Precise Editing of the OsPYL9 Gene by RNA-Guided Cas9 Nuclease Confers Enhanced Drought Tolerance and Grain Yield in Rice (Oryza sativa L.) by Regulating Circadian Rhythm and Abiotic Stress Responsive Proteins



Abstract
:1. Introduction
2. Results
2.1. Assembly of Targets in Vector
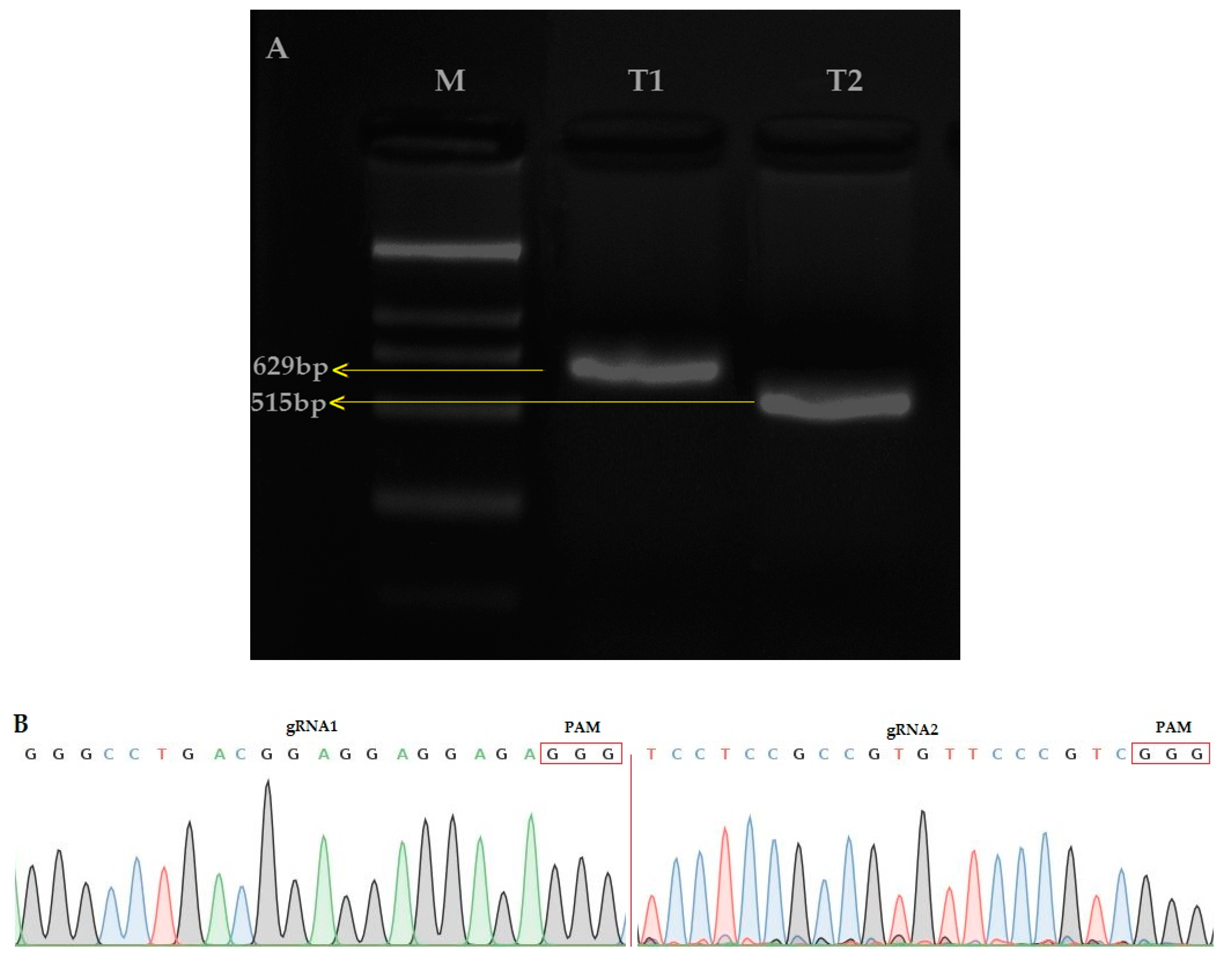
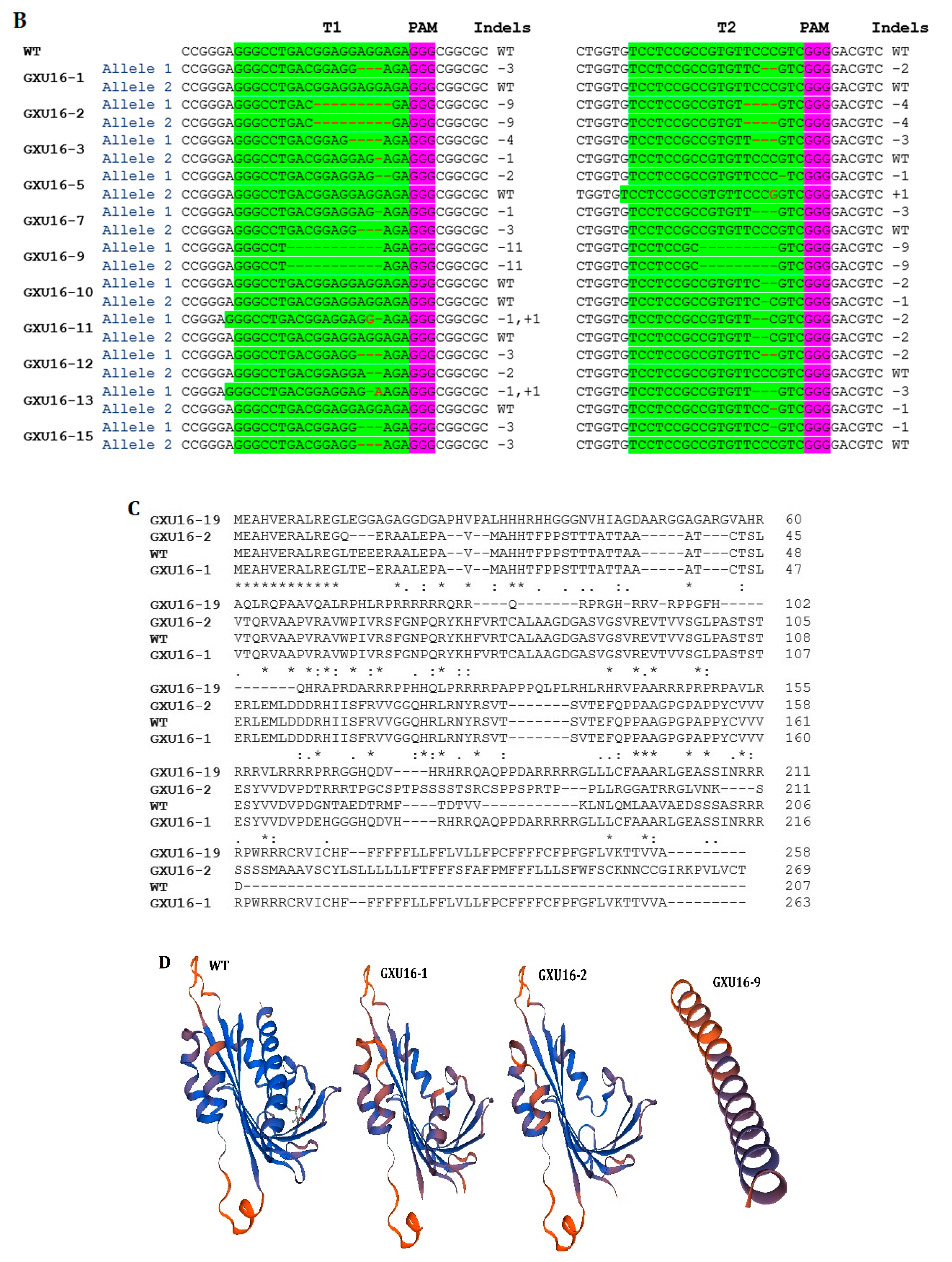
2.2. Editing of OsPYL9 and Analysis of Unmarked T0 Generation
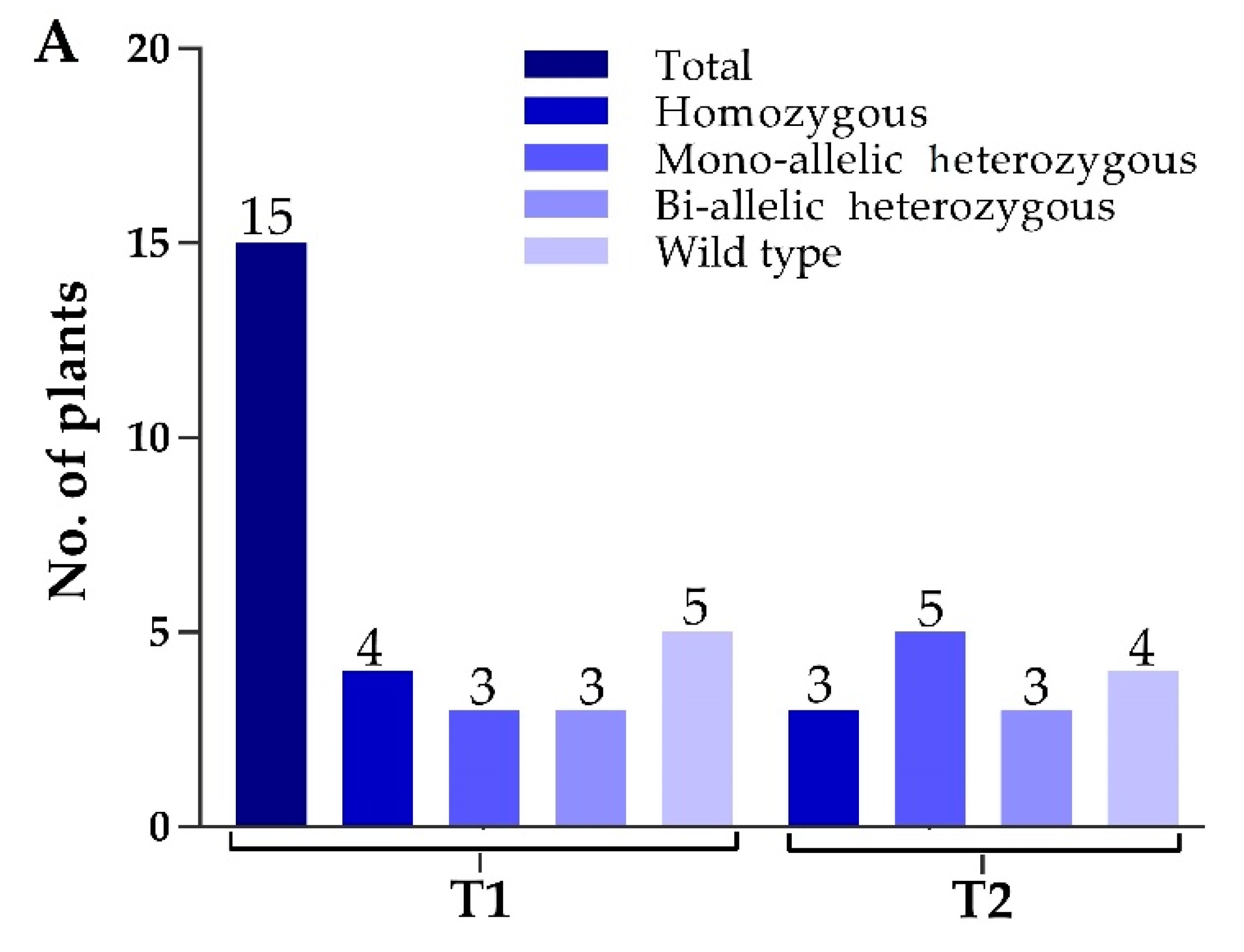
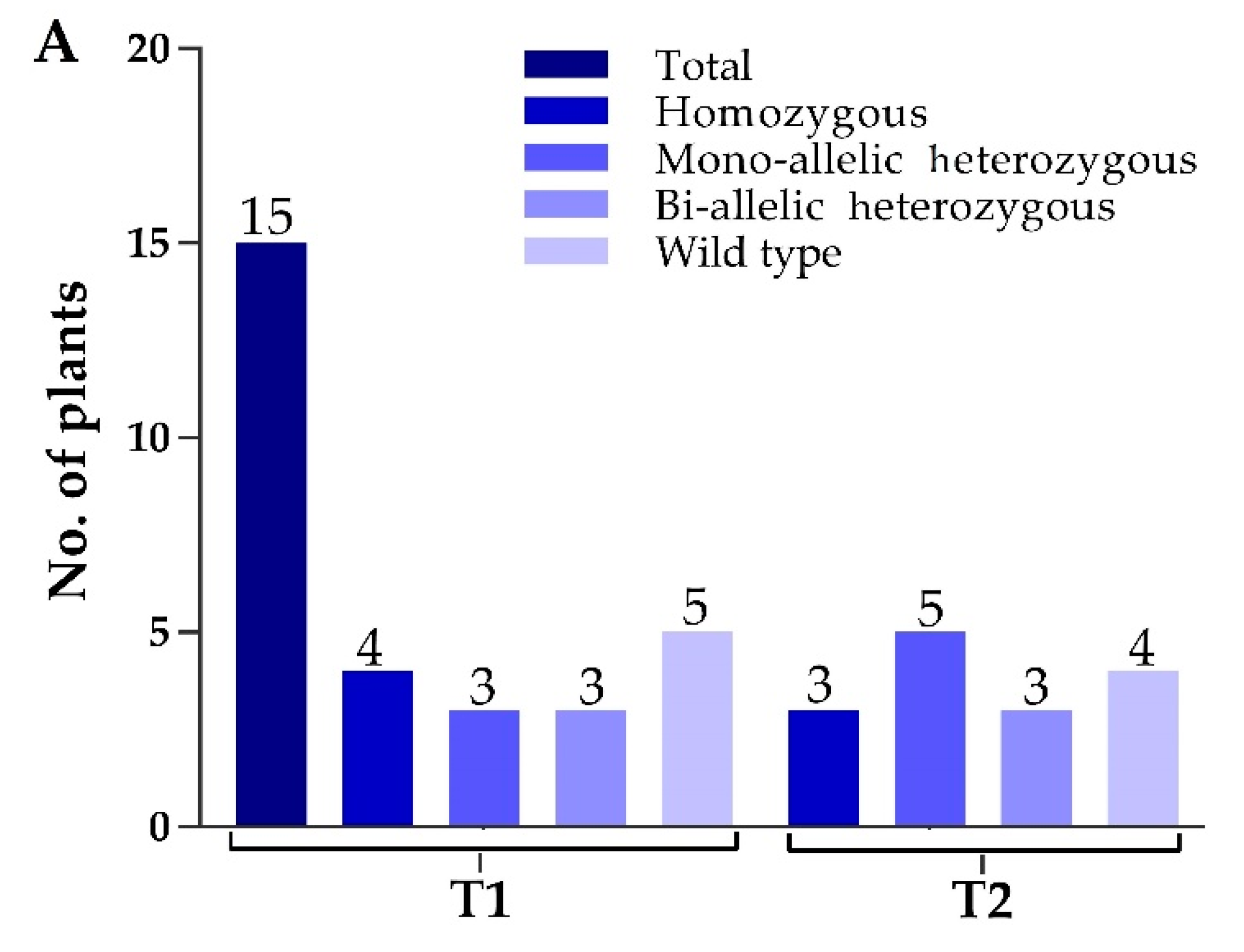
2.3. Screening of Transgene-Free Plants and Segregation Analysis in T1 Generation
2.4. Agronomic Traits Evaluation under Normal and Drought Conditions
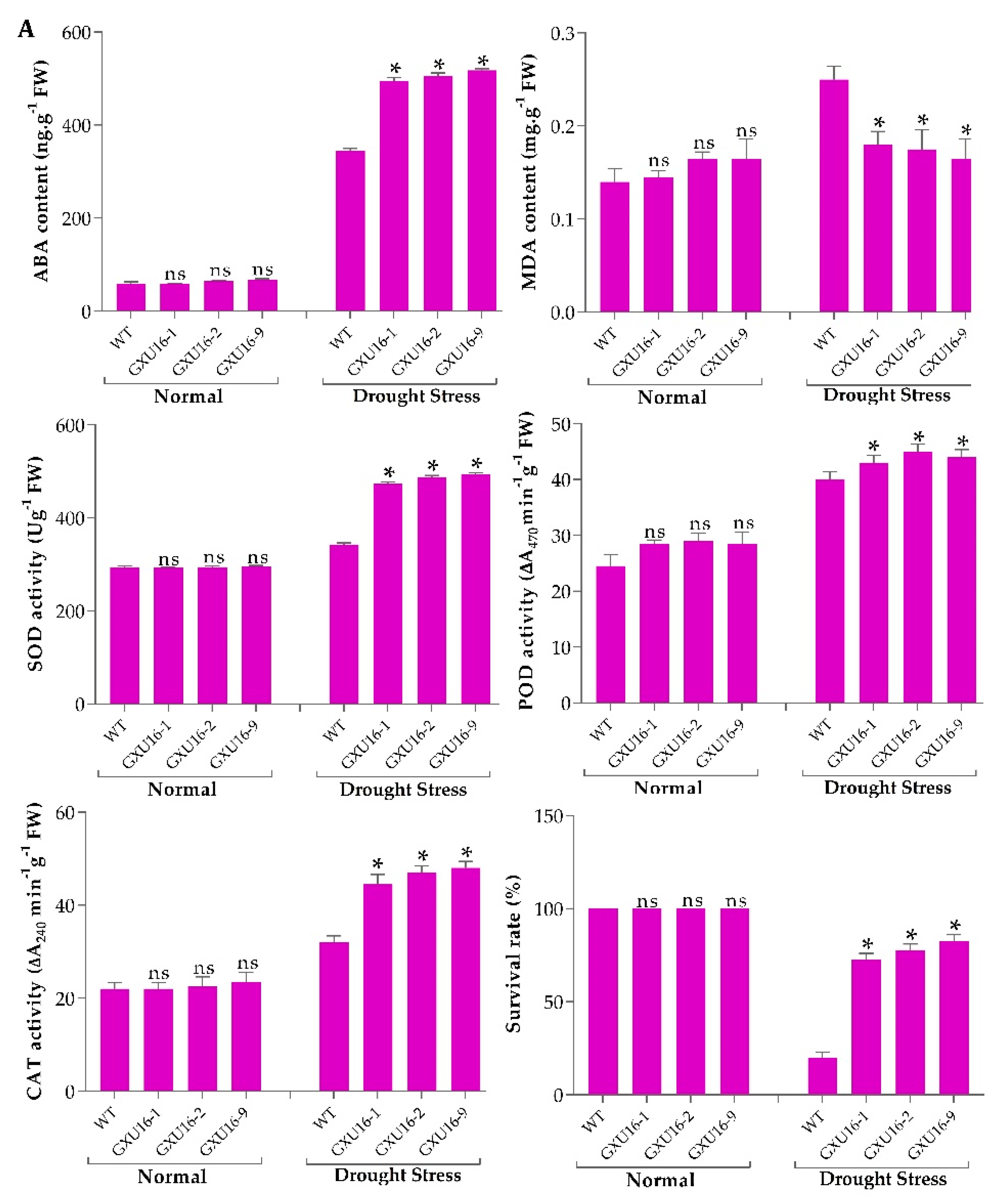
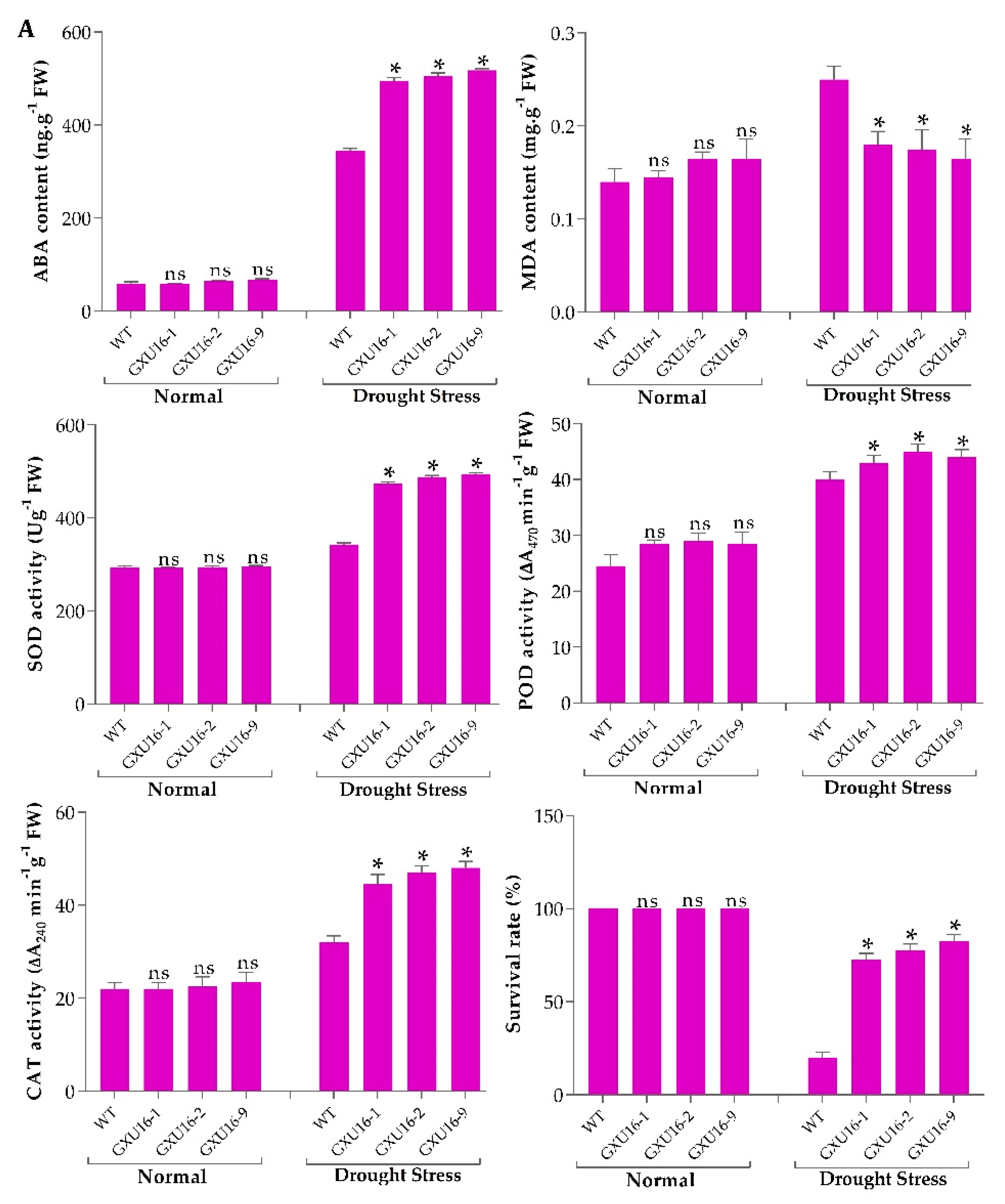
2.5. The Effect of OsPYL9 Mutagenesis on Abscisic Acid (ABA), Malondialdehyde (MDA) and Enzymatic Activities under Normal Conditions and Drought Stress
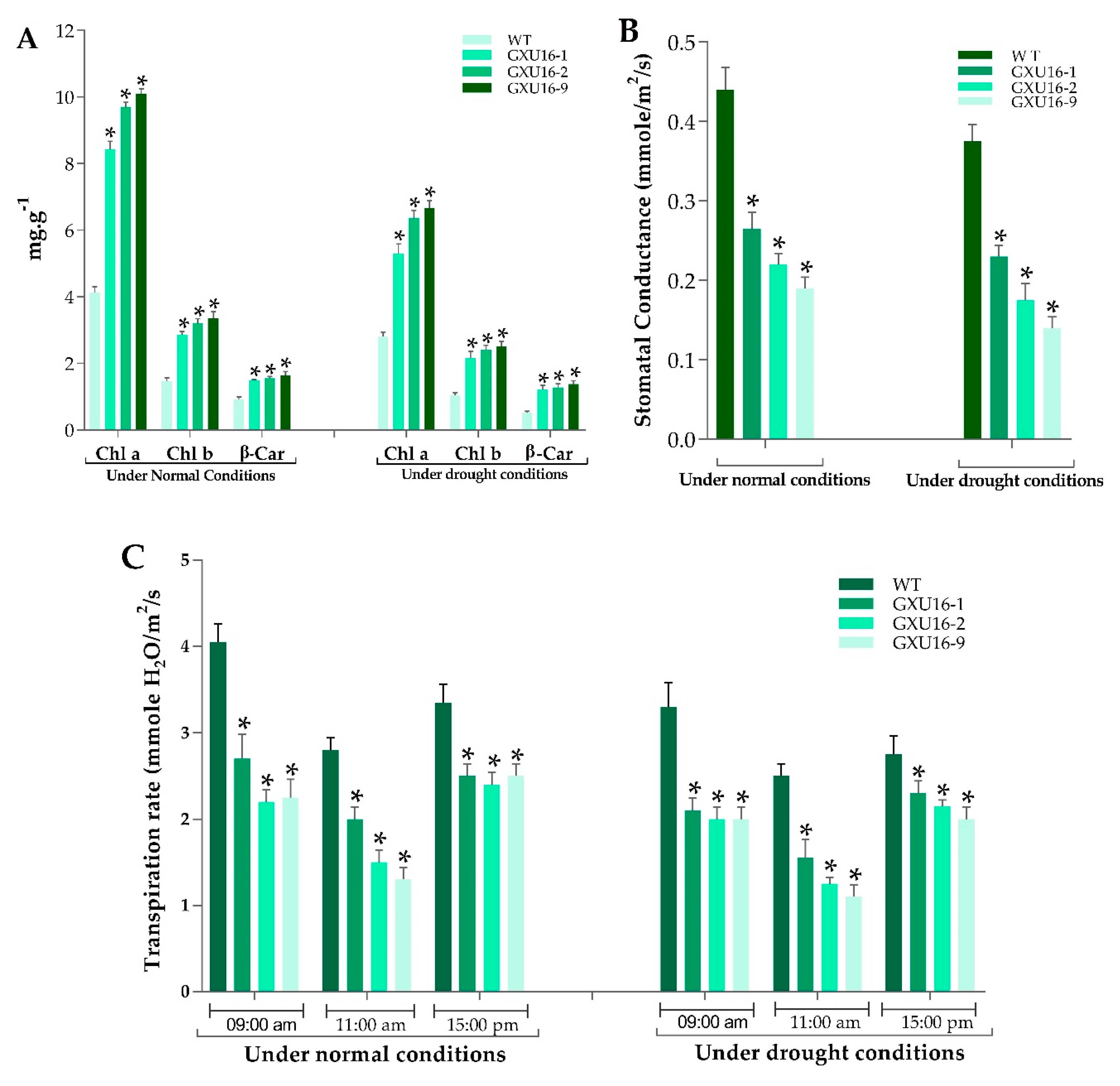
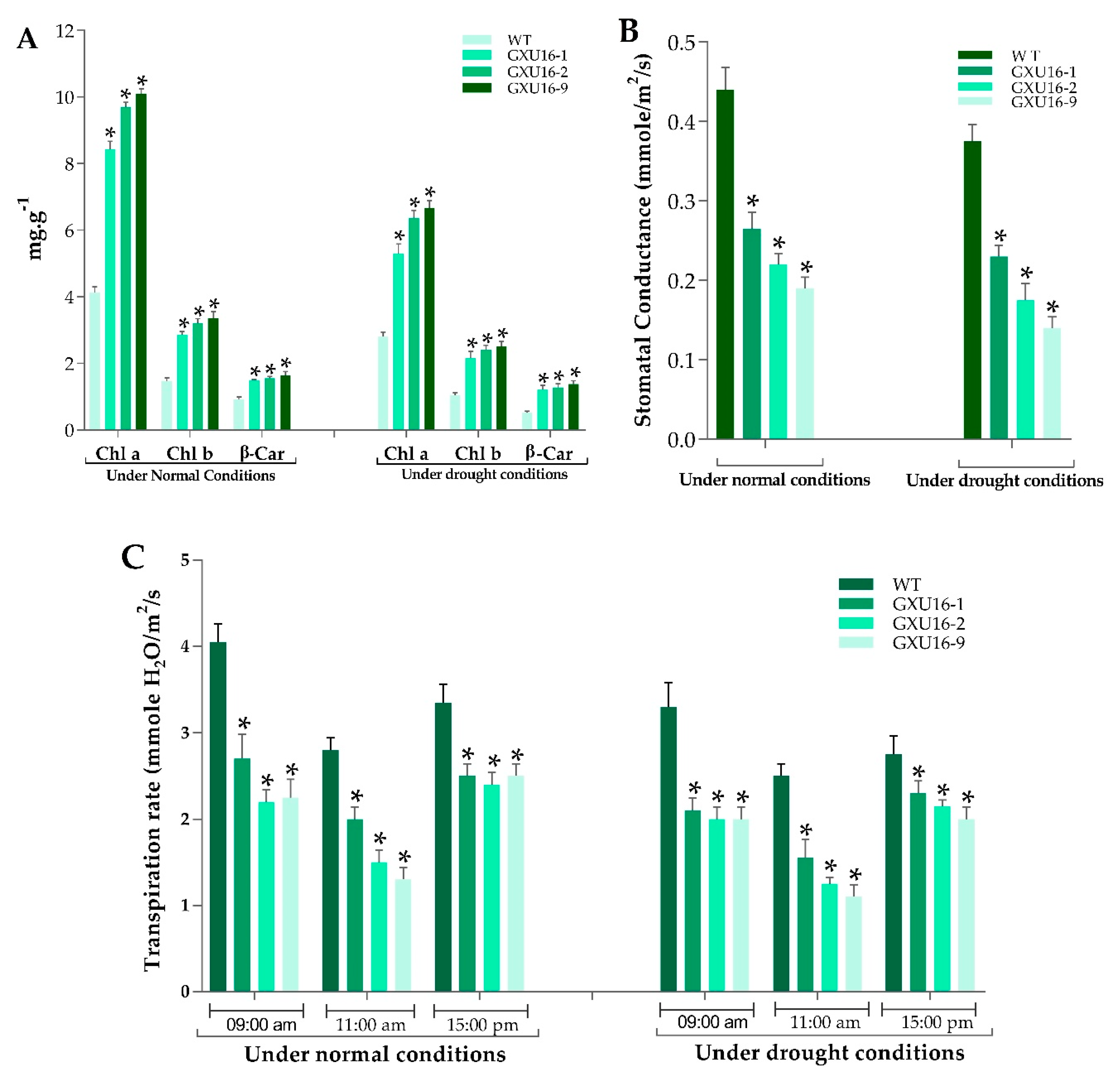
2.6. Measurement of Chlorophyll Content, Transpiration Rate, and Stomatal Conductance
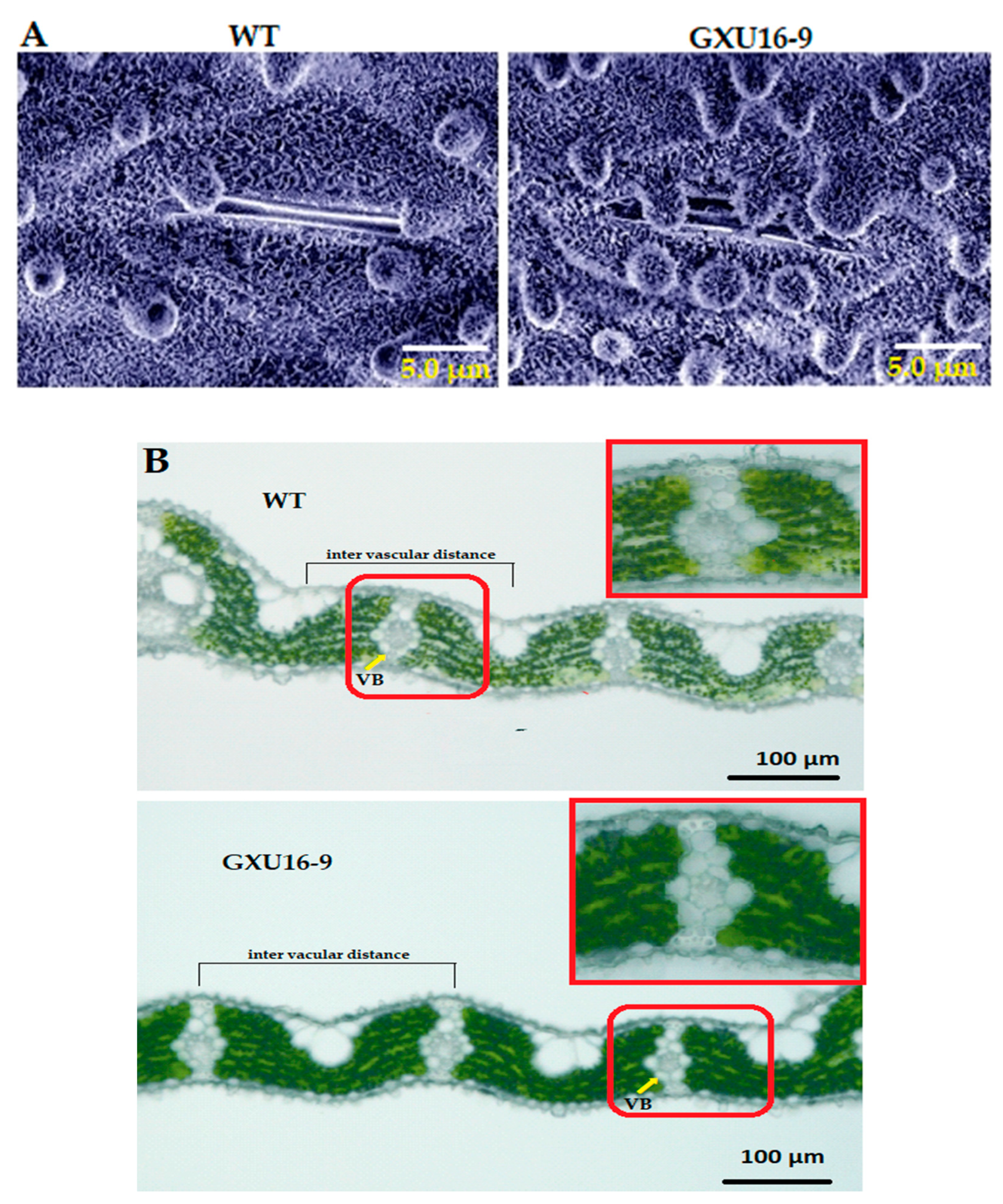
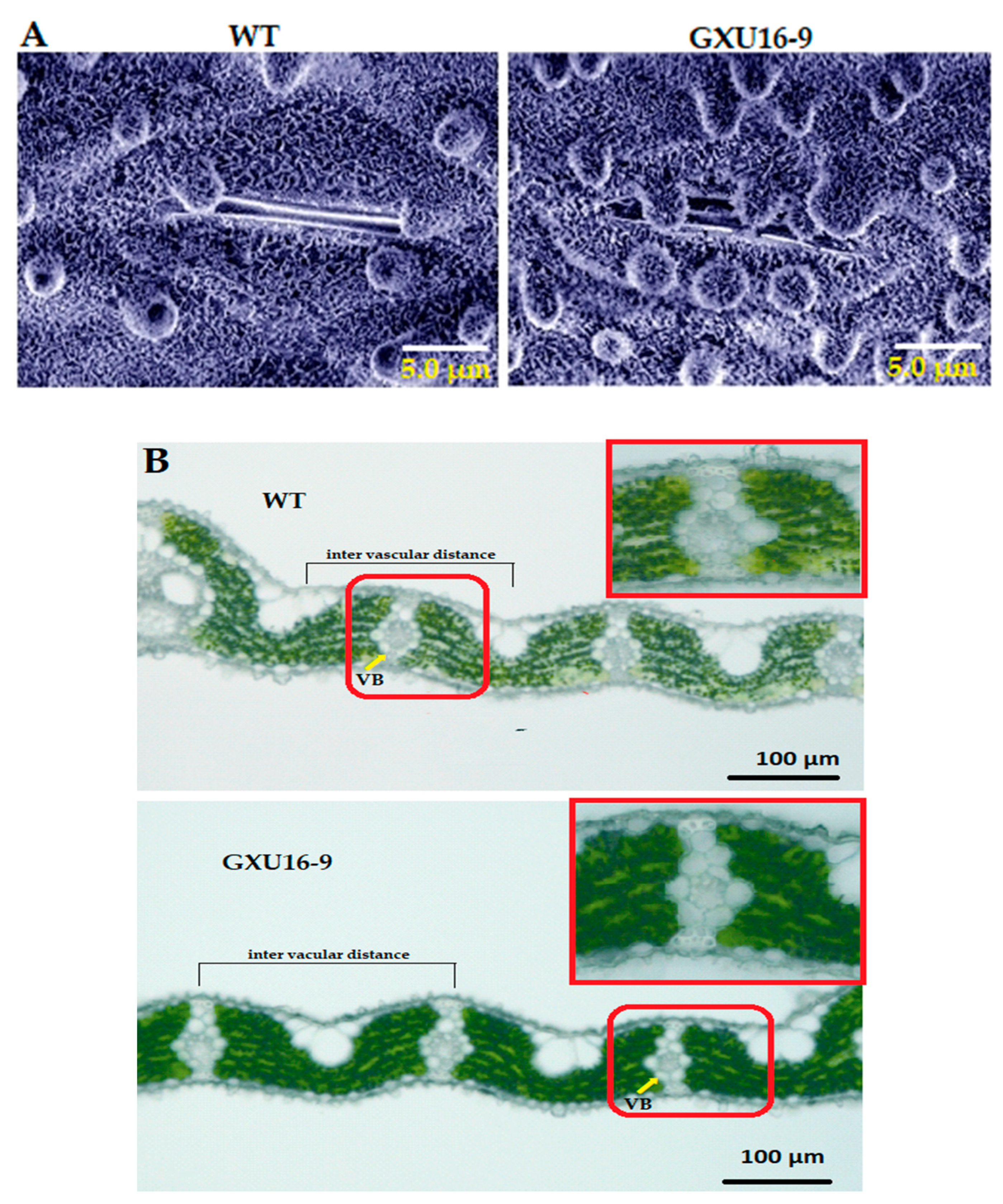
2.7. OsPYL9 Mutant Accumulated More Waxy Crystals on the Leaf Epidermis and Showed Decreased Vascular Bundles
2.8. Peptide/Protein Identification and Absolute Quantitation
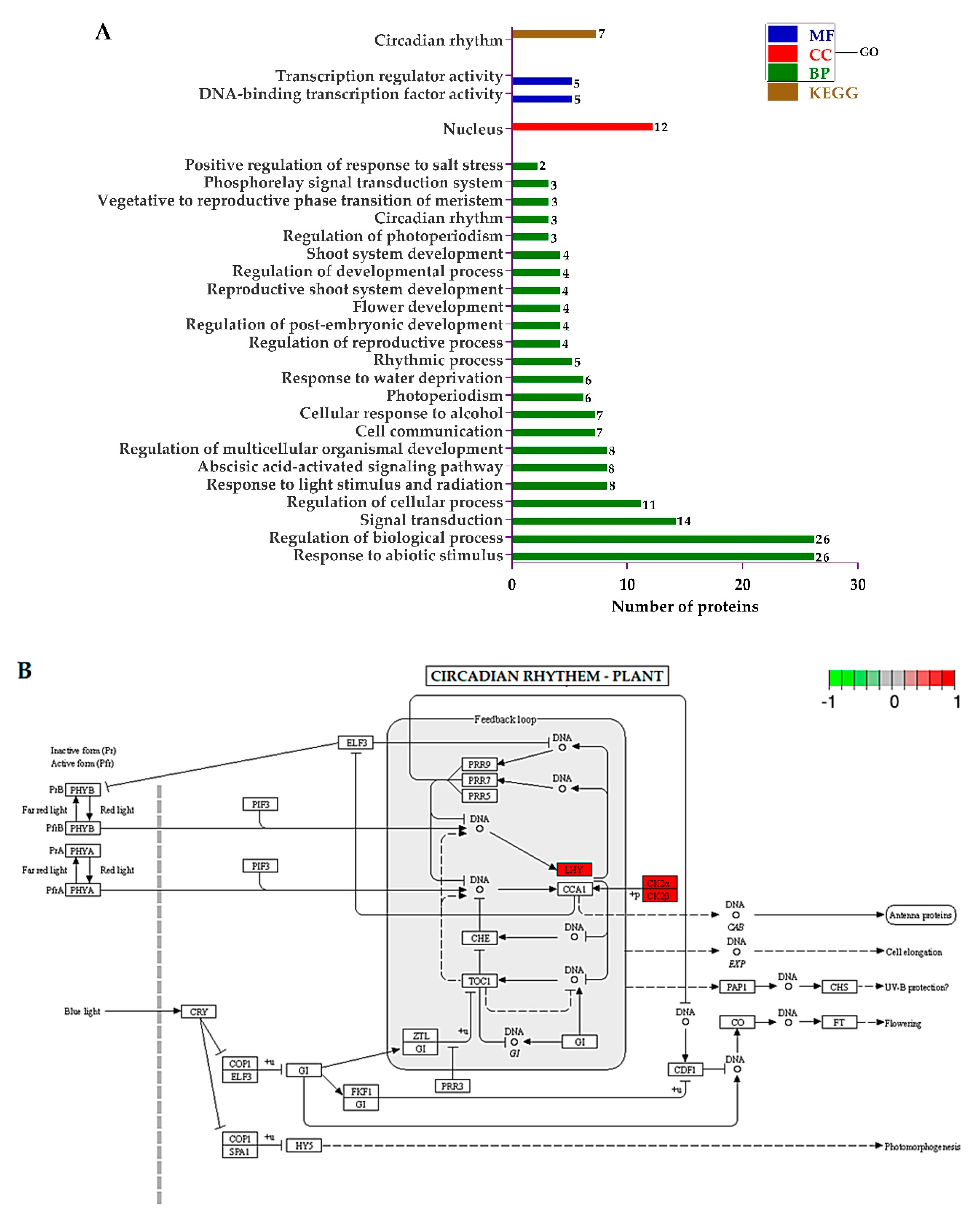
2.9. Screening of Differentially Expressed Proteins (DEPs) and Most Enriched Pathways
2.10. Functional Assignment of the Differentially Expressed Proteins (DEPs)
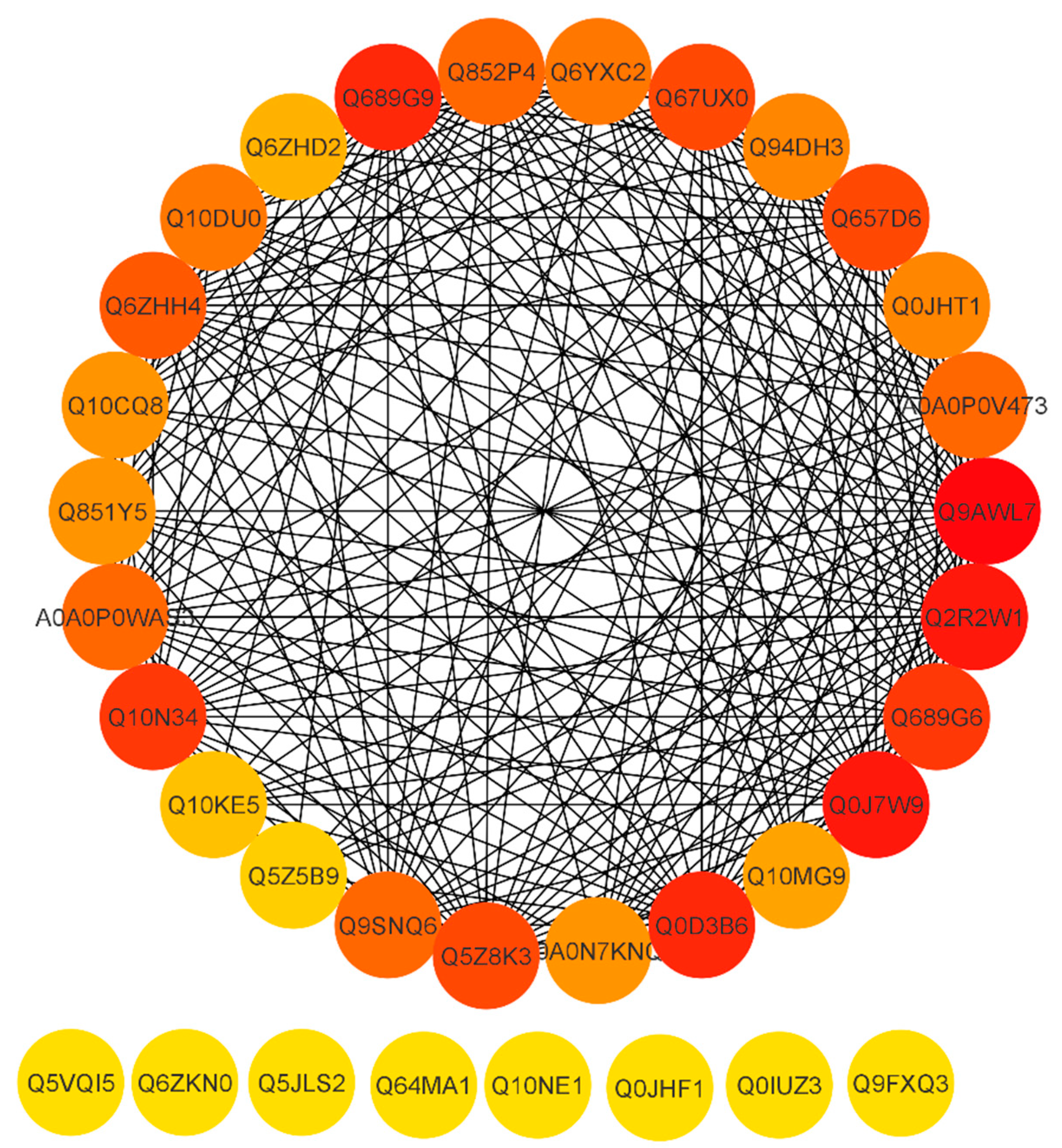
2.11. Functional Interaction Networks of the Differentially Expressed Proteins
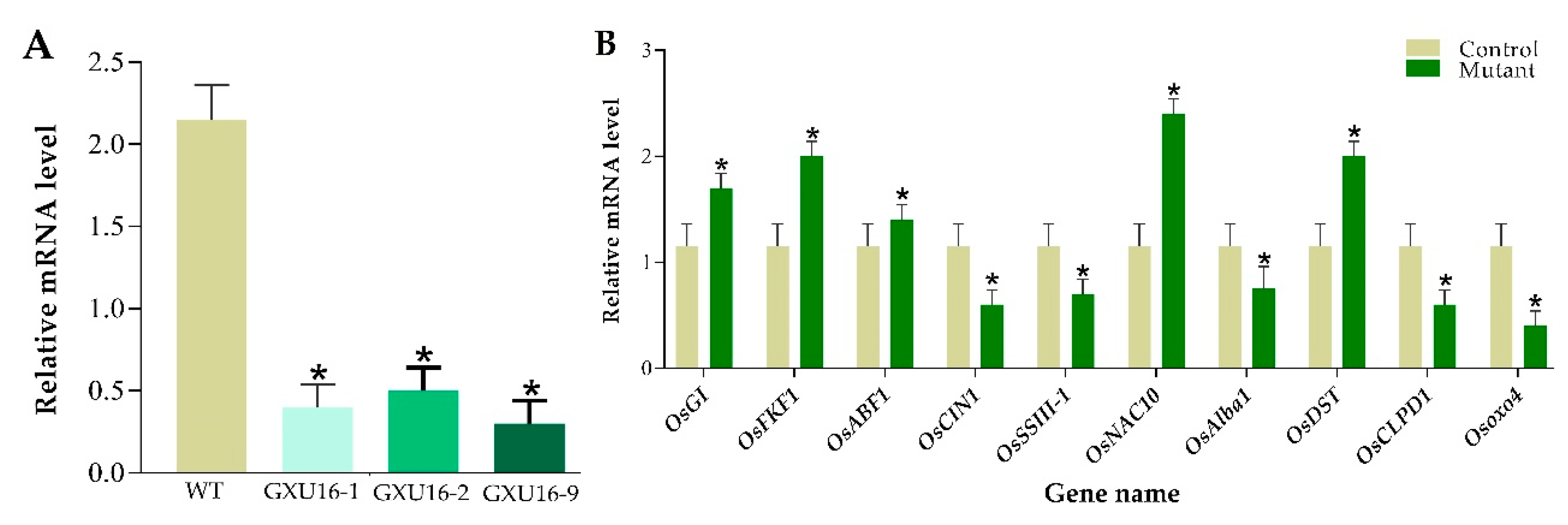
2.12. Quantitative Real-Time-qPCR-Based Assessment of OsPYL9 Expression Level and Validation of DEPs
3. Discussion
4. Materials and Methods
4.1. Experimental Material
4.2. Targets Selection, Generation of the Guiding RNAs Expression Cassettes and Construction of CRISPR-Cas9 Binary Vector
4.3. Rice Transformation and Genotyping of Mutant Plants
4.4. Drought Assays at Seedling and Mature Stage
4.5. Biochemical Assays under Normal Conditions and Drought Stress
4.6. Protein Extraction, Digestion, and Labelling
4.7. Proteomic Data Analysis
4.8. RT-qPCR Analyses
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| Cas9 | CRISPR-associated protein 9 |
| ABA | Abscisic acid |
| MDA | Malondialdehyde |
| CAT | Catalase |
| POD | Peroxidase |
| SOD | Superoxide dismutase |
| ROS | Reactive oxygen species |
| iTRAQ | Isobaric tags for relative and absolute quantitation |
| DEPs | Differentially expressed proteins |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| GO | Gene Ontology |
| PPI | Protein–protein interaction |
References
- Mishra, R.; Joshi, R.K.; Zhao, K. Genome editing in rice: Recent advances, challenges, and future implications. Front. Plant Sci. 2018, 9, 1361. [Google Scholar] [CrossRef]
- Wassmann, R.; Jagadish, S.; Sumfleth, K.; Pathak, H.; Howell, G.; Ismail, A.; Serraj, R.; Redona, E.; Singh, R.; Heuer, S. Regional vulnerability of climate change impacts on Asian rice production and scope for adaptation. Adv. Agron. 2009, 102, 91–133. [Google Scholar] [CrossRef]
- Hu, H.; Xiong, L. Genetic engineering and breeding of drought-resistant crops. Annu. Rev. Plant Biol. 2014, 65, 715–741. [Google Scholar] [CrossRef]
- Zhang, Q. Strategies for developing green super rice. Proc. Natl. Acad. Sci. USA 2007, 104, 16402–16409. [Google Scholar] [CrossRef] [Green Version]
- Kamanga, R.; Mbega, E.; Ndakidemi, P. Drought tolerance mechanisms in plants: Physiological responses associated with water deficit stress in Solanum lycopersicum. Adv. Crop. Sci. Technol. 2018, 6, 1–8. [Google Scholar] [CrossRef]
- Mushtaq, M.; Bhat, J.A.; Mir, Z.A.; Sakina, A.; Ali, S.; Singh, A.K.; Tyagi, A.; Salgotra, R.K.; Dar, A.A.; Bhat, R. CRISPR/Cas approach: A new way of looking at plant-abiotic interactions. J. Plant Physiol. 2018, 224, 156–162. [Google Scholar] [CrossRef]
- Nakashima, K.; Suenaga, K. Toward the genetic improvement of drought tolerance in crops. JARQ 2017, 51, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Gaffney, J.; Schussler, J.; Löffler, C.; Cai, W.; Paszkiewicz, S.; Messina, C.; Groeteke, J.; Keaschall, J.; Cooper, M. Industry-scale evaluation of maize hybrids selected for increased yield in drought-stress conditions of the US corn belt. Crop Sci. 2015, 55, 1608–1618. [Google Scholar] [CrossRef]
- Adee, E.; Roozeboom, K.; Balboa, G.R.; Schlegel, A.; Ciampitti, I. Drought-tolerant corn hybrids yield more in drought-stressed environments with no penalty in non-stressed environments. Front. Plant Sci. 2016, 7, 1534. [Google Scholar] [CrossRef] [Green Version]
- Ashraf, M. Inducing drought tolerance in plants: Recent advances. Biotech. Adv. 2010, 28, 169–183. [Google Scholar] [CrossRef]
- Castiglioni, P.; Warner, D.; Bensen, R.J.; Anstrom, D.C.; Harrison, J.; Stoecker, M.; Abad, M.; Kumar, G.; Salvador, S.; D’Ordine, R. Bacterial RNA chaperones confer abiotic stress tolerance in plants and improved grain yield in maize under water-limited conditions. Plant Physiol. 2008, 147, 446–455. [Google Scholar] [CrossRef] [Green Version]
- Haake, V.; Cook, D.; Riechmann, J.; Pineda, O.; Thomashow, M.F.; Zhang, J. Transcription factor CBF4 is a regulator of drought adaptation in Arabidopsis. Plant Physiol. 2002, 130, 639–648. [Google Scholar] [CrossRef] [Green Version]
- Liao, S.; Qin, X.; Luo, L.; Han, Y.; Wang, X.; Usman, B.; Nawaz, G.; Zhao, N.; Liu, Y.; Li, R. CRISPR/Cas9-Induced Mutagenesis of Semi-Rolled Leaf1,2 Confers Curled Leaf Phenotype and Drought Tolerance by Influencing Protein Expression Patterns and ROS Scavenging in Rice (Oryza sativa L.). Agronomy 2019, 9, 728. [Google Scholar] [CrossRef] [Green Version]
- Haque, E.; Taniguchi, H.; Hassan, M.; Bhowmik, P.; Karim, M.R.; Śmiech, M.; Zhao, K.; Rahman, M.; Islam, T. Application of CRISPR/Cas9 genome editing technology for the improvement of crops cultivated in tropical climates: Recent progress, prospects, and challenges. Front. Plant Sci. 2018, 9, 617. [Google Scholar] [CrossRef]
- Jaganathan, D.; Ramasamy, K.; Sellamuthu, G.; Jayabalan, S.; Venkataraman, G. CRISPR for crop improvement: An update review. Front. Plant Sci. 2018, 9, 985. [Google Scholar] [CrossRef]
- Han, Y.; Teng, K.; Nawaz, G.; Feng, X.; Usman, B.; Wang, X.; Luo, L.; Zhao, N.; Liu, Y.; Li, R.; et al. Generation of semi-dwarf rice (Oryza sativa L.) lines by CRISPR/Cas9-directed mutagenesis of OsGA20ox2 and proteomic analysis of unveiled changes caused by mutations. Biotech 2019, 9, 387. [Google Scholar] [CrossRef]
- Nawaz, G.; Usman, B.; Peng, H.; Zhao, N.; Yuan, R.; Liu, Y.; Li, R. Knockout of Pi21 by CRISPR/Cas9 and iTRAQ-Based Proteomic Analysis of Mutants Revealed New Insights into M. oryzae Resistance in Elite Rice Line. Genes 2020, 11, 735. [Google Scholar] [CrossRef]
- Nawaz, G.; Han, Y.; Usman, B.; Liu, F.; Qin, B.; Li, R. Knockout of OsPRP1, a gene encoding proline-rich protein, confers enhanced cold sensitivity in rice (Oryza sativa L.) at the seedling stage. 3 Biotechnology 2019, 9, 254. [Google Scholar] [CrossRef]
- Han, Y.; Luo, D.; Usman, B.; Nawaz, G.; Zhao, N.; Liu, F.; Li, R. Development of High Yielding Glutinous Cytoplasmic Male Sterile Rice (Oryza sativa L.) Lines through CRISPR/Cas9 Based Mutagenesis of Wx and TGW6 and Proteomic Analysis of Anther. Agronomy 2018, 8, 290. [Google Scholar] [CrossRef] [Green Version]
- Usman, B.; Nawaz, G.; Zhao, N.; Liu, Y.; Li, R. Generation of High Yielding and Fragrant Rice (Oryza sativa L.) Lines by CRISPR/Cas9 Targeted Mutagenesis of Three Homoeologs of Cytochrome P450 Gene Family and OsBADH2 and Transcriptome and Proteome Profiling of Revealed Changes Triggered by Mutations. Plants 2020, 9, 788. [Google Scholar] [CrossRef]
- Jacobs, T.B.; Lafayette, P.R.; Schmitz, R.J.; Parrott, W.A. Targeted genome modifications in soybean with CRISPR/Cas9. BMC Biotechnol. 2015, 15. [Google Scholar] [CrossRef] [Green Version]
- Nawaz, G.; Usman, B.; Zhao, N.; Han, Y.; Li, Z.; Wang, X.; Liu, Y.; Li, R. CRISPR/Cas9 Directed Mutagenesis of OsGA20ox2 in High Yielding Basmati Rice (Oryza sativa L.) Line and Comparative Proteome Profiling of Unveiled Changes Triggered by Mutations. Int. J. Mol. Sci. 2020, 21, 6170. [Google Scholar] [CrossRef]
- Shen, L.; Hua, Y.; Fu, Y.; Li, J.; Liu, Q.; Jiao, X.; Xin, G.; Wang, J.; Wang, X.; Yan, C. Rapid generation of genetic diversity by multiplex CRISPR/Cas9 genome editing in rice. Sci. China Life Sci. 2017, 60, 506–515. [Google Scholar] [CrossRef]
- Osakabe, Y.; Watanabe, T.; Sugano, S.S.; Ueta, R.; Ishihara, R.; Shinozaki, K.; Osakabe, K. Optimization of CRISPR/Cas9 genome editing to modify abiotic stress responses in plants. Sci. Rep. 2016, 6, 26685. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Gao, H.; Wang, H.; Lafitte, H.R.; Archibald, R.L.; Yang, M.; Hakimi, S.M.; Mo, H.; Habben, J. ARGOS 8 variants generated by CRISPR-Cas9 improve maize grain yield under field drought stress conditions. Plant Biotech. J. 2017, 15, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Evans, C.; Noirel, J.; Ow, S.Y.; Salim, M.; Pereira-Medrano, A.G.; Couto, N.; Pandhal, J.; Smith, D.; Pham, T.K.; Karunakaran, E.; et al. An insight into iTRAQ: Where do we stand now? Anal. Bioanal. Chem. 2012, 404, 1011–1027. [Google Scholar] [CrossRef]
- Zou, Y.; Dai, W.; Lei, W.; Su, S.; Huang, Q.; Zhou, Z.; Chen, C.; Li, Z. Identification of proteins interacting with pORF5 in the pathogenesis of C. trachomatis. Am. J. Trans. Res. 2018, 10, 1633. [Google Scholar]
- Cai, Q.; Wang, G.; Li, Z.; Zhang, L.; Fu, Y.; Yang, X.; Lin, W.; Lin, X. SWATH based quantitative proteomics analysis reveals Hfq2 play an important role on pleiotropic physiological functions in Aeromonas hydrophila. J. Proteom. 2019, 195, 1–10. [Google Scholar] [CrossRef]
- Cutler, S.R.; Rodriguez, P.L.; Finkelstein, R.R.; Abrams, S. Abscisic acid: Emergence of a core signaling network. Ann. Rev. Plant Biol. 2010, 61, 651–679. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Szostkiewicz, I.; Korte, A.; Moes, D.; Yang, Y.; Christmann, A.; Grill, E. Regulators of PP2C phosphatase activity function as abscisic acid sensors. Science 2009, 324, 1064–1068. [Google Scholar] [CrossRef]
- Park, S.Y.; Fung, P.; Nishimura, N.; Jensen, D.R.; Fujii, H.; Zhao, Y.; Lumba, S.; Santiago, J.; Rodrigues, A.; Tsz-fung, F. Abscisic acid inhibits type 2C protein phosphatases via the PYR/PYL family of START proteins. Science 2009, 324, 1068–1071. [Google Scholar] [CrossRef] [Green Version]
- Klingler, J.P.; Batelli, G.; Zhu, J.K. ABA receptors: The START of a new paradigm in phytohormone signalling. J. Exp. Bot. 2010, 61, 3199–3210. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Hwang, H.; Hong, J.W.; Lee, Y.N.; Ahn, I.P.; Yoon, I.S.; Yoo, S.D.; Lee, S.; Lee, S.C.; Kim, B.G. A rice orthologue of the ABA receptor, OsPYL/RCAR5, is a positive regulator of the ABA signal transduction pathway in seed germination and early seedling growth. J. Exp. Bot. 2012, 63, 1013–1024. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Hao, Q.; Li, W.; Yan, C.; Yan, N.; Yin, P. Identification and characterization of ABA receptors in Oryza sativa. PLoS ONE 2014, 9, e95246. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Wang, Z.; Li, X.; Lv, T.; Liu, H.; Wang, L.; Niu, H.; Bu, Q. Characterization and functional analysis of pyrabactin resistance-like abscisic acid receptor family in rice. Rice 2015, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Lee, K.; Hwang, H.; Bhatnagar, N.; Kim, D.Y.; Yoon, I.S.; Byun, M.O.; Kim, S.T.; Jung, K.H.; Kim, B.G.; et al. Overexpression of PYL5 in rice enhances drought tolerance, inhibits growth, and modulates gene expression. J. Exp. Bot. 2014, 65, 453–464. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Kong, L.; Zhou, Y.; Chen, Z.; Tian, D.; Lin, Y.; Wang, F.; Chen, S. Endosperm-specific OsPYL8 and OsPYL9 act as positive regulators of the ABA signaling pathway in rice seed germination. Funct. Plant Biol. 2017, 44, 635–645. [Google Scholar] [CrossRef]
- Jiang, M.; Zhang, J. Water stress-induced abscisic acid accumulation triggers the increased generation of reactive oxygen species and up-regulates the activities of antioxidant enzymes in maize leaves. J. Exp. Bot. 2002, 53, 2401–2410. [Google Scholar] [CrossRef] [Green Version]
- Guo, T.; Wang, D.; Fang, J.; Zhao, J.; Yuan, S.; Xiao, L.; Li, X. Mutations in the Rice OsCHR4 Gene, Encoding a CHD3 Family Chromatin Remodeler, Induce Narrow and Rolled Leaves with Increased Cuticular Wax. Int. J. Mol. Sci. 2019, 20, 2567. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Zhang, H.; Srivastava, A.K.; Pan, Y.; Bai, J.; Fang, J.; Shi, H.; Zhu, J.K. Knockdown of rice microRNA166 confers drought resistance by causing leaf rolling and altering stem xylem development. Plant Physiol. 2018, 176, 2082–2094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millar, A.A.; Clemens, S.; Zachgo, S.; Giblin, E.M.; Taylor, D.C.; Kunst, L. CUT1, an Arabidopsis gene required for cuticular wax biosynthesis and pollen fertility, encodes a very-long-chain fatty acid condensing enzyme. Plant Cell 1999, 11, 825–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiebig, A.; Mayfield, J.A.; Miley, N.L.; Chau, S.; Fischer, R.L.; Preuss, D. Alterations in CER6, a gene identical to CUT1, differentially affect long-chain lipid content on the surface of pollen and stems. Plant Cell 2000, 12, 2001–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Dietrich, C.R.; Lessire, R.; Nikolau, B.J.; Schnable, P.S. The endoplasmic reticulum-associated maize GL8 protein is a component of the acyl-coenzyme A elongase involved in the production of cuticular waxes. Plant Physiol. 2002, 128, 924–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aarts, M.; Keijzer, C.J.; Stiekema, W.J.; Pereira, A. Molecular characterization of the CER1 gene of Arabidopsis involved in epicuticular wax biosynthesis and pollen fertility. Plant Cell 1995, 7, 2115–2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, D.; Ranathunge, K.; Huang, H.; Pei, Z.; Franke, R.; Schreiber, L.; He, C. Wax Crystal-Sparse Leaf1 encodes a β–ketoacyl CoA synthase involved in biosynthesis of cuticular waxes on rice leaf. Planta 2008, 228, 675–685. [Google Scholar] [CrossRef]
- Broun, P.; Poindexter, P.; Osborne, E.; Jiang, C.Z.; Riechmann, J.L. WIN1, a transcriptional activator of epidermal wax accumulation in Arabidopsis. Proc. Natl. Acad. Sci. USA 2004, 101, 4706–4711. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.Y.; Broeckling, C.D.; Blancaflor, E.B.; Sledge, M.K.; Sumner, L.W.; Wang, Z. Overexpression of WXP1, a putative Medicago truncatula AP2 domain-containing transcription factor gene, increases cuticular wax accumulation and enhances drought tolerance in transgenic alfalfa (Medicago sativa). Plant J. 2005, 42, 689–707. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Goodwin, S.M.; Boroff, V.L.; Liu, X.; Jenks, M.A. Cloning and characterization of the WAX2 gene of Arabidopsis involved in cuticle membrane and wax production. Plant Cell 2003, 15, 1170–1185. [Google Scholar] [CrossRef] [Green Version]
- Sturaro, M.; Hartings, H.; Schmelzer, E.; Velasco, R.; Salamini, F.; Motto, M. Cloning and characterization of GLOSSY1, a maize gene involved in cuticle membrane and wax production. Plant Physiol. 2005, 138, 478–489. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.A.; Du, H.; Ning, J.; Ye, H.; Xiong, L. Characterization of Glossy1-homologous genes in rice involved in leaf wax accumulation and drought resistance. Plant Mol. Biol. 2009, 70, 443–456. [Google Scholar] [CrossRef]
- Schnurr, J.; Shockey, J. The acyl-CoA synthetase encoded by LACS2 is essential for normal cuticle development in Arabidopsis. Plant Cell 2004, 16, 629–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawrath, C. Unraveling the complex network of cuticular structure and function. Curr. Opin. Plant Biol. 2006, 9, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Li, L. Carotenoid metabolism: Biosynthesis, regulation, and beyond. J. Integr. Plant Biol. 2008, 50, 778–785. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Sola, M.Á.; Rodríguez-Concepción, M. Carotenoid biosynthesis in Arabidopsis: A colorful pathway. Arab. Book/Am. Soc. Plant Biol. 2012, 10. [Google Scholar] [CrossRef] [Green Version]
- Nisar, N.; Li, L.; Lu, S.; Khin, N.C.; Pogson, B. Carotenoid metabolism in plants. Mol. Plant 2015, 8, 68–82. [Google Scholar] [CrossRef] [Green Version]
- Avendaño-Vázquez, A.O.; Cordoba, E.; Llamas, E.; San Román, C.; Nisar, N.; De la Torre, S.; Ramos-Vega, M.; de la Luz Gutiérrez-Nava, M.; Cazzonelli, C.I.; Pogson, B. An uncharacterized apocarotenoid-derived signal generated in ζ-carotene desaturase mutants regulates leaf development and the expression of chloroplast and nuclear genes in Arabidopsis. Plant Cell 2014, 26, 2524–2537. [Google Scholar] [CrossRef] [Green Version]
- Suh, M.C.; Samuels, A.L.; Jetter, R.; Kunst, L.; Pollard, M.; Ohlrogge, J.; Beisson, F. Cuticular lipid composition, surface structure, and gene expression in Arabidopsis stem epidermis. Plant Physiol. 2005, 139, 1649–1665. [Google Scholar] [CrossRef] [Green Version]
- Reicosky, D.A.; Hanover, J.W. Physiological effects of surface waxes: I. Light reflectance for glaucous and nonglaucous Picea pungens. Plant Physiol. 1978, 62, 101–104. [Google Scholar] [CrossRef] [Green Version]
- Jenks, M.A.; Joly, R.J.; Peters, P.J.; Rich, P.J.; Axtell, J.D.; Ashworth, E.N. Chemically induced cuticle mutation affecting epidermal conductance to water vapor and disease susceptibility in Sorghum bicolor (L.) Moench. Plant Physiol. 1994, 105, 1239–1245. [Google Scholar] [CrossRef] [Green Version]
- McWatters, H.G.; Roden, L.C.; Staiger, D. Picking out parallels: Plant circadian clocks in context. Philos. Trans. R. Soc. Lond. 2001, 356, 1735–1743. [Google Scholar] [CrossRef] [Green Version]
- Miglani, G.S. Genome editing in crop improvement: Present scenario and future prospects. J. Crop. Improv. 2017, 31, 453–559. [Google Scholar] [CrossRef]
- Somers, D. The physiology and molecular bases of the plant circadian clock. Plant Physiol. 1999, 121, 9–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litchfield, D.W. Protein kinase CK2: Structure, regulation and role in cellular decisions of life and death. Biochem. J. 2003, 369, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Salinas, P.; Fuentes, D.; Vidal, E.; Jordana, X.; Echeverria, M.; Holuigue, L.; Physiology, C. An extensive survey of CK2 α and β subunits in Arabidopsis: Multiple isoforms exhibit differential subcellular localization. Plant Cell Physiol. 2006, 47, 1295–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olesen, S.H.; Ingles, D.J.; Zhu, J.Y.; Martin, M.P.; Betzi, S.; Georg, G.I.; Tash, J.S.; Schönbrunn, E. Stability of the human Hsp90-p50Cdc37 chaperone complex against nucleotides and Hsp90 inhibitors, and the influence of phosphorylation by casein kinase 2. Molecules 2015, 20, 1643–1660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Li, H.; Yuan, C.; Liu, S.; Li, M.; Zhu, J.; Lin, X.; Lu, Y.; Guo, X. CKB 1 regulates expression of ribosomal protein L10 family gene and plays a role in UV-B response. Plant Biol. 2020, 22, 143–152. [Google Scholar] [CrossRef]
- Nagatoshi, Y.; Fujita, M.; Fujita, Y. Casein kinase 2 α and β subunits inversely modulate ABA signal output in Arabidopsis protoplasts. Planta 2018, 248, 571–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Yue, W.; Wang, M.; Qiu, W.; Zhou, L.; Shou, H. Mutation of OsGIGANTEA leads to enhanced tolerance to polyethylene glycol-generated osmotic stress in rice. Front. Plant Sci. 2016, 7, 465. [Google Scholar] [CrossRef] [Green Version]
- Fu, C.; Yang, X.; Chen, X.; Chen, W.; Ma, Y.; Hu, J.; Li, S. OsEF3, a homologous gene of Arabidopsis ELF3, has pleiotropic effects in rice. Plant Biol. 2009, 11, 751–757. [Google Scholar] [CrossRef]
- Gao, H.; Jin, M.; Zheng, X.M.; Chen, J.; Yuan, D.; Xin, Y.; Wang, M.; Huang, D.; Zhang, Z.; Zhou, K. Days to heading 7, a major quantitative locus determining photoperiod sensitivity and regional adaptation in rice. Proc. Natl. Acad. Sci. USA 2014, 111, 16337–16342. [Google Scholar] [CrossRef] [Green Version]
- Han, S.H.; Yoo, S.C.; Lee, B.D.; An, G.; Paek, N.C. Rice FLAVIN-BINDING, KELCH REPEAT, F-BOX 1 (OsFKF 1) promotes flowering independent of photoperiod. Plant Cell Environ. 2015, 38, 2527–2540. [Google Scholar] [CrossRef] [PubMed]
- Saijo, Y.; Hata, S.; Kyozuka, J.; Shimamoto, K.; Izui, K. Over-expression of a single Ca2+-dependent protein kinase confers both cold and salt/drought tolerance on rice plants. Plant J. 2000, 23, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Huang, Y.; Xiong, L. Characterization of stress-responsive CIPK genes in rice for stress tolerance improvement. Plant Physiol. 2007, 144, 1416–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, H.; Liu, C.; Zhang, Y.; Meng, X.; Zhou, X.; Chu, C.; Wang, X. OsWRKY30 is activated by MAP kinases to confer drought tolerance in rice. Plant Mol. Biol. 2012, 80, 241–253. [Google Scholar] [CrossRef]
- Cui, M.; Zhang, W.; Zhang, Q.; Xu, Z.; Zhu, Z.; Duan, F.; Wu, R. Biochemistry, Induced over-expression of the transcription factor OsDREB2A improves drought tolerance in rice. Plant Physiol. Biochem. 2011, 49, 1384–1391. [Google Scholar] [CrossRef]
- Joo, J.; Lee, Y.H.; Song, S.I. Overexpression of the rice basic leucine zipper transcription factor OsbZIP12 confers drought tolerance to rice and makes seedlings hypersensitive to ABA. Plant Biotech. Rep. 2014, 8, 431–441. [Google Scholar] [CrossRef]
- Gao, T.; Wu, Y.; Zhang, Y.; Liu, L.; Ning, Y.; Wang, D.; Tong, H.; Chen, S.; Chu, C.; Xie, Q. OsSDIR1 overexpression greatly improves drought tolerance in transgenic rice. Plant Mol. Biol. 2011, 76, 145–156. [Google Scholar] [CrossRef]
- Jeong, J.S.; Kim, Y.S.; Baek, K.H.; Jung, H.; Ha, S.H.; Do Choi, Y.; Kim, M.; Reuzeau, C.; Kim, J.K. Root-specific expression of OsNAC10 improves drought tolerance and grain yield in rice under field drought conditions. Plant Physiol. 2010, 153, 185–197. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Sheng, P.; Tan, J.; Chen, X.; Lu, G.; Ma, W.; Heng, Y.; Lin, Q.; Zhu, S.; Wang, J. Plasma membrane receptor-like kinase leaf panicle 2 acts downstream of the DROUGHT AND SALT TOLERANCE transcription factor to regulate drought sensitivity in rice. J. Exp. Bot. 2015, 66, 271–281. [Google Scholar] [CrossRef] [Green Version]
- Dubouzet, J.G.; Sakuma, Y.; Ito, Y.; Kasuga, M.; Dubouzet, E.G.; Miura, S.; Seki, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. OsDREB genes in rice, Oryza sativa L., encode transcription activators that function in drought-, high-salt-and cold-responsive gene expression. Plant J. 2003, 33, 751–763. [Google Scholar] [CrossRef]
- Nakamichi, N.; Takao, S.; Kudo, T.; Kiba, T.; Wang, Y.; Kinoshita, T.; Sakakibara, H. Improvement of Arabidopsis biomass and cold, drought and salinity stress tolerance by modified circadian clock-associated PSEUDO-RESPONSE REGULATORs. Plant Cell Physiol. 2016, 57, 1085–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, W.; Liu, H.; Zhou, X.; Li, Q.; Zhang, J.; Lu, L.; Liu, T.; Liu, H.; Zhang, C.; Zhang, Z. Natural variation in Ghd7. 1 plays an important role in grain yield and adaptation in rice. Cell Res. 2013, 23, 969–971. [Google Scholar] [CrossRef] [PubMed]
- Koo, B.H.; Yoo, S.C.; Park, J.W.; Kwon, C.T.; Lee, B.D.; An, G.; Zhang, Z.; Li, J.; Li, Z.; Paek, N.C. Natural variation in OsPRR37 regulates heading date and contributes to rice cultivation at a wide range of latitudes. Mol. Plant 2013, 6, 1877–1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Zhang, Q.; Zhu, Q.; Liu, W.; Chen, Y.; Qiu, R.; Wang, B.; Yang, Z.; Li, H.; Lin, Y.; et al. A robust CRISPR/Cas9 system for convenient, high-efficiency multiplex genome editing in monocot and dicot plants. Mol. Plant 2015, 8, 1274–1284. [Google Scholar] [CrossRef] [PubMed]
- Hiei, Y.; Ohta, S.; Komari, T.; Kumashiro, T. Efficient transformation of rice (Oryza sativa L.) mediated by Agrobacterium and sequence analysis of the boundaries of the T-DNA. Plant J. 1994, 6, 271–282. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Xie, X.; Ma, X.; Li, J.; Chen, J.; Liu, Y.G. DSDecode: A web-based tool for decoding of sequencing chromatograms for genotyping of targeted mutations. Mol. Plant 2015, 8, 1431–1433. [Google Scholar] [CrossRef]
- Higgins, D.G.; Sharp, P.M. Fast and sensitive multiple sequence alignments on a microcomputer. Comput. Appl. Biosci. 1989, 5, 151–153. [Google Scholar] [CrossRef]
- Zhu, G.; Ye, N.; Zhang, J. Glucose-induced delay of seed germination in rice is mediated by the suppression of ABA catabolism rather than an enhancement of ABA biosynthesis. Plant Cell Physiol. 2009, 50, 644–651. [Google Scholar] [CrossRef]
- Heath, R.L.; Packer, L. Photoperoxidation in isolated chloroplasts: II. Role of electron transfer. Arch. Biochem. Biophys. 1968, 125, 850–857. [Google Scholar] [CrossRef]
- Giannopolitis, C.N.; Ries, S.K. Superoxide dismutases: I. Occurrence in higher plants. Plant Physiol. 1977, 59, 309–314. [Google Scholar] [CrossRef]
- Aebi, H. Catalase in vitro. Methods Enzym. 1984, 105, 121–126. [Google Scholar] [CrossRef]
- Lichtenthaler, H.K. Chlorophylls and carotenoids: Pigments of photosynthetic biomembranes. Methods Enzym. 1987, 148, 350–382. [Google Scholar] [CrossRef]
- Wang, C.; Wang, G.; Gao, Y.; Lu, G.; Habben, J.E.; Mao, G.; Chen, G.; Wang, J.; Yang, F.; Zhao, X.; et al. A cytokinin-activation enzyme-like gene improves grain yield under various field conditions in rice. Plant Mol. Biol. 2020, 102, 373–388. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | Genotypes | PH | PN | PL | FLL | FLW | GNPP | GWT | GL | GWD | YPP |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Normal (T0) | WT | 119.3 ± 3.4 | 7.4 ± 1.6 | 26.3 ± 1.3 | 53.1 ± 2.2 | 2.1 ± 0.3 | 136 ± 09 | 29.4 ± 1.3 | 9.2 ± 0.3 | 3.1 ± 0.3 | 30.8 ± 2.5 |
| GXU16-1 | 123.5 ± 3.8 ns | 7.6 ± 1.5 ns | 26.6 ± 1.6 ns | 39.7 ± 3.3 * | 1.6 ± 0.3 * | 141 ± 10 ns | 37.8 ± 1.3 * | 11.2 ± 0.2 * | 3.6 ± 0.2 * | 38.7 ± 1.7 * | |
| GXU16-2 | 124.6 ± 3.6 ns | 7.8 ± 2.2 ns | 26.5 ± 1.6 ns | 41.0 ± 2.5 * | 1.5 ± 0.3 * | 142 ± 11 ns | 38.4 ± 1.2 * | 11.3 ± 0.3 * | 3.7 ± 0.2 * | 38.9 ± 1.4 * | |
| GXU16-9 | 125.4 ± 4.3 ns | 7.7 ± 1.4 ns | 25.9 ± 1.7 ns | 38.4 ± 1.6 * | 1.5 ± 0.2 * | 142 ± 11 ns | 38.6 ± 1.4 * | 11.4 ± 0.5 * | 3.7 ± 0.4 * | 40.6 ± 1.5 * | |
| Drought (T0) | WT | 107.3 ± 3.2 | 7.7 ± 1.9 | 19.6 ± 1.5 | 47.2 ± 2.3 | 2.0 ± 0.3 | 84 ± 14 | 22.8 ± 1.2 | 7.1 ± 0.4 | 2.5 ± 0.2 | 21.4 ± 1.5 |
| GXU16-1 | 123.3 ± 3.3 * | 7.7 ± 2.4 ns | 26.9 ± 1.6 * | 37.8 ± 1.6 * | 1.6 ± 0.4 * | 112 ± 11 * | 30.6 ± 1.3 * | 8.8 ± 0.3 * | 3.0 ± 0.2 * | 26.8 ± 1.6 * | |
| GXU16-2 | 123.6 ± 4.2 * | 7.6 ± 1.3 ns | 26.9 ± 1.2 * | 39.3 ± 1.7 * | 1.6 ± 0.2 * | 113 ± 11 * | 33.6 ± 1.2 * | 9.5 ± 0.4 * | 3.1 ± 0.4 * | 31.6 ± 1.4 * | |
| GXU16-9 | 124.2 ± 4.1 * | 7.9 ± 1.3 ns | 27.5 ± 1.4 * | 36.4 ± 2.2 * | 1.5 ± 0.3 * | 115 ± 10 * | 34.7 ± 1.6 * | 10.1 ± 0.3 * | 3.1 ± 0.3 * | 32.6 ± 1.5 * | |
| Normal (T1) | WT | 1201 ± 2.4 | 7.6 ± 1.4 | 25.2 ± 1.2 | 52.0 ± 3.6 | 2.2 ± 0.1 | 137 ± 12 | 29.5 ± 1.2 | 9.0 ± 0.1 | 3.0 ± 0.2 | 31.3 ± 1.5 |
| GXU16-1-1 | 122.4 ± 3.6 ns | 7.5 ± 1.8 ns | 26.3 ± 1.4 ns | 39.5 ± 2.1 * | 1.6 ± 0.2 * | 140 ± 11 ns | 37.9 ± 1.5 * | 11.0 ± 0.3 * | 3.5 ± 0.1 * | 38.5 ± 1.3 * | |
| GXU16-2-1 | 124.2 ± 3.4 ns | 7.7 ± 2.1 ns | 26.4 ± 1.3 ns | 40.3 ± 1.6 * | 1.5 ± 0.4 * | 139 ± 10 ns | 38.6 ± 1.3 * | 11.1 ± 0.2 * | 3.6 ± 0.3 * | 39.1 ± 1.2 * | |
| GXU16-9-1 | 124.2 ± 5.4 ns | 7.8 ± 1.2 ns | 25.6 ± 1.2 ns | 38.5 ± 1.2 * | 1.5 ± 0.3 * | 140 ± 10 ns | 38.7 ± 1.1 * | 11.3 ± 0.6 * | 3.7 ± 0.5 * | 40.2 ± 1.2 * | |
| Drought (T1) | WT | 108.1 ± 2.4 | 7.6 ± 1.7 | 19.3 ± 1.4 | 48.5 ± 2.6 | 2.0 ± 0.2 | 85 ± 15 | 23.5 ± 1.3 | 7.0 ± 0.3 | 2.6 ± 0.1 | 21.2 ± 1.0 |
| GXU16-1-1 | 125.2 ± 3.1 * | 7.6 ± 2.1 ns | 26.4 ± 1.3 * | 38.2 ± 1.9 * | 1.6 ± 0.6 * | 111 ± 10 * | 29.4 ± 1.2 * | 8.6 ± 0.2 * | 3.0 ± 0.3 * | 26.2 ± 1.4 * | |
| GXU16-2-1 | 124.1 ± 5.1 * | 7.7 ± 1.0 ns | 27.4 ± 1.3 * | 39.5 ± 1.4 * | 1.6 ± 0.3 * | 112 ± 10 * | 34.5 ± 1.3 * | 9.3 ± 0.3 * | 3.1 ± 0.5 * | 31.2 ± 1.2 * | |
| GXU16-9-1 | 123.1 ± 3.3 * | 7.7 ± 1.2 ns | 27.6 ± 1.5 * | 36.0 ± 3.6 * | 1.5 ± 0.1 * | 114 ± 12 * | 34.5 ± 1.1 * | 10.0 ± 0.2 * | 3.1 ± 0.2 * | 32.1 ± 1.3 * |
| Proteins | Annotation | Regulation | Fold Change |
|---|---|---|---|
| Circadian Clock DEPs | |||
| Q9AWL7 | GIGANTEA | Up | 3.59 |
| Q657D6 | Early flowering protein | Up | 1.62 |
| Q689G9 | Pseudo-response regulator 1 | Up | 1.55 |
| A0A0N7KPA9 | MYB family transcription factor | Up | 1.56 |
| Q0D3B6 | Days to heading 7 | Up | 1.96 |
| Q2R2W1 | FLAVIN-BINDING, KELCH REPEAT, F-BOX 1 | Up | 1.77 |
| C6F1N5 | Pseudo-response regulator 59 | Up | 3.36 |
| Q689G6 | Pseudo-response regulator 95 | Up | 1.70 |
| Drought Responsive DEPs | |||
| Q9FXQ3 | Calcium-dependent protein kinase 13 | Up | 2.08 |
| Q5JLS2 | CBL-interacting protein kinase 12 | Up | 1.68 |
| Q6ZKN0 | WRKY transcription factor 30 | Up | 1.57 |
| Q0JQF7 | AP2/EREBP transcription factor | Up | 1.81 |
| Q0JHF1 | ABA responsive element binding factor 1 | Up | 2.29 |
| Q10NE1 | SALT-AND DROUGHT-INDUCED RING FINGER 1 | Up | 1.56 |
| Q0IUZ3 | NAC protein | Up | 2.78 |
| Q0DMY6 | DROUGHT AND SALT TOLERANCE | Up | 2.15 |
| Q64MA1 | Dehydration-responsive element-binding protein 1A | Up | 3.16 |
| Q6IEN1 | WRKY transcription factor | Up | 1.87 |
| DEPs Related to Reactive Oxygen Species | |||
| A3REN3 | Catalase (CAT) | Up | 1.68 |
| B8ARU3 | Peroxidase (POD) | Up | 1.91 |
| A2YPX2 | POD | Up | 1.59 |
| A2X2T0 | POD | Up | 1.83 |
| B8A755 | POD | Up | 1.58 |
| B8B5W7 | POD | Up | 1.99 |
| A2X822 | POD | Down | 0.56 |
| B8B3L5 | POD | Up | 1.62 |
| B8ASV8 | POD | Up | 2.09 |
| A2Z4F1 | POD | Up | 1.65 |
| B8B653 | POD | Up | 1.61 |
| B8B5W6 | POD | Up | 1.73 |
| Q01MI9 | POD | Down | 0.50 |
| A2WPA1 | POD | Up | 1.74 |
| A2WNR8 | POD | Up | 1.50 |
| B8APG3 | POD | Up | 1.92 |
| B8ARU4 | POD | Up | 1.62 |
| A2Z9R2 | POD | Up | 3.19 |
| B8BM92 | POD | Up | 1.86 |
| A2ZAQ7 | POD | Up | 1.55 |
| B8AWM4 | Superoxide dismutase (SOD) | Up | 1.56 |
| A2YY59 | SOD | Up | 2.08 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Usman, B.; Nawaz, G.; Zhao, N.; Liao, S.; Liu, Y.; Li, R. Precise Editing of the OsPYL9 Gene by RNA-Guided Cas9 Nuclease Confers Enhanced Drought Tolerance and Grain Yield in Rice (Oryza sativa L.) by Regulating Circadian Rhythm and Abiotic Stress Responsive Proteins. Int. J. Mol. Sci. 2020, 21, 7854. https://doi.org/10.3390/ijms21217854
Usman B, Nawaz G, Zhao N, Liao S, Liu Y, Li R. Precise Editing of the OsPYL9 Gene by RNA-Guided Cas9 Nuclease Confers Enhanced Drought Tolerance and Grain Yield in Rice (Oryza sativa L.) by Regulating Circadian Rhythm and Abiotic Stress Responsive Proteins. International Journal of Molecular Sciences. 2020; 21(21):7854. https://doi.org/10.3390/ijms21217854
Chicago/Turabian StyleUsman, Babar, Gul Nawaz, Neng Zhao, Shanyue Liao, Yaoguang Liu, and Rongbai Li. 2020. "Precise Editing of the OsPYL9 Gene by RNA-Guided Cas9 Nuclease Confers Enhanced Drought Tolerance and Grain Yield in Rice (Oryza sativa L.) by Regulating Circadian Rhythm and Abiotic Stress Responsive Proteins" International Journal of Molecular Sciences 21, no. 21: 7854. https://doi.org/10.3390/ijms21217854