PARP Inhibitor Olaparib Causes No Potentiation of the Bleomycin Effect in VERO Cells, Even in the Presence of Pooled ATM, DNA-PK, and LigIV Inhibitors

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
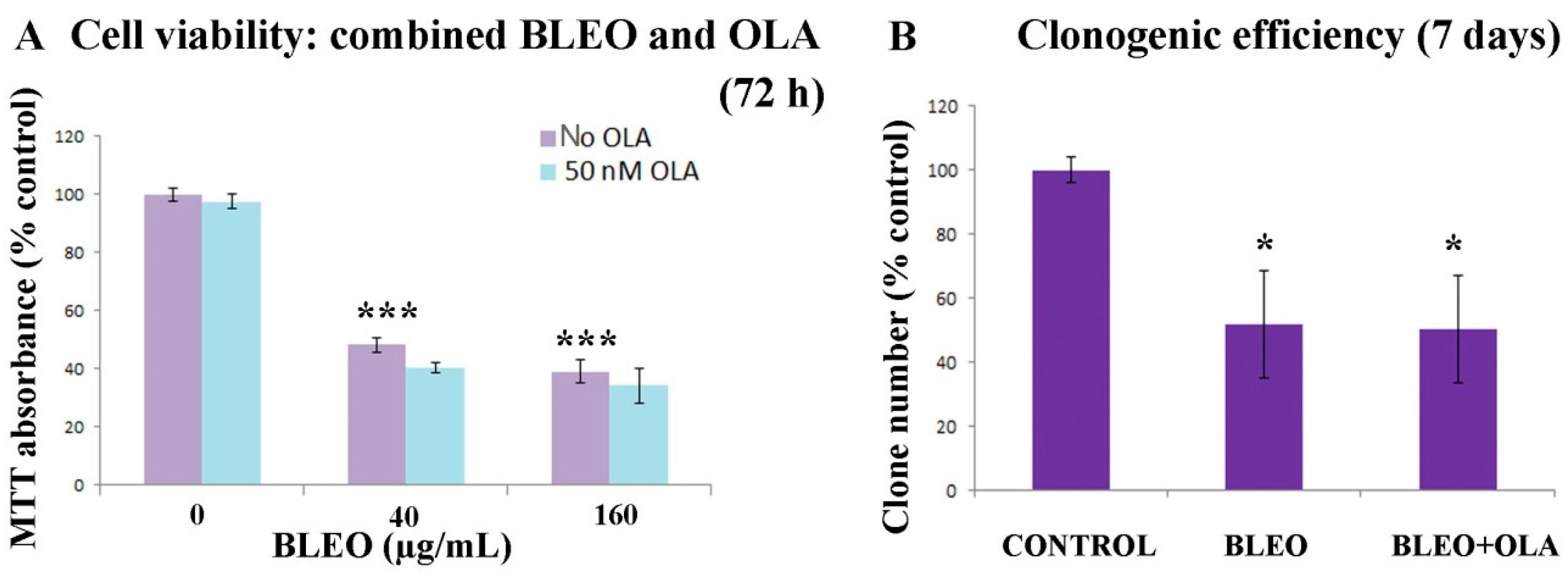
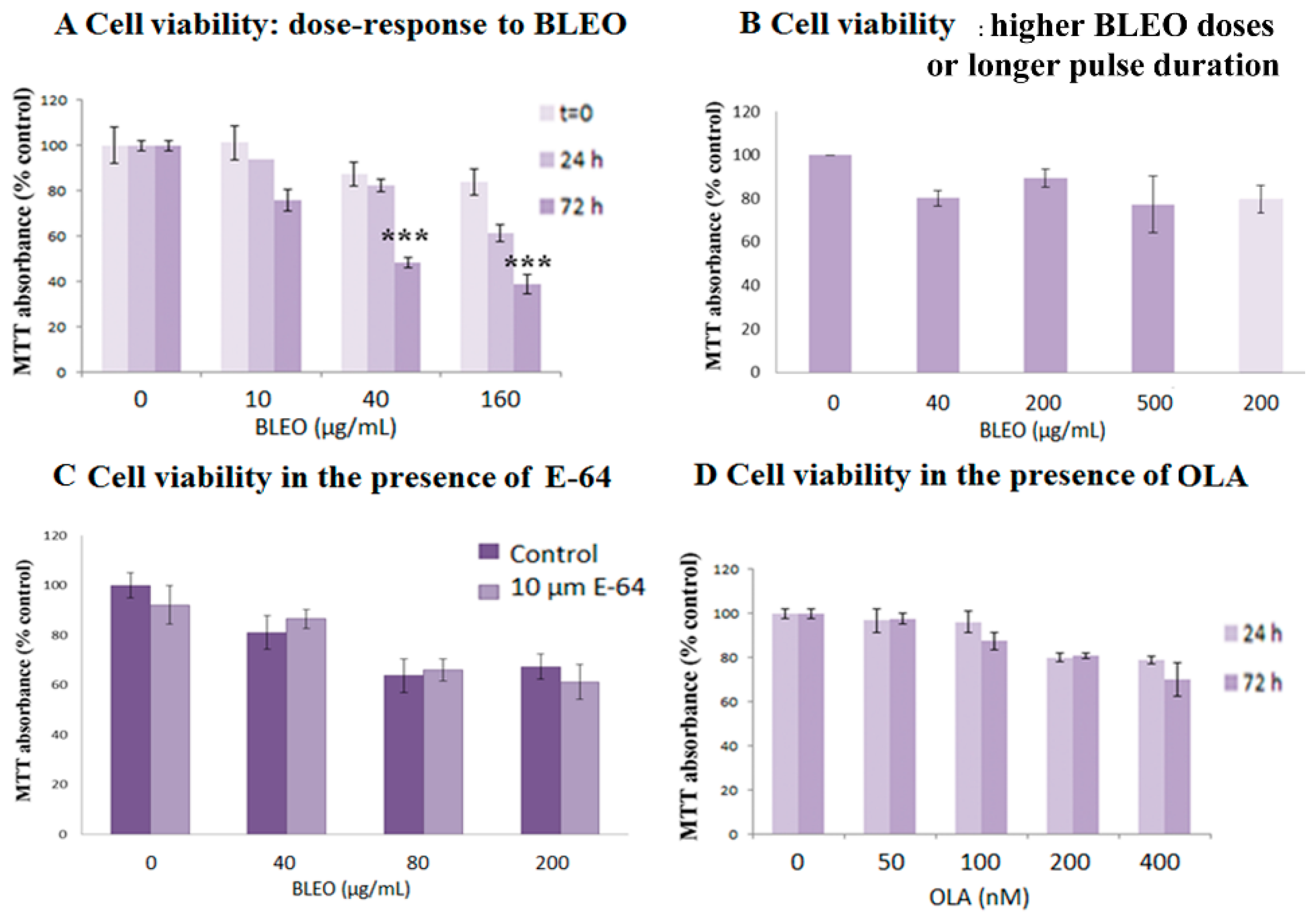
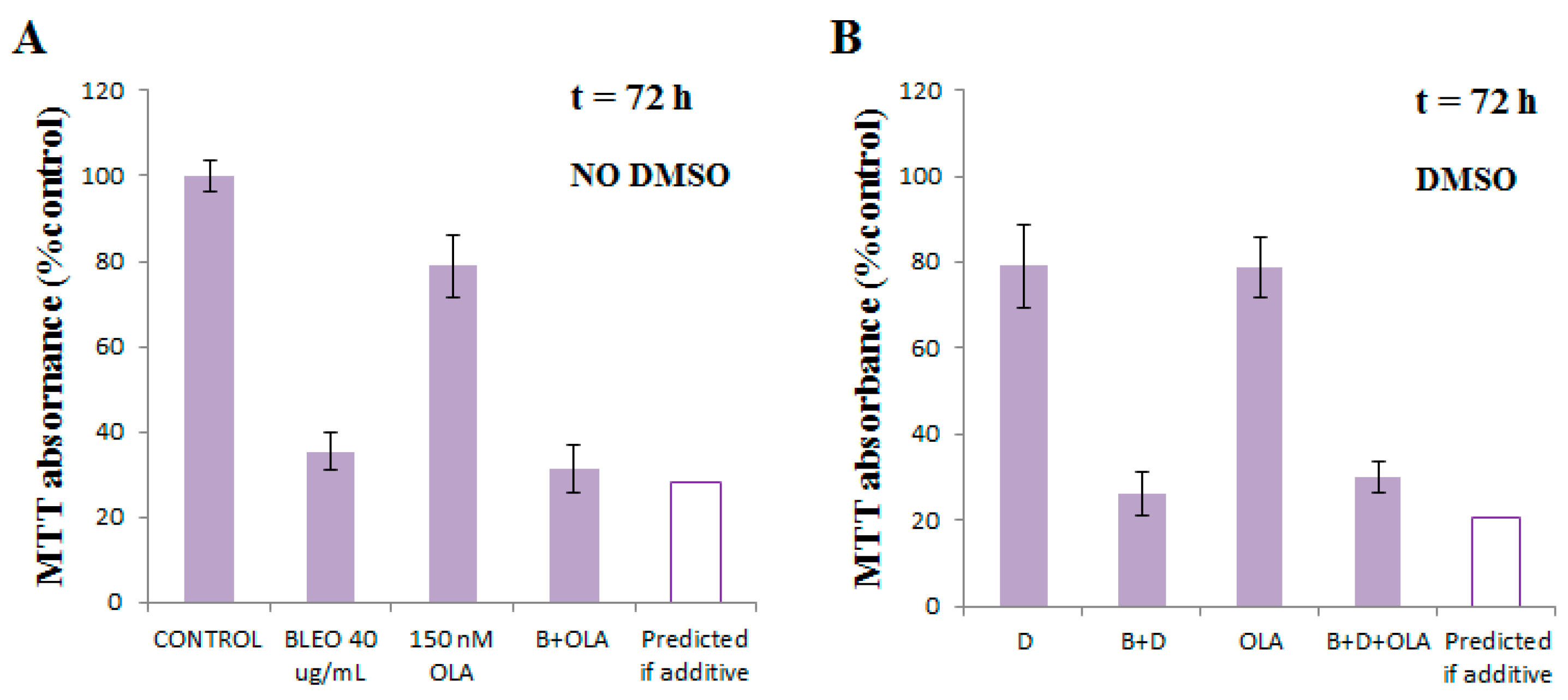
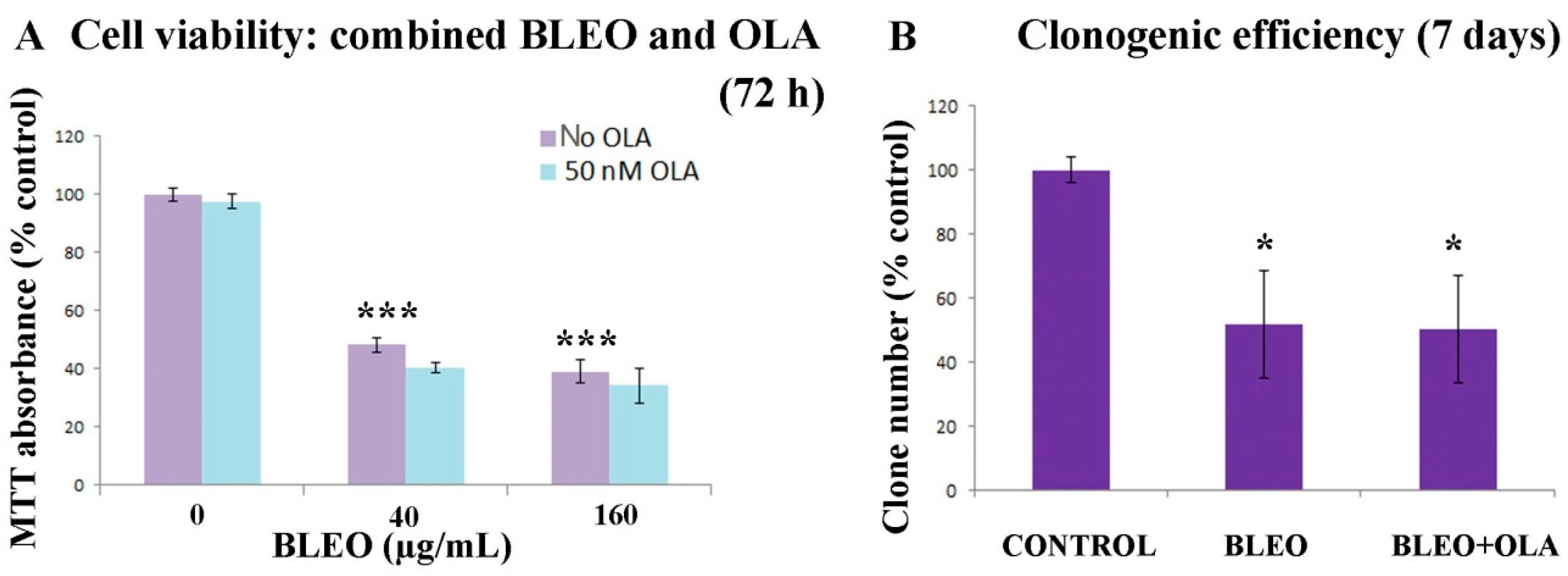
2.1. OLA Did Not Potentiate BLEO Effects on VERO Cells
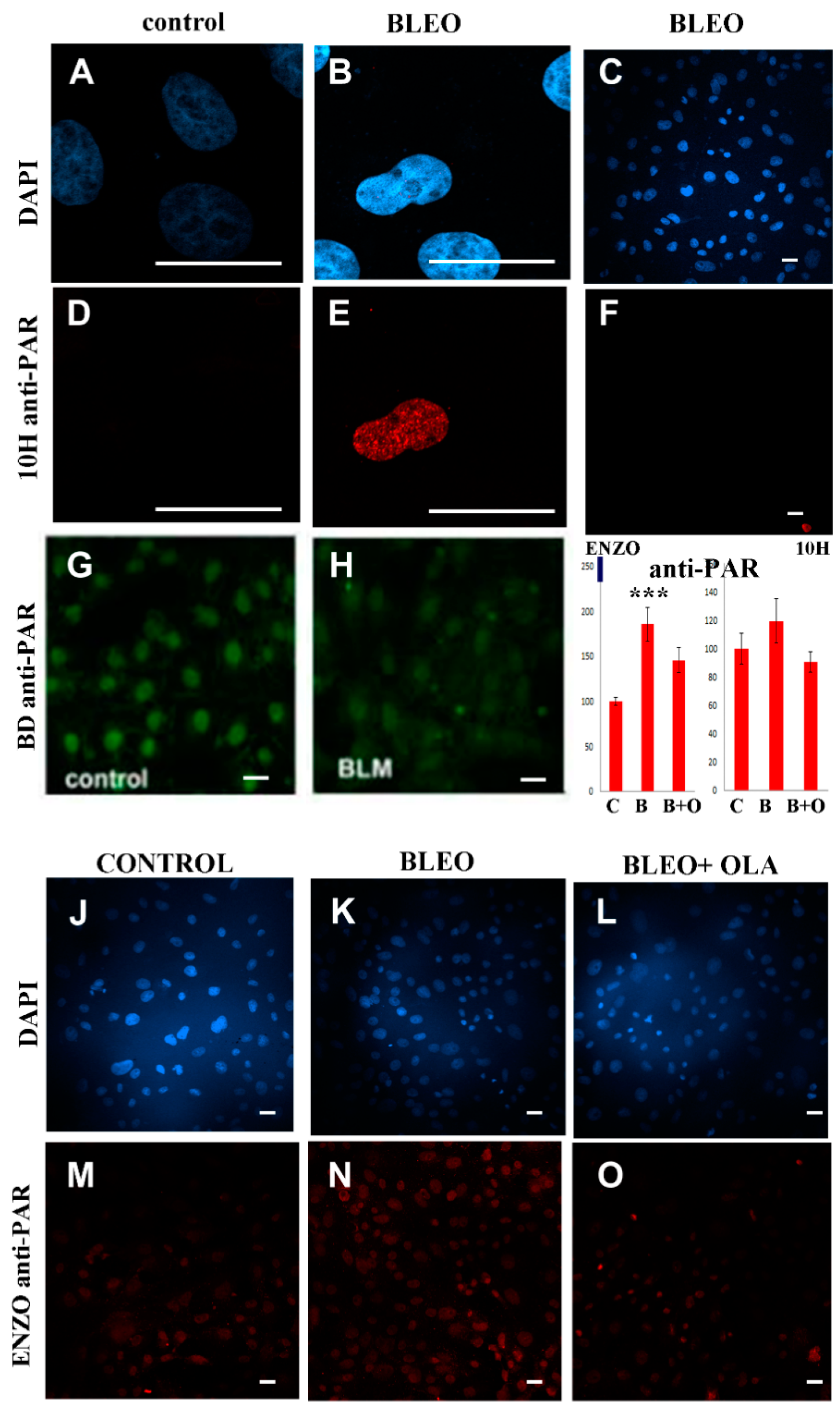
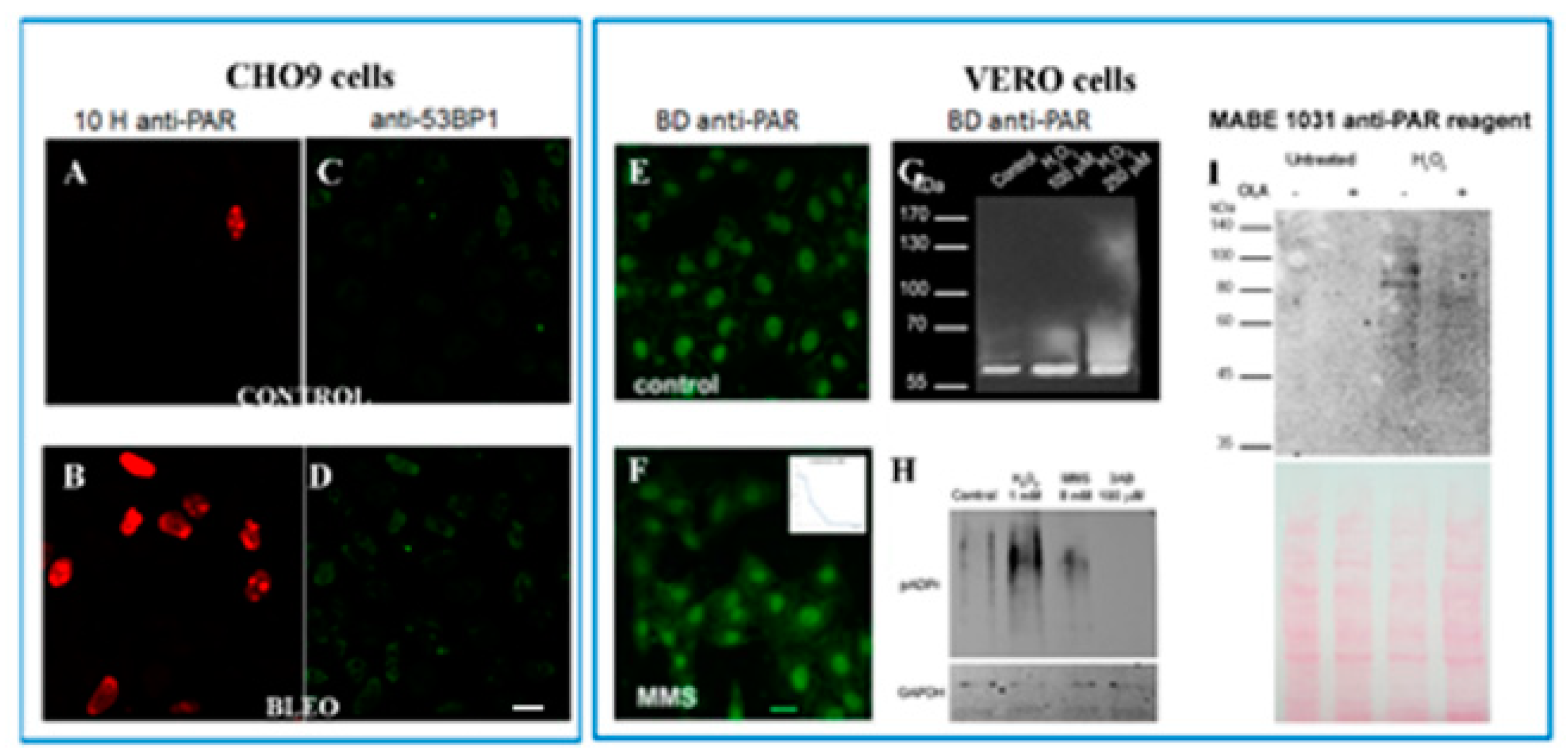
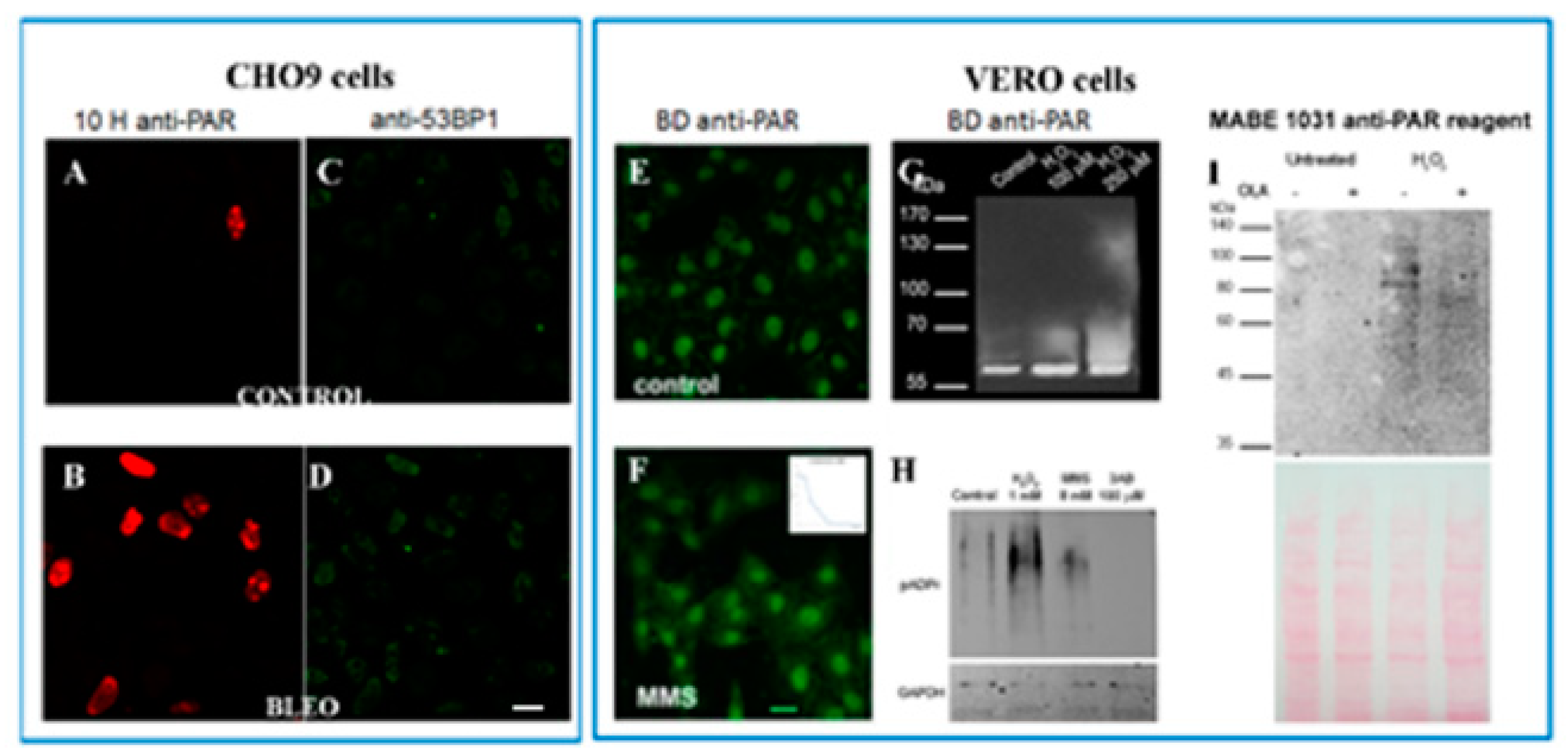
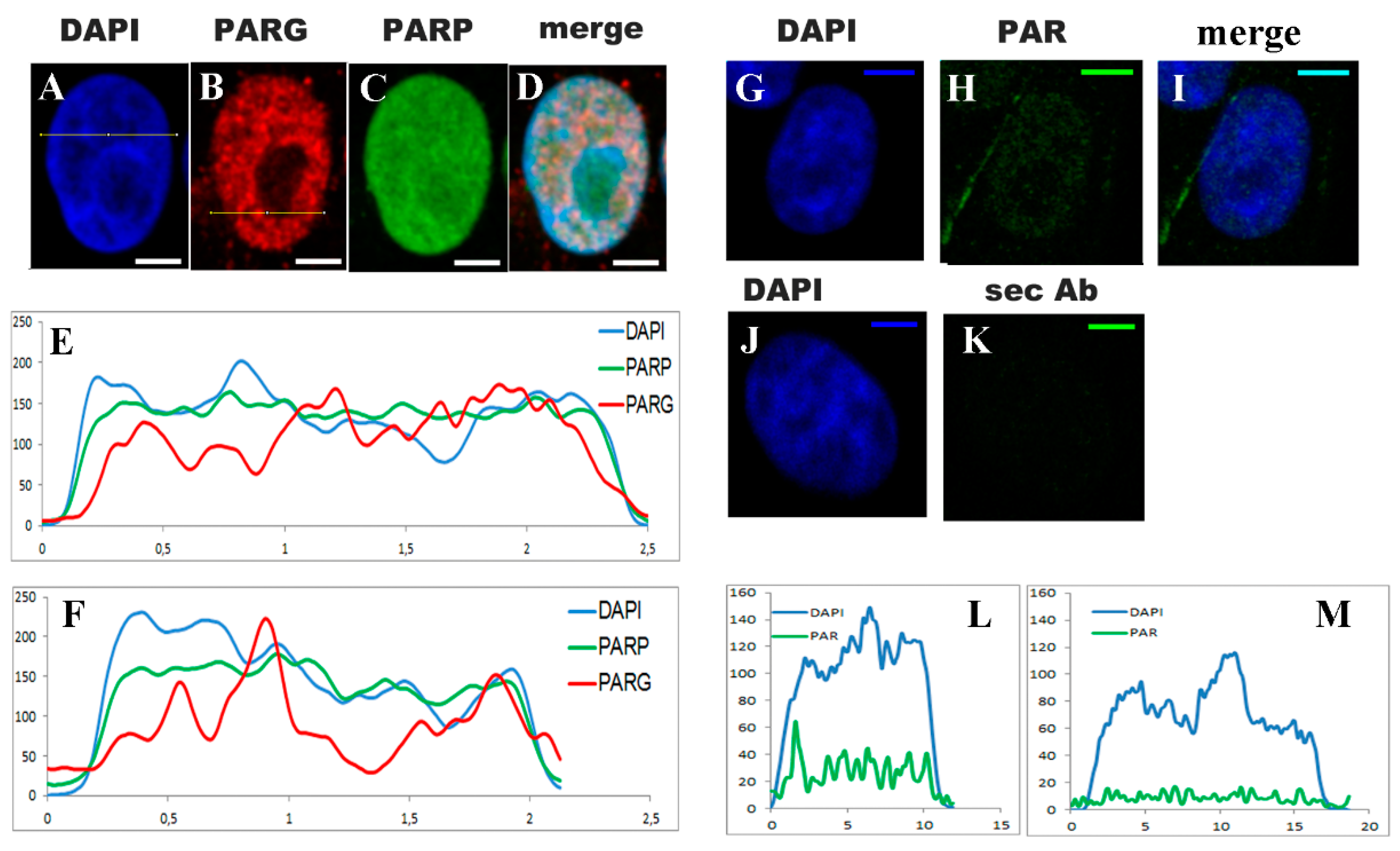
2.2. Untreated VERO Cell Nuclei Harbor PARP, PARG, and PAR
2.3. No Sharp PAR Increase Could Be Detected Immediately after the 45 min Pulse of BLEO
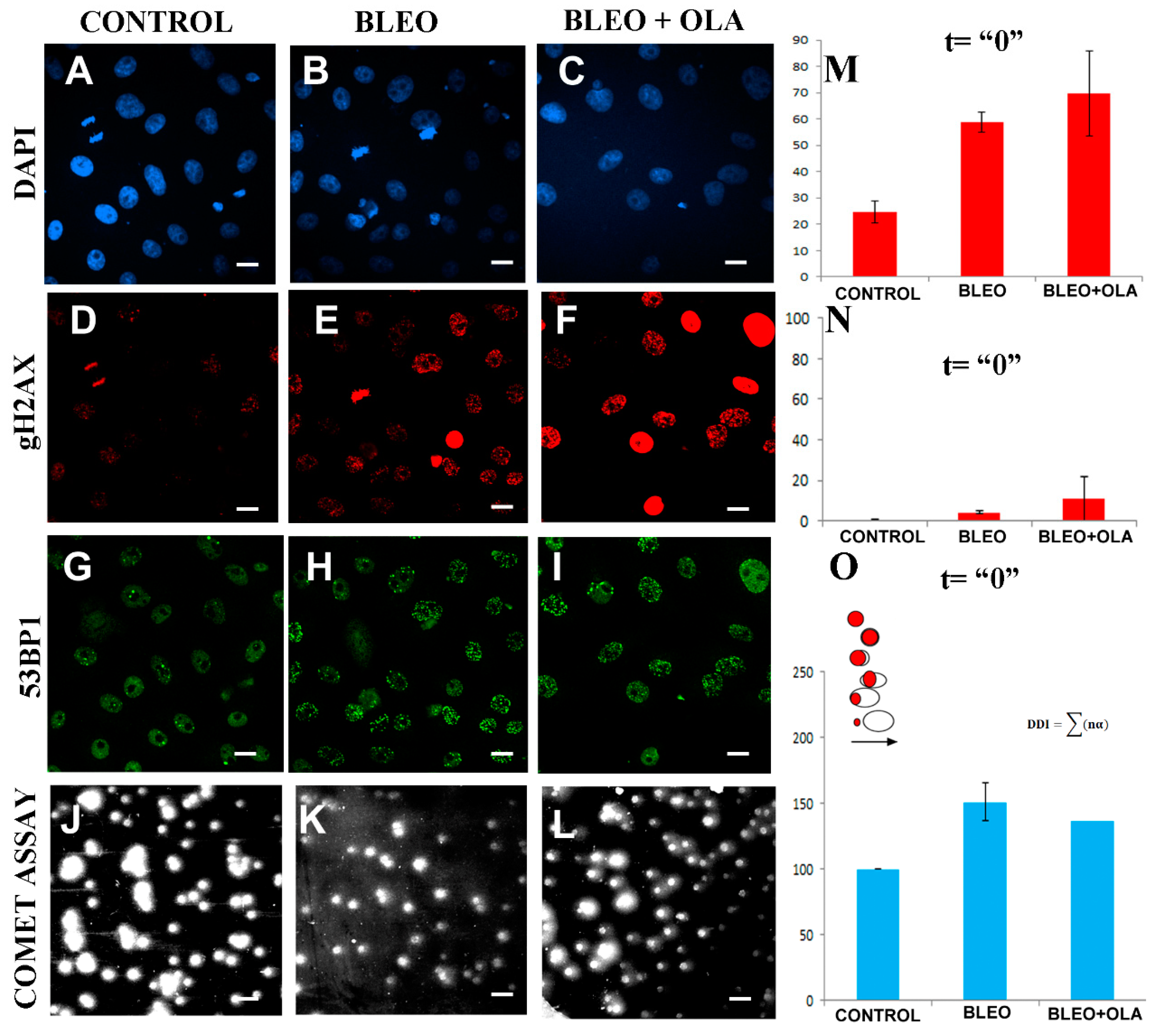
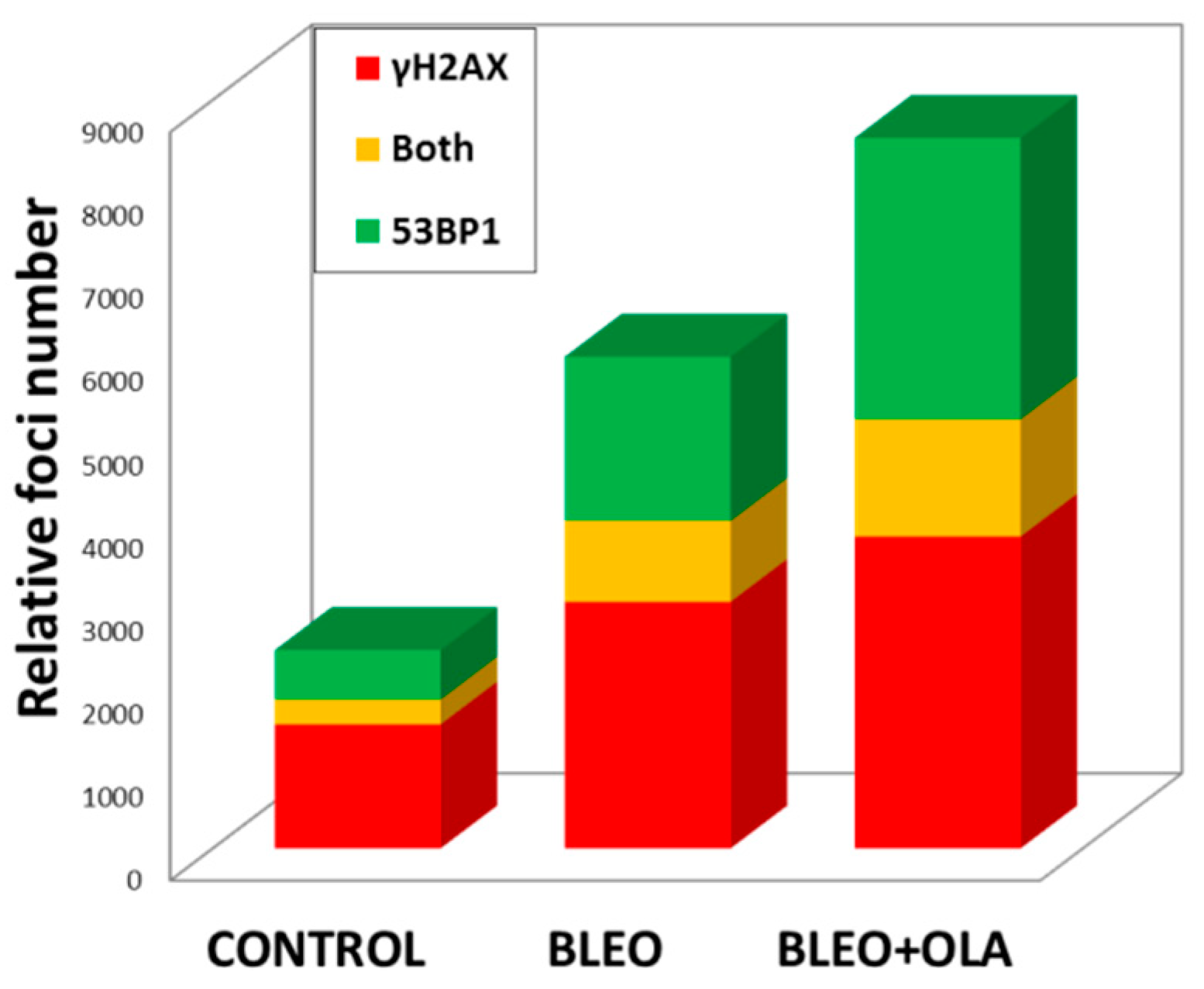
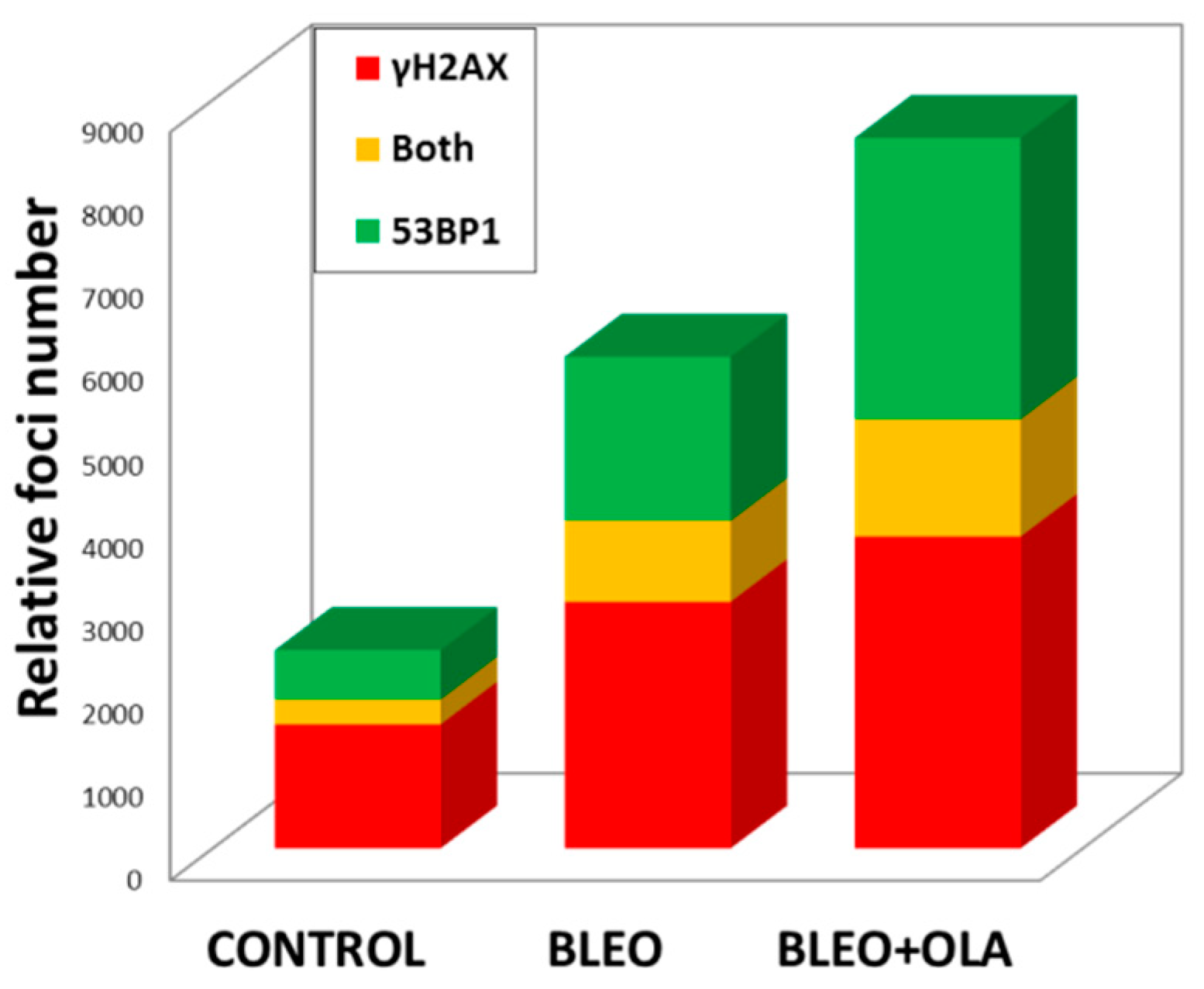
2.4. OLA Did Not Hamper the DNA Damage Induction by BLEO
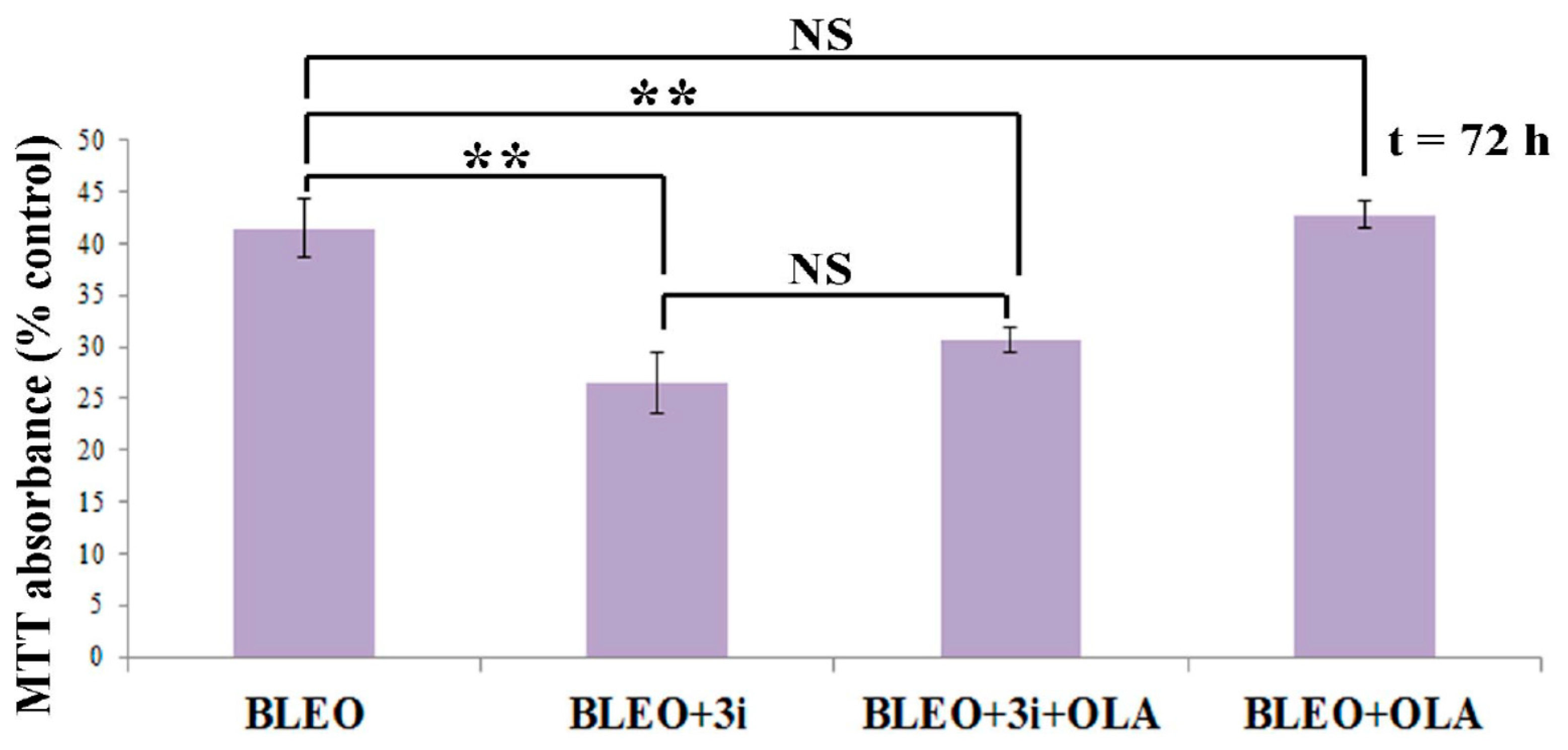
2.5. OLA Did Not Potentiate BLEO, Even in the Presence of a Pool of DNA Repair Enzyme Inhibitors
3. Discussion
4. Materials and Methods
4.1. VERO Cell Culture
4.2. Treatments with BLEO and Inhibitors
4.3. Verification of the PARPis and PARGi Effects on VERO Basal PAR Pool
4.4. Cell Viability Assay (MTT)
4.5. Clonogenic Assay
4.6. Comet Assay
4.7. Indirect Inmunocytofluorescence and Image Acquisition
4.8. Cell Counting Using Low-Magnification Fields
4.9. Relative PAR Quantification
4.10. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| γH2AX | Histone H2AX phosphorylated on Ser 139 |
| 10H | Clone mouse monoclonal anti-PAR antibody (Tulip #1020) |
| 3AB | 3-Aminobenzamide (PARP inhibitor) |
| 3i | Inhibition of DNA-PK, LigIV, and ATM |
| 4-ANI | 4-Amino-1,8-naphthalimide |
| 53BP1 | p53 binding protein 1 |
| AG14361 | 2-[4-[(dimethylamino)methyl]phenyl]-1,3,10-triazatricyclo[6.4.1.04,13]trideca-2,4,6,8(13)-tetraen-9-one (PARP-1i) |
| ATM | Ataxia telangiectasia mutated |
| ATMi | ATM inhibitor |
| ATR | Ataxia telangiectasia and Rad3-related protein |
| ATRi | ATR inhibitor |
| AZ20 | 4{4-[(3R)3-Methylmorpholin-4-yl]-6-[1-(methylsulfonyl)cyclopropyl]pyrimidin-2-yl}-1H-indole |
| BER | Base excision repair |
| BLEO | Bleomycin |
| BRCA1/2 | Breast cancer gene 1 or 2 |
| CDKN2 | Cyclin-dependent kinase inhibitor 2 |
| CHK1 | Checkpoint kinase 1 |
| CHO9 | Chinese hamster ovary 9 cell line |
| C-NHEJ | Canonic-non-homologous End Joining |
| DAPI | 4′,6-diamidino-2-phenylindole (fluorescent DNA marker) |
| DDI | DNA damage index |
| DDR | DNA damage response |
| DEA | 6,9-Diamino-2-ethoxyacridine-DL-lactate monohydrate |
| DMSO | Dimethyl sulfoxide |
| DNA-PK | DNA-dependent protein kinase |
| DNA-PKi | DNA-PK inhibitor |
| DSBs | Double-strand breaks |
| E-64 | trans-Poxysuccinyl-l-leucylamido-(4-guanidino) butane |
| E7016 | 10-((4-Hydroxypiperidin-1-yl)methyl)chromeno[4,3,2-de]phthalazin-3(2H)-one (PARPi) |
| EB | 5′-deoxy-5′-[4-[2-[(2,3-dihydro-1oxo-1H-isoindol-4-yl)amino]-2-oxoethyl]-1-piperazinyl]-5′-oxoadenosine dihydrochloride (PARPi) |
| FBS | Fetal bovine serum |
| HeLa | Henrietta Lacks human epithelial carcinoma cell line |
| HEp-2 | Human epithelial type 2 cells |
| HNSCC | Head and neck squamous cell carcinomas |
| HR | Homologous recombination |
| IC50 | The half-maximal inhibitory concentration |
| ICF | Inmunocytofluorescence |
| INO1001 | 3-Aminobenzamide (PARP inhibitor) |
| IR | Ionizing radiation |
| KU-0060648 | 4-Ethyl-N-[4-[2-(4-morpholinyl)-4-oxo-4H-1-benzopyran-8-yl]-1-dibenzothienyl]-1-piperazineacetamide (DNA-PKi) |
| KU55933 | 2-Morpholin-4-yl-6-thianthren-1-yl-pyran-4-one |
| KU70/80 | Ku heterodimer: Ku70 (XRCC6) and Ku80 (XRCC5) |
| LigIII | DNA ligase 3 |
| LigIV | DNA ligase 4 |
| LigIVi | LigIV inhibitor |
| MAPK | Mitogen-activated protein kinase |
| MEM | Minimum essential medium |
| MMEJ | Microhomology-mediated end joining |
| MMS | Methylmethanosulfonate |
| MRE11 | Complex consisting of meiotic recombination 11 |
| Mst-1 | Macrophage-stimulating 1 (mitogen-activated protein kinase) |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NAD+ | Nicotinamide adenine dinucleotide |
| Nam | Nicotinamide |
| NMuMG | Nontransformed mouse mammary gland epithelial cell line |
| OLA | Olaparib (Lynparza, AZD-2281; PARP-1/2 & 3 inhibitor); IUPAC name:4-[[3-[4-(cyclopropanecarbonyl)piperazine-1-carbonyl]-4-fluorophenyl]methyl]-2H-phthalazin-1-one |
| p38 | A class of MAPK |
| PAR | Poly(ADP-ribose) |
| PARG | Poly-ADP-glicohidrolase |
| PARP | Poly(ADP-ribosyl) polymerase |
| PARPis | PARP inhibitors |
| PARylation | Poly(ADP-ribosylation) |
| PBS | Phosphate-buffered saline |
| PFA | Paraformaldehyde |
| Polβ | DNA polymerase β |
| Prexacertib | 5-[[5-[2-(3-aminopropoxy)-6-methoxyphenyl]-1H-pyrazol-3-yl]amino]pyrazine-2-carbonitrile (CHK1 inhibitor) |
| PtK2 | Male rat kangaroo kidney epithelial cell |
| RT | Room temperature |
| SSBs | Single-strand breaks |
| TNKS | Tankyrase |
| UV-C | Ultraviolet C radiation |
| VE-821 | 3-amino-6-(4-methylsulfonylphenyl)-N-phenylpyrazine-2-carboxamide (ATRi) |
| VE-822 | 3-[3-[4-(methylaminomethyl)phenyl]-1,2-oxazol-5-yl]-5-(4-propan-2-ylsulfonylphenyl)pyrazin-2-amine (ATRi) |
| VERO | African green monkey kidney epithelial cell line |
| VRK1 | Vaccinia-related kinase 1 |
| WBWEE1 | Western BlotsG2 checkpoint kinase |
| XRCC1 | X-ray repair cross-complementing protein 1 |
Appendix A

Appendix B

Appendix C

Appendix D
Appendix E
References
- Teloni, F.; Altmeyer, M. Survey and summary readers of poly(ADP-ribose): Designed to be fit for purpose. Nucleic Acids Res. 2016, 44, 993–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Althaus, F.R.; Kleczkowska, H.E.; Malanga, M.; Müntener, C.R.; Pleschke, J.M.; Ebner, M.; Auer, B. Poly ADP-ribosylation: A DNA break signal mechanism. Proc. Mol. Cell. Biochem. 1999, 193, 5–11. [Google Scholar] [CrossRef]
- Gagné, J.P.; Pic, É.; Isabelle, M.; Krietsch, J.; Éthier, C.; Paquet, É.; Kelly, I.; Boutin, M.; Moon, K.M.; Foster, L.J.; et al. Quantitative proteomics profiling of the poly(ADP-ribose)-related response to genotoxic stress. Nucleic Acids Res. 2012, 40, 7788–7805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [Green Version]
- Salzano, M.; Sanz-García, M.; Monsalve, D.M.; Moura, D.S.; Lazo, P.A. VRK1 chromatin kinase phosphorylates H2AX and is required for foci formation induced by DNA damage. Epigenetics 2015, 10, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Maréchal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Dueva, R.; Iliakis, G. Alternative pathways of non-homologous end joining (NHEJ) in genomic instability and cancer. Transl. Cancer Res. 2013, 2, 163–177. [Google Scholar] [CrossRef]
- Mladenov, E.; Iliakis, G. The Pathways of Double-Strand Break Repair. In DNA Repair—On the Pathways to Fixing DNA Damage and Errors; Storici, F., Ed.; InTech: London, UK, 2011; pp. 143–168. ISBN 978-953-307-649-2. [Google Scholar]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Hottiger, M.O. SnapShot: ADP-Ribosylation Signaling. Mol. Cell 2015, 58, 1134–1134.e1. [Google Scholar] [CrossRef]
- Virag, L.; Szabo, C. The Therapeutic Potential of Poly(ADP-Ribose) Polymerase Inhibitors. J. Natl. Cancer Inst. 2011, 103, 334–346. [Google Scholar] [CrossRef]
- Kraus, W.L. PARPs and ADP-Ribosylation: 50 Years… and Counting. Mol. Cell 2015, 58, 902–910. [Google Scholar] [CrossRef] [Green Version]
- Thraves, P.; Mossman, K.L.; Brennan, T.; Dritschilo, A. Radiosensitization of human fibroblasts by 3-aminobenzamide: An inhibitor of poly(ADP-ribosylation). Radiat. Res. 1985, 104, 119–127. [Google Scholar] [CrossRef]
- Brock, W.A.; Milas, L.; Bergh, S.; Lo, R.; Szabo, C.; Mason, K.A. Radiosensitization of human and rodent cell lines by INO-1001, a novel inhibitor of poly(ADP-ribose) polymerase. Cancer Lett. 2004, 205, 155–160. [Google Scholar] [CrossRef]
- Arundel-Suto, C.M.; Scavone, S.V.; Turner, W.R.; Suto, M.J.; Sebolt-Leopold, J.S. Effect of PD 128763, a new potent inhibitor of poly(ADP-ribose) polymerase, on X-ray-induced cellular recovery processes in Chinese hamster V79 cells. Radiat. Res. 1991, 126, 367–371. [Google Scholar] [CrossRef]
- Oleinick, N.L.; Evans, H.H. Poly(ADP-ribose) and the response of cells to ionizing radiation. Radiat. Res. 1985, 101, 29–46. [Google Scholar] [CrossRef]
- Russo, A.L.; Kwon, H.-C.; Burgan, W.E.; Carter, D.; Beam, K.; Weizheng, X.; Zhang, J.; Slusher, B.S.; Chakravarti, A.; Tofilon, P.J.; et al. In vitro and In vivo Radiosensitization of Glioblastoma Cells by the Poly (ADP-Ribose) Polymerase Inhibitor E7016. Clin. Cancer Res. 2009, 15, 607–612. [Google Scholar] [CrossRef] [Green Version]
- Veuger, S.J.; Curtin, N.J.; Richardson, C.J.; Smith, G.C.M.; Durkacz, B.W. Radiosensitization and DNA repair inhibition by the combined use of novel inhibitors of DNA-dependent protein kinase and poly(ADP-ribose) polymerase-1. Cancer Res. 2003, 63, 6008–6015. [Google Scholar]
- Noel, G.; Godon, C.; Fernet, M.; Giocanti, N.; Megnin-Chanet, F.; Favaudon, V. Radiosensitization by the poly(ADP-ribose) polymerase inhibitor 4-amino-1,8-naphthalimide is specific of the S phase of the cell cycle and involves arrest of DNA synthesis. Mol. Cancer Ther. 2006, 5, 564–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huet, J.; Laval, F. Potentiation of Cell Killing by Inhibitors of Poly (Adenosine Diphosphate-Ribose) Synthesis in Bleomycin-treated Chinese Hamster Ovary Cells. Cancer Res. 1985, 45, 987–991. [Google Scholar]
- Kelland, L.R.; Burgess, L.; Steel, G.G. Radiation damage repair capacity of a human germ-cell tumour cell line: Inhibition by 3-aminobenzamide. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1987, 51, 227–241. [Google Scholar] [CrossRef]
- Horton, J.K.; Stefanick, D.F.; Naron, J.M.; Kedar, P.S.; Wilson, S.H. Poly(ADP-ribose) polymerase activity prevents signaling pathways for cell cycle arrest after DNA methylating agent exposure. J. Biol. Chem. 2005, 280, 15773–15785. [Google Scholar] [CrossRef] [Green Version]
- Cleaver, J.E.; Bodello, W.J.; Morgan, W.F.; Zellell, B. Poly (ADP-ribose) of Repair of DNA Differences in the Regulation by and Ultraviolet Light According to Damage from Alkylating Agents Cell Type. J. Biol. Chem. 1983, 258, 9059–9068. [Google Scholar]
- Vodenicharov, M.D.; Ghodgaonkar, M.M.; Halappanavar, S.S.; Shah, R.G.; Shah, G.M. Mechanism of early biphasic activation of poly(ADP-ribose) polymerase-1 in response to ultraviolet B radiation. J. Cell Sci. 2005, 118, 589–599. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.-C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef] [PubMed]
- Saha, T.; Smulson, M.; Rosen, E.M. BRCA1 regulation of base excision repair pathway. Cell Cycle 2010, 9, 2471–2472. [Google Scholar] [CrossRef] [Green Version]
- Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef] [Green Version]
- Ström, C.E.; Johansson, F.; Uhlén, M.; Szigyarto, C.A.K.; Erixon, K.; Helleday, T. Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res. 2011, 39, 3166–3175. [Google Scholar] [CrossRef]
- Horton, J.K.; Stefanick, D.F.; Prasad, R.; Gassman, N.R.; Kedar, P.S.; Wilson, S.H. Base excision repair defects invoke hypersensitivity to PARP inhibition. Mol. Cancer Res. 2014, 12, 1128–1139. [Google Scholar] [CrossRef] [Green Version]
- Czyż, M.; Toma, M.; Gajos-Michniewicz, A.; Majchrzak, K.; Hoser, G.; Szemraj, J.; Nieborowska-Skorska, M.; Cheng, P.; Gritsyuk, D.; Levesque, M.; et al. PARP1 inhibitor olaparib (Lynparza) exerts synthetic lethal effect against ligase 4-deficient melanomas. Oncotarget 2016, 7, 75551–75560. [Google Scholar] [CrossRef] [Green Version]
- Ko, H.L.; Ren, E.C. Functional aspects of PARP1 in DNA repair and transcription. Biomolecules 2012, 2, 524–548. [Google Scholar] [CrossRef] [Green Version]
- Feng, F.Y.; de Bono, J.S.; Rubin, M.A.; Knudsen, K.E. Chromatin to Clinic: The Molecular Rationale for PARP1 Inhibitor Function. Mol. Cell 2015, 58, 925–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, P. Review Biology of Poly (ADP-Ribose) Polymerases: The Factotums of Cell Maintenance. Mol. Cell 2015, 58, 947–958. [Google Scholar] [CrossRef] [Green Version]
- Berger, N.A.; Besson, V.C.; Boulares, A.H.; Bürkle, A.; Chiarugi, A.; Clark, R.S.; Curtin, N.J.; Cuzzocrea, S.; Dawson, T.M.; Dawson, V.L.; et al. Opportunities for the repurposing of PARP inhibitors for the therapy of non-oncological diseases. Br. J. Pharmacol. 2018, 175, 192–222. [Google Scholar] [CrossRef]
- Sleijfer, S. Bleomycin-induced pneumonitis. Chest 2001, 120, 617–624. [Google Scholar] [CrossRef] [Green Version]
- Beck, C.; Robert, I.; Reina-San-Martin, B.; Schreiber, V.; Dantzer, F. Poly(ADP-ribose) polymerases in double-strand break repair: Focus on PARP1, PARP2 and PARP3. Exp. Cell Res. 2014, 329, 18–25. [Google Scholar] [CrossRef]
- Sukhanova, M.V.; Abrakhi, S.; Joshi, V.; Pastre, D.; Kutuzov, M.M.; Anarbaev, R.O.; Curmi, P.A.; Hamon, L.; Lavrik, O.I. Single molecule detection of PARP1 and PARP2 interaction with DNA strand breaks and their poly(ADP-ribosyl)ation using high-resolution AFM imaging. Nucleic Acids Res. 2015. [Google Scholar] [CrossRef] [Green Version]
- Fouquin, A.; Guirouilh-Barbat, J.; Lopez, B.; Hall, J.; Amor-Guéret, M.; Pennaneach, V. PARP2 controls double-strand break repair pathway choice by limiting 53BP1 accumulation at DNA damage sites and promoting end-resection. Nucleic Acids Res. 2017, 45, 12325–12339. [Google Scholar] [CrossRef] [Green Version]
- Rulten, S.L.; Fisher, A.E.O.; Robert, I.; Zuma, M.C.; Rouleau, M.; Ju, L.; Poirier, G.; Reina-San-Martin, B.; Caldecott, K.W. PARP-3 and APLF function together to accelerate nonhomologous end-joining. Mol. Cell 2011, 41, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Grundy, G.J.; Polo, L.M.; Zeng, Z.; Rulten, S.L.; Hoch, N.C.; Paomephan, P.; Xu, Y.; Sweet, S.M.; Thorne, A.W.; Oliver, A.W.; et al. PARP3 is a sensor of nicked nucleosomes and monoribosylates histone H2B Glu2. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oplustil O’Connor, L.; Rulten, S.L.; Cranston, A.N.; Odedra, R.; Brown, H.; Jaspers, J.E.; Jones, L.; Knights, C.; Evers, B.; Ting, A.; et al. The PARP Inhibitor AZD2461 Provides Insights into the Role of PARP3 Inhibition for Both Synthetic Lethality and Tolerability with Chemotherapy in Preclinical Models. Cancer Res. 2016, 76, 6084–6094. [Google Scholar] [CrossRef] [Green Version]
- Nagy, Z.; Kalousi, A.; Furst, A.; Koch, M.; Fischer, B.; Soutoglou, E. Tankyrases Promote Homologous Recombination and Check Point Activation in Response to DSBs. PLoS Genet. 2016, 12, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Vilchez Larrea, S.C.; Haikarainen, T.; Narwal, M.; Schlesinger, M.; Venkannagari, H.; Flawiá, M.M.; Villamil, S.H.F.; Lehtiö, L. Inhibition of poly(ADP-ribose) Polymerase Interferes with Trypanosoma cruzi Infection and Proliferation of the Parasite. PLoS ONE 2012, 7, e46063. [Google Scholar] [CrossRef]
- Thorsell, A.G.; Ekblad, T.; Karlberg, T.; Löw, M.; Pinto, A.F.; Trésaugues, L.; Moche, M.; Cohen, M.S.; Schüler, H. Structural Basis for Potency and Promiscuity in Poly(ADP-ribose) Polymerase (PARP) and Tankyrase Inhibitors. J. Med. Chem. 2017, 60, 1262–1271. [Google Scholar] [CrossRef]
- Sharif-Askari, B.; Amrein, L.; Aloyz, R.; Panasci, L. PARP3 inhibitors ME0328 and olaparib potentiate vinorelbine sensitization in breast cancer cell lines. Breast Cancer Res. Treat. 2018, 172, 23–32. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [Green Version]
- Terasima, T.; Takabe, Y.; Katsumata, T.; Watanabe, M.; Umezawa, H. Effect of Bleomycin on Mammalian Cell Survival. J. Natl. Cancer Inst. 1972, 49, 1093–1100. [Google Scholar]
- Sheets, R. History and Characterization of the Vero Cell Line. Open Sess. 2000, 1–12. [Google Scholar]
- Naoki, O.; Arihiro, K.; Toshiyuki, Y.; Noriko, H.; Fumio, K.; Suyoshi, S.; Makoto, K.; Kentaro, H.; Hattori, M. The genome landscape of the African Green Monkey kidney-derived vero cell line. DNA Res. 2014, 21, 673–683. [Google Scholar] [CrossRef]
- Ammerman, A. NIH Public Access. Curr. Protoc. Microbiol. 2009, 1–10. [Google Scholar] [CrossRef]
- Duran-Rehbein, G.A.; Vargas-Zambrano, J.C.; Cuéllar, A.; Puerta, C.J.; Gonzalez, J.M. Mammalian cellular culture models of trypanosoma cruzi infection: A review of the published literature. Parasite 2014, 21. [Google Scholar] [CrossRef] [Green Version]
- Schacke, M.; Kumar, J.; Colwell, N.; Hermanson, K.; Folle, G.A.; Nechaev, S.; Dhasarathy, A.; Lafon-Hughes, L. PARP-1/2 inhibitor olaparib prevents or partially reverts EMT induced by TGF-β in NMuMG cells. Int. J. Mol. Sci. 2019, 20, 518. [Google Scholar] [CrossRef] [Green Version]
- Kawamitsu, H.; Hoshino, H.; Okada, H.; Miwa, M.; Momoi, H.; Sugimura, T. Monoclonal antibodies against poly(ADP-ribose) recognize different structures of poly(ADP-ribose). Biochemistry 1984, 23, 41–47. [Google Scholar] [CrossRef]
- Gagné, J.P.; Isabelle, M.; Lo, K.S.; Bourassa, S.; Hendzel, M.J.; Dawson, V.L.; Dawson, T.M.; Poirier, G.G. Proteome-wide identification of poly(ADP-ribose) binding proteins and poly(ADP-ribose)-associated protein complexes. Nucleic Acids Res. 2008, 36, 6959–6976. [Google Scholar] [CrossRef] [Green Version]
- Fahrer, J.; Kranaster, R.; Altmeyer, M.; Marx, A.; Bürkle, A. Quantitative analysis of the binding affinity of poly(ADP-ribose) to specific binding proteins as a function of chain length. Nucleic Acids Res. 2007, 35. [Google Scholar] [CrossRef] [PubMed]
- Lafon-Hughes, L. Localización Preferencial del Daño Genético en Regiones Eucromáticas y Replicantes de los Cromosomas y los Núcleos de Mamíferos. Roles de la Poli-ADP-Ribosa en Presencia y Ausencia de Daño Genético Inducido en Células VERO. Ph.D. Thesis, Facultad de Ciencias—PEDECIBA, IIBCE, INGEBI, Universidad de la República, Montevideo, Uruguay, 2014. Available online: http://www.bib.fcien.edu.uy/files/etd/biol/uy24-17142.pdf (accessed on 3 November 2020).
- Lafon-Hughes, L.; Vilchez Larrea, S.C.; Kun, A.; Fernández Villamil, S.H. VERO cells harbor a poly-ADP-ribose belt partnering their epithelial adhesion belt. PeerJ 2014, 2, e617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Wu, W.; Wu, W.; Rosidi, B.; Zhang, L.; Wang, H.; Iliakis, G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006, 34, 6170–6182. [Google Scholar] [CrossRef] [PubMed]
- Burger, R.M. Cleavage of nucleic acids by bleomycin. Chem. Rev. 1998, 98, 1153–1169. [Google Scholar] [CrossRef]
- Lafon-Hughes, L.; Di Tomaso, M.V.; Liddle, P.; Toledo, A.; Reyes-Ábalos, A.L.; Folle, G.A. Preferential localization of γh2AX foci in euchromatin of retina rod cells after DNA damage induction. Chromosome Res. 2013, 21. [Google Scholar] [CrossRef]
- Xia, C.; Wolf, J.J.; Sun, C.; Xu, M.; Studstill, C.J.; Chen, J.; Ngo, H.; Zhu, H.; Hahm, B. PARP1 Enhances Influenza A Virus Propagation by Facilitating Degradation of Host Type I Interferon Receptor. J. Virol. 2020, 94, e01572-19. [Google Scholar] [CrossRef]
- Nguyen, M.L.; Kraft, R.M.; Blaho, J.A. African green monkey kidney Vero cells require de novo protein synthesis for efficient herpes simplex virus 1-dependent apoptosis. Virology 2005, 336, 274–290. [Google Scholar] [CrossRef] [Green Version]
- The UniProt Consortium UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [CrossRef] [Green Version]
- Cruz, G.M.S.; Kong, X.; Silva, B.A.; Khatibzadeh, N.; Thai, R.; Berns, M.W.; Yokomori, K. Femtosecond near-infrared laser microirradiation reveals a crucial role for PARP signaling on factor assemblies at DNA damage sites. Nucleic Acids Res. 2015, 44, 1–17. [Google Scholar] [CrossRef]
- Hochegger, H.; Dejsuphong, D.; Fukushima, T.; Morrison, C.; Sonoda, E.; Schreiber, V.; Zhao, G.Y.; Saberi, A.; Masutani, M.; Adachi, N.; et al. Parp-1 protects homologous recombination from interference by Ku and Ligase IV in vertebrate cells. EMBO J. 2006, 25, 1305–1314. [Google Scholar] [CrossRef] [Green Version]
- Scully, R.; Xie, A. Double strand break repair functions of histone H2AX. Mutat. Res.-Fundam. Mol. Mech. Mutagen. 2013, 750, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Orr, K.S.; Savage, K.I. The BRCA1 and BRCA2 Breast and Ovarian Cancer Susceptibility Genes—Implications for DNA Damage Response, DNA Repair and Cancer Therapy. In Advances in DNA Repair; InTech: London, UK, 2015. [Google Scholar]
- Kawahara, N.; Ogawa, K.; Nagayasu, M.; Kimura, M.; Sasaki, Y.; Kobayashi, H. Candidate synthetic lethality partners to parp inhibitors in the treatment of ovarian clear cell cancer (Review). Biomed. Rep. 2017, 7, 391–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Andrea, A.D. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair 2018, 71, 172–176. [Google Scholar] [CrossRef]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Dréan, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef]
- Mweempwa, A.; Wilson, M.K. Mechanisms of resistance to PARP inhibitors—An evolving challenge in oncology. Cancer Drug Resist. 2019, 608–617. [Google Scholar] [CrossRef] [Green Version]
- Haynes, B.; Murai, J.; Lee, J.M. Restored replication fork stabilization, a mechanism of PARP inhibitor resistance, can be overcome by cell cycle checkpoint inhibition. Cancer Treat. Rev. 2018, 71, 1–7. [Google Scholar] [CrossRef]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.M.; Nguyen, H.D.; Ho, C.K.; Kwan, T.T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef]
- Young, L.A.; O’Connor, L.O.; de Renty, C.; Veldman-Jones, M.H.; Dorval, T.; Wilson, Z.; Jones, D.R.; Lawson, D.; Odedra, R.; Maya-Mendoza, A.; et al. Differential activity of ATR and Wee1 inhibitors in a highly sensitive subpopulation of DLBCL linked to replication stress. Cancer Res. 2019, 79, 3762–3775. [Google Scholar] [CrossRef] [Green Version]
- Gadhikar, M.A.; Zhang, J.; Shen, L.; Rao, X.; Wang, J.; Zhao, M.; Kalu, N.N.; Johnson, F.M.; Byers, L.A.; Heymach, J.; et al. CDKN2A/p16 Deletion in Head and Neck Cancer Cells Is Associated with CDK2 Activation, Replication Stress, and Vulnerability to CHK1 Inhibition. Cancer Res. 2018, 78, 781–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MedlinePlus, National Library of Medicine. Available online: https://medlineplus.gov/genetics/gene/cdkn2a/ (accessed on 22 October 2020).
- Bakr, A.; Oing, C.; Köcher, S.; Borgmann, K.; Dornreiter, I.; Petersen, C.; Dikomey, E.; Mansour, W.Y. Involvement of ATM in homologous recombination after end resection and RAD51 nucleofilament formation. Nucleic Acids Res. 2015, 43, 3154–3166. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, M.; Nambiar, M.; Sharma, S.; Karki, S.S.; Goldsmith, G.; Hegde, M.; Kumar, S.; Pandey, M.; Singh, R.K.; Ray, P.; et al. An inhibitor of nonhomologous end-joining abrogates double-strand break repair and impedes cancer progression. Cell 2012, 151, 1474–1487. [Google Scholar] [CrossRef] [Green Version]
- Denizot, F.; Lang, R. Rapid colorimetric assay for cell growth and survival. Modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods 1986, 89, 271–277. [Google Scholar] [CrossRef]
- Klaude, M.; Eriksson, S.; Nygren, J.; Ahnstrom, G. The comet assay: Mechanisms and technical considerations. Mutat. Res. 1996, 363, 89–96. [Google Scholar] [CrossRef]
- Tice, R.R.; Andrews, P.W.; Singh, N.P. The single cell gel assay: A sensitive technique for evaluating intercellular differences in DNA damage and repair. Basic Life Sci. 1990, 53, 291–301. [Google Scholar] [CrossRef]
- Olive, P.L.; Banáth, J.P. The comet assay: A method to measure DNA damage in individual cells. Nat. Protoc. 2006, 1, 23–29. [Google Scholar] [CrossRef]
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; DeZonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinform. 2017, 18, 529. [Google Scholar] [CrossRef]
- Kurt De Vos; University of Sheffield, Academic Neurology Image J, Cell Counter Plugin. Available online: https://imagej.nih.gov/ij/plugins/cell-counter.html (accessed on 22 October 2020).
- Available online: https://worddisk.com/wiki/Microsoft_Excel/ (accessed on 4 November 2020).
- Navendu, V. Online Web Statistical Calculators for Categorical Data Analysis. 2016. Available online: astatsa.com (accessed on 22 October 2020).


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perini, V.; Schacke, M.; Liddle, P.; Vilchez-Larrea, S.; Keszenman, D.J.; Lafon-Hughes, L. PARP Inhibitor Olaparib Causes No Potentiation of the Bleomycin Effect in VERO Cells, Even in the Presence of Pooled ATM, DNA-PK, and LigIV Inhibitors. Int. J. Mol. Sci. 2020, 21, 8288. https://doi.org/10.3390/ijms21218288
Perini V, Schacke M, Liddle P, Vilchez-Larrea S, Keszenman DJ, Lafon-Hughes L. PARP Inhibitor Olaparib Causes No Potentiation of the Bleomycin Effect in VERO Cells, Even in the Presence of Pooled ATM, DNA-PK, and LigIV Inhibitors. International Journal of Molecular Sciences. 2020; 21(21):8288. https://doi.org/10.3390/ijms21218288
Chicago/Turabian StylePerini, Valentina, Michelle Schacke, Pablo Liddle, Salomé Vilchez-Larrea, Deborah J. Keszenman, and Laura Lafon-Hughes. 2020. "PARP Inhibitor Olaparib Causes No Potentiation of the Bleomycin Effect in VERO Cells, Even in the Presence of Pooled ATM, DNA-PK, and LigIV Inhibitors" International Journal of Molecular Sciences 21, no. 21: 8288. https://doi.org/10.3390/ijms21218288