The HGF/Met/NF-κB Pathway Regulates RANKL Expression in Osteoblasts and Bone Marrow Stromal Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
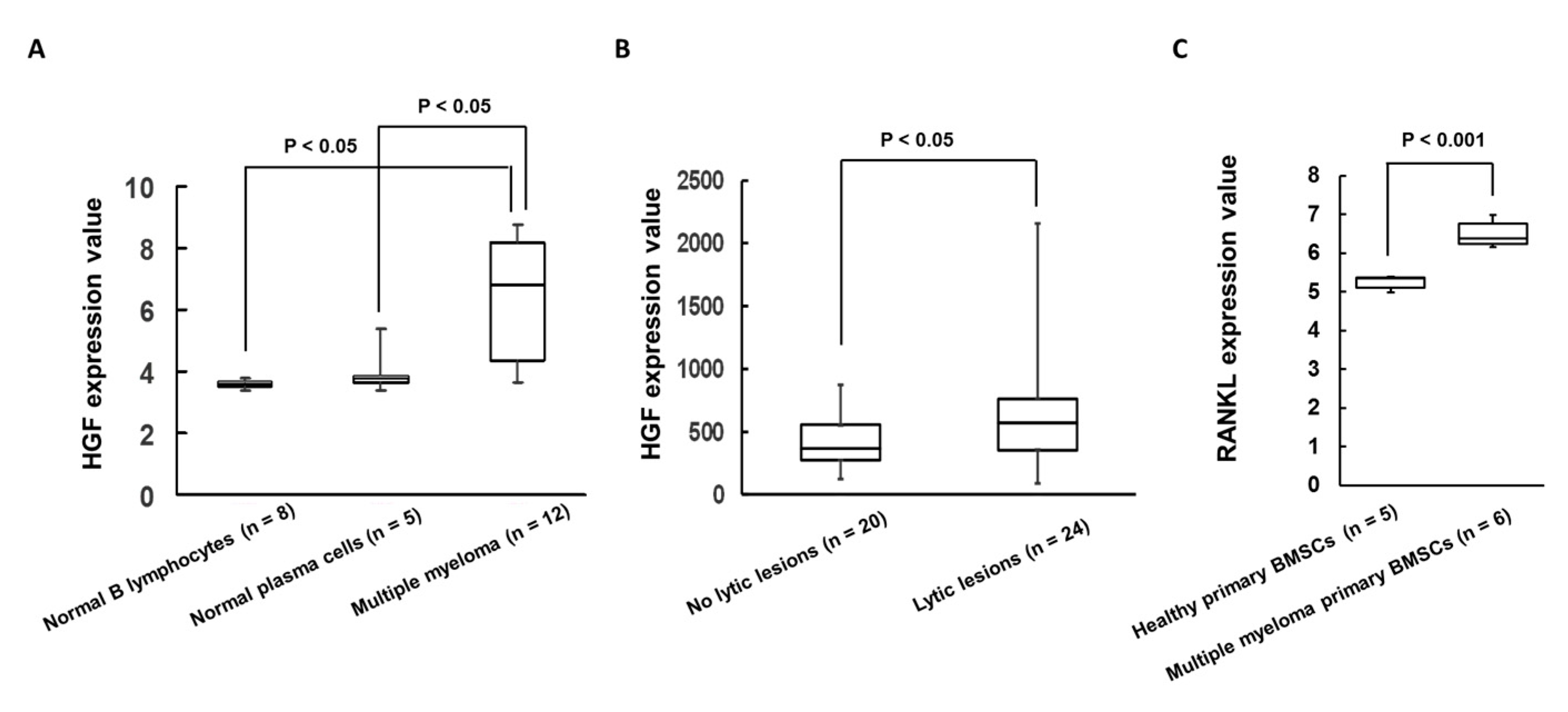
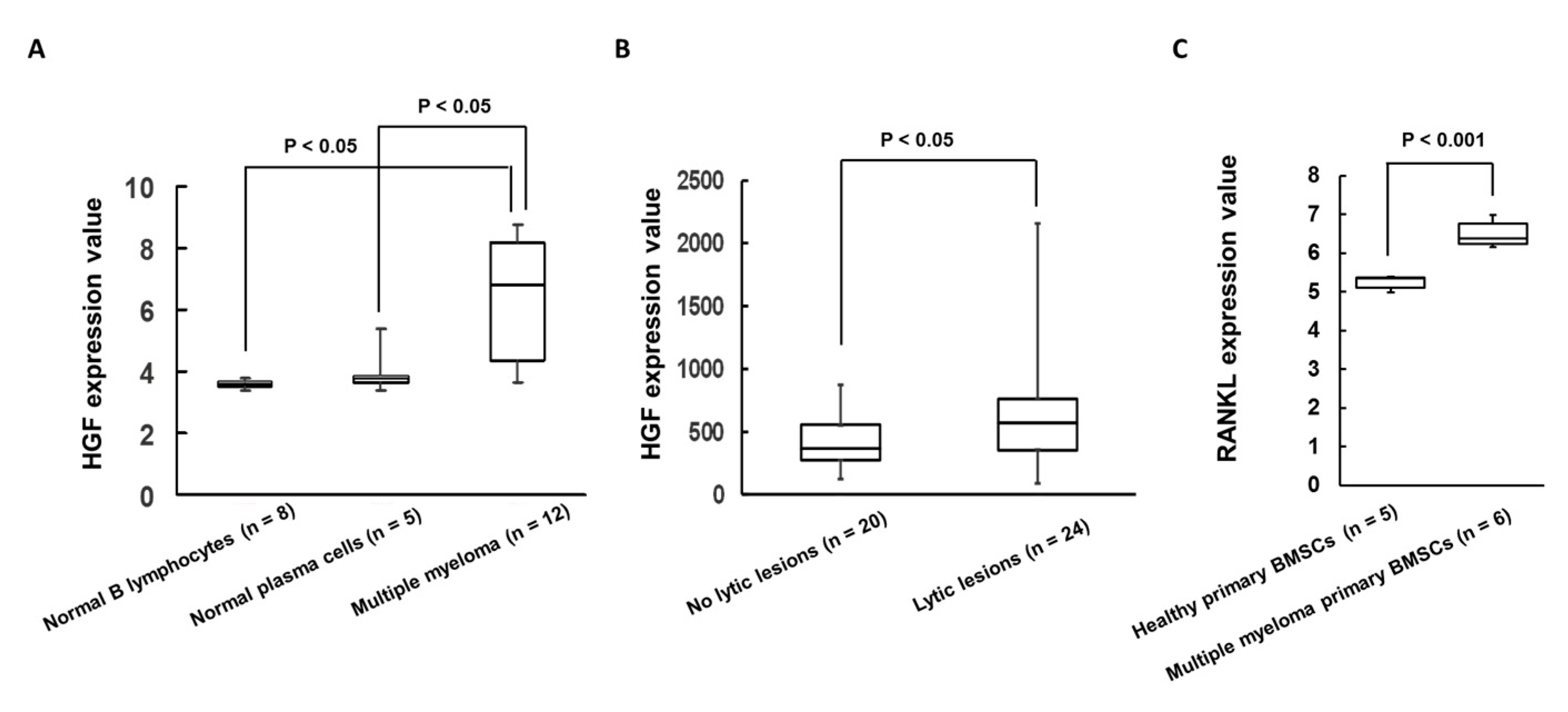
2.1. Expression of HGF Was Increased in Patients with MM Cells and Bone Lytic Lesions in MM Patients
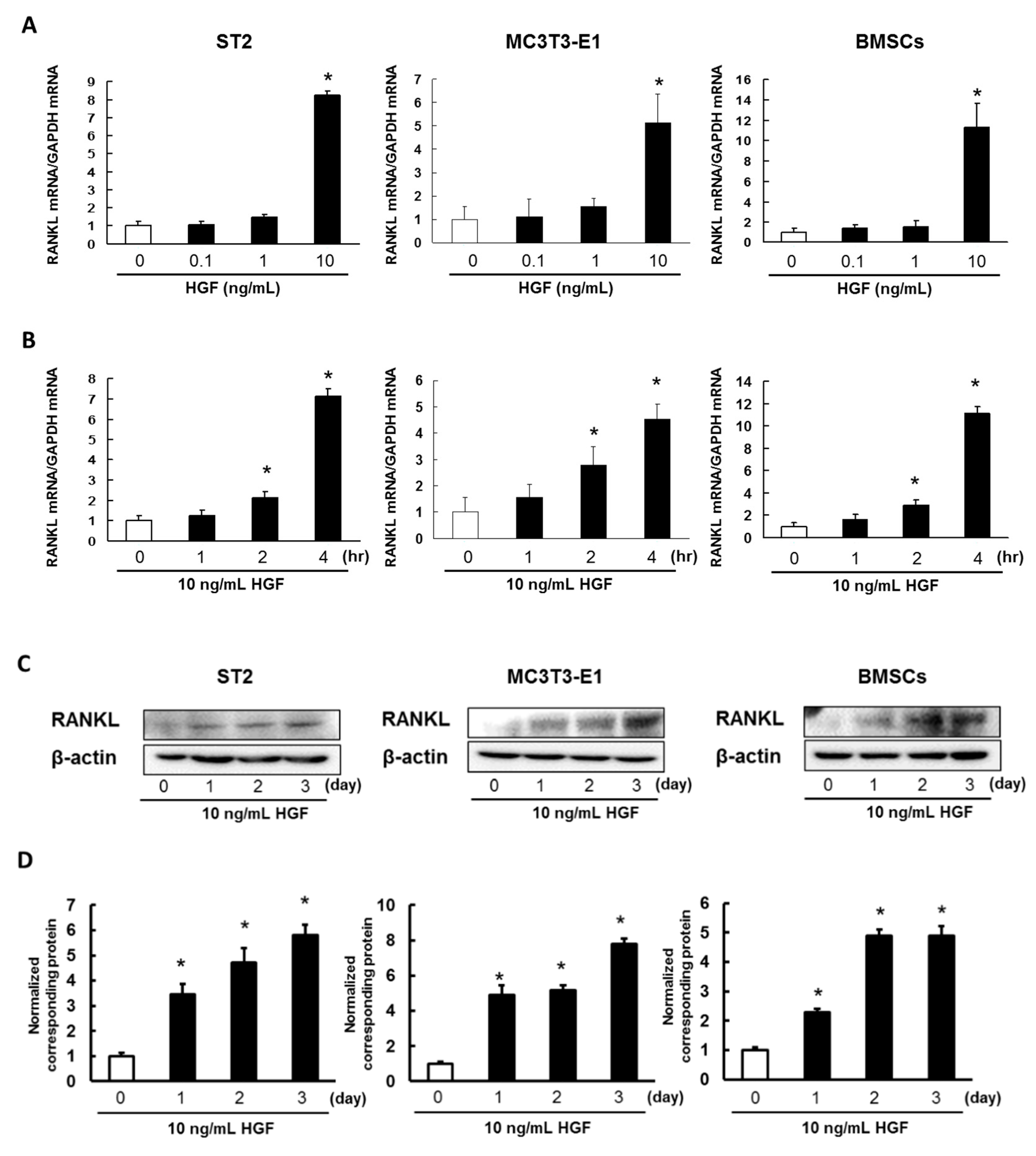
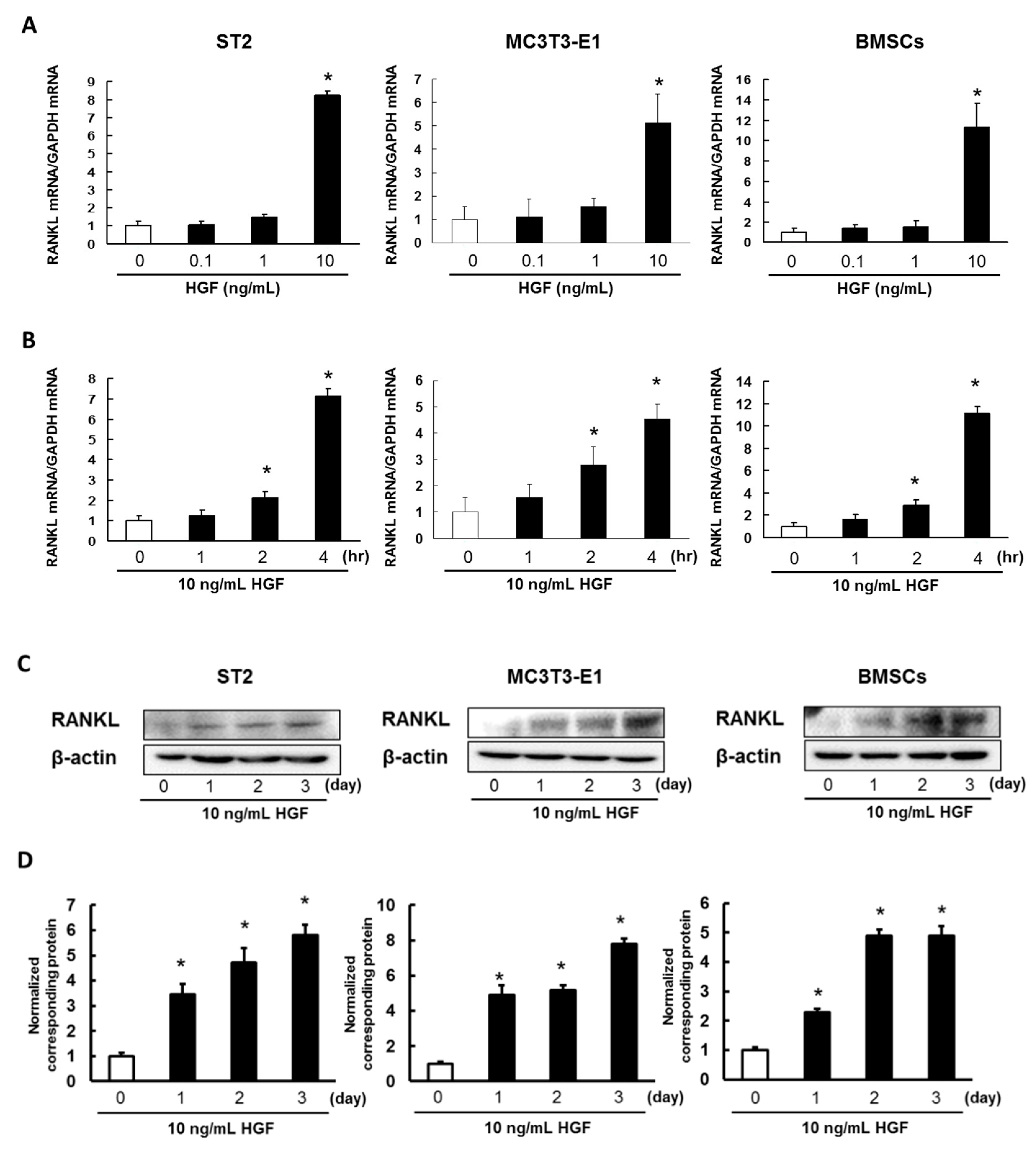
2.2. HGF Promotes Expression of RANKL in ST2 Cells, MC3T3-E1 Cells, and Mouse BMSCs
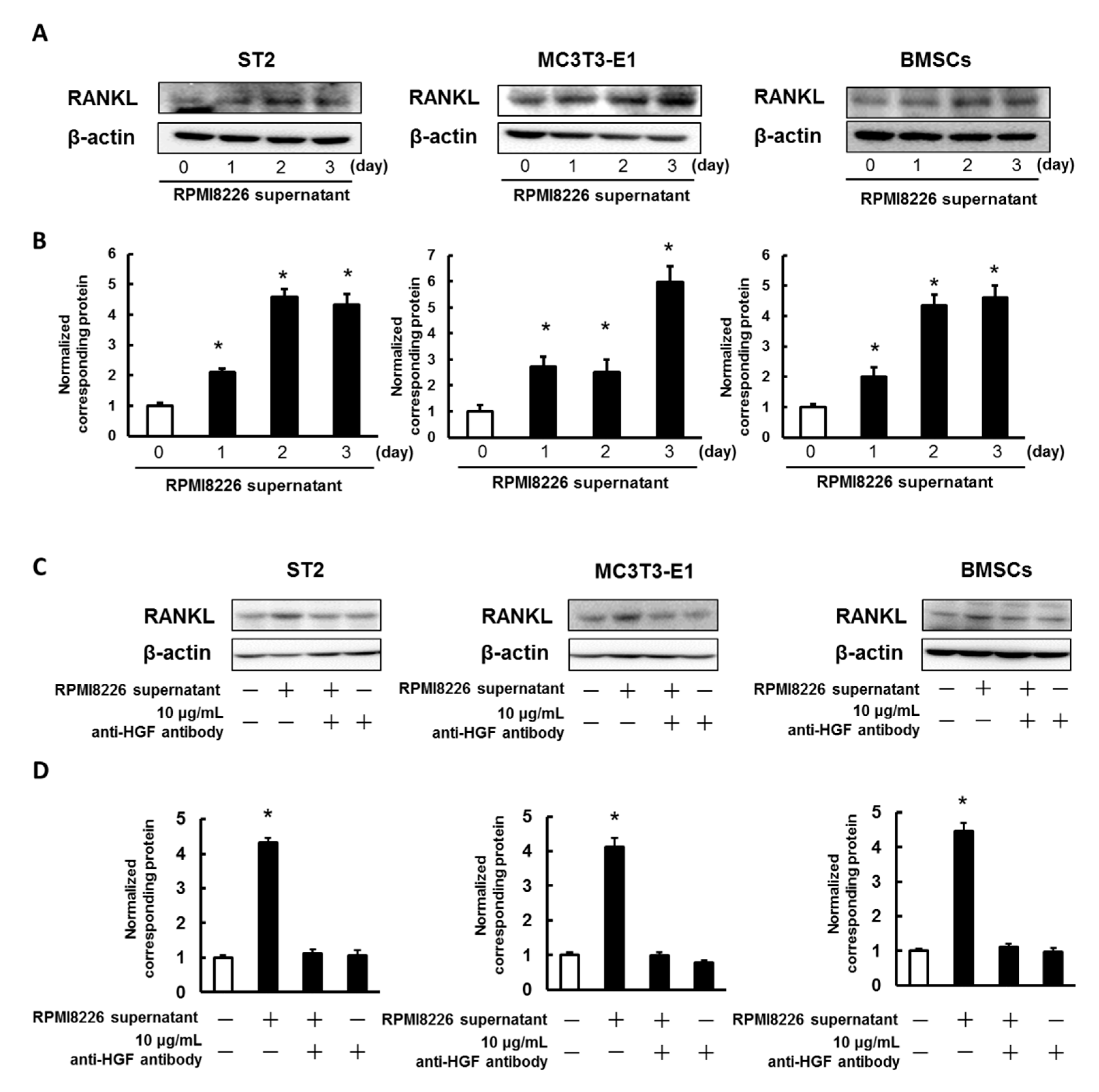
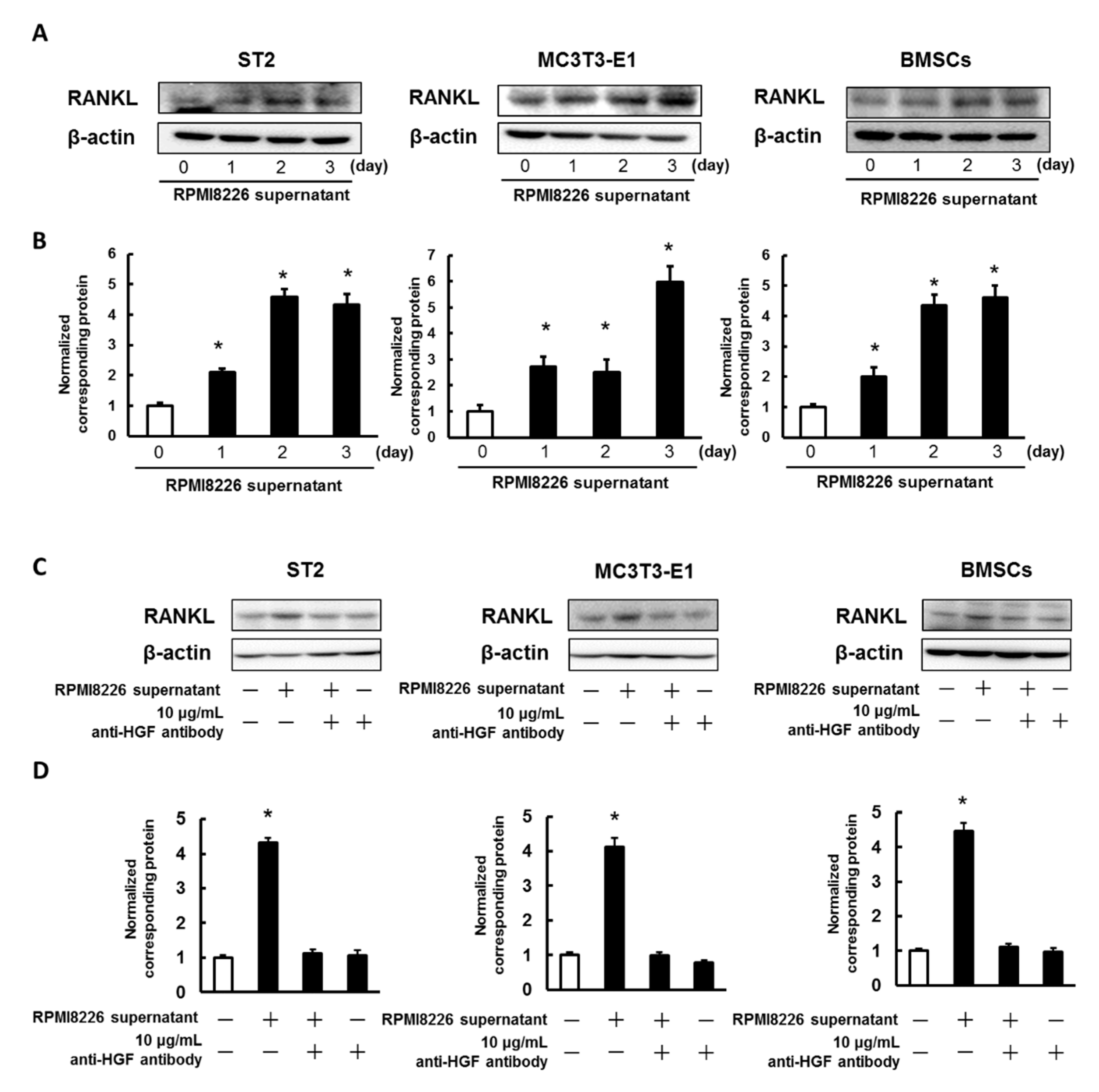
2.3. Enhancing the Expression of RANKL in ST2 Cells, MC3T3-E1 Cells, and Mouse BMSCs Using the Culture Media Collected from Multiple Myeloma Cells
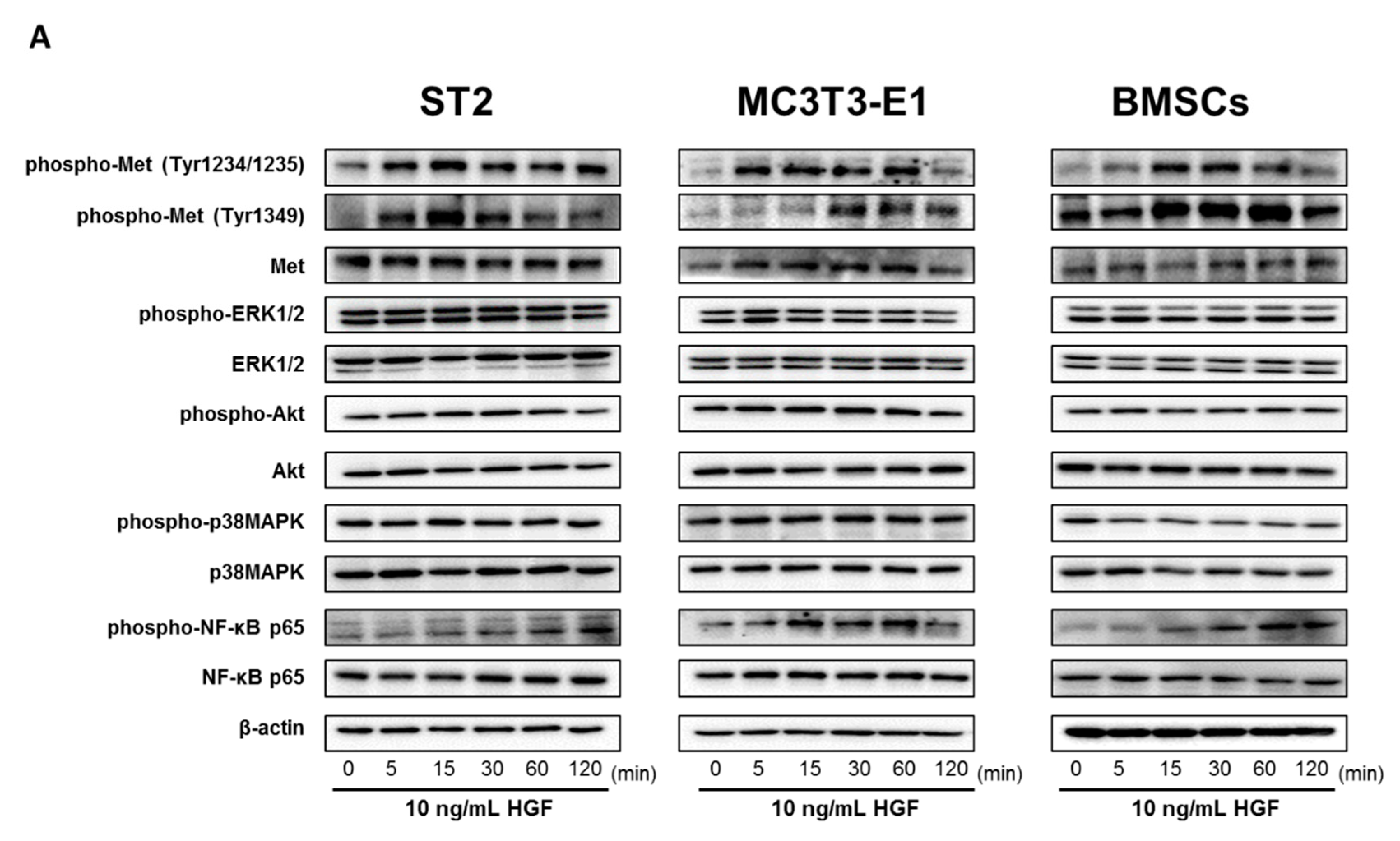
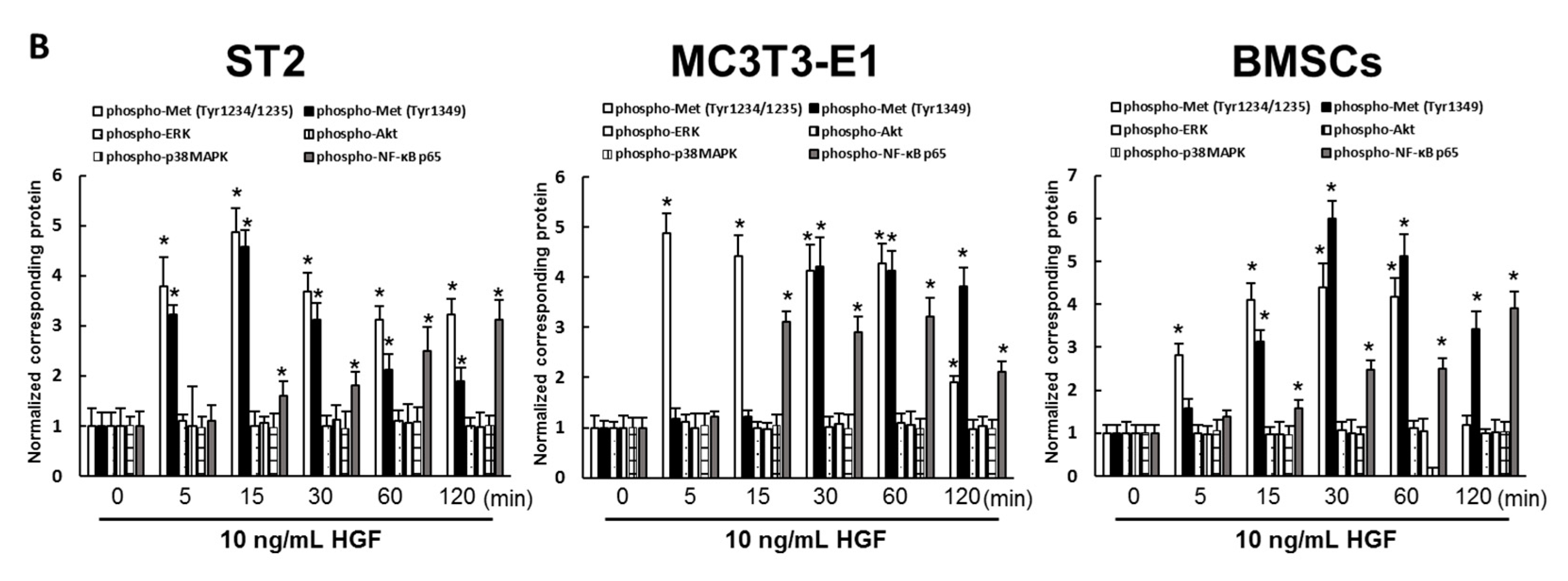
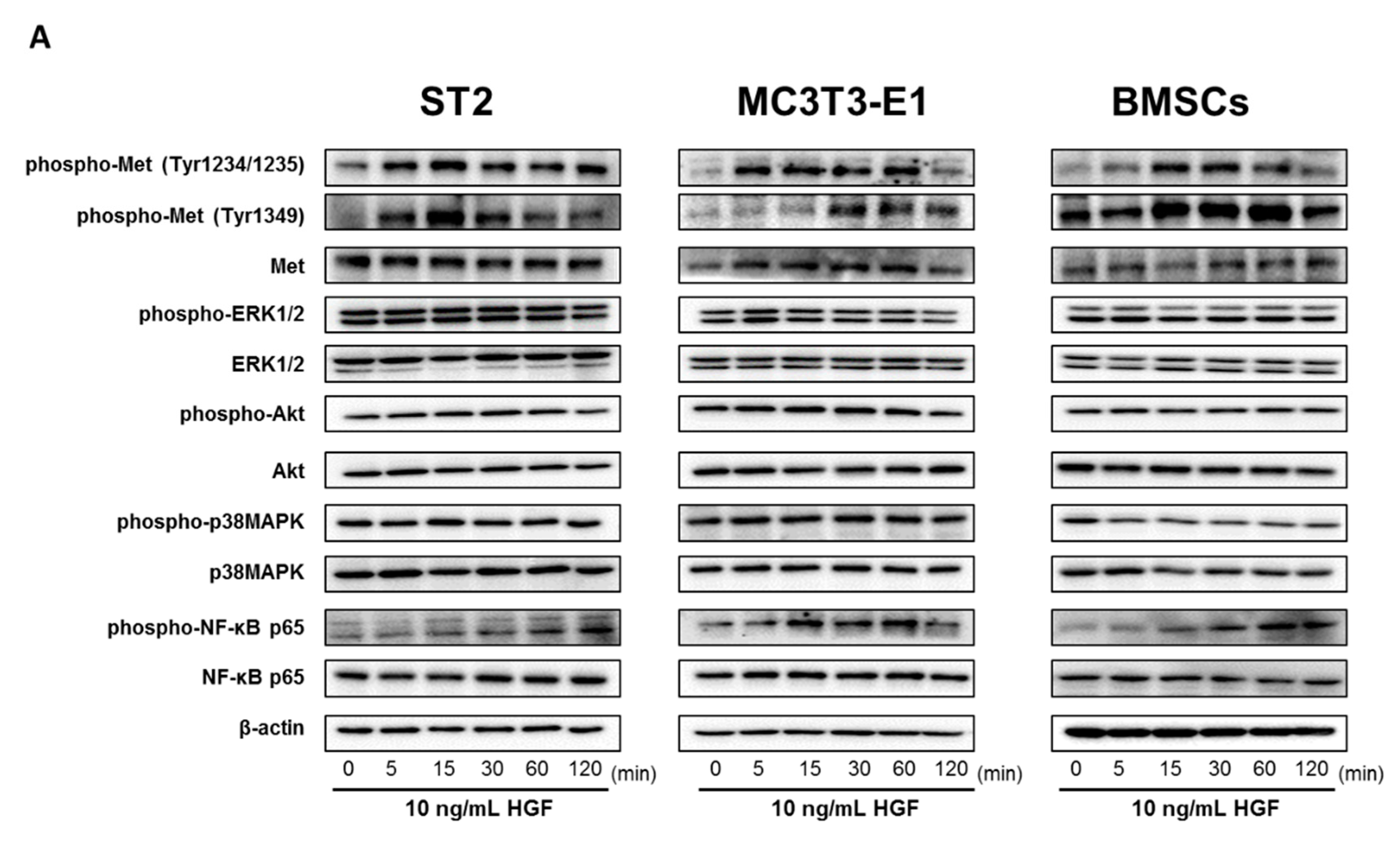
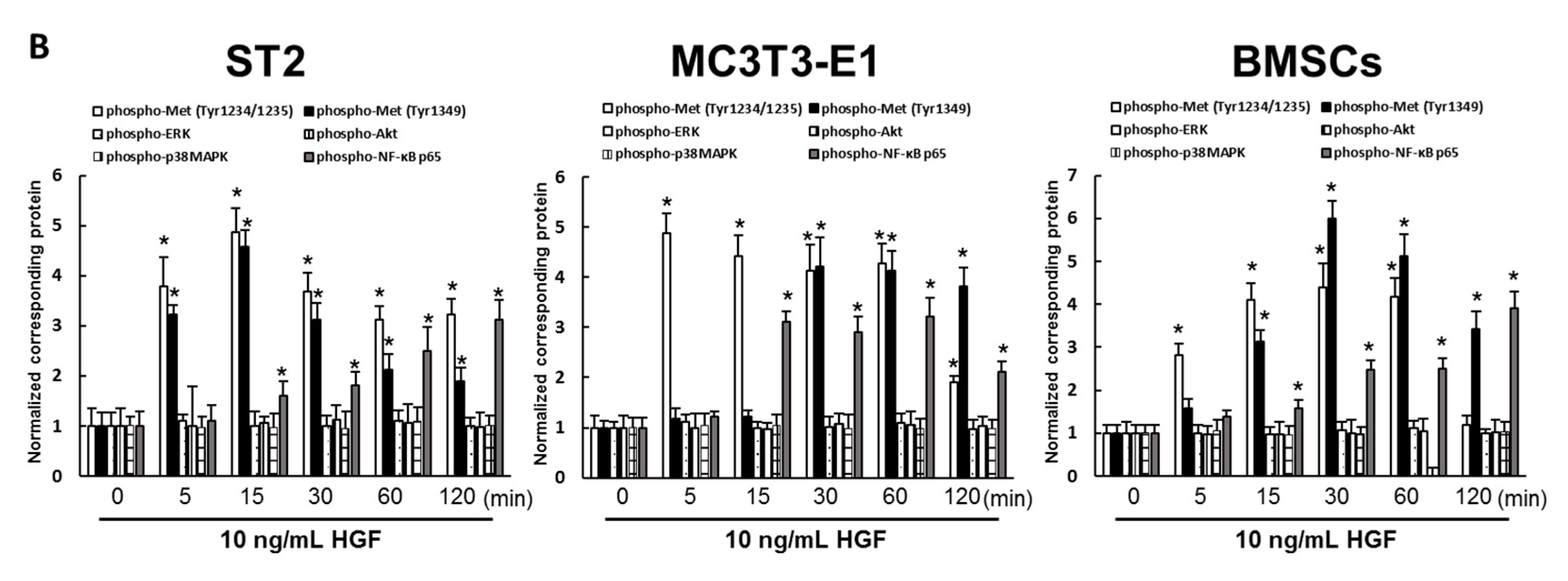
2.4. HGF Activated the Met/NF-κB Pathway in ST2 Cells, MC3T3-E1 Cells, and Mouse BMSCs
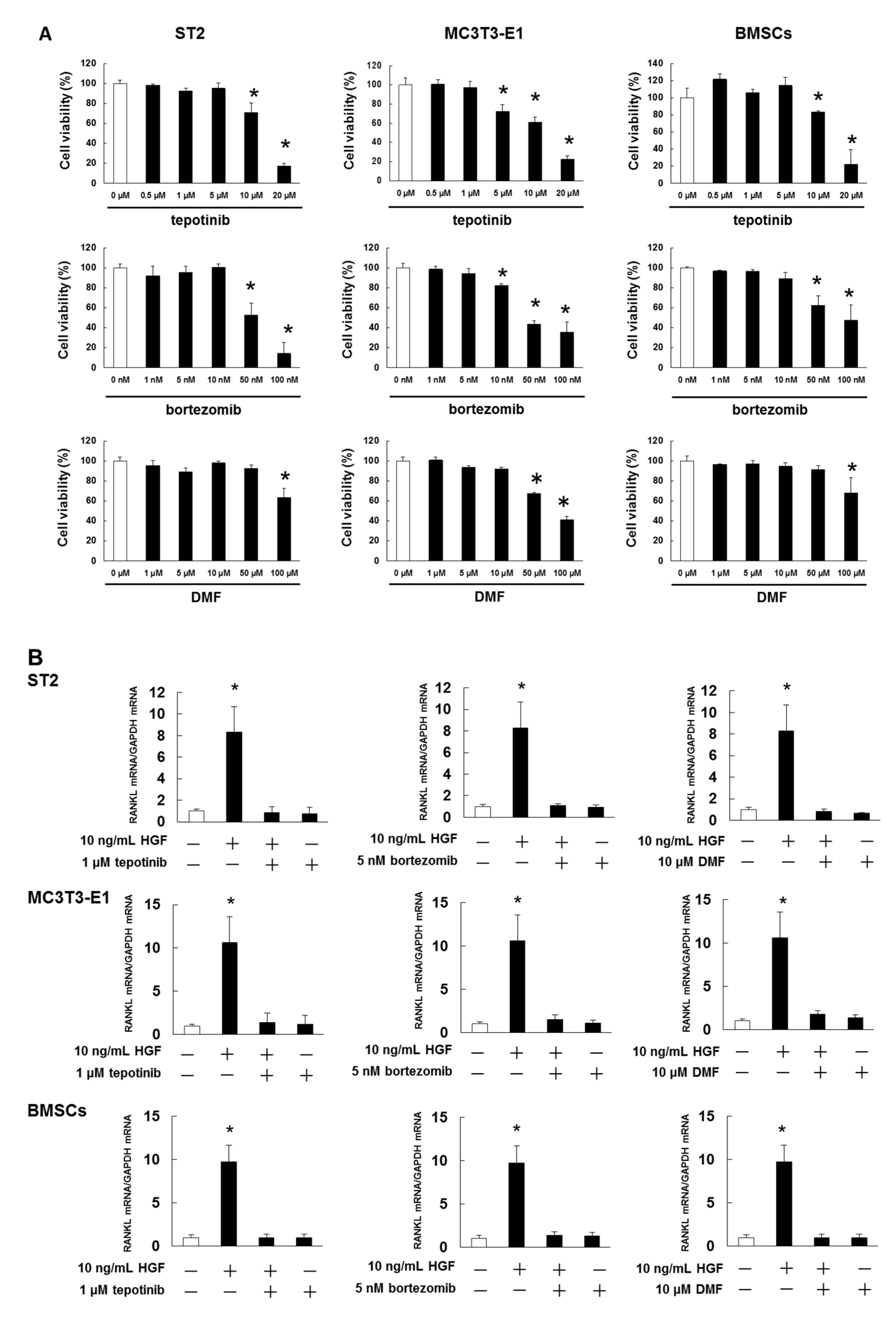
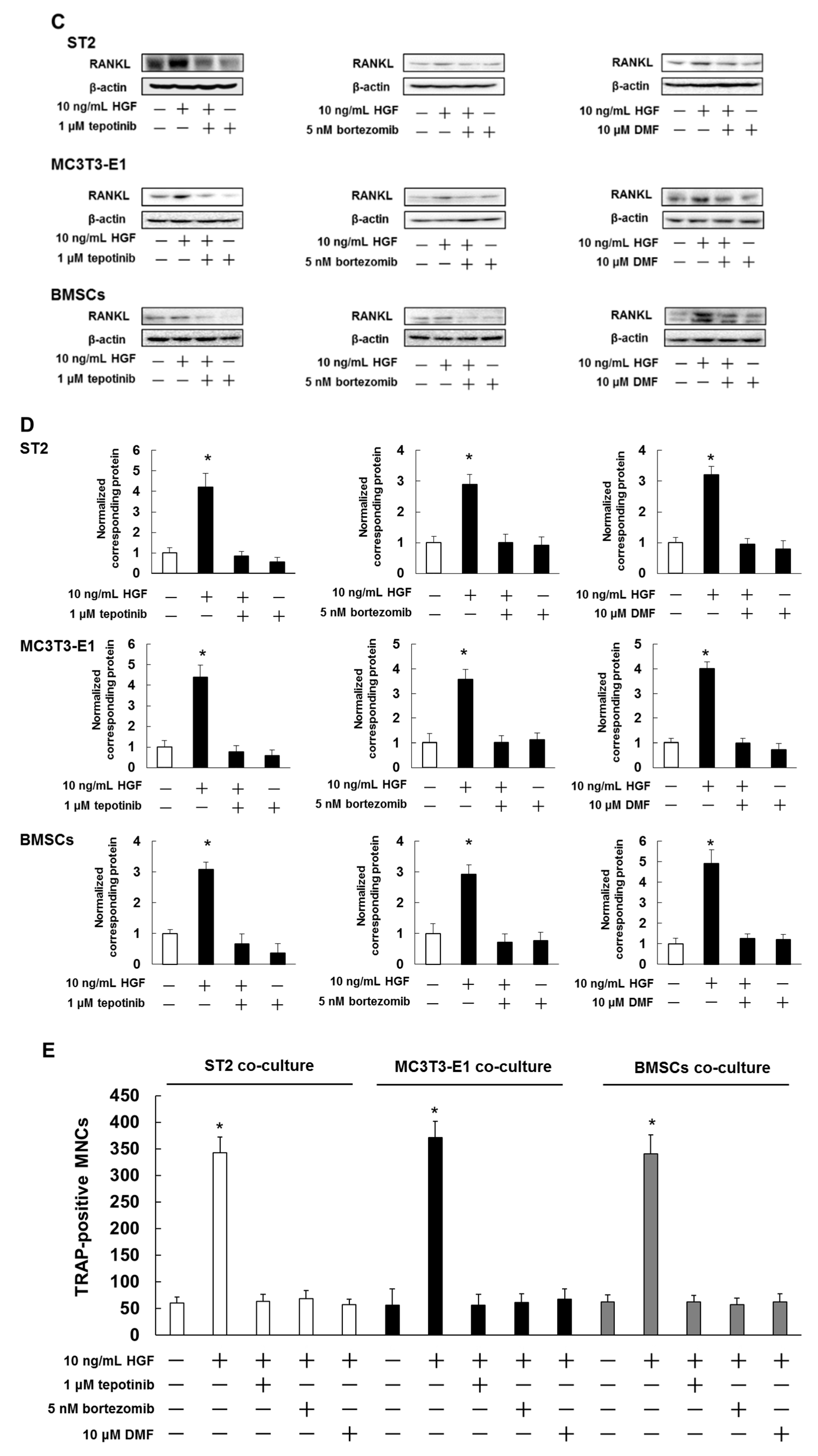
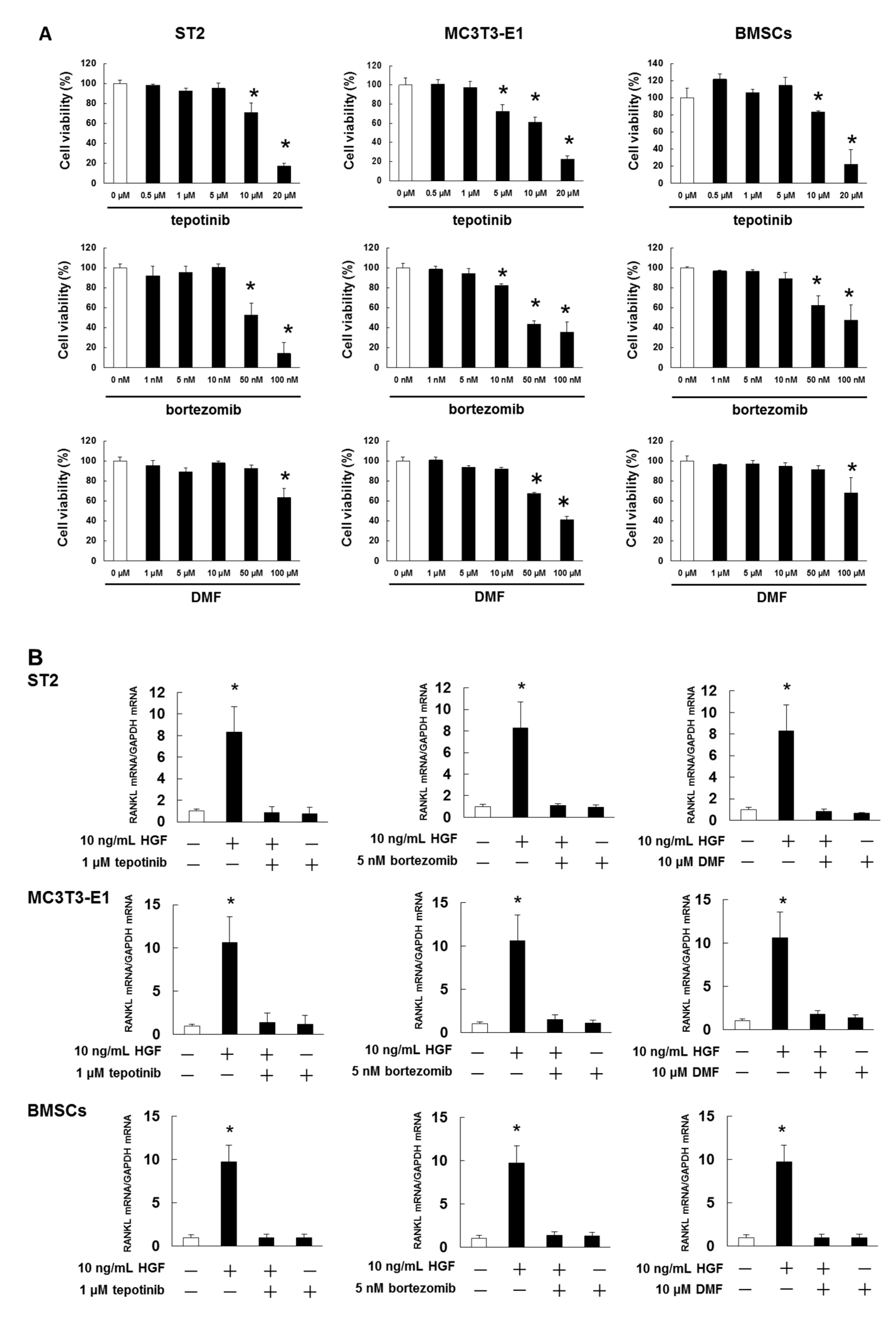
2.5. Met and NF-κB Inhibitors Suppressed HGF-Induced RANKL Expression
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Quantitative Real-Time Polymerase Chain Reaction (PCR)
4.3. Western Blotting
4.4. Trypan Blue Dye Exclusion Assay
4.5. Microarray Dataset Resources
4.6. TRAP Staining
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MM | Multiple myeloma |
| RANKL | Receptor activator of nuclear factor κB ligand |
| BMSCs | Bone marrow stromal cells |
| HGF | Hepatocyte growth factor |
| NF-κB | Nuclear factor κB |
| RANK | Receptor activator of nuclear factor κB |
| IL-6 | Interleukin-6 |
| TNFα | Tumor necrosis factor alpha |
| MIP-1α | Macrophage inflammatory protein-1α |
| bFGF | Basic fibroblast growth factor |
| ERK1/2 | Extracellular regulated protein kinase 1/2 |
| p38MAPK | p38 mitogen activated protein kinase |
| M-CSF | Macrophage-colony stimulating factor |
| VEGF | Vascular endothelial growth factor |
References
- Ring, E.S.; Lawson, M.A.; Snowden, J.A.; Jolley, I.; Chantry, A.D. New agents in the Treatment of Myeloma Bone Disease. Calcif. Tissue Int. 2018, 102, 196–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, M.M.; Reagan, M.R.; Youlten, S.E.; Mohanty, S.T.; Seckinger, A.; Terry, R.L.; Pettitt, J.A.; Simic, M.K.; Cheng, T.L.; Morse, A.; et al. Inhibiting the osteocyte-specific protein sclerostin increases bone mass and fracture resistance in multiple myeloma. Blood 2017, 129, 3452–3464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida, S.; Fujii, Y.; Yoshioka, S.; Kikuichi, S.; Tsubaki, M.; Irimajiri, K. A new bisphosphonate, YM529 induces apoptosis in HL60 cells by decreasing phosphorylation of single survival signal ERK. Life Sci. 2003, 73, 2655–2664. [Google Scholar] [CrossRef]
- Nishida, S.; Kikuichi, S.; Haga, H.; Yoshioka, S.; Tsubaki, M.; Fujii, K.; Irimajiri, K. Apoptosis-inducing effect of a new bisphosphonate, YM529, on various hematopoietic tumor cell lines. Biol. Pharm. Bull. 2003, 26, 96–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsubaki, M.; Itoh, T.; Satou, T.; Imano, M.; Komai, M.; Ogawa, N.; Mukai, J.; Nishida, S. Nitrogen-containing bisphosphonates induce apoptosis of hematopoietic tumor cells via inhibition of Ras signaling pathways and Bim-mediated activation of the intrinsic apoptotic pathway. Biochem. Pharmacol. 2013, 85, 163–172. [Google Scholar] [CrossRef]
- Tsubaki, M.; Komai, M.; Itoh, T.; Imano, M.; Sakamoto, K.; Shimaoka, H.; Takeda, T.; Ogawa, N.; Mashimo, K.; Fujiwara, D.; et al. Nitrogen-containing bisphosphonates inhibit RANKL- and M-CSF-induced osteoclast formation through the inhibition of ERK1/2 and Akt activation. J. Biomed. Sci. 2014, 21, 10. [Google Scholar] [CrossRef] [Green Version]
- Terpos, E.; Morgan, G.; Dimopoulos, M.A.; Drake, M.T.; Lentzsch, S.; Raje, N.; Sezer, O.; García-Sanz, R.; Shimizu, K.; Turesson, I.; et al. International Myeloma Working Group recommendations for the treatment of multiple myeloma-related bone disease. J. Clin. Oncol. 2013, 31, 2347–2357. [Google Scholar] [CrossRef] [Green Version]
- Terpos, E.; Ntanasis-Stathopoulos, I.; Gavriatopoulou, M.; Dimopoulos, M.A. Pathogenesis of bone disease in multiple myeloma: From bench to bedside. Blood Cancer J. 2018, 8, 7. [Google Scholar] [CrossRef] [Green Version]
- Heusschen, R.; Muller, J.; Duray, E.; Withofs, N.; Bolomsky, A.; Baron, F.; Beguin, Y.; Menu, E.; Ludwig, H.; Caers, J. Molecular mechanisms, current management and next generation therapy in myeloma bone disease. Leuk. Lymphoma 2018, 59, 14–28. [Google Scholar] [CrossRef]
- Silvestris, F.; Lombardi, L.; De Matteo, M.; Bruno, A.; Dammacco, F. Myeloma bone disease: Pathogenetic mechanisms and clinical assessment. Leuk Res. 2007, 31, 129–138. [Google Scholar] [CrossRef]
- Nishida, S.; Tsubaki, M.; Hoshino, M.; Namimatsu, A.; Uji, H.; Yoshioka, S.; Tanimori, Y.; Yanae, M.; Iwaki, M.; Irimajiri, K. Nitrogen-containing bisphosphonate, YM529/ONO-5920 (a novel minodronic acid), inhibits RANKL expression in a cultured bone marrow stromal cell line ST2. Biochem. Biophys. Res. Commun. 2005, 328, 91–97. [Google Scholar] [CrossRef]
- Tsubaki, M.; Satou, T.; Itoh, T.; Imano, M.; Yanae, M.; Kato, C.; Takagoshi, R.; Komai, M.; Nishida, S. Bisphosphonate- and statin-induced enhancement of OPG expression and inhibition of CD9, M-CSF, and RANKL expressions via inhibition of the Ras/MEK/ERK pathway and activation of p38MAPK in mouse bone marrow stromal cell line ST2. Mol. Cell. Endocrinol. 2012, 361, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Udagawa, N.; Takahashi, N.; Jimi, E.; Matsuzaki, K.; Tsurukai, T.; Itoh, K.; Nakagawa, N.; Yasuda, H.; Goto, M.; Tsuda, E.; et al. Osteoblasts/stromal cells stimulate osteoclast activation through expression of osteoclast differentiation factor/RANKL but not macrophage colony-stimulating factor: Receptor activator of NF-kappa B ligand. Bone 1999, 25, 517–523. [Google Scholar] [CrossRef]
- Tsubaki, M.; Kato, C.; Manno, M.; Ogaki, M.; Satou, T.; Itoh, T.; Kusunoki, T.; Tanimori, Y.; Fujiwara, K.; Matsuoka, H.; et al. Macrophage inflammatory protein-1alpha (MIP-1alpha) enhances a receptor activator of nuclear factor kappaB ligand (RANKL) expression in mouse bone marrow stromal cells and osteoblasts through MAPK and PI3K/Akt pathways. Mol. Cell. Biochem. 2007, 304, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Tsubaki, M.; Kato, C.; Nishinobo, M.; Ogaki, M.; Satou, T.; Ito, T.; Kusunoki, T.; Fujiwara, K.; Yamazoe, Y.; Nishida, S. Nitrogen-containing bisphosphonate, YM529/ONO-5920, inhibits macrophage inflammatory protein 1 alpha expression and secretion in mouse myeloma cells. Cancer Sci. 2008, 99, 152–158. [Google Scholar]
- Tsubaki, M.; Kato, C.; Isono, A.; Kaneko, J.; Isozaki, M.; Satou, T.; Itoh, T.; Kidera, Y.; Tanimori, Y.; Yanae, M.; et al. Macrophage inflammatory protein-1α induces osteoclast formation by activation of the MEK/ERK/c-Fos pathway and inhibition of the p38MAPK/IRF-3/IFN-β pathway. J. Cell. Biochem. 2010, 111, 1661–1672. [Google Scholar] [CrossRef]
- Tsubaki, M.; Komai, M.; Itoh, T.; Imano, M.; Sakamoto, K.; Shimaoka, H.; Ogawa, N.; Mashimo, K.; Fujiwara, D.; Takeda, T.; et al. Inhibition of the tumour necrosis factor-alpha autocrine loop enhances the sensitivity of multiple myeloma cells to anticancer drugs. Eur. J. Cancer 2013, 49, 3708–3717. [Google Scholar] [CrossRef]
- Tsubaki, M.; Takeda, T.; Sakamoto, K.; Shimaoka, H.; Fujita, A.; Itoh, T.; Imano, M.; Mashimo, K.; Fujiwara, D.; Sakaguchi, K.; et al. Bisphosphonates and statins inhibit expression and secretion of MIP-1α via suppression of Ras/MEK/ERK/AML-1A and Ras/PI3K/Akt/AML-1A pathways. Am. J. Cancer Res. 2014, 5, 168–179. [Google Scholar]
- Tsubaki, M.; Mashimo, K.; Takeda, T.; Kino, T.; Fujita, A.; Itoh, T.; Imano, M.; Sakaguchi, K.; Satou, T.; Nishida, S. Statins inhibited the MIP-1α expression via inhibition of Ras/ERK and Ras/Akt pathways in myeloma cells. Biomed. Pharm. 2016, 78, 23–29. [Google Scholar] [CrossRef]
- Tsubaki, M.; Takeda, T.; Tomonari, Y.; Mashimo, K.; Koumoto, Y.I.; Hoshida, S.; Itoh, T.; Imano, M.; Satou, T.; Sakaguchi, K.; et al. The MIP-1α autocrine loop contributes to decreased sensitivity to anticancer drugs. J. Cell. Physiol. 2018, 233, 4258–4271. [Google Scholar] [CrossRef]
- Kuku, I.; Bayraktar, M.R.; Kaya, E.; Erkurt, M.A.; Bayraktar, N.; Cikim, K.; Aydogdu, I. Serum proinflammatory mediators at different periods of therapy in patients with multiple myeloma. Mediators Inflamm. 2005, 2005, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Lauta, V.M. A review of the cytokine network in multiple myeloma: Diagnostic, prognostic, and therapeutic implications. Cancer 2003, 97, 2440–2452. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Luetkens, T.; Kobold, S.; Hildebrandt, Y.; Gordic, M.; Lajmi, N.; Meyer, S.; Bartels, K.; Zander, A.R.; Bokemeyer, C.; et al. The cytokine/chemokine pattern in the bone marrow environment of multiple myeloma patients. Exp. Hematol. 2010, 38, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Rampa, C.; Tian, E.; Våtsveen, T.K.; Buene, G.; Slørdahl, T.S.; Børset, M.; Waage, A.; Sundan, A. Identification of the source of elevated hepatocyte growth factor levels in multiple myeloma patients. Biomark. Res. 2014, 2, 8. [Google Scholar] [CrossRef] [Green Version]
- Phillip, C.J.; Zaman, S.; Shentu, S.; Balakrishnan, K.; Zhang, J.; Baladandayuthapani, V.; Taverna, P.; Redkar, S.; Wang, M.; Stellrecht, C.M.; et al. Targeting MET kinase with the small-molecule inhibitor amuvatinib induces cytotoxicity in primary myeloma cells and cell lines. J. Hematol. Oncol. 2013, 6, 92. [Google Scholar] [CrossRef] [Green Version]
- Rocci, A.; Gambella, M.; Aschero, S.; Baldi, I.; Trusolino, L.; Cavallo, F.; Gay, F.; Larocca, A.; Magarotto, V.; Omedè, P.; et al. MET dysregulation is a hallmark of aggressive disease in multiple myeloma patients. Br. J. Haematol. 2014, 164, 841–850. [Google Scholar] [CrossRef]
- Taylor, R.M.; Kashima, T.G.; Knowles, H.J.; Athanasou, N.A. VEGF, FLT3 ligand, PlGF and HGF can substitute for M-CSF to induce human osteoclast formation: Implications for giant cell tumour pathobiology. Lab. Investig. 2012, 92, 1398–1406. [Google Scholar] [CrossRef]
- Standal, T.; Abildgaard, N.; Fagerli, U.M.; Stordal, B.; Hjertner, O.; Borset, M.; Sundan, A. HGF inhibits BMP-induced osteoblastogenesis: Possible implications for the bone disease of multiple myeloma. Blood 2007, 109, 3024–3030. [Google Scholar] [CrossRef] [Green Version]
- Lath, D.L.; Buckle, C.H.; Evans, H.R.; Fisher, M.; Down, J.M.; Lawson, M.A.; Chantry, A.D. ARQ-197, a small-molecule inhibitor of c-Met, reduces tumour burden and prevents myeloma-induced bone disease in vivo. PLoS ONE 2018, 13, e0199517. [Google Scholar] [CrossRef] [Green Version]
- Boissy, P.; Andersen, T.L.; Lund, T.; Kupisiewicz, K.; Plesner, T.; Delaissé, J.M. Pulse treatment with the proteasome inhibitor bortezomib inhibits osteoclast resorptive activity in clinically relevant conditions. Leuk Res. 2008, 32, 1661–1668. [Google Scholar] [CrossRef]
- Takeda, T.; Tsubaki, M.; Asano, R.; Itoh, T.; Imano, M.; Satou, T.; Nishida, S. Dimethyl fumarate suppresses metastasis and growth of melanoma cells by inhibiting the nuclear translocation of NF-κB. J. Derm. Sci. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Tsubaki, M.; Ogawa, N.; Takeda, T.; Sakamoto, K.; Shimaoka, H.; Fujita, A.; Itoh, T.; Imano, M.; Satou, T.; Nishida, S. Dimethyl fumarate induces apoptosis of hematopoietic tumor cells via inhibition of NF-κB nuclear translocation and down-regulation of Bcl-xL and XIAP. Biomed. Pharmacother. 2014, 68, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Tsubaki, M.; Komai, M.; Fujimoto, S.; Itoh, T.; Imano, M.; Sakamoto, K.; Shimaoka, H.; Takeda, T.; Ogawa, N.; Mashimo, K.; et al. Activation of NF-κB by the RANKL/RANK system up-regulates snail and twist expressions and induces epithelial-to-mesenchymal transition in mammary tumor cell lines. J. Exp. Clin. Cancer Res. 2013, 32, 62. [Google Scholar] [CrossRef] [Green Version]
- Adamopoulos, I.E.; Xia, Z.; Lau, Y.S.; Athanasou, N.A. Hepatocyte growth factor can substitute for M-CSF to support osteoclastogenesis. Biochem. Biophys. Res. Commun. 2006, 350, 478–483. [Google Scholar] [CrossRef]
- Lean, J.M.; Fuller, K.; Chambers, T.J. FLT3 ligand can substitute for macrophage colony-stimulating factor in support of osteoclast differentiation and function. Blood 2001, 98, 2707–2713. [Google Scholar] [CrossRef] [PubMed]
- Niida, S.; Kondo, T.; Hiratsuka, S.; Hayashi, S.; Amizuka, N.; Noda, T.; Ikeda, K.; Shibuya, M. VEGF receptor 1 signaling is essential for osteoclast development and bone marrow formation in colony-stimulating factor 1-deficient mice. Proc. Natl. Acad. Sci. USA 2005, 102, 14016–14021. [Google Scholar] [CrossRef] [Green Version]
- Jurczyszyn, A.; Czepiel, J.; Biesiada, G.; Gdula-Argasińska, J.; Cibor, D.; Owczarek, D.; Perucki, W.; Skotnicki, A.B. HGF, sIL-6R and TGF-β1 Play a Significant Role in the Progression of Multiple Myeloma. J. Cancer 2014, 5, 518–524. [Google Scholar] [CrossRef] [Green Version]
- Sfiridaki, K.; Pappa, C.A.; Tsirakis, G.; Kanellou, P.; Kaparou, M.; Stratinaki, M.; Sakellaris, G.; Kontakis, G.; Alexandrakis, M.G. Angiogenesis-related cytokines, RANKL, and osteoprotegerin in multiple myeloma patients in relation to clinical features and response to treatment. Mediators Inflamm. 2011, 2011, 867576. [Google Scholar] [CrossRef] [Green Version]
- Tsirakis, G.; Roussou, P.; Pappa, C.A.; Kolovou, A.; Vasilokonstantaki, C.; Miminas, I.; Kyriakaki, S.; Alegakis, A.; Alexandrakis, M.G. Increased serum levels of MIP-1alpha correlate with bone disease and angiogenic cytokines in patients with multiple myeloma. Med. Oncol. 2014, 31, 778. [Google Scholar] [CrossRef]
- Mondello, P.; Cuzzocrea, S.; Navarra, M.; Mian, M. Bone marrow micro-environment is a crucial player for myelomagenesis and disease progression. Oncotarget 2017, 8, 20394–20409. [Google Scholar] [CrossRef] [Green Version]
- Kwan Tat, S.; Padrines, M.; Théoleyre, S.; Heymann, D.; Fortun, Y. IL-6, RANKL, TNF-alpha/IL-1: Interrelations in bone resorption pathophysiology. Cytokine Growth Factor Rev. 2004, 15, 49–60. [Google Scholar] [PubMed]
- Gupta, D.; Treon, S.P.; Shima, Y.; Hideshima, T.; Podar, K.; Tai, Y.T.; Lin, B.; Lentzsch, S.; Davies, F.E.; Chauhan, D.; et al. Adherence of multiple myeloma cells to bone marrow stromal cells upregulates vascular endothelial growth factor secretion: Therapeutic applications. Leukemia 2001, 15, 1950–1961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaman, S.; Shentu, S.; Yang, J.; He, J.; Orlowski, R.Z.; Stellrecht, C.M.; Gandhi, V. Targeting the pro-survival protein MET with tivantinib (ARQ 197) inhibits growth of multiple myeloma cells. Neoplasia 2015, 17, 289–300. [Google Scholar] [CrossRef] [Green Version]
- Tsubaki, M.; Takeda, T.; Kino, T.; Sakai, K.; Itoh, T.; Imano, M.; Nakayama, T.; Nishio, K.; Satou, T.; Nishida, S. Contributions of MET activation to BCR-ABL1 tyrosine kinase inhibitor resistance in chronic myeloid leukemia cells. Oncotarget 2017, 8, 38717–38730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Blair, H.C.; Shapiro, I.M.; Wang, B. The Proteasome Inhibitor Carfilzomib Suppresses Parathyroid Hormone-induced Osteoclastogenesis through a RANKL-mediated Signaling Pathway. J. Biol. Chem. 2015, 290, 16918–16928. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Chen, Y.; Usmani, S.Z.; Ye, S.; Qiang, W.; Papanikolaou, X.; Heuck, C.J.; Yaccoby, S.; Williams, B.O.; Van Rhee, F.; et al. Characterization of the molecular mechanism of the bone-anabolic activity of carfilzomib in multiple myeloma. PLoS ONE 2013, 8, e74191. [Google Scholar] [CrossRef] [Green Version]
- Obri, A.; Makinistoglu, M.P.; Zhang, H.; Karsenty, G. HDAC4 integrates PTH and sympathetic signaling in osteoblasts. J. Cell. Biol. 2014, 205, 771–780. [Google Scholar] [CrossRef]
- von Metzler, I.; Krebbel, H.; Hecht, M.; Manz, R.A.; Fleissner, C.; Mieth, M.; Kaiser, M.; Jakob, C.; Sterz, J.; Kleeberg, L.; et al. Bortezomib inhibits human osteoclastogenesis. Leukemia 2007, 21, 2025–2034. [Google Scholar] [CrossRef]
- Mohty, M.; Malard, F.; Mohty, B.; Savani, B.; Moreau, P.; Terpos, E. The effects of bortezomib on bone disease in patients with multiple myeloma. Cancer 2014, 120, 618–623. [Google Scholar] [CrossRef] [Green Version]
- Terpos, E.; Christoulas, D.; Katodritou, E.; Bratengeier, C.; Gkotzamanidou, M.; Michalis, E.; Delimpasi, S.; Pouli, A.; Meletis, J.; Kastritis, E.; et al. Elevated circulating sclerostin correlates with advanced disease features and abnormal bone remodeling in symptomatic myeloma: Reduction post-bortezomib monotherapy. Int. J. Cancer 2012, 131, 1466–1471. [Google Scholar] [CrossRef]
- Mazière, C.; Salle, V.; Gomila, C.; Mazière, J.C. Oxidized low density lipoprotein enhanced RANKL expression in human osteoblast-like cells. Involvement of ERK, NFkappaB and NFAT. Biochim. Biophys. Acta 2013, 1832, 1756–1764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slørdahl, T.S.; Denayer, T.; Moen, S.H.; Standal, T.; Børset, M.; Ververken, C.; Rø, T.B. Anti-c-MET Nanobody—A new potential drug in multiple myeloma treatment. Eur. J. Haematol. 2013, 91, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Ramani, V.C.; Yang, Y.; Ren, Y.; Nan, L.; Sanderson, R.D. Heparanase plays a dual role in driving hepatocyte growth factor (HGF) signaling by enhancing HGF expression and activity. J. Biol. Chem. 2011, 286, 6490–6499. [Google Scholar] [CrossRef] [Green Version]
- Kido, S.; Kuriwaka-Kido, R.; Imamura, T.; Ito, Y.; Inoue, D.; Matsumoto, T. Mechanical stress induces Interleukin-11 expression to stimulate osteoblast differentiation. Bone 2009, 45, 1125–1132. [Google Scholar] [CrossRef]
- Nakchbandi, I.A.; Mitnick, M.A.; Masiukiewicz, U.S.; Sun, B.H.; Insogna, K.L. IL-6 negatively regulates IL-11 production in vitro and in vivo. Endocrinology 2001, 142, 3850–3856. [Google Scholar] [CrossRef] [PubMed]
- Peister, A.; Mellad, J.A.; Larson, B.L.; Hall, B.M.; Gibson, L.F.; Prockop, D.J. Adult stem cells from bone marrow (MSCs) isolated from different strains of inbred mice vary in surface epitopes, rates of proliferation, and differentiation potential. Blood 2004, 103, 1662–1668. [Google Scholar] [CrossRef] [Green Version]
- Maggini, J.; Mirkin, G.; Bognanni, I.; Holmberg, J.; Piazzón, I.M.; Nepomnaschy, I.; Costa, H.; Cañones, C.; Raiden, S.; Vermeulen, M.; et al. Mouse bone marrow-derived mesenchymal stromal cells turn activated macrophages into a regulatory-like profile. PLoS ONE 2010, 5, e9252. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsubaki, M.; Seki, S.; Takeda, T.; Chihara, A.; Arai, Y.; Morii, Y.; Imano, M.; Satou, T.; Shimomura, K.; Nishida, S. The HGF/Met/NF-κB Pathway Regulates RANKL Expression in Osteoblasts and Bone Marrow Stromal Cells. Int. J. Mol. Sci. 2020, 21, 7905. https://doi.org/10.3390/ijms21217905
Tsubaki M, Seki S, Takeda T, Chihara A, Arai Y, Morii Y, Imano M, Satou T, Shimomura K, Nishida S. The HGF/Met/NF-κB Pathway Regulates RANKL Expression in Osteoblasts and Bone Marrow Stromal Cells. International Journal of Molecular Sciences. 2020; 21(21):7905. https://doi.org/10.3390/ijms21217905
Chicago/Turabian StyleTsubaki, Masanobu, Shiori Seki, Tomoya Takeda, Akiko Chihara, Yuuko Arai, Yuusuke Morii, Motohiro Imano, Takao Satou, Kazunori Shimomura, and Shozo Nishida. 2020. "The HGF/Met/NF-κB Pathway Regulates RANKL Expression in Osteoblasts and Bone Marrow Stromal Cells" International Journal of Molecular Sciences 21, no. 21: 7905. https://doi.org/10.3390/ijms21217905