2025. [Google Scholar] [CrossRef]
- López-Mateo, I.; Arruabarrena-Aristorena, A.; Artaza-Irigaray, C.; López, J.A.; Calvo, E.; Belandia, B. HEY1 functions are regulated by its phosphorylation at Ser-68. Biosci. Rep. 2016, 36, e00343. [Google Scholar] [CrossRef]
- Festuccia, C. Investigational serine/threonine kinase inhibitors against prostate cancer metastases. Expert. Opin. Investig. Drugs 2017, 26, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Devis-Jauregui, L.; Eritja, N.; Davis, M.L.; Matias-Guiu, X.; Llobet-Navàs, D. Autophagy in the physiological endometrium and cancer. Autophagy 2021, 17, 1077–1095. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Zhou, Y.H.; Zhao, J.; Su, X.L.; Chen, N.X.; Zhao, Y.Q.; Ye, Q.; Hu, J.; Ou-Yang, Z.Y.; Zhong, M.M.; et al. Prognostic biomarker GSTK1 in head and neck squamous cell carcinoma and its correlation with immune infiltration and DNA methylation. Front. Genet. 2023, 14, 1041042. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, K.; Liu, T.; Zhao, H.; Jamal, M.A.; Chen, G.; Huo, X.; Yang, C.; Jiao, D.; Wei, T.; et al. Production and Functional Verification of 8-Gene (GGTA1, CMAH, β4GalNT2, hCD46, hCD55, hCD59, hTBM, hCD39)-Edited Donor Pigs for Xenotransplantation. Cell Prolif. 2025, e70028. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Forward | Reverse |
|---|---|---|
| ATG10 | TTCTGAAGTGACGAGACCTGC | AGCCTCGGCTTATAGCACTCA |
| FAS | TATCAAGGAGGCCCATTTTGC | TGTTTCCACTTCTAAACCATGCT |
| DRAM2 | GCTGTCCTTGCCTTTAGTATGG | AGATAACCAACAGTAGTCGGACC |
| GSTK1 | GGTCCTATGCAGATACCAACAC | GTACTGGCCTTTTCGGGGAA |
| MYCBP | GCTGGACACGCTGACGAAA | TCTAGGCGAAGCAGCTCTATTT |
| STK4 | AGCCCTCACGTAGTCAAGTAT | TCTTGTTCCGTAGCCGAATGATA |
| STK38 | GCAACCTTATCGCTCAACATGA | CGCGGATCTTCTGAGTCGT |
| VPS29 | GCACCAAGGAGAGCTACGAC | TCAGACCGATCTTGAACTGGC |
| VPS50 | TATTGTGGAAGGATACGCCAATG | GCTTCGTTGAGTATTCCCTGTG |
| CASP1 | CTTGGAGACATCCTGTCAGGG | AGTCACAAGACCAGGCATATTCT |
| CASP2 | TACTCCCACCGTTGAGCTGT | CCGTAGCATCTGTGGATAGGC |
| CASP8 | CAACTTCCTAGACTGCAACCG | TCCAACTCGCTCACTTCTTCT |
| GAPDH | TGACCTCAACTACATGGTCTACA | CTTCCCATTCTCGGCCTTG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Xiong, Z.; Liu, Y.; Jamal, M.A.; Wang, X.; Yang, C.; Gu, Z.; Chen, X.; Xiong, J.; Qing, Y.; et al. Enhancement of 3-MA in Paclitaxel Treatment of MDA-MB-231 Tumor-Bearing Nude Mice and Its Mechanisms. Int. J. Mol. Sci. 2025, 26, 6191. https://doi.org/10.3390/ijms26136191
Wang J, Xiong Z, Liu Y, Jamal MA, Wang X, Yang C, Gu Z, Chen X, Xiong J, Qing Y, et al. Enhancement of 3-MA in Paclitaxel Treatment of MDA-MB-231 Tumor-Bearing Nude Mice and Its Mechanisms. International Journal of Molecular Sciences. 2025; 26(13):6191. https://doi.org/10.3390/ijms26136191
Chicago/Turabian StyleWang, Jing, Zhe Xiong, Yaowen Liu, Muhammad Ameen Jamal, Xia Wang, Chang Yang, Ziyi Gu, Xiaojing Chen, Jingjing Xiong, Yubo Qing, and et al. 2025. "Enhancement of 3-MA in Paclitaxel Treatment of MDA-MB-231 Tumor-Bearing Nude Mice and Its Mechanisms" International Journal of Molecular Sciences 26, no. 13: 6191. https://doi.org/10.3390/ijms26136191
APA StyleWang, J., Xiong, Z., Liu, Y., Jamal, M. A., Wang, X., Yang, C., Gu, Z., Chen, X., Xiong, J., Qing, Y., Li, H., Xu, K., Wei, H.-J., & Zhao, H.-Y. (2025). Enhancement of 3-MA in Paclitaxel Treatment of MDA-MB-231 Tumor-Bearing Nude Mice and Its Mechanisms. International Journal of Molecular Sciences, 26(13), 6191. https://doi.org/10.3390/ijms26136191