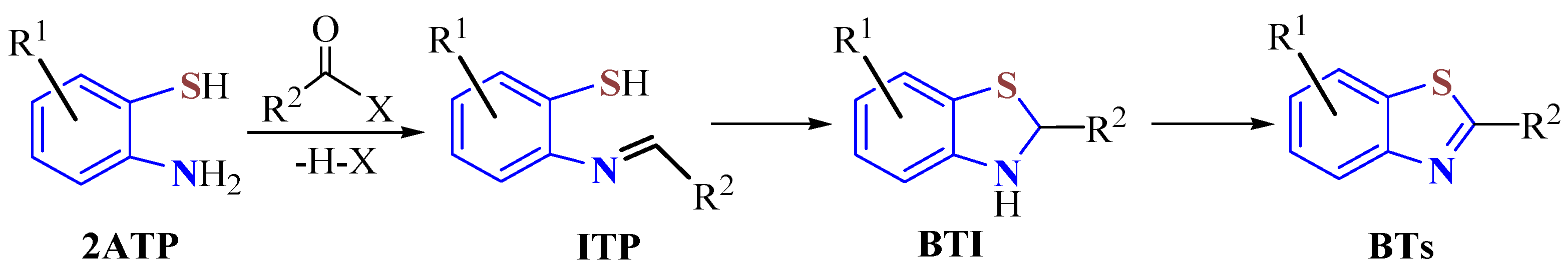
2.1. Condensation with Aldehydes
In one step, the condensation reaction of 2ATP with several benzaldehydes using sodium hydrosulfite (Na
2S
2O
4) as an oxidizing agent in refluxing a mixture of water–ethanol as the dissolvent for 12 h was carried out to afford 2-arylBTs
1a–
k in 51–82% yields (
Table 1) [
85]. On the other hand, the condensation of 5-substituted 2ATPs as hydrochlorides or disulfides with benzaldehydes under refluxion for 36 h afforded the BTs
2b–
k in 53–82% yields and
3a–
k in 51–84% yields.
Compounds were evaluated as sun-filtering, antioxidant, antifungal, and antiproliferative agents [
85]. Compound
1g was the most effective blocker of hERG potassium channels expressed in HEK 293 cells, resulting in 60.32% inhibition, with an IC
50 = 4.79 μM. Compounds
1g,
1h,
1k,
2d,
2g,
3d, and
3g were considered broad-spectrum and ultraviolet A filters (UVA filters) because their λc ≥ 370 nm and the ultraviolet A-protector factor (UVA-PF) values were greater than one-third of their sun protection factor (SPF) values. Compounds
1d and
1j were regarded as candidates for UVB protection. All compounds were photostable, especially
2e, which showed a degradation rate of less than 2%.
It was found that the wavelength of maximum absorption (λmax) was related to stereo electronic effects by substituents on the 2-aryl or BT moiety, which alter the electron density of compounds. The -OH group at the para position of the 2-aryl group leads the resonance interaction of the lone pair of oxygen and the π cloud of 2-aryl and the BT group. This shift increases if -OH is at the ortho position, due to the formation of an intramolecular hydrogen bond between the nitrogen of BT and the hydrogen of the -OH group. On the other hand, the order of λmax along the series was -H ≪ -SO2NH2 < -COOH. Additionally, the -COOH or -SO2NH2 groups on the BT ring increased the photostability of the compounds.
Compounds 1i,j, 2g,h, and 3r,s showed a good antioxidant profile. Compound 1h had the highest antifungal activity against the dermatophytes Trichophyton tonsurans, Epidermophyton floccosum, and Microsporum gypsum. Significant activities were found for compounds 1f and 1g against Epidermophyton floccosum and Microsporum gypseum. Compounds 1g and 1k exhibited high antiproliferative activity against CEM and SK-Mel 5 tumor cells, 1g (6-fold) and 1k (25-fold) being more selective for CEM and SK-Mel 5 cells, respectively. Because of their various biological activities, some 2-arylBTs could be applied as drugs in the treatment of neoplastic diseases such as melanoma, childhood leukemia, and pancreatic cancer. However, based on the experimental results and SAR studies on positions 2 and 6 of 2-arylBTs, compounds 1g and 1k were proposed as powerful candidates for designing multifunctional drugs.
2ATP was condensed with substituted benzaldehydes by using biodegradable rice husk chemically activated carbon (RHCAC) as the catalyst at room temperature in ethanol–water to afford 2-arylBTs
4a–
f in 93–98% yields (
Table 2) [
86]. The catalyst was recovered by filtration, washed, dried, and recycled for eight runs, unlike a corrosive and toxic acid catalyst that was difficult to recover and separate.
No significant effects of electron donation were observed, while OMe, CH3, and Cl groups and electron-withdrawing NO2 groups were found, and the reactions were completed in short times (5–10 min), giving high yields of the desired products (93–98%).
A series of 2-arylBTs
5a–
f were synthesized from the condensation reaction of 2ATP with substituted benzaldehydes in 1,4-dioxane under an O
2 atmosphere in the presence of [PhI(OH)OTs] (Koser’s reagent) as a catalyst at room temperature (
Table 3) [
87]. The features of this protocol included short reaction times (15 min), a broad substrate scope, 80–90% yields, low catalyst loading, and scalability.
Six
p-substituted 2-arylBTs
6a–
f were obtained in 70–90% yields from the condensation of 2ATP with
p-benzaldehydes catalyzed with Cu
2O and DMSO as oxidants in string at room temperature for 3–5 h (
Table 4) [
88]. The reaction tolerated a wide range of functional groups. The compounds
6c and
6e showed equal antifungal activity against
A. niger and the highest activity against
C. albicans, compared to voriconazole as a reference.
Three green synthetic protocols were used to synthesize 6-substituted 2-arylBTs,
7a–
f and
8a–
f, in 62–89% yields (
Table 5) [
89]. Method A: A stirred suspension of 4-substituted-2ATP and the 2-hydroxybenzaldehyde was heated in glycerol at 110–170 °C for 1 to 24 h to afford compounds
7a–
c in 62–71% yields. Method B: A stirred suspension of the 5-substituted 2ATP disulfide and 2-hydroxybenzaldehyde or 2-methoxybenzaldehyde was heated in glycerol at 160–175 °C for 30–60 min to afford compounds
7a–
c in 74–80% yields and
8a–
c in 70–79% yields. Method C: The corresponding 2-substituted benzaldehyde was added to a stirred solution of 4-amidino-2ATPate in glacial acetic acid, and the mixture was heated under nitrogen for 4 h, then alkalinized with NaOH (pH 10–11); the resulting free base was mixed with 2-propanol and methanesulfonic acid and stirred at room temperature for 2 h to afford compounds
7d,
7e,
8d, and
8e in 62–83% yields. Method D: A solution of SnCl
2 dihydrate in conc. HCl and methanol was added to the corresponding 6-nitro-substituted BTs
7b and
8b, then refluxed for 30 min. The mixture was alkalinized with 20% NaOH (pH 8–9) to afford compounds
7f and
8f in 89.3 and 81.6% yields, respectively. The influence of the hydroxy and methoxy groups and substituents on C6 on the 2-arylBT scaffold against antibacterial, antitumor, and antioxidant activities was studied. The amidino derivatives
7d and
8d showed modest activity against Gram-negative and Gram-positive bacterial strains. At the same time, compounds
7b,
7c,
7e,
7f, and
8f resulted in potent and selective antiproliferative activity towards tumor cells, without activity against human skin fibroblasts. Compounds
7f and
8f were the most selective against the growth of HeLa cell lines. Compound
7f resulted in the most promising radical scavenging activity. HIF-1α hydroxylated protein was upregulated by treatment of HeLa cells with compounds
7f,
8c, and
8f, which were considered potent suppressors of the hypoxia-induced HIF-1 protein. Compounds
7f and
8f were proposed to lead compounds for further rationalized design of the BT skeleton. Compound
7f with the OH group on the 2-arylBT core had the most promising antioxidative activity as evaluated by DPPH, ABTS, and FRAP in vitro assays. The presence of the amino protonated group attached at the BT moiety was essential for the antiproliferative and antioxidant activities observed, exerted through a change in the levels of the reactive oxygen species-modulated HIF-1 protein. The BTs
7a–
f and
8f showed good antioxidant activity (38 to 117 µM), while BTs
8a–
e had a low ability for stabilization of ABTS
•+ radicals (>200 µM).
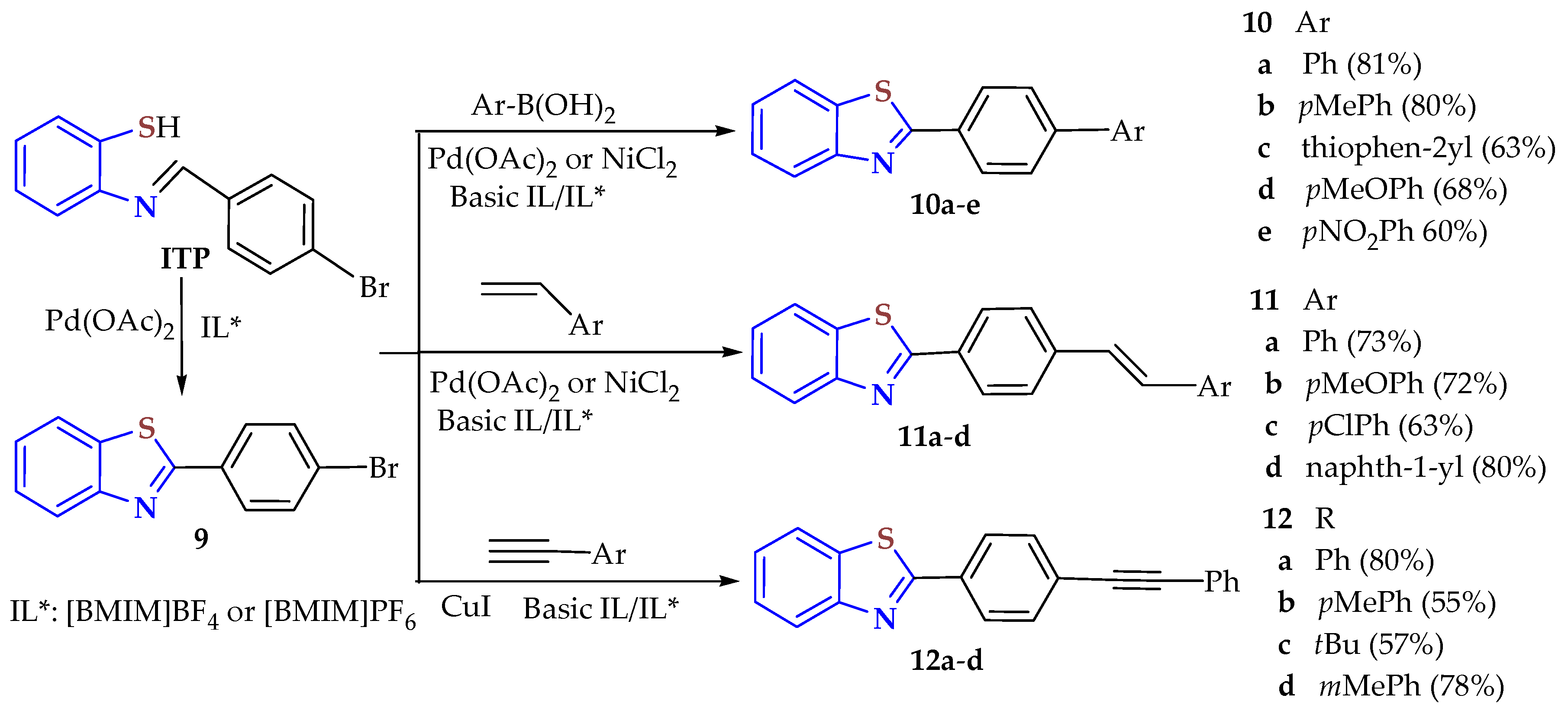
In a two-step protocol, the corresponding
p-bromophenyl-ITP intermediate, accessed from the condensation of 2ATP and
p-bromobenzaldehyde, suffered a Pd-catalyzed cyclization to 2-bromophenyl-BT
9 in [BMIM][BF
4] or [BMIM][PF
6] as a recycled IL solvent. Compound
9 was functionalized by cross-coupling reactions (Suzuki, Heck, or Sonogashira) and catalyzed by Ni or Pd to afford a series of substituted 2-aryl-/heteroaryl-BTs,
10a–
e,
11a–
d, and
12a–
d, in acceptable yields (55–86%) under mild reaction conditions (
Scheme 2) [
90]. The yields were from moderate to good for
10a–
e (60–81%, 12–14.5 h),
11a–
d (63–80%, 16.5–17 h), and
12a–
d (55–80%, 14.5–16 h).
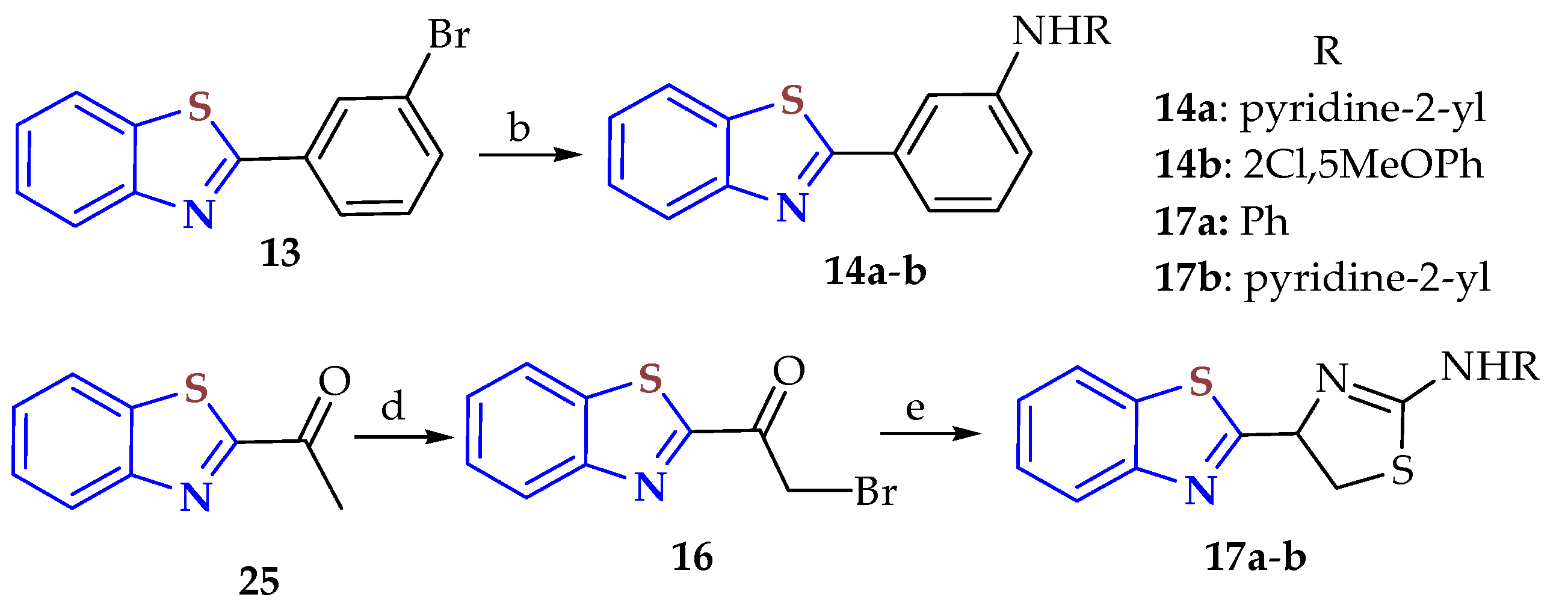
2-substituted BTs,
14a,
b (25, 34%) and
17a,
b (52, 60%), were designed and synthesized from BTs
13 (60%) and
15 (34%), which were obtained from the condensation of 2-aminobenzothiazole (2ABT) with 3-bromobenzaldehyde and lactic acid, then oxidation of the derived alcohol with MnO
2, to be tested as antiproliferative cancer cell lines (
Scheme 3) [
91]. Compounds
217a–
b were obtained from compound
15 by (a) cyclization of 2ABT with 3-bromobenzaldehyde to afford compound
13, and the BTs
14a,
b were obtained from compound
13 by (b) Buchwald–Hartwig amination with anilines. Compounds
17a–
b were obtained from (d) α-bromination with CuBr
2, then (e) reaction with the corresponding substituted thiourea. Antiproliferative assays using cancer cells from the breast (MCF7) and prostate (22Rv1 and PC3) were carried out for all synthesized BTs. However, these compounds had worse activity than JG-98 as a reference (IC
50 values from ~0.7 to 13 µM).
A photo-assisted radical cyclo-condensation of 2ATP with a series of aromatic and aliphatic aldehydes was designed using wosin Y as a photocatalyst and K
2CO
3 or Et
3N as basic media and
tert-butyl hydroperoxide (TBHP) (70% in water) in acetonitrile to afford the 2-aryl(alkyl)BTs
18a–
j in 70–92% yields (
Table 6) [
92]. Unfortunately, the designed method required harsh reaction conditions, a long reaction time (24–36 h), and flash column chromatography and allowed no recovery of the catalyst.
A reasonable mechanistic process has been proposed beginning with the in situ formation of the corresponding imine and the excitation of eosin Y under blue LED irradiation, then the reaction of eosin Y* with TBHP to generate an (eosin y).-radical anion and a t-Bu. radical, which undergo hydrogen atom abstraction with the imine to produce the Ar-O. imine-fenoxy radical. Then, there occurs an intramolecular cycloaddition to the corresponding benzothiazoline, which is transformed to a benzothiazoline.+ radical cation that reacts with the eosin Y.-radical anion to afford the corresponding BT. This method tolerates several aldehydes, including aliphatic, aromatic, cyclic, and heteroaromatic aldehydes, but has several limitations, such as the requirement of a transition metal, a high temperature, and a pre-synthesized catalyst/ligand scaffold.
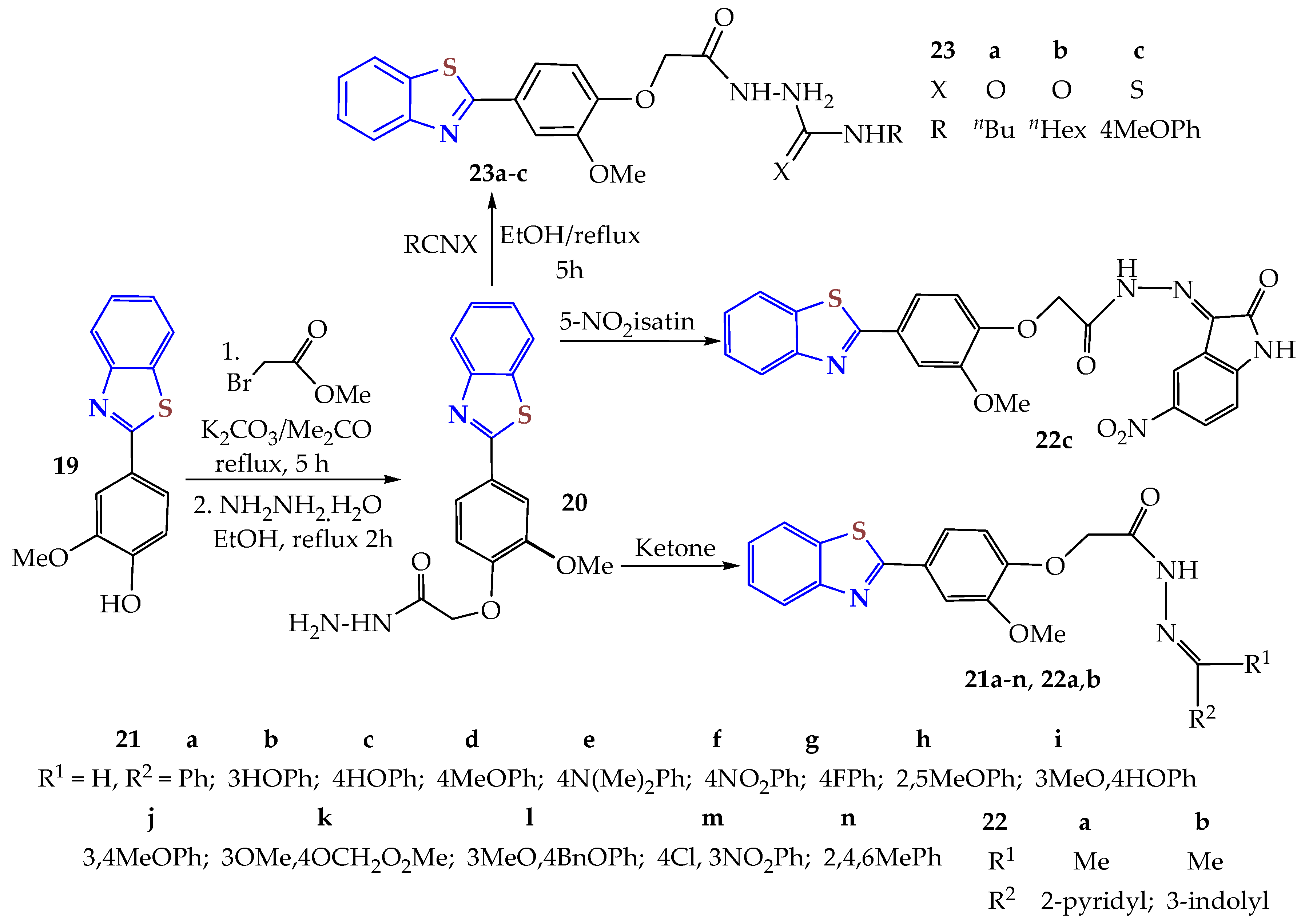
As previously reported, three series of substituted 2-arylBTs,
21–
23, were synthesized as VEGFR-2/FGFR-1/PDGFR-β multi-angiokinase inhibitors targeting breast cancer (
Scheme 4) [
93]. The compounds
21 and
22 were obtained from the condensation of 2ATP and vanillin in refluxing DMF to afford the 2-arylBT
19, which was subsequently reacted with methyl bromoacetate, followed by hydrazinolysis with hydrazine hydrate in refluxing ethanol to afford the hydrazide
20, which through acid catalysis with aldehydes or ketones afforded the Schiff bases
21a–
n and
22a–
c in 40% to 88% yields. On the other hand, the reaction of hydrazide
20 with isocyanates and isothiocyanate in refluxing ethanol afforded the corresponding ureas
23a,
b in 84 and 72% yields and thiourea
23c in a 61% yield.
Compounds 21d, 21f, 21i, and 21k showed high inhibitory activity against multiple kinases: VEGFR-2 (IC50s of 0.19, 0.18, 0.17, and 0.13 μM, respectively), FGFR-1 (IC50s of 0.28, 0.37, 0.19, and 0.27 μM, respectively), and PDGFR-β (IC50s of 0.07, 0.04, 0.08, and 0.14 μM, respectively). Additionally, the substituted 2-arylBTs exhibited promising cytotoxic activity against the MCF-7 cell line comparable to that of sorafenib as a reference drug (IC50 = 4.33 μM). The most potent arylBTs were 21d and 21i (IC50s of 7.83 and 6.58 μM). Also, 21d and 21i had VEGFR-2 inhibitory activity in MCF-7 cells of 81% and 83% compared with sorafenib (88%). Molecular docking of compounds in the VEGFR-2 and FGFR-1 active sites showed that the 2-phenylBT moiety was placed in the hinge region of the tyrosine kinase (RTK)-binding receptor site. In contrast, the amide moiety showed hydrogen bonding interactions with the amino acids, directing the aryl group to the hydrophobic allosteric back pocket of the RTKs in a type II-like binding mode. Additionally, the aryl BTs exhibited good ADME properties, facilitating further optimization in drug discovery.
An acetic acid-mediated three-component condensation reaction of 2ATPs and
α,
β-unsaturated aldehydes in the presence of thiophenols was developed to afford 2-thioalkyl BTs
24a–
t (
Figure 4) [
94]. Metal-free conditions were used in the reaction, with oxygen as an oxidant. This method was regioselective, tolerant of several functional groups, and provided access to various kinds of BTs with modest to good yields (34–81%).
On comparing the yields of compounds 24a (70%), 24b (42%), and 24d, the position of the methyl/phenyl group (R4) on the α, β-unsaturated aldehydes affected the reaction yield. However, α, β-unsaturated aldehydes bearing an electron-donating group or electron-withdrawing group like Me, Cl, or Br at the para position of the phenyl ring produced the compounds 24e–g in moderate yields (54–57%). Also, moderate yields (51–59%) were observed when Me, Cl, and F substituents were located at different positions on the benzene of the BT ring. Thiophenols containing methyl groups at the para, meta, or ortho positions did not show an influence and efficiently reacted in moderate yields. However, electron-donating groups such as tBu and OMe at the para position of thiophenols afforded BTs in 71% (24k) and 80% (24l) yields, respectively. Halogen-containing thiophenols such as F, Cl, and Br successfully afforded the thioalkyl-substituted BTs 24a, 24m, and 24n in 53–70% yields. Various disubstituted thiophenols resulted in good substrates for this reaction to afford the BTs 24o–q in 56, 80, and 81% yields, respectively.
The eco-friendly biocatalytic oxidant system Laccase/DDQ (2,3-dichloro-5,6-dicyano-1,4-benzoquinone) was applied to condense 2ATP with aromatic aldehydes to afford 2-arylBTs
25a–
x in 65–98% yields using O
2 pure or from the air as an oxidant in aqueous media at room temperature in 1 h (
Table 7) [
95]. Two steps were required in the aerobic oxidative cyclization: (1) chemical cyclization and (2) chemoenzymatic oxidation. Benzaldehydes with electron-donating and electron-withdrawing groups, heterocyclic and α,β-unsaturated aldehydes, 1-naphthaldehyde, 2-naphthaldehyde, 9-anthraldehyde, and terephthaldehyde were successfully applied to prepare the corresponding BTs. The advantages of this method are as follows: (1) the use of air or O
2 as an environmentally benign, inexpensive, and abundant oxidant agent and the formation of H
2O as a non-toxic by-product; (2) the synthesis of BTs in good to high yields in aqueous media at room temperature; (3) its superiority with respect to other available methods and its attractiveness to the pharmaceutical industry owing to its being free from any toxic and expensive transition metals and halide catalysts; (4) its conforming to several of the guiding principles of green chemistry.
In a one-pot reaction, 2ATP was condensed with benzaldehydes and aromatic heterocyclic aldehydes under solvent-free conditions using ZnCl
2 nano-flakes supported on nano-hydroxyapatite (HAp) as a catalyst to afford the 2-arylBTs
26a–
l in 65–95% yields in 15–90 min (
Table 8) [
96]. The reaction was fast, eco-friendly, and efficient. The molecular docking validation with transpeptidase and 14 α-demethylase enzyme inhibitors showed that five 2-substituted-BTs gave good energy values. Also, antioxidant studies showed that four BTs were promising in relation to ABTS (inhibition %) compared with ascorbic acid. Substituted and disubstituted benzaldehydes with electron-donating groups on R
2, such as 4MeO and OH groups, and electron-withdrawing groups, such as F, Cl, and NO
2 groups, gave BTs
26a,
b,
d–
h with excellent yields (75–80%); however, 1,2,3-OMe-substituted and 3-pyridil-substituted benzaldehydes gave BTs
26c and
26j in 67 and 65% yields, respectively.
The condensation reaction of substituted 2ATPs with several aromatic and aliphatic aldehydes was catalyzed with sulfated tungstate (ST) under solvent-free, room temperature, and ultrasound irradiation conditions to afford 2-substituted BTs,
27a–
z and
28a–
d, in excellent yields (90–98%) (
Table 9) [
97]. ST is considered a mildly acidic, easy-to-prepare, non-toxic, recyclable, efficient, and heterogeneous green catalyst. This method is very good for the synthesis of 2-substituted-BTs from several functionalized aromatic, aliphatic, and heteroaromatic aldehydes. The advantages of this method are as follows: easy handling, functional group compatibility, short reaction times (5 min), high catalytic activity and recyclability, chemoselectivity, very good yields, no column purification, low corrosiveness, and environmental compatibility.
2ATP and substituted aryl aldehydes were condensed using ruthenium silicate (RS-1) zeolite as a catalyst in a hydrothermal process to afford the 2-arylBTs
29a–
i in 85–93% yields (
Table 10) [
98]. The benefits of this protocol include mild reaction conditions, short reaction times (30 min), and high thermal catalyst and catalysis recyclability.
Substituted BTs
30a–
t were synthesized in 82–94% yields in 3 h by the visible-light-induced condensation–cyclization reaction of substituted 2ATP with substituted aromatic aldehydes (
Table 11) [
99]. Fluorescein was used as a photocatalyst, a blue LED lamp was used as the light source, and atmospheric molecular oxygen was required for the reaction to proceed.
Substituted aromatic aldehydes, such as 4-Br, 4-Cl, and 4-F (electron-withdrawing) and 2-Me, 4-Me and 4-OMe (electron-donating) substituents, afforded BTs in high to excellent yields: 30d–f (88–94%) and 30g–i (87–92%). Notably, the position of the substituents on the aromatic aldehydes had little effect on the yield of the BTs 30b–d (88–91%) and 30g,h (91, 92%). In addition, two electron-donating groups on the aromatic aldehydes gave a lower yield (82%) of the BT 3j. The reactions were smooth, with good functional group tolerance. Additionally, the catalytic system eliminates the need for an oxidant or metal catalyst, aligning with the principles of green chemistry.
The 2-aryl-BTs
31a–
t were synthesized in 64–99% yields in 10 min by combining enzymatic (trypsin) and visible-light (450 nm) catalysis under an air atmosphere (
Table 12) [
100]. The method consisted of biocatalytic condensation of 2ATP with aromatic or aliphatic aldehydes to afford the corresponding benzothiazoline
BTI as an intermediate. Subsequent visible-light-induced oxidization produced the 2-arylBTs in approximately 10 min. Additional oxidants or metals were not required in this protocol, which was environmentally benign.
It was found that the electronic effect on the substituents on the benzene ring influences the reaction. Aldehydes containing electron-deficient groups afforded the BTs 31a, 31g, and 31j, which were obtained in 98–99% yields, while an electron-rich pair group afforded the BT 31m in 75% yields. If the electron-richness group of the aldehyde is increased, the yield of BTs decreases, as in the series of BTs 31g–i, whose yields were 98%, 95%, and 89%, and the yield in BT 31p was reduced to 64%. In the case of steric hindrance of methyl-substituted benzaldehydes, the yield is negligible, as in the BTs 31c–e (96–98%). With a larger volume of 1-naphthaldehyde, the yield of the BT 31s decreases to 78%.
An ionic liquid immobilized on silica-coated cobalt–ferrite magnetic nanoparticles (CoFe
2O
4@SiO
2@PAF-IL) was formed. This hybrid nanostructure was used as a catalyst in the condensation reaction of 2ATP with aromatic aldehydes to synthesize the 2-aryl-BTs
32a–
i in 83–91% yields (
Table 13) [
101]. The reaction was carried out under heating in solvent-free conditions at 70 °C for 10 min. The advantages of this procedure included the use of solvent-free conditions, the simple work-up, the short reaction times, and the environmentally benign conditions. The nano-catalyst could be reused several times without loss of catalytic activity and was easily separated.
Substituted 2ATP disulfides or dihydrochlorides were condensed with the corresponding aldehydes in refluxing glycerol without a catalyst for 45 min to afford the previously designed 6-cyano- (
33a, 83%;
33d, 64%) and 6-amidino- (
33b,
c,
e,
f, 22–56%) 2,5-disubstituted furane-BTs
33a–
f to be screened for antimicrobial and antitumor activities (
Figure 5) [
102]. The antitumor activity was tested on the human lung cancer cell lines A549, HCC827, and NCI-H358, with MTS cytotoxicity and in vitro BrdU proliferation assays performed using 2D and 3D cell culture methods. Compounds
33b,
c,
e resulted in possible antitumor activity to stop the proliferation of cells. Broth microdilution testing was used to evaluate the antimicrobial activity on Gram-negative
E. coli and Gram-positive
S. aureus and
S. cerevisiae as eukaryotic model organisms, according to Clinical Laboratory Standards Institute (CLSI) guidelines. The BTs
33b,
c,
e and 233f showed promising antibacterial activity. All compounds were more activated in the 2D than in the 3D assays on the three cancer cell lines and in the antimicrobial assays. Compounds with the amidine group at the 6-position of the benzothiazole ring,
33b and
33e, gave low yields (22 and 28%, respectively).
Compound 4-(BT-2-yl)-2-methoxyphenol
34, obtained from the condensation of 2ATP with the corresponding benzaldehyde, was transformed to compound 2-[4-BT-2-yl)-2-methoxyphenoxy]acetohydrazide
36 through ethyl 2-(4-BT-2-yl)-2-methoxyphenoxy) acetate
35, then reacted with chlorine acetyl chloride to afford the 2-(4-BT-2-yl)-2-methoxy-phenoxy)-N′-(2-chloroacetyl)acetohydrazide
38 (
Scheme 5) [
103]. The acetohydrazides
36 and
38 were reacted with anhydrides and amino acids to afford the BT derivatives
37a–f in 65–90% yields and
39a–k in 65–90% yields, respectively. The in vitro anticancer activity of all the compounds exhibited promising potency against hepatocellular carcinoma HepG-2 (IC
50s from 0.7 ± 0.4 to 1.0 ± 0.7 μM) and very good potency against breast cancer cells MCF-7 (IC
50s from 2.5 ± 2.5 to 3.5 ± 3.4 μM) compared with the standard drug doxorubicin (IC
50s = 1.0 ± 0.8 μM and 2.9 ± 1.9 μM, respectively). The highest cytotoxic activity was observed for compounds
37a–
c,
39c, and
39f. Also, these compounds were further evaluated for their EGFR inhibitory activity compared with the reference drug erlotinib. Molecular docking studies of the promising compounds
39a–
c were carried out to interpret their enzymatic activities. Moreover, compounds
39a and
39b exhibited considerable pre-G1 and G2/M cell cycle arrest compared with the untreated MCF-7 cells. Additionally,
39a and
39b increased the levels of Bax, p53, and caspase-3 levels while decreasing the levels of Bcl-2, which are oncogenic parameters. The results of these BT-based derivatives proposed an excellent framework for detecting new potent antitumor leads.
The condensation reaction of (un)substituted 2ATP, 2ATP-disulfide, or 2ATP-hydrochloride with furfural, pyrrole-2-carboxaldehyde, or 2-thiophenecarboxaldehyde occurred under stirring in ethanol at room temperature, then unsubstituted 2ATP was reacted under refluxion for 12 h and substituted under refluxion for 36 h in the presence of an aqueous solution of sodium hydrosulfite as a catalyst to afford the 2-substituted-BTs
40a–
i in 69, 48, 87, 34, 30, 48, 31, 29, and 54% yields (
Figure 6) [
76]. The synthesized BTs were investigated for their photoprotective, antioxidant, antiproliferative, and antifungal activities. Compounds
40d and
40g exhibited a multifunctional profile with an excellent filtering UVB capacity, which was higher than that of PBSA, which served as a reference and is currently used as a UV sunscreen filter. These compounds were the best at inhibiting the growth of dermatophytes and
C. albicans, and
40g showed good antioxidant activity. Furthermore,
40d was effective on melanoma tumor cells (SK-Mel 5). These compounds are proposed as new skin preventive and protective agents.
2ATP was cyclo-condensed with a series of aromatic/aliphatic aldehydes in the presence of bovine serum albumin (BSA) as a biocatalyst in water under stirring at room temperature for 8 h to afford a series of 2-substituted BTs,
41a–
v, in 79–93% yields (
Table 14) [
104]. The advantages of this protocol were its excellent yields, high atom economy, gram scalability, operational simplicity, and recyclability.
The condensation–cyclization reaction of 2ATP with aromatic benzaldehydes was carried out in a self-neutralizing acidic CO
2–alcohol system to afford the BTs
42a–
n in 55–87% yields. Practicality, economic viability, and environmental friendliness were the advantages of this protocol (
Table 15) [
105]. The alkyl carbonic acid formed CO
2, and methanol was proposed as an intermediate in the reaction mechanism, generating hydrogen cations to catalyze the reaction. 2-PhenylBT
42a gave the highest yield (87%), while in the case of the 2-substituted BTs
42b–
m the yields decreased to 55–62%. However, in 5-chloro-2-phenylbenzothiazole
42n the yield increased to 72%.
2ATP was condensed with aromatic aldehydes in the presence of 0.05 mol% of bacterium-derived hemoglobin
Vitreoscilla (VHb) as a biocatalyst,
tert-buthyl hydroperoxide (TBHP) as an oxidant, and dimethyl sulfoxide (DMSO) as a solvent at room temperature to afford 2-arylBTs
43a–
n in only 5 min with excellent yields (85–97%) (
Table 16) [
106]. The advantages of this protocol were its mild reaction conditions and high efficiency, with a wide substrate scope.
It was found that electron-rich groups on R2 resulted in decreased yields (43b,c,g,i; 92, 88, 89, and 87% yields, respectively) compared with 2PhBT (43a), whereas electron-poor moieties within the aromatic aldehydes (43d–f,h,j; 91–97%) were the best substrates. Aldehydes with heteroaromatic groups produced the BTs 43k–m in high yields (93–95%) with a longer reaction time (20 min). The yield decreased significantly when the benzaldehyde contained the naphthalyl group, affording the BT 43n in an 85% yield, due to the steric effect.
Substituted 2ATPs were condensed with a series of aldehydes at 80 °C using an environmentally friendly, inexpensive, and easily accessible Zn(OAc)
2·2H
2O (5 mol%) in a solvent-free reaction to afford a series of substituted 2-arylBTs,
44a–
p, in 67–96% yields in 30–60 min (
Table 17) [
107]. The reaction involving heterocyclic aldehydes interestingly afforded the corresponding BTs
44m–
o in good yields (79–84%), and with alkyl aldehydes, such as isobutyraldehyde, the corresponding BT
44p was produced in a moderate yield (67%). The BTs
44a–
o were obtained in 79–96% yields.
The nanocomposite MNPs-phenanthroline-Pd (Fe
3O
4@SiO
2-diPy-Pd) was synthesized to be used as a catalyst for the cyanation of aryl halides at 80 °C for 6 h in the synthesis of BTs
45a–
h in 89–98% yields (
Table 18) [
108]. The optimal conditions for the reaction were 20 mg of the MNPs-phenanthroline-Pd nano-catalyst and K
2CO
3 in DMF under reflux conditions. The catalyst was highly efficient and easily recovered and reused, without losing the morphology and dispersion of the particles. Aryl halide compounds with electron-donating groups at the
p-position of the aryl group afforded the BTs
45c,
d,
f in higher yields (96%, 98%, and 94%) than that of aryl with electron-withdrawing groups (BT
45e, 92% yield), whereas aryl bromide in the
m-position decreased the yield (BT
45h, 89% yield).
The eco-friendly DABCO-based dicationic acidic ionic liquid [C
4H
10-DABCO][HSO
4]
2 (DABCO = 1,4-diazabicyclooctane) was synthesized, characterized, and successfully used as a catalyst in the condensation of aryl aldehydes under H
2O reflux for 30–50 min to afford 2-phenylBTs
46a–
h in 85–91% yields (
Table 19) [
109]. The advantages of this protocol were its short reaction time; easy work-up; moderate to excellent yields; the use of green solvents; the use of non-metal, inexpensive catalysts; and catalyst recyclability. Aldehydes with electron-withdrawing groups at the
m- or
p-position of the aryl group afforded BTs
46b,
e,
f in higher yields (87%, 90%, and 91%) than that of aldehyde with an electron-donating group (BT
45c, 82%).
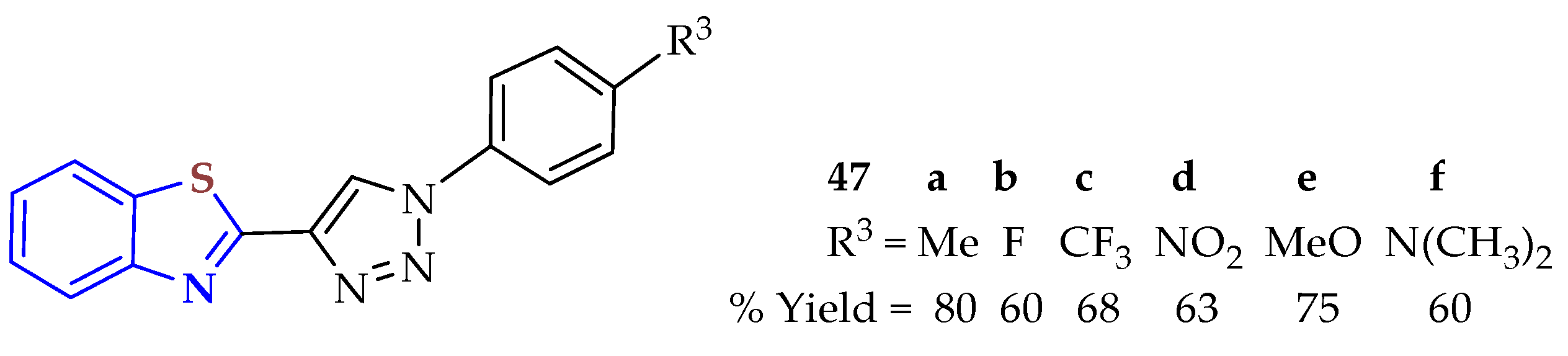
Six 1-aryl-4-BTyl-1,2,3-triazoles
47a–
f were prepared from condensation of 2ATP with aldehydes (
Figure 7) [
110]. The synthesized donor–acceptor molecules exhibited optical properties based on charge transfer emission depending on the substituent in the 1,2,3-triazole moiety. Compounds
47a and
47e with moderately electron-donating groups (CH
3 and OCH
3) or 4
7b and
47c with electron-withdrawing substituent groups (F and CF
3) in the triazole fragment were blue-shifted, in contrast to compounds
47d and
47f with strong electron-withdrawing (-NO
2) and electron-donating (-N(CH
3)
2) substituents, respectively, which showed red-shifted maxima. Compounds
5a–
d had low fluorescence quantum yields,
5d being almost non-emissive with a ΦF less than 0.01 due to the –NO
2 group. However, compound
5f had the highest quantum yield, which increased with the decreasing polarity of the solvent.
Some of these compounds were active in human A2780, HeLa, and A549 cancer cell lines, exhibiting IC50 values in the low micromolar range and low cytotoxicity in healthy CHO cells. Interestingly, compound 47f showed cytoplasmic staining determined by confocal fluorescent microscopy.
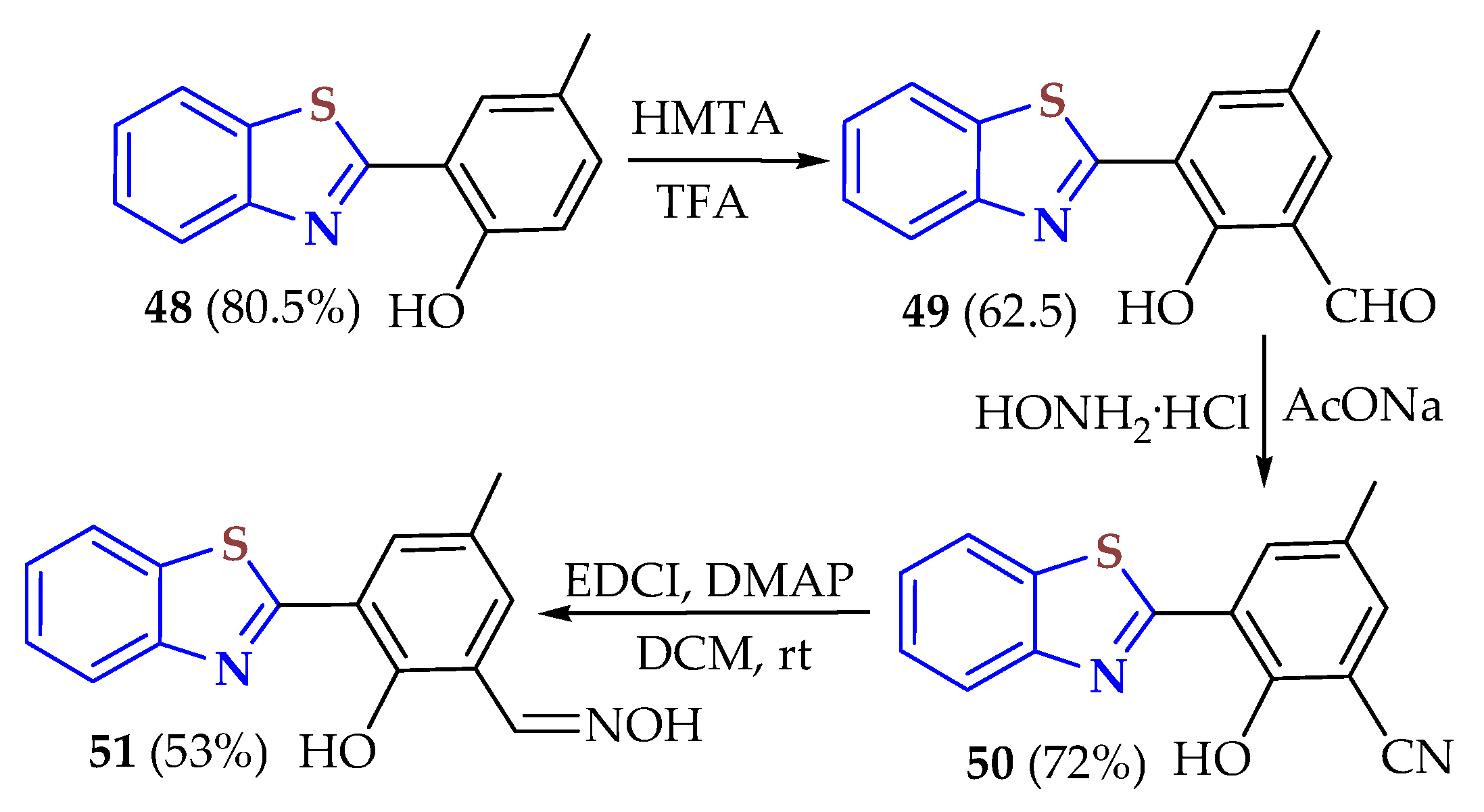
The fluorescent probe
51 based on BT was synthesized in a 53% yield by condensing 2ATP with 5-methyl salicylaldehyde in DMF and Na
2S
2O
5 as an oxidant to afford BT
48 in an 80.5% yield, followed by three steps through BT
49 in a 62.5% yield and
50 in a 72% yield (
Scheme 6) [
111]. The excellent selectivity enabled the detection of Cu
2+. The probe exhibited a cyan blue fluorescence emission peak at 487 nm under 360 nm UV excitation with fluorescence quenching after adding Cu
2+. The detection limit of Cu
2+ of the probe was determined as 3.08 × 10
−7 M. According to a Job plot and high-resolution mass spectrometry, the complexation ratio of the probe to Cu
2+ was 2:1. An excited intramolecular proton transfer (ESIPT) was confirmed as the sensor mechanism. Under alkaline conditions, the probe showed cyan blue and green fluorescence under acidic conditions. On the other hand, due to the excellent membrane permeability and low cytotoxicity, the probe could detect Cu
2+ in water samples and recognize Cu
2+ in human breast cancer cells (MCF-7).
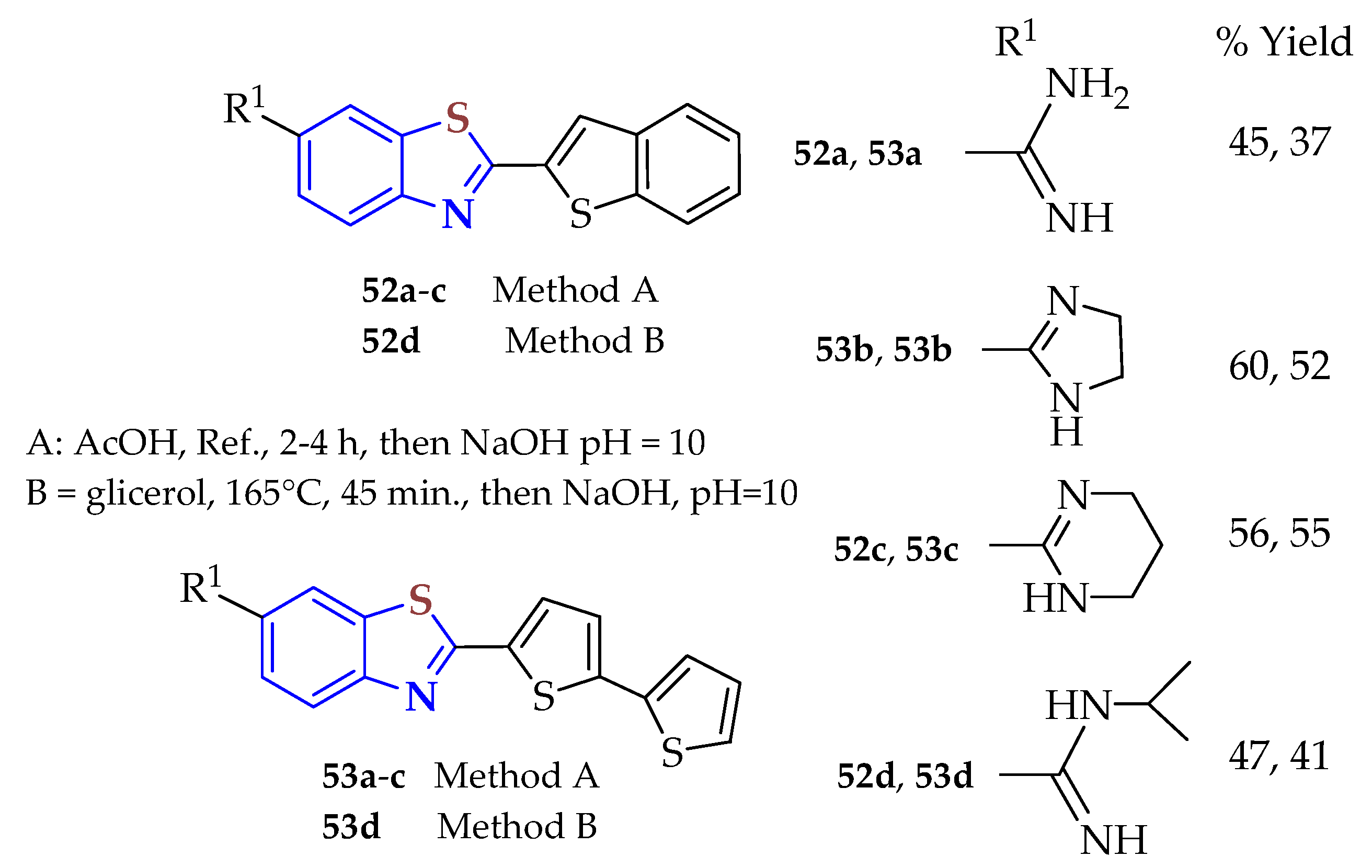
2ATPolate, 2ATP-disulfide, or 2ATP-hydrochloride were condensed with the corresponding aldehyde using two methods to afford the series of benzo[b]thienyl and 2,2′-bithienyl-BTs
52a–
d and
53a–
d to study the in vitro antitrypanosomal and antiproliferative activities (
Figure 8) [
112]. Specifically, the amidine group substitutions and the type of thiophene backbone impacted the biological activity. All synthesized BTs were active as both antiproliferative and antitrypanosomal agents.
The 2,2′-bithienyl-BTs with unsubstituted and 2-imidazolinyl amidine showed the most selective antitrypanosomal activity. The 2,2′-bithiophene BTs showed the most selective antiproliferative activity, whereas all 2,2′-bithienyl BTs were selectively active against lung carcinoma. The BT with an unsubstituted amidine group also produced strong antiproliferative effects. The pronounced antiproliferative activity of the BTs was attributed to different cytotoxicity mechanisms. Cell cycle analysis and DNA binding experiments provided evidence that BTs are localized in the cytoplasm and do not interact with DNA.
All amidino-substituted BTs, 52a–d and 53a–d, were tested in vitro for their antiproliferative activity against human cancer cell lines, including HCT116 and SW620 (colon carcinomas), H460 (lung carcinoma), MCF-7 (breast carcinoma), PC3 (prostate carcinoma) and HeLa (cervical carcinoma), and HEK 293 (human embryonic kidney) cells. The 2,2′-bithiophene-BT 53a displayed very potent and selective activity against H460 cells, with an IC50 value of 0.02 μM, which was twice the magnitude of the IC50 values for the other cancer cells tested. All other 2,2′-bithienyl-BTs, 53b, 53c, and 53d, also showed selective activity against H460 cells, with IC50 values of 0.2 μM, compared with doxorubicin (IC50 = 0.04 ± 0.01).
4NO
2-2ATP was condensed with 4-N-substituted benzaldehydes to afford the targeting fluorescent 2-(
N,
N-dimethyl/diphenyl-aminophenyl)-5-substituted-BTs
54a,
b in 73.5% and 69.1% yields, respectively. Then, the nitro group of compounds
54a,
b was reduced to the amine derivatives
55a,
b (81.3 and 78.8%, respectively) with Pd(OAc)
2 in ethanol and reacted with the corresponding aldehyde under stirring in methanol to afford the imines
56a,
b in 73.8 and 70.2% yields, respectively (
Scheme 7) [
113]. The compounds were analyzed for their photophysical properties, absorbance, and fluorescence in different organic solvents. The photophysical properties of BTs
54a,
b and
56a,
b were strongly impacted by the solvent used. In DMSO, all BTs had higher fluorescence intensities, showing that solvent polarity has a great impact on their excited-state properties. For BT
54a, the λmax values ranged from 337 nm to 389 nm, and BT
54b showed λmax values ranging from 373 nm to 394 nm, with the highest absorption observed in DMSO, whereas BTs
56a and
56b presented λmax values ranging from 387 to 417 nm and 393 nm to 423 nm, respectively. For BT
54a,
b, the fluorescence emission maxima ranged from 495 to 522 nm and 512 to 531 nm, respectively, with their highest values observed in ethanol. Conversely, BTs
56a,
b displayed emission maxima ranging from 518 to 537 nm and 518 to 571 nm, respectively, with the highest fluorescence observed in DMSO and ethanol, respectively.
All BTs were evaluated for cytotoxicity across anticancer cell lines and inhibition against VEGFR-2 kinase using an anti-phosphotyrosine antibody test. BT 54a showed moderate inhibitory activity with little variability (IC50 = 0.38 ± 0.13 μM). Conjugate 54b had a higher potency, with an IC50 of 0.29 ± 0.16 μM, with lower variability. The BT 56a showed considerable inhibitory activity, with an IC50 of 0.31 ± 0.08 μM, with the lowest variability, whereas BT 56b showed an IC50 value of 0.23 ± 0.20 μM. Compounds 54b and 56b resulted in potent cytotoxic activity against MCF-7 cells (IC50 values of 7.06 ± 0.29 and 8.82 ± 0.37 μM, respectively), whereas compound 56b exhibited the greatest potency in VEGFR-2 inhibition (IC50 of 0.23 ± 0.20 μM). Molecular docking studies indicated BT 54b to have a stronger interaction. An ADME study showed BTs 54b and 56b to have enhanced lipophilicity and decreased solubility, which could influence their pharmacokinetic behavior.
2ATP was condensed with aromatic aldehydes in the presence of the prepared Co/Niacin-MOF as an economic, efficient, sustainable, and green stable catalyst under stirring at 60–70 °C for the appropriate length of time (30–60 min) to afford 2-heteroaryl-BTs
57a–
f in 75–95% yields (
Table 20) [
73]. All compounds were screened as acetylcholine esterase (AChE) inhibitors. Validation in molecular docking studies showed that BTs
57e and
57f gave good results in binding with acetylcholine esterase. The data from in vitro studies showed that compound
57e had a promising value (59.8% inhibition, IC
50 = 5.25 μg/mL) compared with the Alzheimer’s reference drug donepezil (74.89% inhibition, IC
50 = 4.19 mg/μL).
2.2. Condensation with Carboxylic Acids and Their Derivatives
In a simple trituration method, 2ATP was condensed with N-protected amino acids using molecular iodine as a mild Lewis acid catalyst to synthesize the 2-substituted BTs
58a–
f in 66–97% yields and
59a–
f in 54–98% yields (
Figure 9) [
114]. The reaction was carried out in solvent-free conditions for 20–25 min to provide the products with moderate to excellent yields (54–98%).
Graphene oxide (GO) was used as a catalyst in the condensation of 2ATF with α-phenyl glyoxylic acids, and water was used as a solvent; the reaction was conducted at room temperature under heating conditions for 1 h to synthesize the 2-aryl-BTs
60a–
d (
Table 21) [
115]. Their neuroprotective effects were assayed in the U87 MG cell line under a H
2O
2-induced stressed condition and compared with the breast cancer (MCF-7) and macrophage (RAW264.7) cell lines using a cell viability assay. These 2-aryl-BTs enhanced the neuronal cell viability and protected neuronal cells from the ROS-mediated neuronal damage induced by H
2O
2. Furthermore, these compounds modulated catalase and enhanced the catalase activity by up to 90% during the H
2O
2 exposure in the U87MG cell line. Molecular modeling studies in the AutoDockTool-1.5.6. Lig Plot + program showed these compounds to have strong binding energies of −7.39, −7.52, −6.5, and −7.1 Kcal/mol, as observed by using the potent analogs
60b and
60c and the catalase enzyme, which indicated the presence of hydrophobic interactions in the catalytic site. Furthermore, a simulation of the ligand and catalase protein performed using DESMOND software showed further strengthened ligand and enzyme interactions. An in silico ADMET study revealed the drug-likeliness of these analogs. The BTs
60b,
d had potential catalase-modulating activity comparable with valproic acid as a standard drug. These results indicated the use of an in vivo animal model for possible therapy.
Substituted 2ATPs and substituted α-keto acids were reacted by a decarboxylative cross-coupling reaction under blue LED (λ = 435–445 nm) irradiation without using any photocatalyst or metal at room temperature for 8 h to afford unsubstituted and substituted BTs,
61a–
p, in moderate to good yields (40–88%) (
Table 22) [
116]. The reaction was carried out without any photocatalyst or metal. An electron donor–acceptor complex (EDA) formed with α-keto acid, and 2ATP drove the formation of the corresponding BT. The BT
61n had the lowest yield (40%), the 5ClBT
61o had a yield of 61%, and the 2MeBT
61p had a yield of 53%; the 2-phBT
61a had the highest yield (88%) yield. The 2-(
oOHPh)BT
61e had a yield of 73%), the 2-(
pOMePh)BT
61j a yield of 74%, the 2-(
pMePhBT)
61h a yield of 58%, the 2-(
pClPh)BT
61m a yield of 77%, the 2-(
pFPh)BT
61c a yield of 73%, and the 5Cl,2PhBT
61b a yield of 83%).
The 2PhBT 61a had the higher yield (88%), whereas BT 61n had the lowest yield (40%). Substituents in the para position of the phenyl ring just as the compounds 61c,m with strong deactivating effect (F and Cl) with yields of 73 and 77%, and compounds 61h,j with strong activating effect (OH and OMe) with yields of 73 and 77%, respectively), reflected a decrease compared to unsubstituted 2PhBT 61a. Also, the yields of the BTs with activating groups had lower yields than those of BTs with deactivating groups. The 2-alkyl BTs 61n–p had the lowest yields (40, 61, and 53%, respectively). The 4-amino substituted α-keto acid (R3 = p-aminoPh) could not be transformed to the BT 61q in the present reaction condition, whereas from α-ketoglutaric acid, the BT 61s was obtained in traces.
A decarboxylative coupling of α-keto acids with 2ATP was carried out in a one-pot reaction using an aqueous suspension of K
2CO
3 as a base under grinding at room temperature for the appropriate time for the synthesis of the 2-aryl-BTs
62a–
h (
Table 23) [
117]. The protocol was carried out with excellent tolerance of functional groups in yields of 95, 97, 93, 95, 92, 98, 95, and 91%. The advantages of this method were short reaction times, a straightforward work-up, and catalyst-free and chromatography-free purification. On comparing BTs
62e (92%) and
62f (98%), it was clear that an electron-withdrawing group at the meta position of the phenyl ring disfavored the yield, whereas electron-donating groups at the para position favored the yield compared with the 2PhBT
62a (95%).
On comparing 3NO2PhBT 62e (92%) with 2PhBT 62a (95%), it was clear that an electron-withdrawing group at the meta position of the phenyl ring disfavored the yield, whereas an electron-donating in 3MePhBT 62g (95%) favored the yield. However, the best electron-donating groups at the para position were in 4MeOPhBT 62f (98%), the yield of which was more highly favored, surprisingly, and with electron-withdrawing groups at this position in 4FPhBT 62b (97%) the yield also increased, maybe due to the resonance effect of the fluor atom in this case.
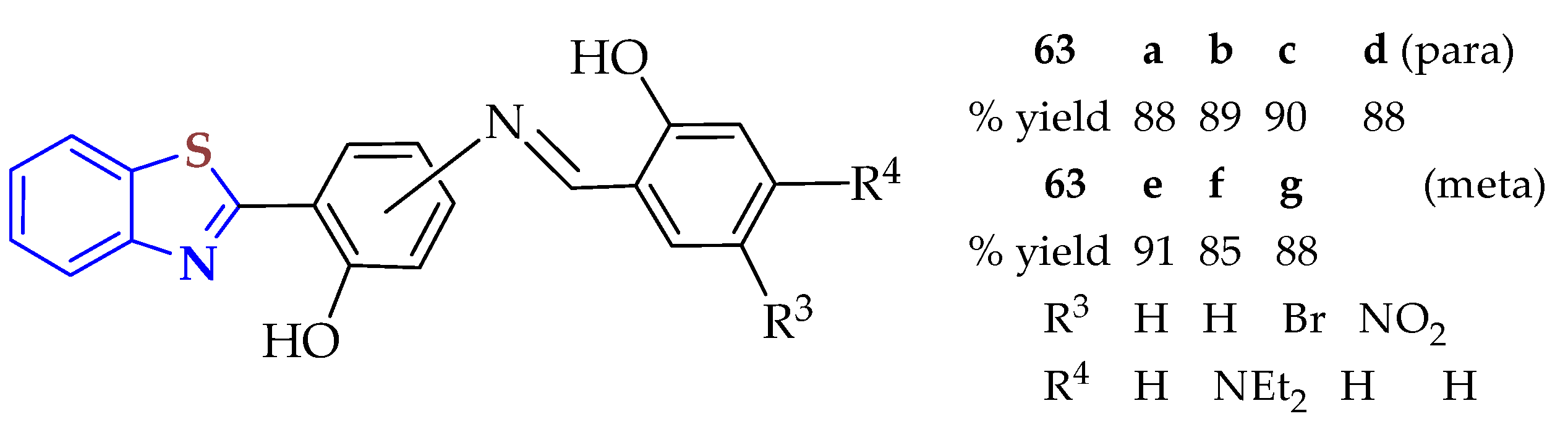
The Schiff base BTs
63a–
g were synthesized in two steps: (a) 4-minosalicylic acid and 5-aminosalicylic acid were reacted with 2ATP in polyphosphoric acid to produce the intermediates 2-(2-hydroxyaniline)BT and 2-(3-hydroxyaniline)BT. Finally, (b) these intermediates were treated with a series of trisubstituted aldehydes by stirring a few drops of acetic acid in ethanol at room temperature for 8 h to produce the
63a–
g BTs in good yields (85–91%) (
Figure 10) [
118]. These compounds were evaluated for in vitro antibacterial activities on
Bacillus sps,
S. aureus,
K. pneumoniae, and
E. Coli strains and antifungal activity on the
C. Albicans strain using the ‘micro-broth dilution method’ (MICs in μg/μL). All the compounds displayed very good antibacterial activity for the
S.
aureus,
E.
coli, and
K. pneumonia strains (MIC = 0.8 μg/mL), compared with ciprofloxacin (2.0 μg/mL), and antifungal activity for
C. albicans, compared with fluconazole (16 μg/mL), as standard drugs. Molecular docking studies were performed to understand the binding mechanism. Molecular electrostatic potential (MEP) was used to evaluate the reactivity of the molecules. Antimicrobial activity was correlated with the calculated HOMO–LUMO gap, chemical hardness, and global softness. BT
63e exhibited very good antimicrobial activity, less toxicity, and more chemical reactivity, as confirmed by a smaller HOMO–LUMO gap, a lesser electrophilicity index, a higher global softness, and a lesser chemical hardness. The evaluation of DNA cleavage of
63e against MCF-7 breast cancer cells revealed 85.82% inhibition of cancer cells at 200 μg/μL. In addition,
63e showed less toxicity to normal cells at the concentration required to produce an anticancer effect (IC
50 = 973 μg/μL).
2ATP was condensed with the corresponding 2-aryl-1
H-benzimidazole-5-carboxylic acid in the presence of polyphosphoric acid (PPA) via heating to 250 °C and 20 bar for 3 min to produce BTs
64a–
h in moderate yields (40–72%) (
Figure 11) [
119]. NIH3T3 (ATCC CRL-1658), Caco-2 (ATCC HTB-37), and A549 (ATCC CCL-185) cell lines were used to test the cytotoxic effects of the BTs. All the BTs exhibited low to moderate activity against the tested cancer cell lines. Compound
64e had the most potent activities against A549 and Caco-2 cells, with IC
50 values of 73.76 ± 2.54 μM and 73.15 ± 2.84 μM, respectively. This result suggested that the methyl group of the phenyl ring group may affect the antiproliferative activity to a certain extent. Also, BT
64e displayed antiproliferative activity similar to cisplatin against NIH3T3. On the other hand, BT
64c displayed significant potency against A549 and Caco-2 cells, with IC
50 values of 98.83 ± 3.71 μM and 77.80 ± 3.19 μM, respectively. In addition, BT
64e showed a similar selectivity to that of cisplatin.
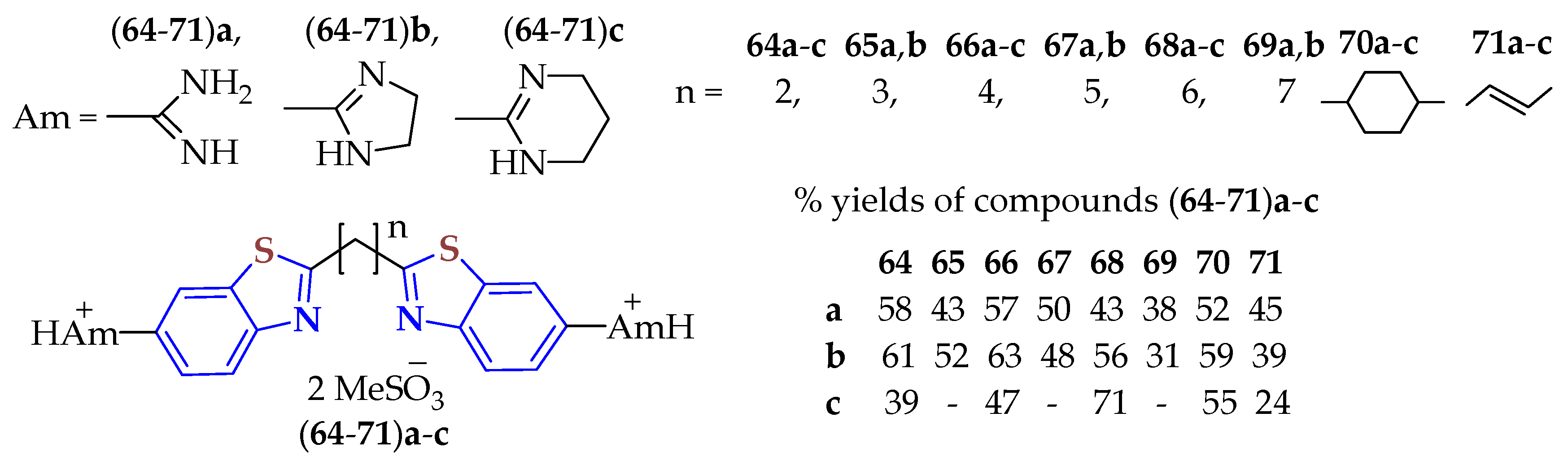
Aliphatic dicarboxylic acids were condensed with 5-amidino-2ATPs in polyphosphoric acid (PPA) at 180 °C to afford a series of symmetric bis-6-amidino-BTs
64–
71 with aliphatic central units, as previously designed, with 24–71% yields (
Figure 12) [
120]. The BTs were isolated as methanesulfonates by an additional acid–base reaction because these salts are more stable and soluble in water. All BTs were evaluated for their efficacy against bloodstream forms of the African trypanosome
Trypanosoma brucei. Most of the tested compounds were more potent than fexinidazole (2.40 μM), with EC
50 values against
T. brucei ranging from 0.51 to 5.39 μM. With the exception of BTs
68a–
c, trypanocidal activity decreased in the following order: unsubstituted amidine > pyrimidine > imidazoline. The aliphatic spacer between the two BTs also influenced the activity. The bis-BTs
70a–
c with a cyclohexyl unit had the most pronounced impact, and bis-BT
70a, which has an unsubstituted amidino moiety, displayed a remarkable potency (EC
50 = 0.51 nM). BTs
71a–
c, which have conformationally constrained ethenyl spacers, showed better activity than the corresponding conformationally unrestricted ethyl spacers
64a–
c. All BTs with conformationally unrestricted alkyl chains, with the exception of the most active BT,
68c (n = 6; EC
50 = 0.028 ± 0.001 μM), and BTs
64–
69, with three to eight different lengths (n = 4 (
66a–
c; 0.063, 1.14, 0.19 μM)), were generally optimal. On the other hand, BT
70a showed sub-nanomolar in vitro potency with good selectivity over mammalian cells (>26,000-fold). In all the experiments, mice treated with a dose of 20 mg kg
−1 were cured of stage 1 trypanosomiasis. Compound
70a displayed a favorable in vitro ADME profile, except for its low membrane permeability, while
70a was also active at low nanomolar concentrations against cultured asexual forms of the malaria parasite
Plasmodium falciparum. On these bases, compound
70a was considered a lead with therapeutic potential.
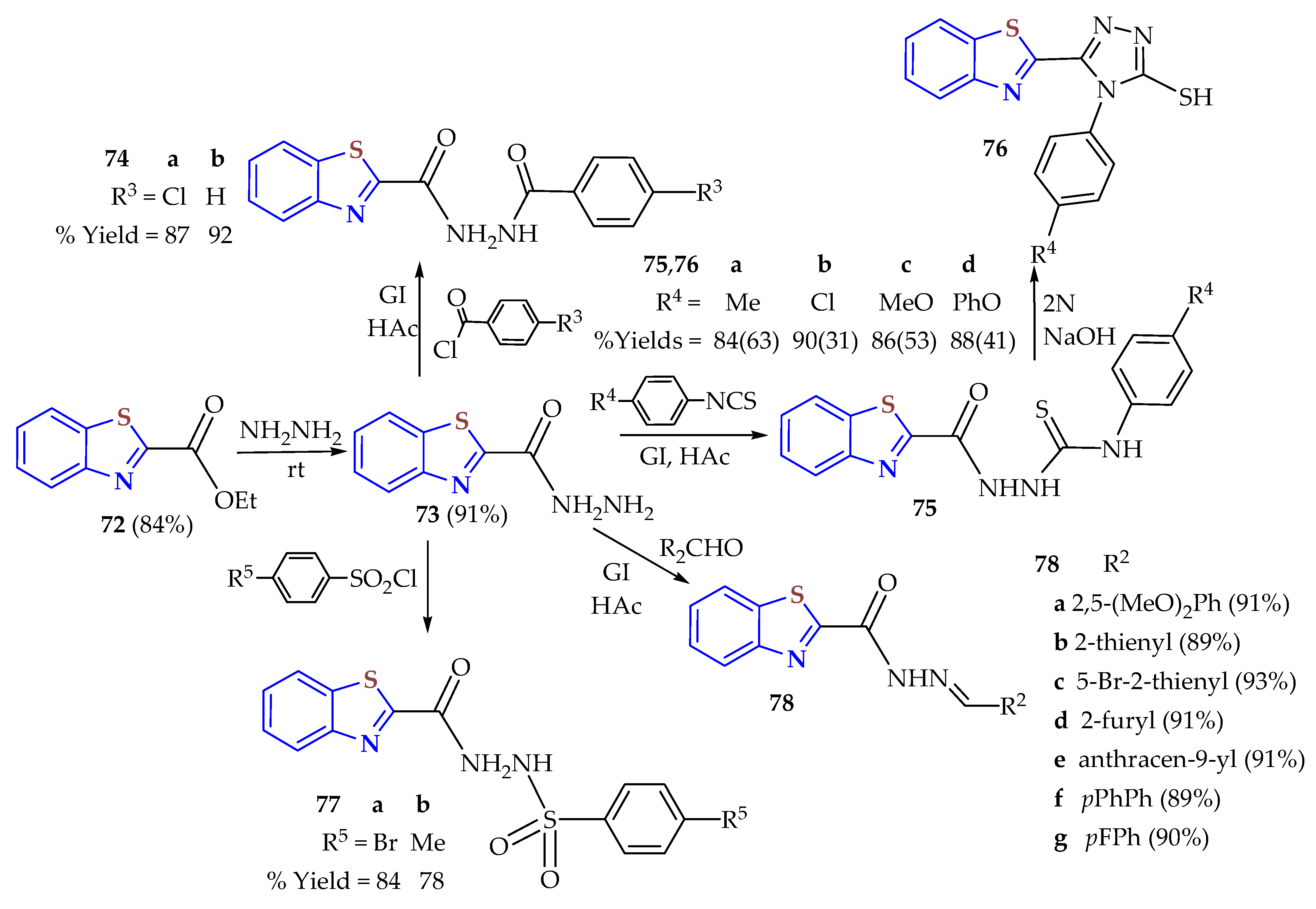
2ATP was condensed with ethyl oxalate to afford the ethyl 2-carboxylateBT
72, which suffered hydrazinolysis to be transformed to the acid hydrazide
73, the precursor used for the preparation of the hydrazone derivatives
72–
76 (
Scheme 8) [
121]. BTs
74a,
b were obtained in 87 and 92% yields, respectively, through the interaction of
73 with either 4-chloro or 4-f luorobenzoyl chloride, respectively, in glacial acetic acid. Also, compound
73 was reacted with
p-substituted isothiocyanates to afford the respective thiosemicarbazides
75a–
d in 84–90% yields. The thiosemicarbazides
75a–
d were refluxed in 2N NaOH to suffer cyclization into their 1,2,4-triazole-3-thiols
76a–
d in 31–63% yields. Also, compound
73 was reacted with 4-bromo benzenesulfonyl chloride or tosyl chloride in glacial acetic acid to afford the sulfonamides
77a,
b in 84 and 78% yields, respectively. Finally, acid hydrazide
73 was used as a precursor for the synthesis of the hydrazones
78a–
g in 88–93% yields through a reaction with aromatic and heteroaromatic aldehydes. The antitumor activities of the synthesized hydrazone BTs
73–
76 were evaluated. Also, they were tested in vitro against colorectal carcinoma (HCT-116), hepatocellular carcinoma (HepG2), prostate cancer (PC-3), mammary gland cancer (MCF-7), and epithelioid carcinoma (HeLa). The most active compounds were
78e,
78f,
75b,
76c, and
77b, exhibiting IC
50 values comparable to that of lapatinib, the reference drug. The most potent compounds for EGFR inhibitory activity were
78e and
78f, with IC
50 values of 24.58 and 30.42 nM, respectively, compared to the standard drug lapatinib (17.38 nM). Molecular modeling studies were conducted, including docking into the EGFR active site and surface mapping for all compounds. The hydrazones
78e and
78f showed superior binding with EGFR, showing them to be excellent candidates for targeted antitumor therapy through EGFR kinase inhibition.
The condensation reaction of 2ATP with acyl chlorides or acid anhydrides was performed in the presence of KF-Al
2O
3 as a heterogeneous base catalyst under stirring with acetonitrile at room temperature for 30 min to afford 2-substituted BTs
79a–
k in 87–97% yields (
Table 24). The catalyst showed good recyclability [
122]. It was observed that electron-withdrawing functionalities gave higher isolated yields (
79a,
b; 97 and 95%). On the other hand, electron-donating groups (
79d–
f; 94, 91, 90%) gave lower isolated yields.
2ATP was condensed with nine pyrrolidinone ester derivatives under microwave irradiation in the presence of a heterogeneous eutectic mixture of (CuCl
2 or NiCl
2/urea) and Cu- or Ni-doped TiO
2 nanoparticles to produce N-aryl pyrrolidinone-BTs
80a–
i in 83–96 and 78–91% yields, respectively (
Figure 13) [
123]. Eco-friendliness and short reaction times (5 min) were the advantages of this protocol. All synthesized BTs were screened for their antibacterial activity against Gram-positive and Gram-negative bacterial strains: compounds
80a and
80b were active against
E. coli and
P. aeruginosa, with an MIC of 12.5 µg/mL. Compounds
80c,
80e, and
80f showed important activity against
MRSA and
BS, with MIC values ranging from 12.5 µg/mL to 25 µg/mL. Also, compounds
80c and
80f were active against the four tested bacteria strains, with MIC values of 12.5 for
EC, 12.5 for
PA, 12.5 for
BS, and 18.75 μg/mL for
MRSA, compared with the antibiotic ciprofloxacin as a standard control.
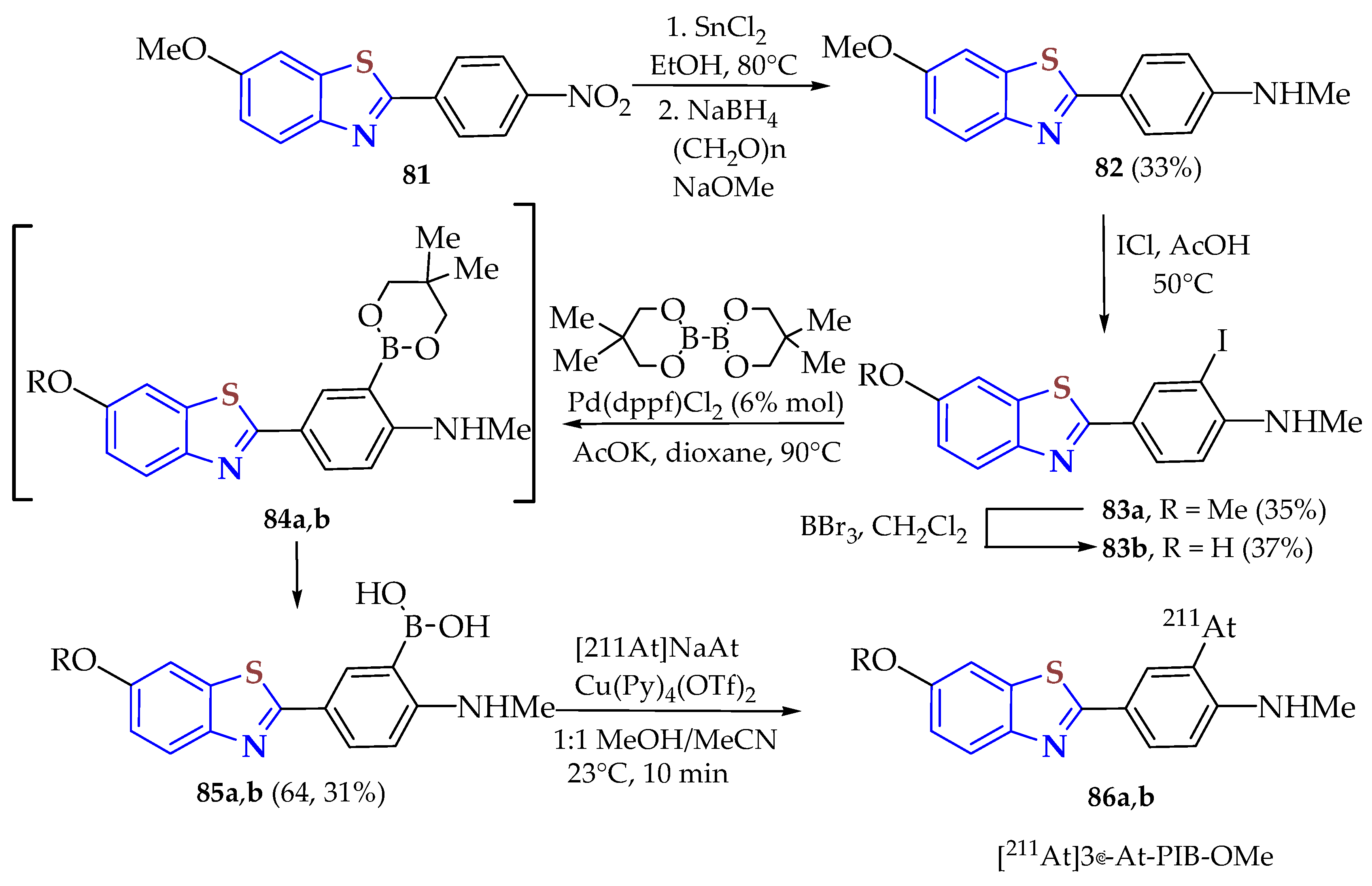
The
p-nitro benzoyl chloride was condensed with 5-methoxy-2ATP in toluene as a dissolvent upon heating for 1 h to afford the BT derivative
81, which was reacted in ethanol with stannous chloride upon heating to 80 °C for 3 h; then, paraformaldehyde dissolved in fresh NaOMe in MeOH solution was mixed and stirred at room temperature for 4 h. After that, sodium boron hydride was added, and the solution was refluxed for 1.5 h to afford BT
82 in a 33% yield. A solution of
82 and ICl in acetic acid was refluxed upon stirring overnight for 10 h to afford BT
83a in a 35% yield, which was cooled to −78 °C and reacted with BBr
3 upon stirring at room temperature for 16 h to afford
83b in a 37% yield. The neopentylboronic ester
84a could be quantitatively formed from
83a, but the isolation of
84a without hydrolysis was challenging. Partial and full hydrolysis of
84a during isolation efforts led to unsatisfactory mixtures of
84a alongside boronic acid
85a and protodeborylated compound
82. The synthesis of BT derivatives with [
211At]3′-At-PIB-OMe
86a,
b was carried out using a Cu-catalyzed astatination protocol via boronic acid precursors
85a,
b (
Scheme 9) [
74]. Compound
86a had a radiochemical yield of 55% and was stable for at least 3 h in phosphate-buffered saline. Studies were carried out to evaluate the binding affinities of
86a and
86b against Aβ(1–40) AD plaques and their in vivo stability, biodistribution, and AD-associated plaque clearance abilities.
A series of substituted 2ATPs were condensed with a variety of aryl/alkyl nitriles under mild reaction conditions (oil bath, 70 °C) using ZnO nanoparticles as a catalyst in solvent-free conditions to produce two series of 2-substituted BTs,
87a–
z and
88a–
d, in 88–96% yields (
Table 25) [
124]. This method was compatible with several functional groups and exhibited excellent catalyst recyclability, easy recovery of materials, and high economy; there is no need to purify the products by recrystallization or column chromatography, and it is an environmentally benign process.
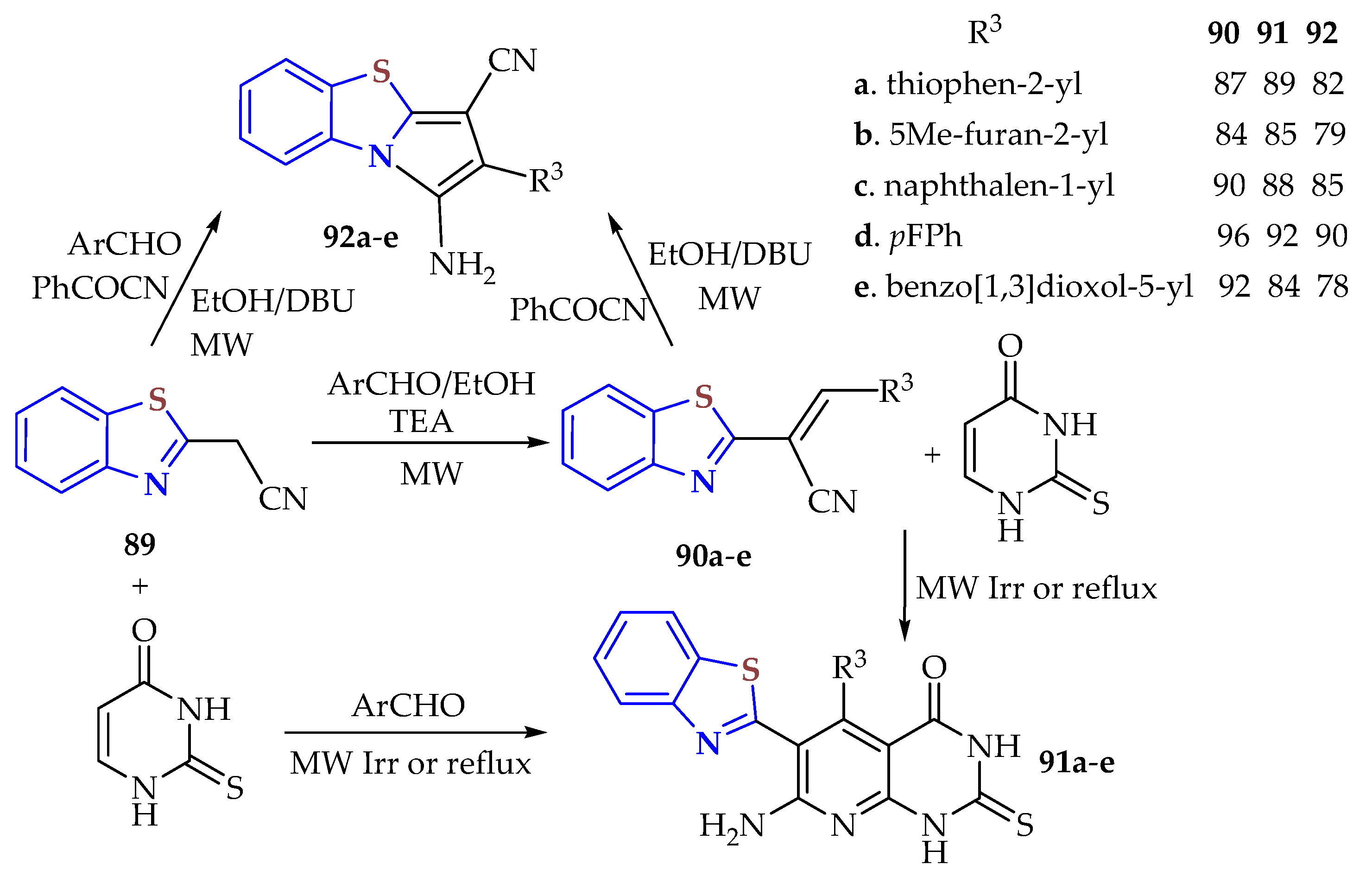
Five 2-[3-(aryl)prop-2-enenitrile]BTs
90a–
e were synthesized in 84–96% (4–8 min) yields by a microwave irradiation method from 2-cyano-methylBT
89 in EtOH/AcOH, obtained in a 92% yield from the condensation of 2ATP and malononitrile under microwave irradiation at 40 °C for 10 min and used as an intermediate in the synthesis of 7-amino-6-(BT-2-yl)-5-(aryl)-2-thioxo-2,3dihydropyrido[2,3-d]pyrimidin-4(1H)-ones
91a–
e obtained in 84–92% yields (25 min) and 1-amino-2-(aryl)pyrrolo[2,1-b][
1,
3]BT-3-carbonitrile derivatives
92a–
e obtained in 78–90% yields (20–25 min) (
Scheme 10) [
125]. These compounds were tested as antioxidant, antimicrobial, and anticancer agents.
It was found that pyrimidine-BTs 91a,d were more potent against bacteria, with MICs from 4 to 12 μmol L–1, than cefotaxime (MIC = 6–12 μmolL–1); pyrimidine-BTs 91bc,e showed good activity against all the bacterial strains. This high efficacy was attributed to the presence of a BT and thiophene moieties, as in pyrimidine-BT 91a, and to the presence of an electron-withdrawing (fluoro) group in the para position of the phenyl ring attached to the pyridopyrimidine, as in pyrimidine-BT 91d. Pyrimidine-BT 91a had equipotent activity with MICs of 6 and 12 μmol L−1 similar to those of cefotoxime against Bacillus subtilis and Chlamydia pneumonia, respectively, whereas 91e was equipotent (MIC = 8 μmol L−1) with respect to cefatoxime against Salmonella typhi. Pyrrolo-BTs 92a–e effectively inhibited the growth of the tested bacteria strains. PyrroloBT 92a was more potent against Staphylococcus aureus and had equipotent efficacy against Bacillus subtilis and Chlamydia pneumonia compared with cefotaxime; this was attributed to the presence of a thiophene moiety. In addition, pyrrolo-BT 92d (MICs from 4 to 10 μmol L–1) had extraordinary activity toward all bacteria due to the presence of a p-fluorophenyl substituent which increased the antibacterial potency compared to the reference drug. Also, pyrrolo-BTs 92b,c,e exhibited good inhibition activity against all bacterial strains. The cytotoxic activity of pyrimidine-BTs 91a–d and pyrrolo-BTs 92a–d against the colon cancer HCT-116, lung cancer NCI-H460, and liver cancer HepG2 human cell lines was observed and compared with that of doxorubicin as a reference drug. The series of pyrimidine-BTs 91a–e exhibited higher cytotoxic activity than the pyrrolo-BTs 92a–e. Pyrrolo-BT 91a containing thiophene (IC50s = 0.66, 0.45, and 0.70 μmol L−1), pyrrolo-BT 91d containing p-fluorophenyl (IC50s = 0.28, 0.39, and 0.14 μmol L−1), pyrrolo-BT 92a with thiophenyl (IC50s = 1.10, 0.49 and, 0.89 μmol L−1), and pyrrolo-BT 92d containing p-fluorophenyl (IC50s = 0.45, 0.47, and 0.59 μmol L−1) were found to be more potent and efficacious than doxorubicin as the reference drug (IC50s = 1.98, 0.52, and 1.12 μmol L−1). It was found that the presence of the p-fluorophenyl group improved the antitumor activity more than thiophene. In addition, pyrrolo-BT 91b (IC50s = 2.56, 2.89, and 2.68), and pyrrolo-BT 91e (2.60, 3.00, and 3.08 μmol L−1) exhibited good cytotoxic effects towards the three tumor cell lines. On the other hand, 5-methylfurany 92b (IC50s = 3.90, 4.08, and 4.50 μmol L−1) showed good antitumor activity against all tumor cell lines. Compounds with 5-naphthalenyl side chains in both pyrimidine-BT 91c and pyrrolo-BT 92c showed moderate antitumor activity. From the above structure–activity relationships (SARs), the most selective pyrimidine BT and pyrrolo-BT on the studied cancer cell lines bore p-fluorophenyl and thiophene moieties. Thus, pyrimidine-BTs 91a,d and pyrrolo-BTs 92a,d were considered promising candidates as pharmacophores against cancer cell lines. A series of pyrimidine-BTs 91a–e and pyrrolo-BTs 91a–e were tested for antioxidant activity, as reflected in the ability to inhibit lipid peroxidation in rat brain and kidney homogenates using (3 ethylbenzthiazoline-6-sulfonicacid) an ABTS free radical scavenging assay. Pyrimidine-BTs 91a,d (91.2 and 92.8% yields, respectively) and pyrrolo-BT 92d (90.4%) showed inhibition levels higher than that of Trolox (89.5%), while pyrrolo-BT 92a (88.7%) showed nearly equipotent inhibition activity.
Scheme 10.
Condensation of 2ATP with malononitrile to afford 2-[3-(aryl)prop-2-enenitrile]BTs 90a–e as intermediates in synthesizing BT derivatives 91,92a–e by the MWI method in % yields.
Scheme 10.
Condensation of 2ATP with malononitrile to afford 2-[3-(aryl)prop-2-enenitrile]BTs 90a–e as intermediates in synthesizing BT derivatives 91,92a–e by the MWI method in % yields.
The nano-catalyst aminopropyl-1,3,5-triazine-2,4-diphosphonium tetrachloroferrate immobilized on halloysite nanotubes [(APTDP)(FeCl
4)
2@HNTs] was prepared to be used as a highly effective agent in the condensation reaction of 2ATP with benzonitriles containing various electron-withdrawing and -donating substituents for the synthesis of several BTs,
93a–
o, in excellent yields (90–96%). The reaction was performed in the absence of a catalyst under solvent-free conditions at 80 °C within 4–5 h (
Table 26) [
126]. Additionally, the use of dinitriles such as 1,3-dicyanobenzene and 1,3-phenylenediacetonitrile, using a 1:1 molar ratio, gave the corresponding mono-benzonitrile-BT
93t and mono-methylphenylacetonitrile-BT
93u in 90 and 85% yields, respectively, using a 1:1 molar ratio. Also, bis- and tris-BTs
93v–
x were smoothly synthesized from dinitrile and trinitrile in those conditions in 78 and 75% yields. The advantages of this protocol were its easy work-up, high purity, high catalytic activity, reusability, and the easy recovery of the catalyst. On the other hand, the competitive condensation of 2ATP with an equimolar mixture of an aromatic nitrile (4-methylbenzonitrile) and an aliphatic nitrile (heptanenitrile) was examined under the same conditions. The results showed that 4-methylbenzonitrile was transformed to the corresponding BT
93j (90%) in the presence of heptanenitrile. This outcome indicated the selectivity performance of the catalyst.
2.4. Miscellaneous Condensations
2ATP and both electron-rich and electron-poor substituted styrenes were condensed in a one-pot methodology for the synthesis of the pharmacologically active 2-arylBTs
95a–
f in 30–66% yields (
Table 27). The reaction was carried out with Pd(OAc)
2 in AcOH as a solvent and heating at 90 °C for 12 h in the presence of O
2. The process involved sequential C-C/C-N bond cleavage followed by C-N/C-S bond formation [
128]. To further the scope of the reaction, 2ATP was reacted with styrene under standard conditions. However, no desired product was obtained, and the starting materials were isolated.
2ATP disulfide was reacted with terminal alkynes under aerobic oxidative C(sp)–S coupling in a visible-light-driven copper-catalyzed reaction (
Table 28) [
129]. The photochemical reaction was achieved using a wide range of thiol dimers and alkynes with good chemoselectivity and excellent conversion. The utility of alkynyl sulfide formations as isolated intermediates (35–92%) was demonstrated in the construction of the 2-phenylBTs
96a–
n in 55–79
% yields via “thia-Wolff rearrangement” in the presence of AgNO
3, using visible light with 9-mesityl-10-methylacridinium ions (Acr
+–Mes) as a photoredox catalyst system. BTs with electron-donating groups on the benzene ring increased the yield, while BTs
96b–
d and BTs with electron-withdrawing groups at the ortho (
96f), meta (
96k), or para (
96e,
g,
h) positions decreased the yields compared with the 2PhBT
96a. The BT with a pyridinyl ring instead of a benzene ring in
96l (59%) decreased the yield. Curiously, napthalenyl and phenanthrenyl groups, instead of the phenyl ring, resulted in high yields of
96m,
n. This reaction failed with aliphatic terminal alkynes.
In a metal-free and environmentally friendly approach, 2ATP was condensed with methyl arenes under visible-light irradiation using eosin Y as a photocatalyst, ethanol–water (1:2) as a green solvent at room temperature, and atmospheric air as an oxidant to synthesize the biologically important BTs
97a–
q (
Table 29) [
130]. This methodology demonstrated a broad substrate scope, yielding the desired products in good to excellent yields (83–92%). The advantages of this procedure included its environmental friendliness, low cost, non-toxicity, green solvent, ease of handling, and use of visible light as a renewable energy source. All the BTs were obtained in good yields when the substituent was in the meta and ortho positions (
97b–
h,
j; 85–92%). When the methyl arene had F, Cl, and Br (
97b–
d; 91, 90, and 90%, respectively) and OMe (
97j, 86%) in the
p-position, the yield obtained with electron-withdrawing groups was higher than that obtained with electron-donating groups. Additionally, methyl arene with multiple substituents yielded 85% (
97i). The use of heteroaromatic methyl arenes (
97n–
q,
v,
w) afforded BTs with good yields (83–84%).
2ATP was condensed by an oxidative coupling with substituted aryl methyl amines in the presence of a Ba-doped CoMoO
4 system as a catalyst to synthesize several 2-aryl BTs
98a–
k under visible-light irradiation (
Table 30) [
131]. The reaction was carried out under atmospheric air and had good to excellent yields (78–98%). The advantages of this protocol were its atom economy, functional group tolerance, a wide range of substrate scopes, and suitability for scale-up reusability. Aryl methyl amines with an OMe as an electron-donating group at the para position of the benzene ring favored the yield, as in
98b, but an
iPr group (
98d) decreased the yield due to steric effects. On the other hand, electron-withdrawing groups, such as Cl or F (
98h and
981), decreased the yield, compared with 2PhBT
98a. The aryl methyl amines with electron-donating or electron-withdrawing substituents at the para position, BTs
98b,
d,
h,
i, were obtained with good to excellent yields. Electron-donating or electron-withdrawing substituents at the ortho position of the benzene ring decreased the yields (
98e,
j), but electron-withdrawing substituents decreased the yields more. The OPh group at the meta opposition of the phenyl ring produced only a little decrease (
98f).
The condensation of 2ATP with aromatic primary alcohols via the acceptorless dehydrogenative coupling (ADC) method was carried out in the presence of Pd(II)N^N^S as a pincer-type catalyst for the synthesis of the pharmaceutically important BT derivatives
98a–
u (
Table 31) [
132]. Several primary aromatic alcohols, such as electron-releasing and -withdrawing alcohols, sterically hindered alcohols, heterocyclic alcohols, polycyclic alcohols, and aliphatic alcohols, were coupled with 2ATP.
p-substituted benzyl alcohols, including electron-donating 4-methyl and 4-isopropyl benzyl alcohol, were coupled with 2ATP to afford the corresponding BTs
99a,
b in 90 and 86% yields, respectively. Interestingly, alcohols containing electron-poor groups, such as 3-fluro, 4-choloro, and 4-nitro benzyl alcohols, were well-tolerated to obtain BTs
99g–
i in 69, 72, and 53% yields, respectively. Benzylic alcohols with sterically hindered 2-substituted alcohols, such as 2-Br, 2,5-Cl
2, 2-amino, and 2-NO
2-benzyl alcohols, gave BTs
99j–
m with 58, 63, 71, and 47% yields, respectively. Next, benzyl alcohols containing electron-rich groups like 2,3-dimethoxy and 2,5-substituted groups were reacted to afford BTs
99n,
o in 74 and 71% yields, respectively. Reactions with heteroaromatic benzyl alcohols such as 2-pyridine methanol, piperonyl alcohol, and thiophene-2-methanol proceeded smoothly to furnish BTs
99p–
r in 69, 73, and 72% yields, respectively. Interestingly, the reaction of cinnamyl alcohol and polycyclic alcohols, such as 4-biphenyl benzyl acohol, 1-naphthalene methanol, and pyrene 2-methanol, afforded the corresponding BTs
99s–
v in 66, 79, 75, and 56% yields, respectively. The use of aliphatic alcohols such as propan-1-ol, 2-methylpropan-1-ol, and hexan-1-ol gave the BTs
99w–
y in 52, 53, and 48% yields, respectively. To understand the electronic effect, an intermolecular competitive experiment involving 4-methyl benzyl alcohol and 4-chloro benzyl alcohol was carried out under the same reaction conditions. It was found that the electron-rich benzyl alcohol is more reactive to give BT
99b with a 53% yield than the electron-deficient benzyl alcohol to give BT
99h with a 32% yield.
The BTs were synthesized via C−S and C−N bond formations, yielding up to 93%. This method was sustainable, with excellent yields, 1 mol% catalyst loading, inexpensive primary alcohol, and only water and hydrogen gas as by-products. A mechanism was proposed via in situ aldehyde formation and dehydrogenation of primary alcohols. The utility of this catalytic procedure was illustrated by the large-scale synthesis of the 2-(4-methoxyphenyl)benzo[d]thiazole 99a.
The treatment of the respective substituted (2-isocyano-phenyl)(alkyl)sulfanes, derived from 2ATP, with a series of diarylphosphine oxides under irradiation with 23W white LED light for 13 h at room temperature in an air atmosphere, using the photocatalyst Rose Bengal and DMF as a solvent, gave the 2-phosphorylBTs
100a–
l in 65–89% yields (
Figure 15) [
133]. On the other hand, electron-poor and electron-rich substituents on the benzene ring in the 2-isocyanoaryl thioethers produced the substituted 2-phosphorylBTs
100m–
t in 64–88% yields.
Several H-phosphorus oxides were well-tolerated, regardless of whether the ring of diarylphosphine oxides was substituted with either electron-deficient, electron-rich, or steric-hindered groups; the reactions proceeded to afford BTs 100a–i in medium to good yields (65–87%). Additionally, dimethyl-substituted H-phosphorus oxide was used to give BT 100j in a moderate yield (58%). The same procedure was used with piperonyl and naphthyl phosphine oxide derivatives, affording the BTs 100k,l (79 and 81% yields, respectively). On the other hand, functional groups with electron-rich (OMe and Me) and electron-poor (F, Cl, Br, and –SO2Me) substituents on the BT ring were also tolerated to afford BTs 100m–t in good yields (64–88%).
A metal-free radical cascade cyclization of substituted 2-isocyanophenyl-methyl-sulfanes with 5.0 mL of the corresponding primary or secondary alcohol in the presence of the radical initiator di-
tert-butyl-peroxide (DTBP) was refluxed for 6 h at 120 °C (
Figure 16) [
134]. The desired 2-hydroxyalkylBTs
101a–
i and
102a–
v were obtained in yields ranging from 34 to 92%. The advantages of this reaction were its mild reaction conditions, its being a metal-free reaction, the good functional group tolerance, and the broad substrate scope.
This cascade cyclization reaction of 2-isocyanoaryl thioethers proceeded smoothly with alcohols such as isopropanol, ethanol, and 2-butyl alcohol to give the desired BTs 101a,c,f in good yields (68.92%). Other alcohols such as methanol, n-propanol, n-butyl alcohol, cyclopentanol, and cyclohexanol afforded BTs 101b,d,e,g,h with moderate yields (51–68%). Additionally, the benzyl alcohol gave BT 101i in a 34% yield. On the other hand, several 5-substituted 2-isocyanoaryl thioethers reacted well with isopropanol to yield BTs 102a–g in 57–91% yields. It was found that substituents at the C5 position of 2-isocyanoaryl thioethers had little effect on the reaction. Other substituents such as phenyl, 4-chlorophenyl, 4-methylphenyl, naphthalen-2-yl, and phenanthren-9-yl also proceeded smoothly to synthesize BTs 102h–j and 102r,s in good yields (53–86% yields). Substituents at the C5 position were also well-tolerated to afford BTs 102k–m in 76–82% yields. C4 substituents were also tolerated to afford BT 102o in a 73% yield. Moreover, it was found that heteroaryl substituents also gave BTs 102t–v in 64–84% yields. 3-chloro, 4,6-dichloro, and 5,6-dimethyl substituents also reacted with isopropanol to give BTs 102p,q in 68 and 84% yields, respectively.
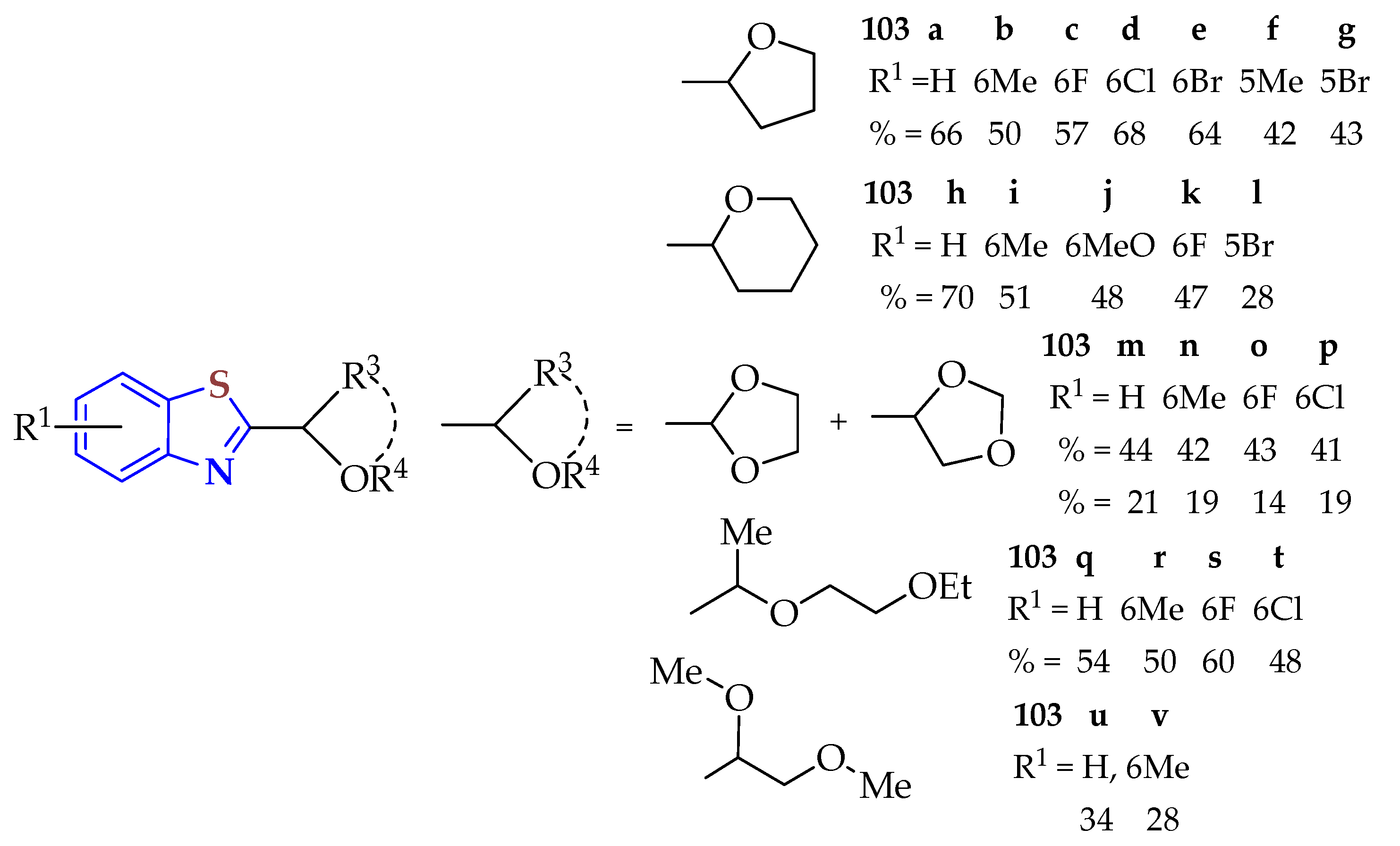
2-Isocyanoaryl thioethers derived from 2ATPs were cyclized with ethers in an oxidant-free and metal-free visible-light-induced reaction to access the ether-functionalized BTs
103a–
v (
Figure 17) [
135]. The cyclization reaction was carried out with 1,2,3,5-tetrakis-(carbazol-9-yl)-4,6-dicyanobenzene (4CzIPN) as a photocatalyst and the irradiation of blue LED light in a nitrogen atmosphere in mild reaction conditions, and the procedure was characterized by operational simplicity. This strategy provides an efficient approach to access various ether-containing BTs in acceptable to good yields.
Both electron-rich groups (−Me and −OMe) and electron-poor groups (−F, −Cl, and −Br) were well-tolerated in the reactions and smoothly afforded BTs 103a–k in moderate to good yields (42–70 %). Notably, the Br substituent at both the 4- and 5-positions of the phenyl rings gave BTs 103e,g,l in relatively low yields (64, 43, and 47% yields, respectively). Moreover, this methodology was extended to 1,3-dioxolane and several substituted isocyanides to afford BTs 103m/m′-p/p′ in 65 (2:1), 61 (2:1), 57 (3:1), and 60% (2:1) yields, respectively. It was observed that the BTs showed a preference for C2 selectivity over the C4 position. The synthesized compounds combine BT with an ether functional group representing key structural motifs of various biologically active molecules of interest in medicinal chemistry.
A photo-induced cascade sulfone alkylation/cyclization of 2-isocyanoaryl thioethers and α-iodosulfones was carried out to afford alkyl/benzyl-sulphonyl BTs in up to 97% yields
104a–
zf (
Figure 18) [
136]. This visible-light-triggered reaction not only occurs under extremely mild reaction conditions but also does not require the presence of a photosensitizer. The photocatalytic process is triggered by the photochemical activity of in situ-generated electron donor–acceptor complexes arising from the association of 2-isocyanoaryl thioethers and α-iodosulfones. The radical pathway was confirmed by UV–vis spectroscopy, radical trapping, a Job plot, and on/off irradiation experiments.
A visible-light-induced radical addition/cyclization cascade reaction of 2-isocyanoaryl thioethers was achieved under metal-free and mild conditions (
Table 32) [
137]. This method utilizes hydrazines as radical precursors and efficiently accesses a series of 2-aryl and 2-alkyl BTs,
105a–
r and
106a–
k, in good yields (71–83% and 70–81%).
In the reaction of 2-isocyanoaryl thioethers, the groups on the benzene ring, electron-donating (Me and MeO) and electron-withdrawing groups (halogens, CF3, CN, and NO2), were compatible with the process to afford BTs 105b–k in good yields (71–83% yields). No influence of the position of the substituents was observed on the yields. In addition, the disubstituted substrate reacted well to afford BT 105l in a 79% yield. Instead of aryl rings, β-naphthyl-, 4-pyridyl-, and 2-furanyl-substituted hydrazines were used in this method to afford BTs 105m–o in 74–80% yields. It is worth mentioning that benzylhydrazine, isopropylhydrazine, and cyclopropylhydrazine could furnish BTs 105p–r in good yields (70–75%) when a series of substituted (2-isocyanophenyl)(methyl)sulfanes were used. In general, substrates bearing electron-rich (Me, Et, and MeO) or electron-deficient groups (halogens, CF3, and CN) were tolerable and afforded BTs 106a–j in good yields (71–82% yields). When a disubstituted substrate was used, BT 106k was formed in a 73% yield.
The 2-guanidinoBT
107, obtained from condensation of 2ATP with cyanoguanidine, was used for the development of a range of synthetic methods with various reagents, such as α,β-unsaturated carbonyl, 2-cyano-three-(dimethylamino)-
N-acrylamide, β-diketones, β-keto esters, and (
S,
S)-ketene dithioacetals, to afford pyrimidine-based BTs
108–
115 in 66–72% yields as novel anticancer agents (
Scheme 11) [
138].
The docking analysis revealed that the most active BTs, 7b, 7c, 13b, 13c, 15c, 17d, 18, and 24, fit inside the active site within the protein tyrosine kinase (PTK). It was observed that these compounds formed a hydrogen donor bond with Met318, except for 15c. Among these compounds, BTs 15c, 17d, 18, and 24 resulted in binding energies closer to that of the cocrystallized ligand 1N1, with values of −7.729, −7.321, −7.681, and −7.5790 kcal mol−1, respectively. Compound 13c had a binding energy of −6.963 kcal mol−1, but it had four more interactions with the active site, including one arene–H interaction with Leu248 and three H-bond donors with Met318 and Thr319. Furthermore, the in silico studies and ADMET properties of the most potent BTs suggested them as promising candidates for further development, with favorable bioavailability and pharmacokinetic profiles. In vitro cytotoxicity studies were carried out against HepG2, HCT116, and MCF7 human tumor cell lines at a concentration 100 μmol mL−1 using the standard 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) bioassay. BTs 108c, 109b, 111d, and 112 showed strong efficacy against the HepG2 cell line, exhibiting cell viability percentages of 61.29, 68.18, 61.04, and 66.85, respectively, compared with the standard drug (64.41%). BTs 108b, 109a, 110c, and 111b exhibited moderate activities against the same cell line, with cell viabilities of 72.70, 72.35, 72.69, and 76.02%. In addition, BTs 108b, 109c, and 110c showed slightly strong efficacy against the HCT116 cell line, with viabilities of 70.62, 67.11, and 65.68%, respectively, in contrast to the standard drug with a cell viability percentage of 55.96. Furthermore, the singular BT 24 exhibited efficacy against the MCF7 cell line, with a cell viability of 69.98%, compared with the standard drug (62.76%). From the IC50 results, it was found that the pyrimidine-based BT 111d (IC50 = 0.41 ± 0.01 μmol mL−1) was the most potent of the tested BTs against HepG2. Also, BT 112 (IC50 = 0.53 ± 0.05 μmol mL−1) was the second most potent compound against HepG2, followed by compound 109b (IC50 of 0.56 ± 0.03 μmol mL−1). Notably, BTs 111d, 112, and 109b showed higher potency, based on their IC50 data, compared with 5-fluorouracil (IC50 of 1.03 μmol mL−1) as the reference drug. Surprisingly, BT 110c (IC50 of 0.02 ± 0.001 μmol mL−1) showed higher efficacy against HCT116 compared with 5-fluorouracil (IC50 of 9 ± 1.7 μmol mL−1). BT 110c not only showed heightened potency related to 5-fluorouracil but also showed notable efficacy together with BTs 108b and 109c (IC50 values of 2.95 ± 0.26 and 1.033 ± 0.06, respectively). In addition, BT 115 showed an IC50 value of 1.485 ± 0.15 μmol mL−1, lower than that of the 5-fluorouracil (IC50 of 7.12 μmol mL−1), against MCF7. These results suggest that the investigated BTs exhibit potential as anticancer agents. The findings revealed that the inclusion of halogen groups, such as F and Cl, on the aryl group bonded with pyrimidine-based BTs 108a–d resulted in an increase in their activity. In addition, the incorporation of CO2Et, as in 110c, enhanced the potency in comparison to its analogs, 110a,b, containing COCH3 and COPh, respectively. In the context of pyrimidine-based BTs 111a–d, the presence of a OMe group amplifies the compound’s potency more significantly than those possessing halogen substituents. Notably, BTs containing SCH3 exhibited the lowest activity levels across the three tested cell lines.

 ,
, 

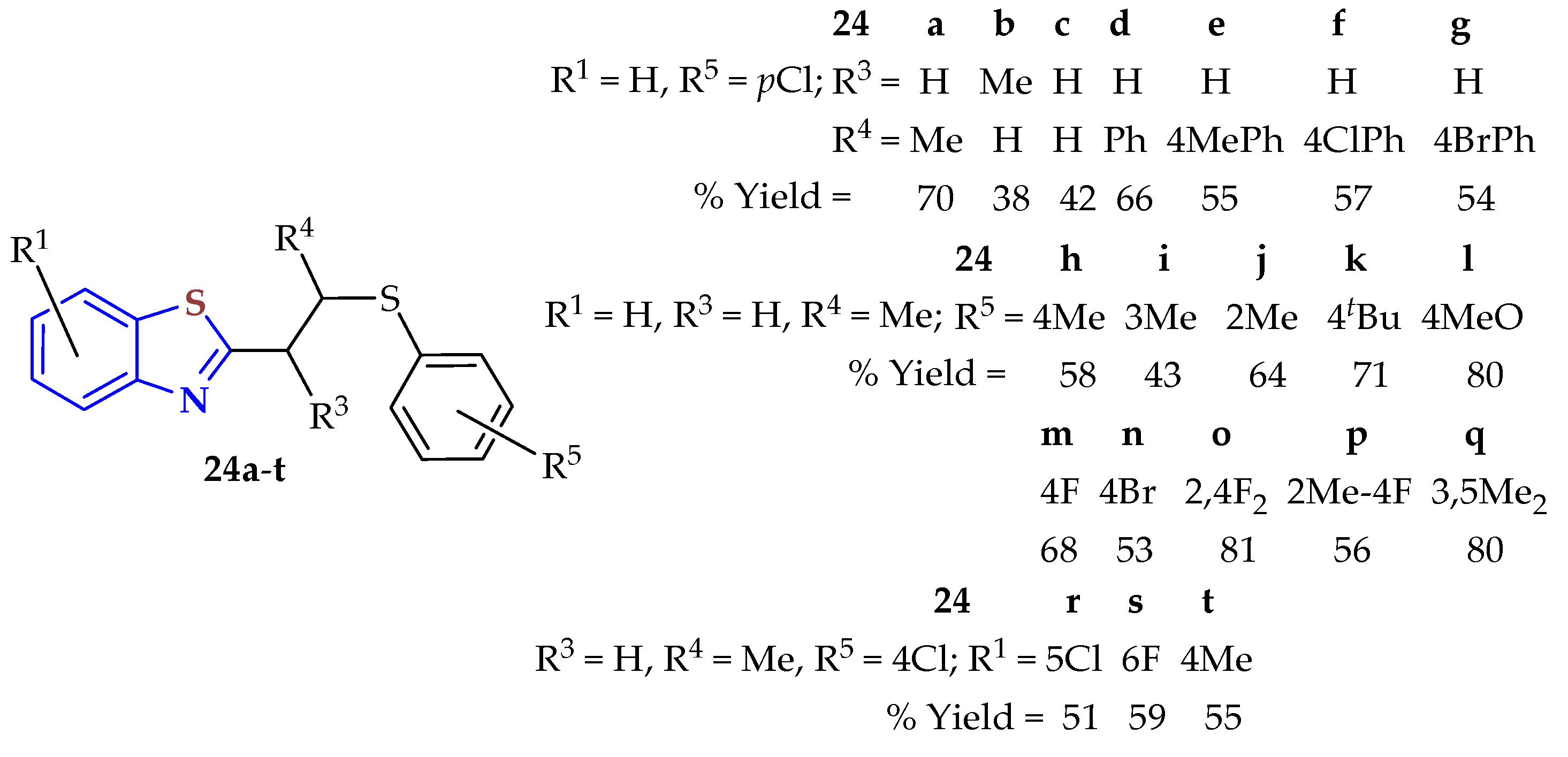
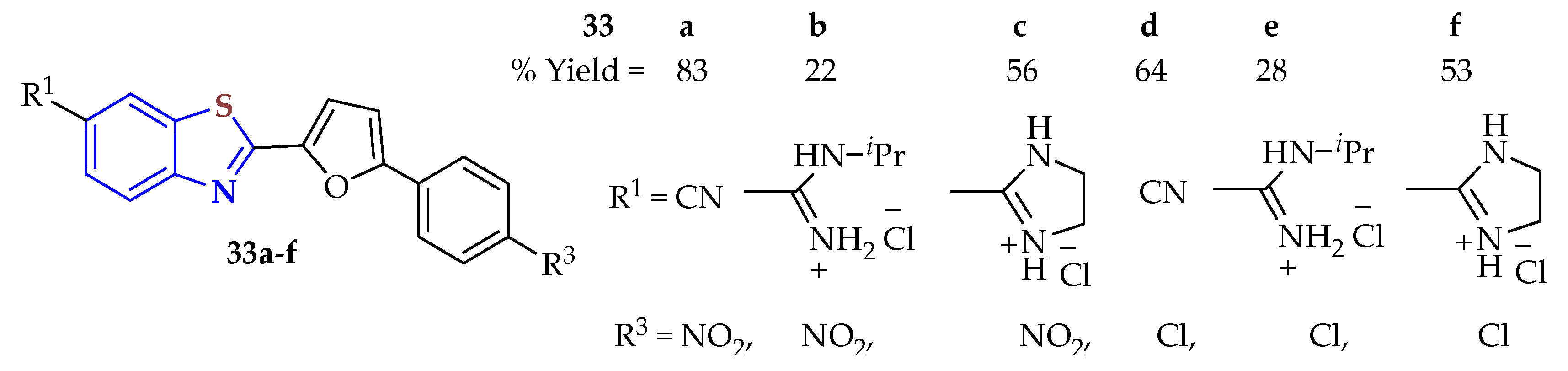

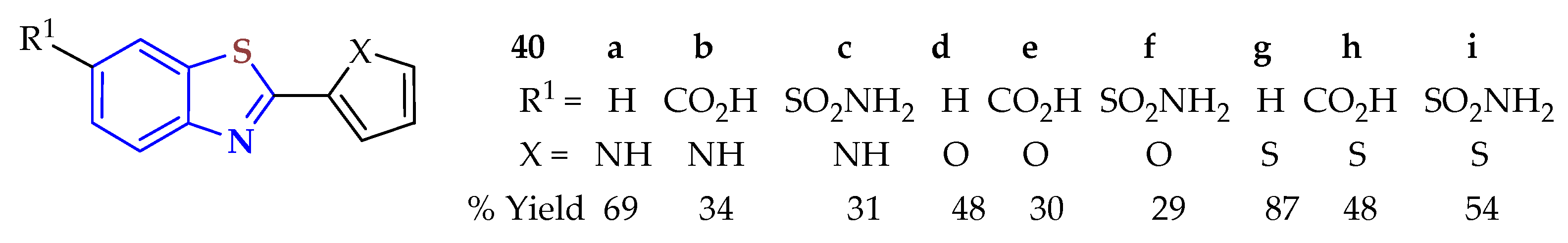

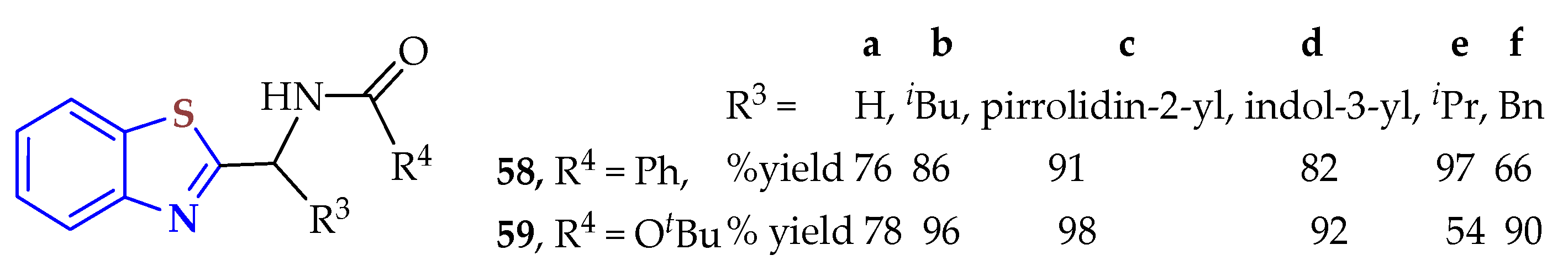
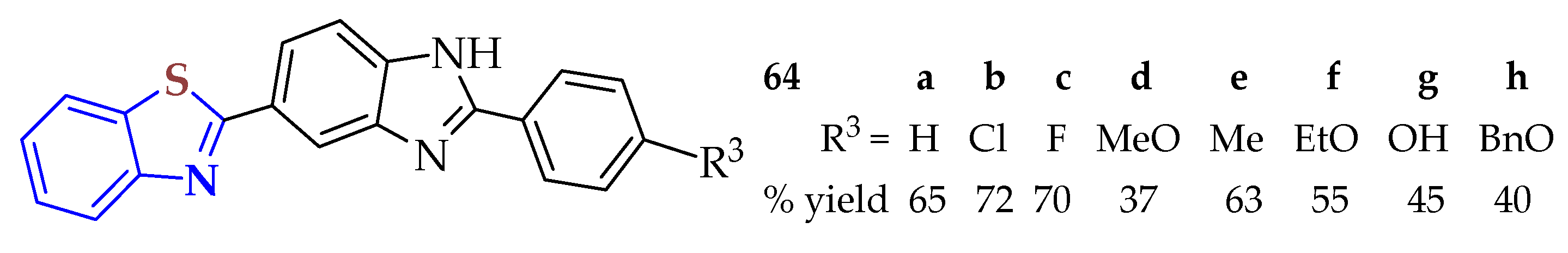
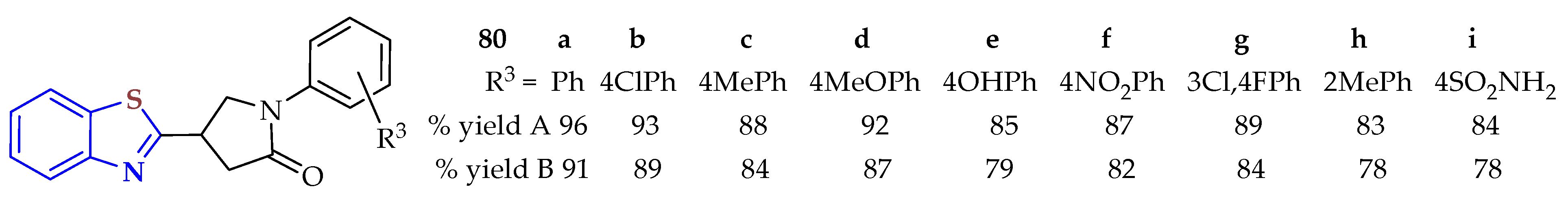

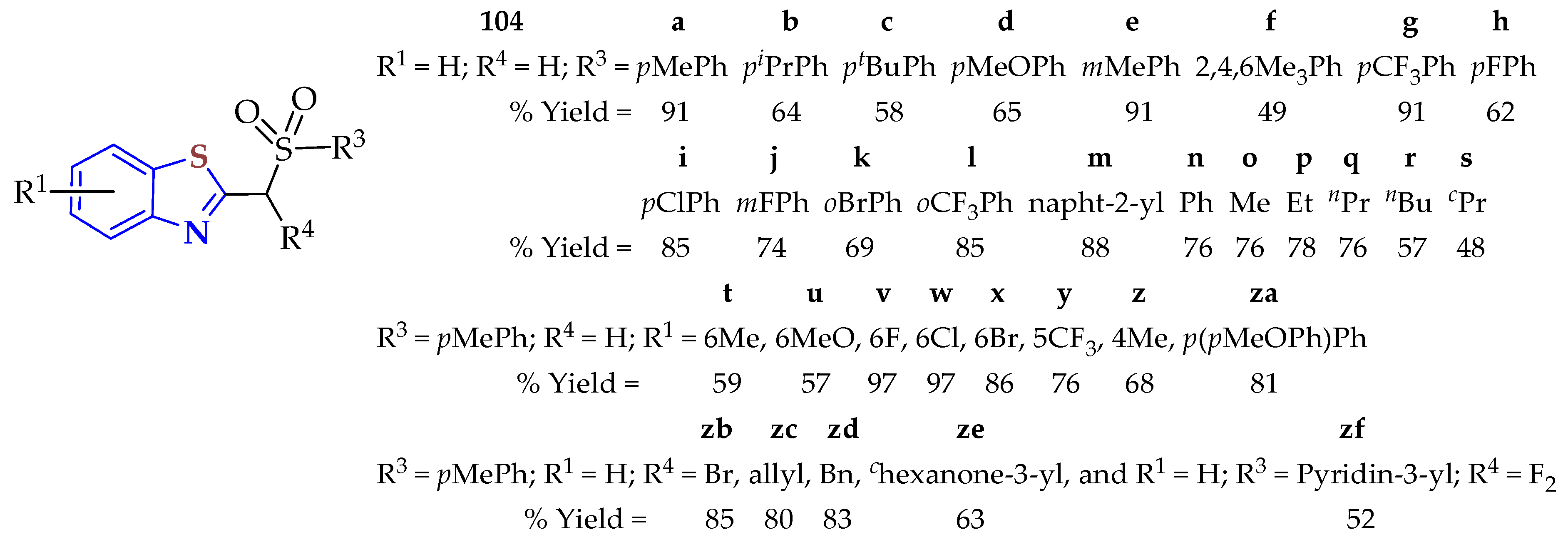


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}