Cellular Processes Involved in Jurkat Cells Exposed to Nanosecond Pulsed Electric Field


Abstract
:1. Introduction
2. Results
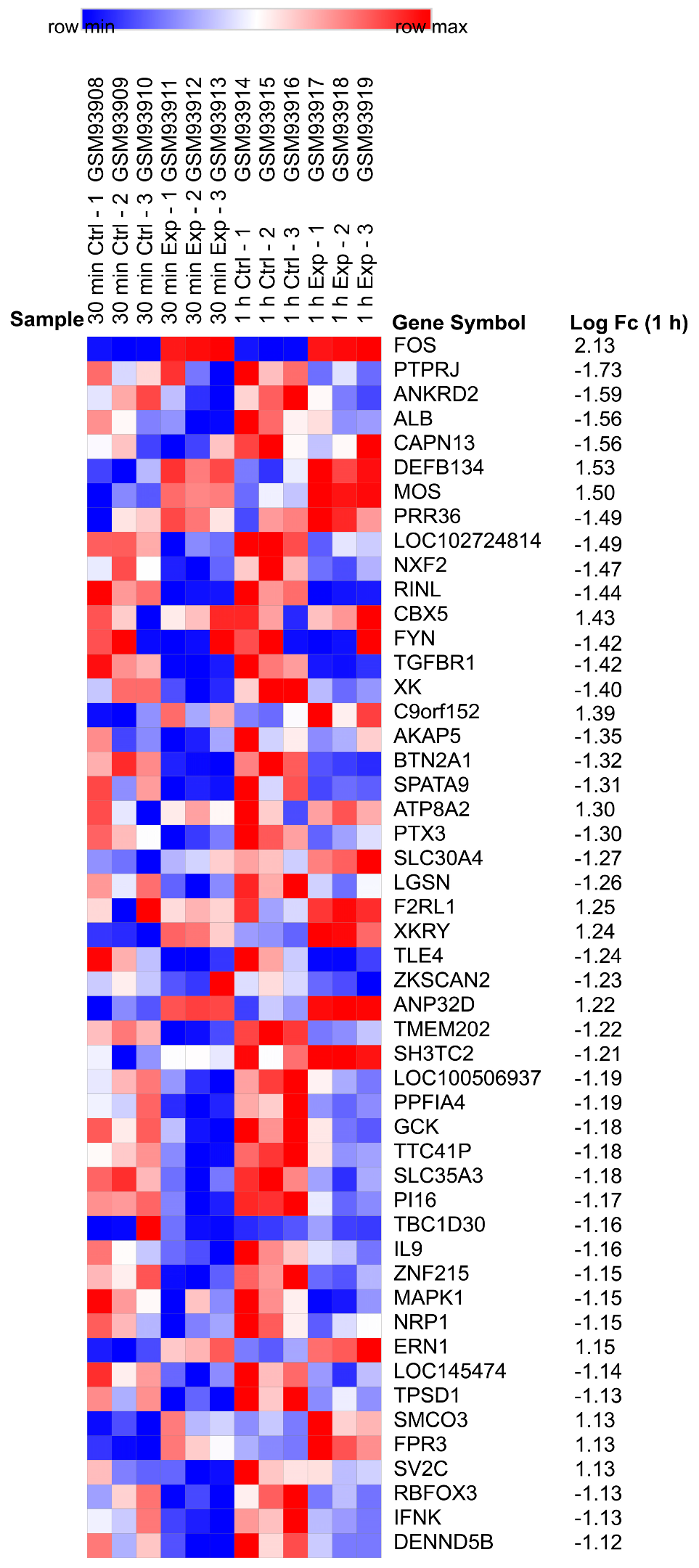
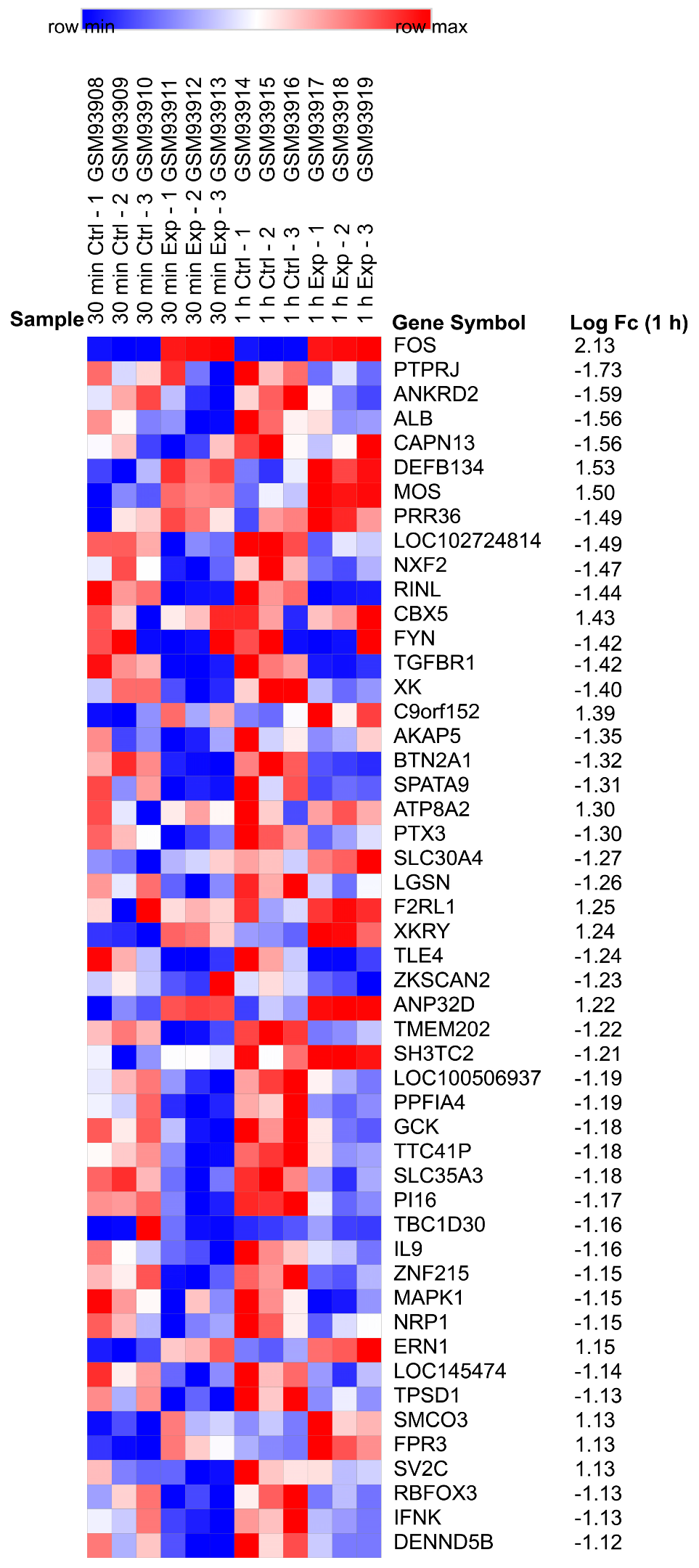
2.1. Identification of DEGs
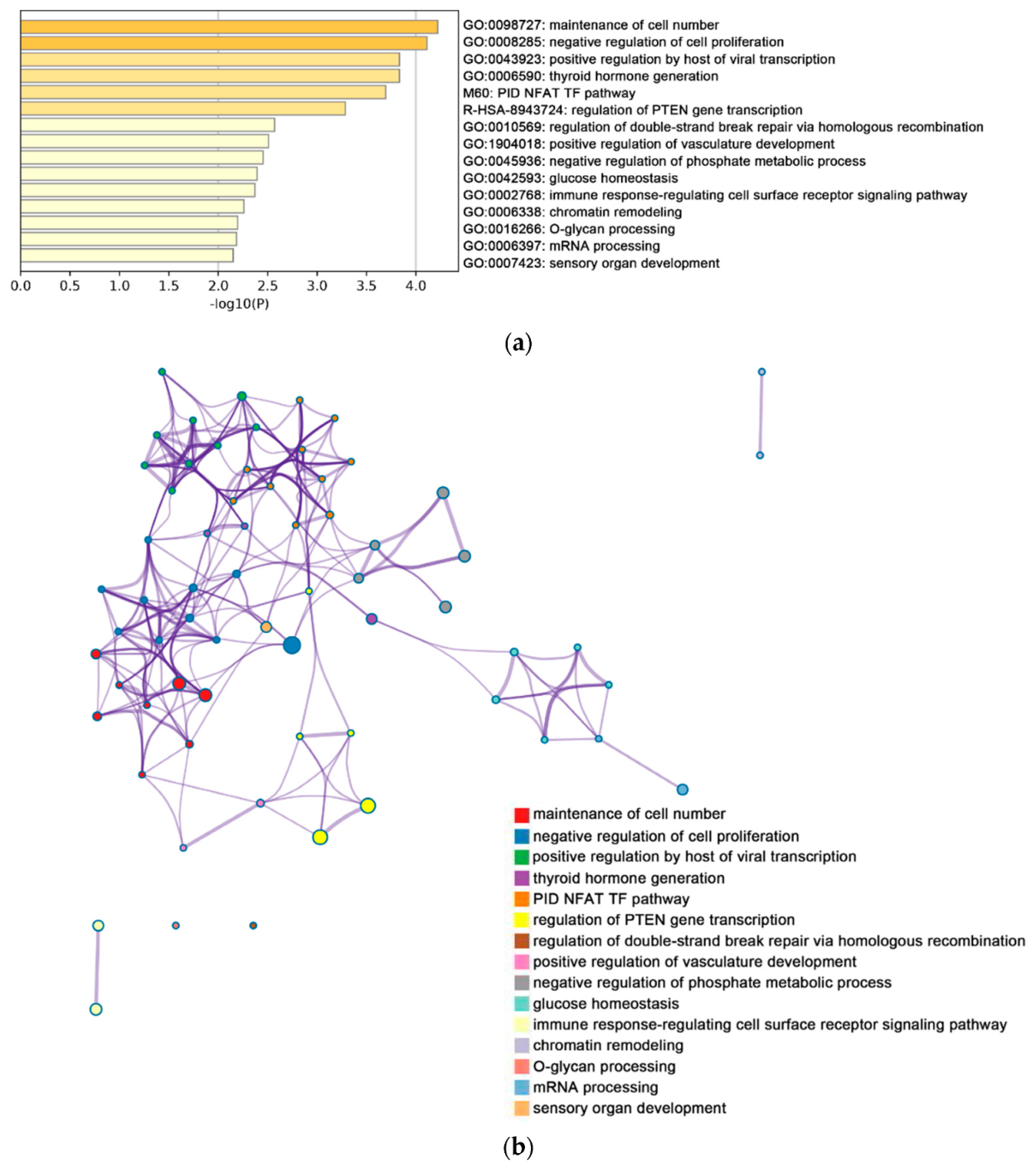
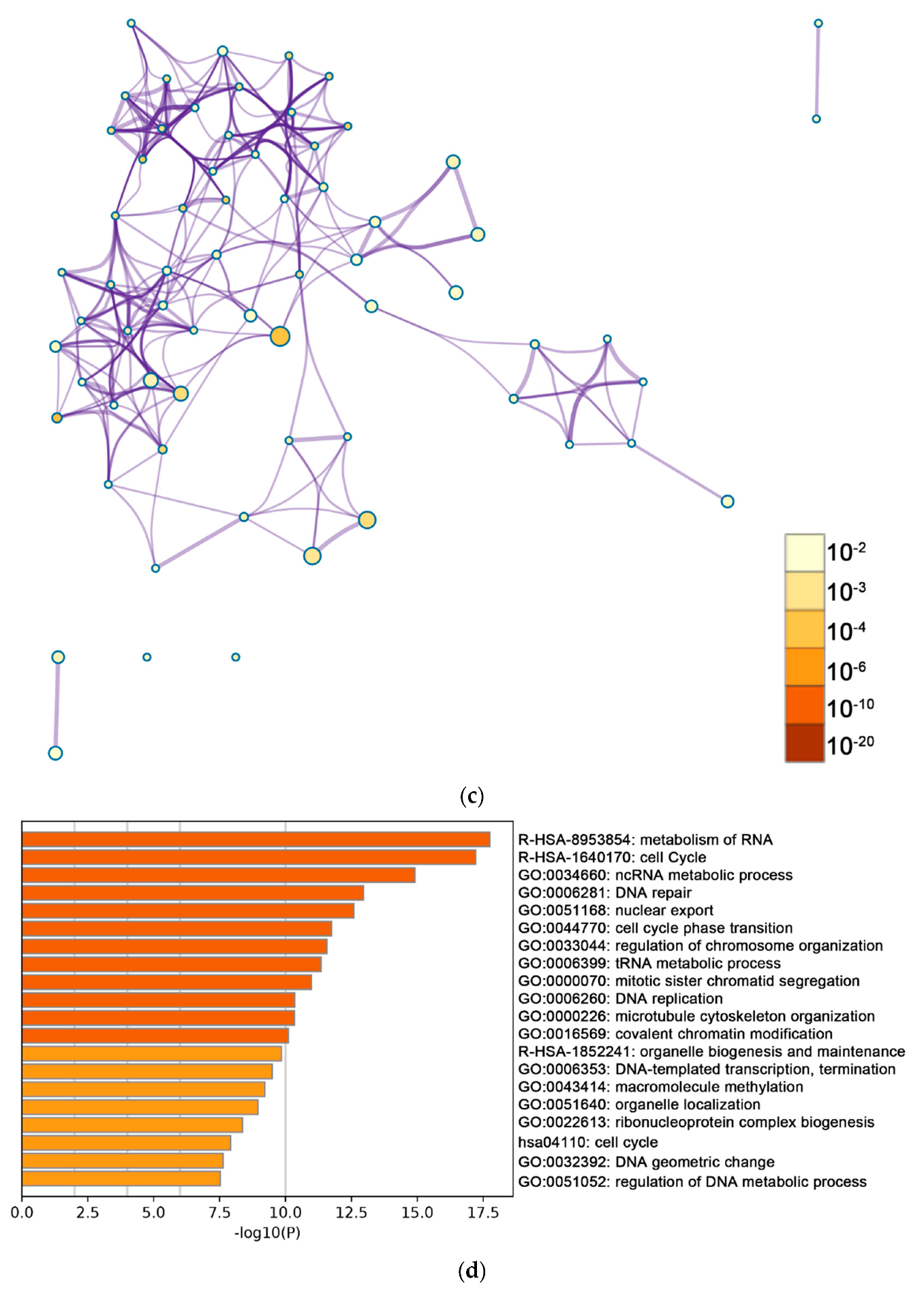
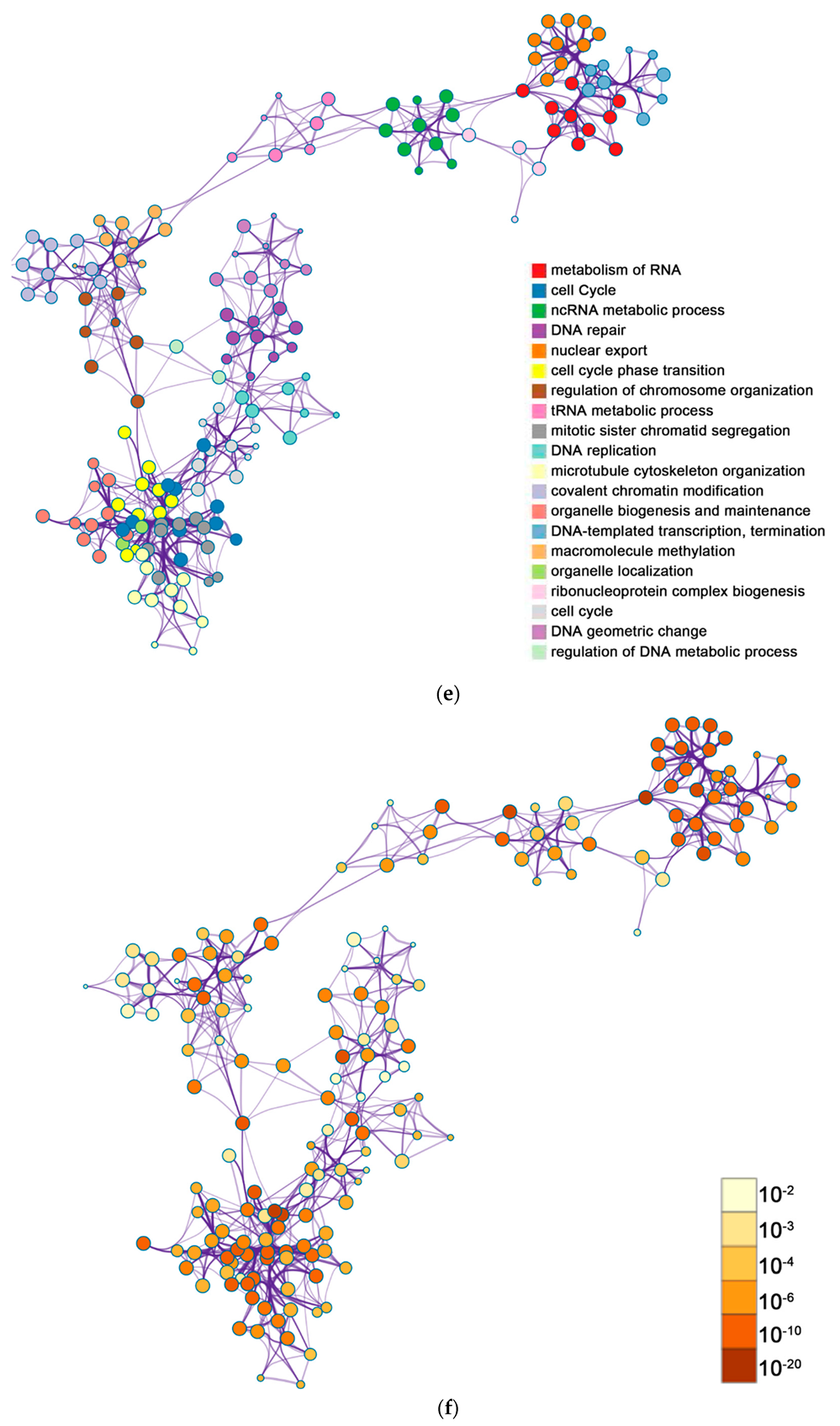
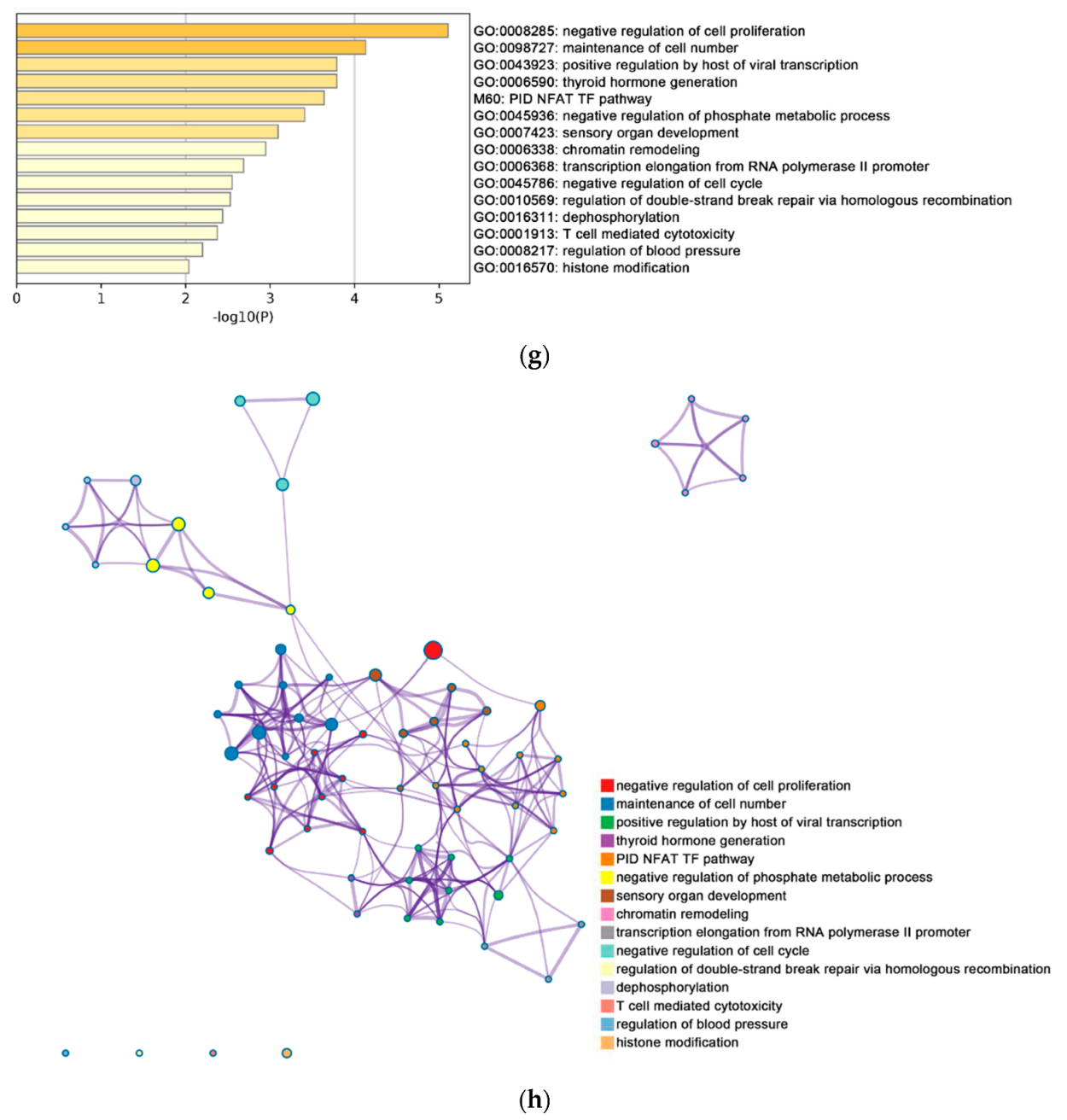
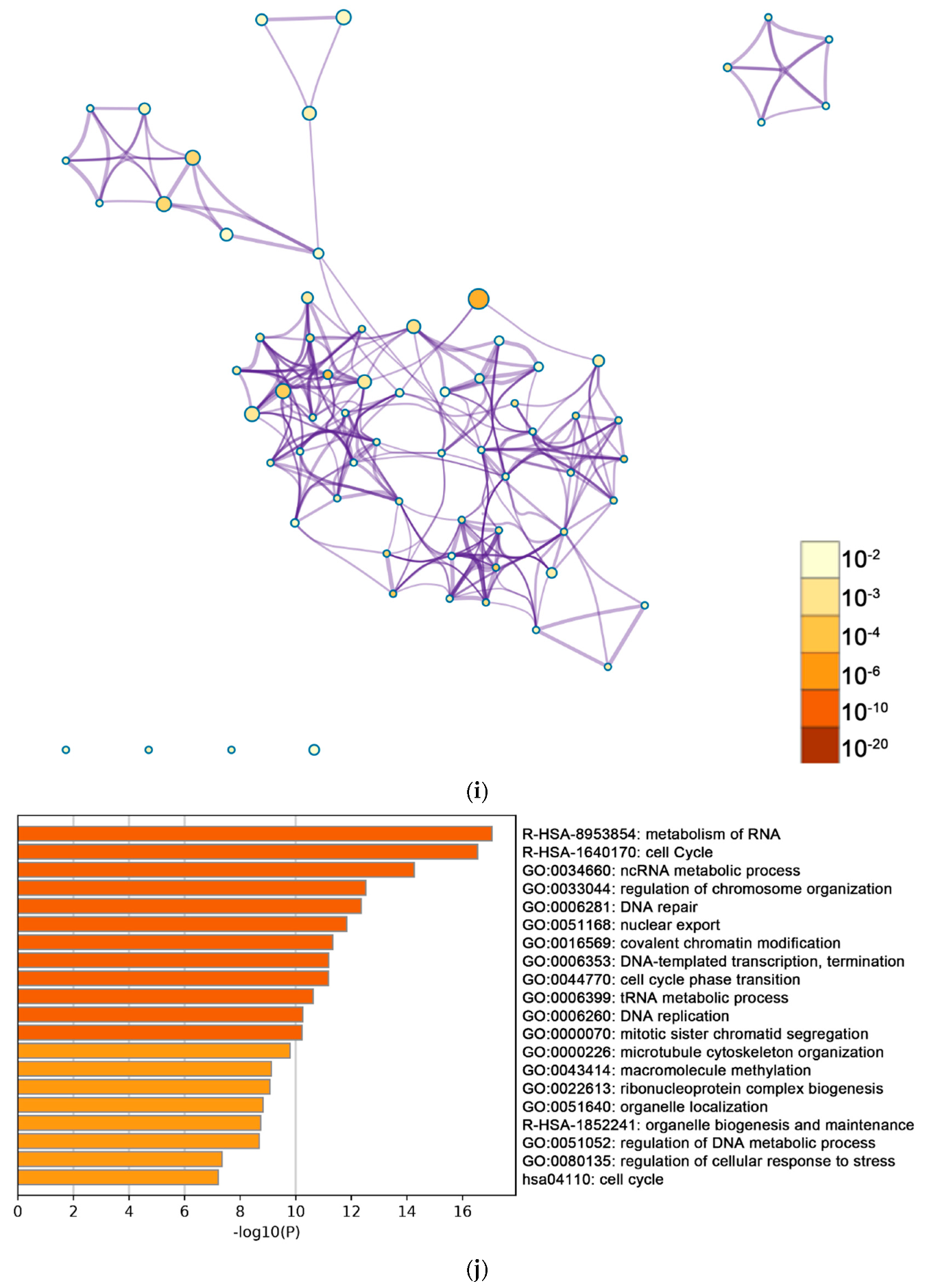
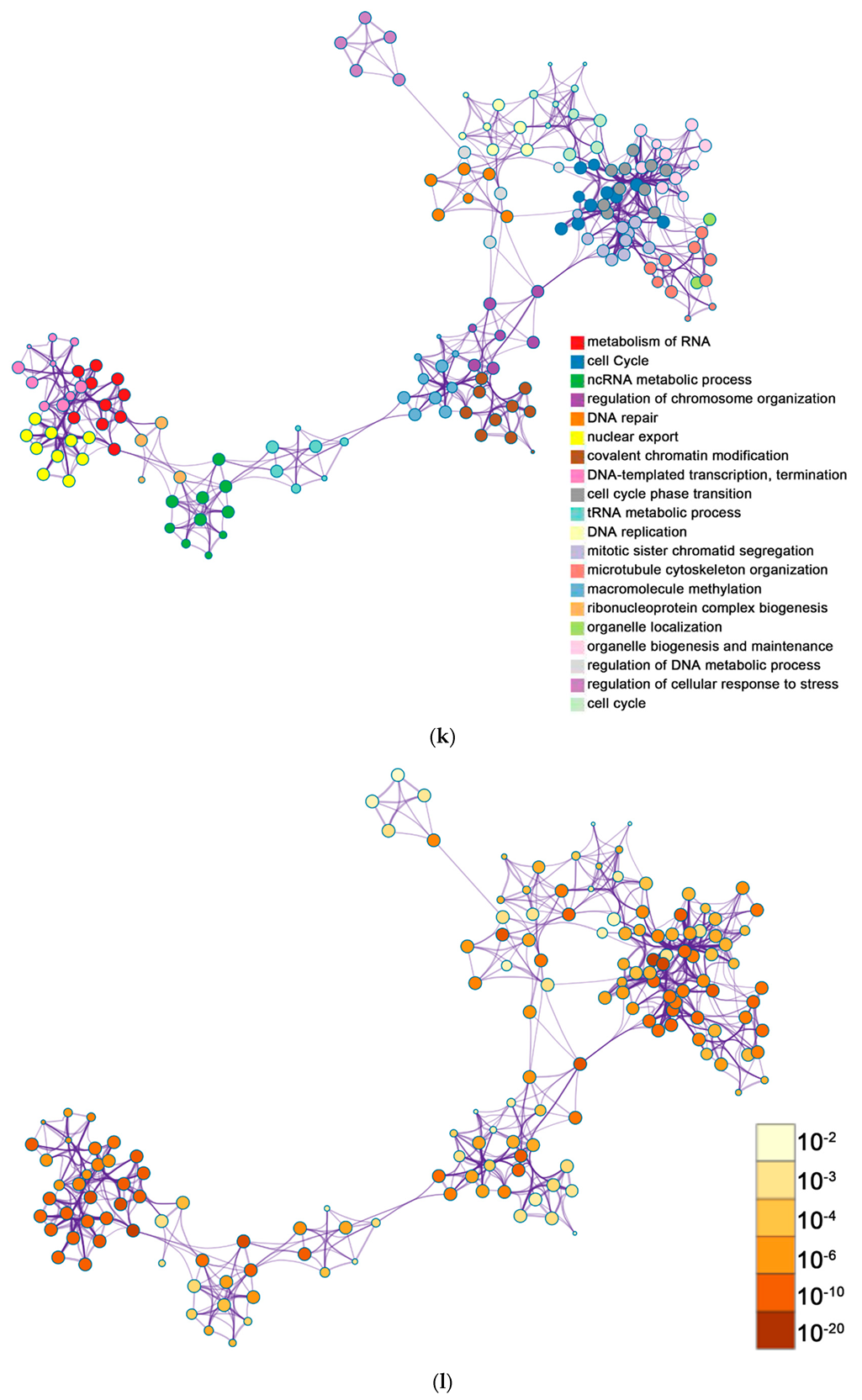
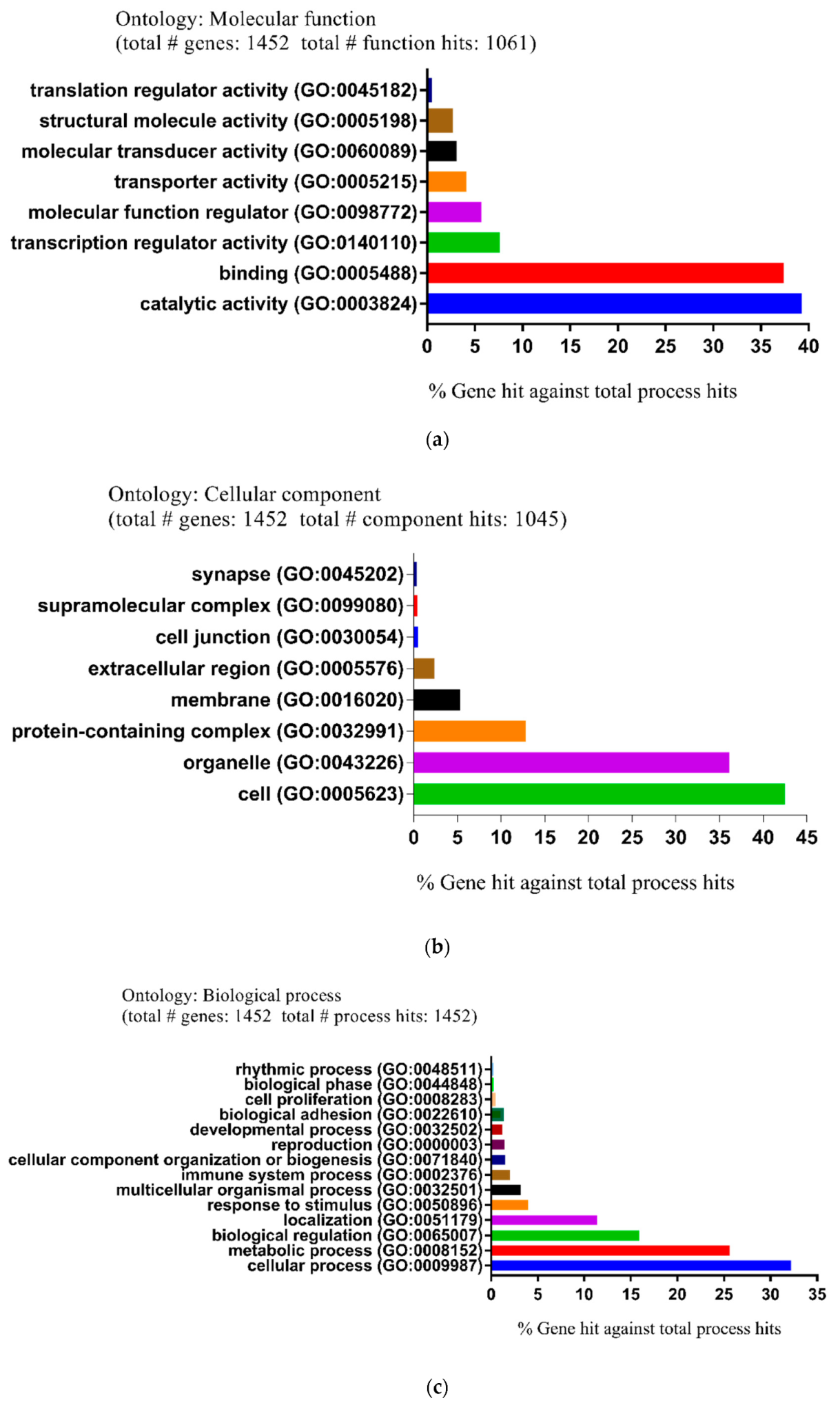
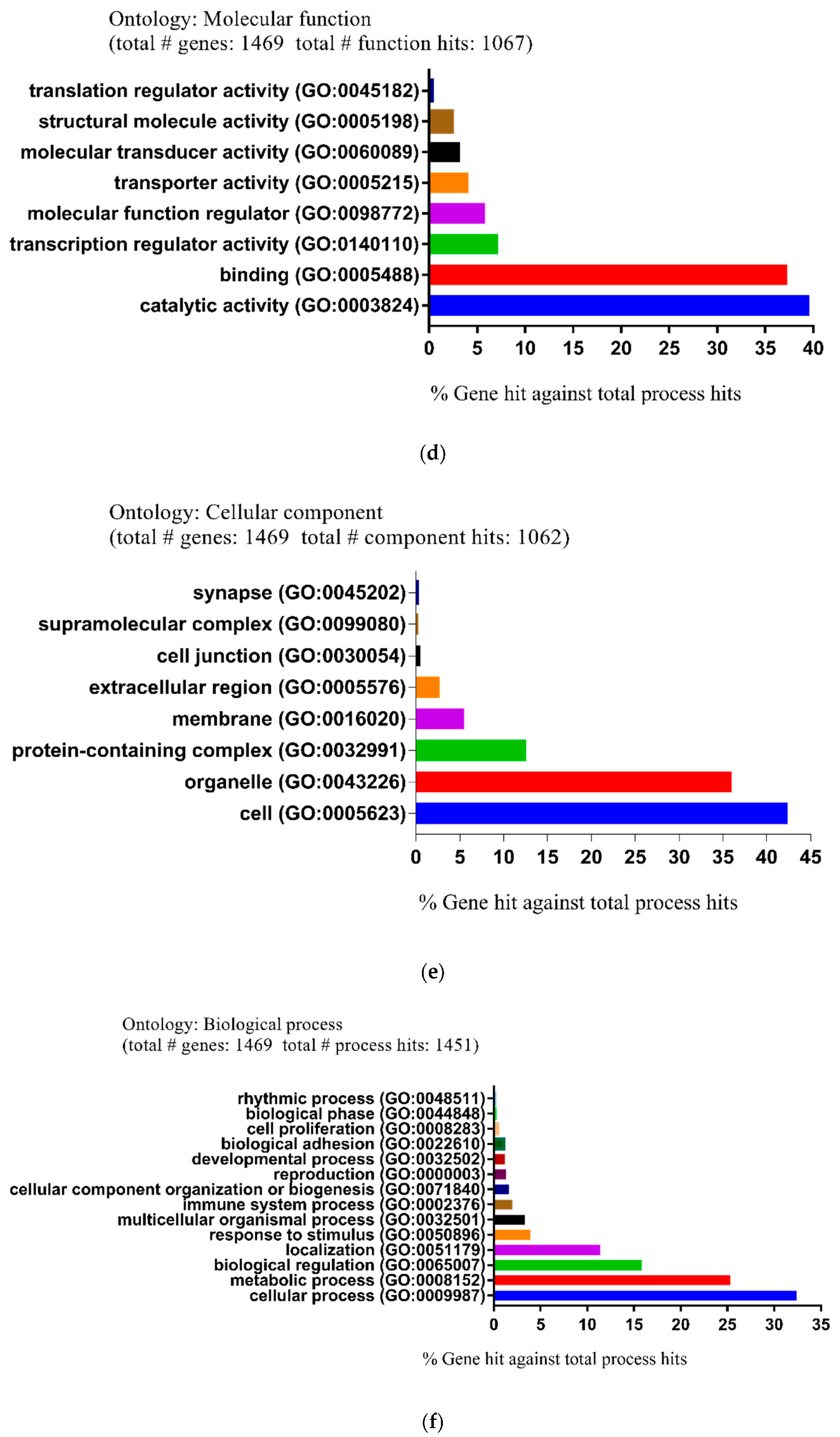
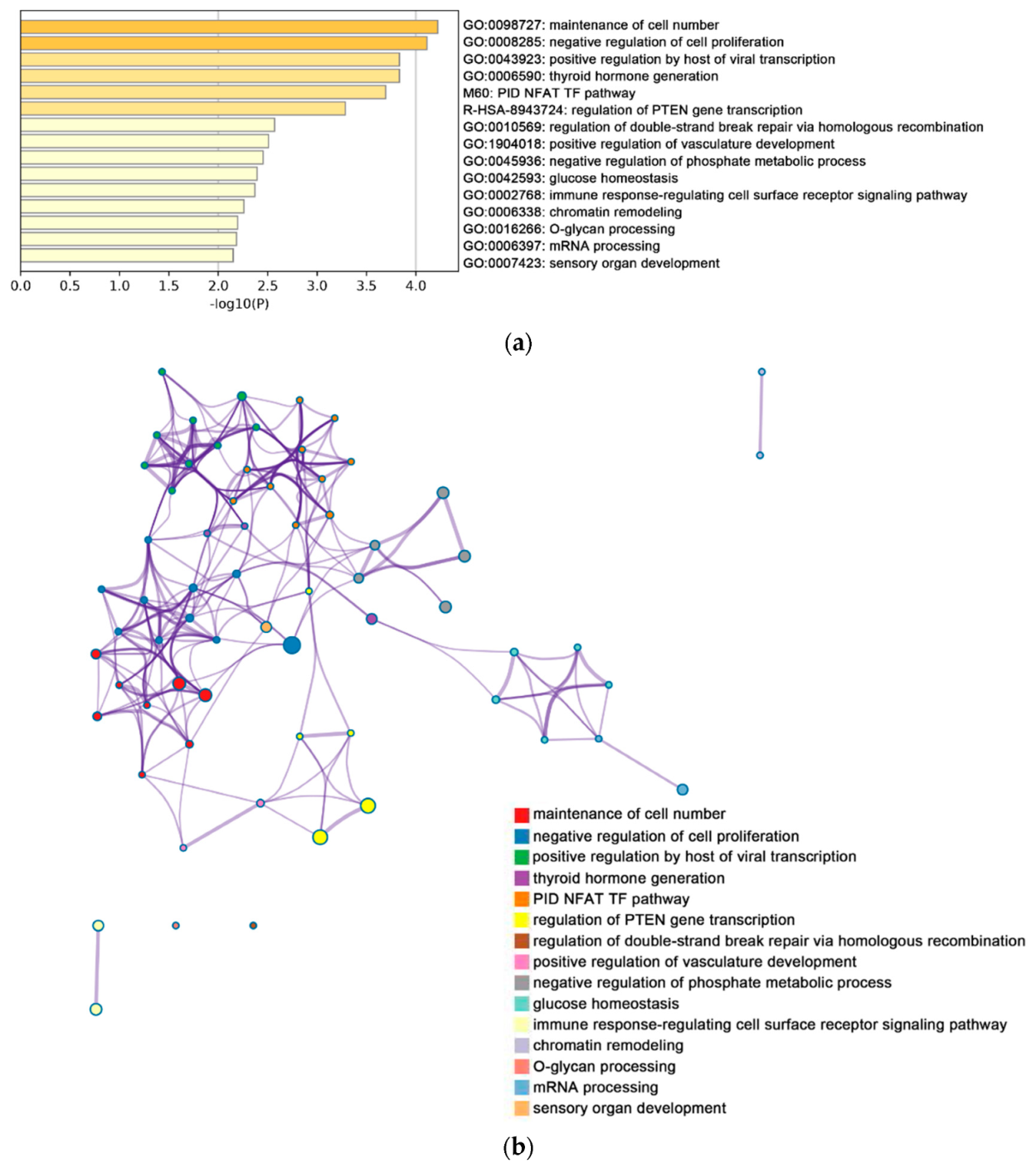
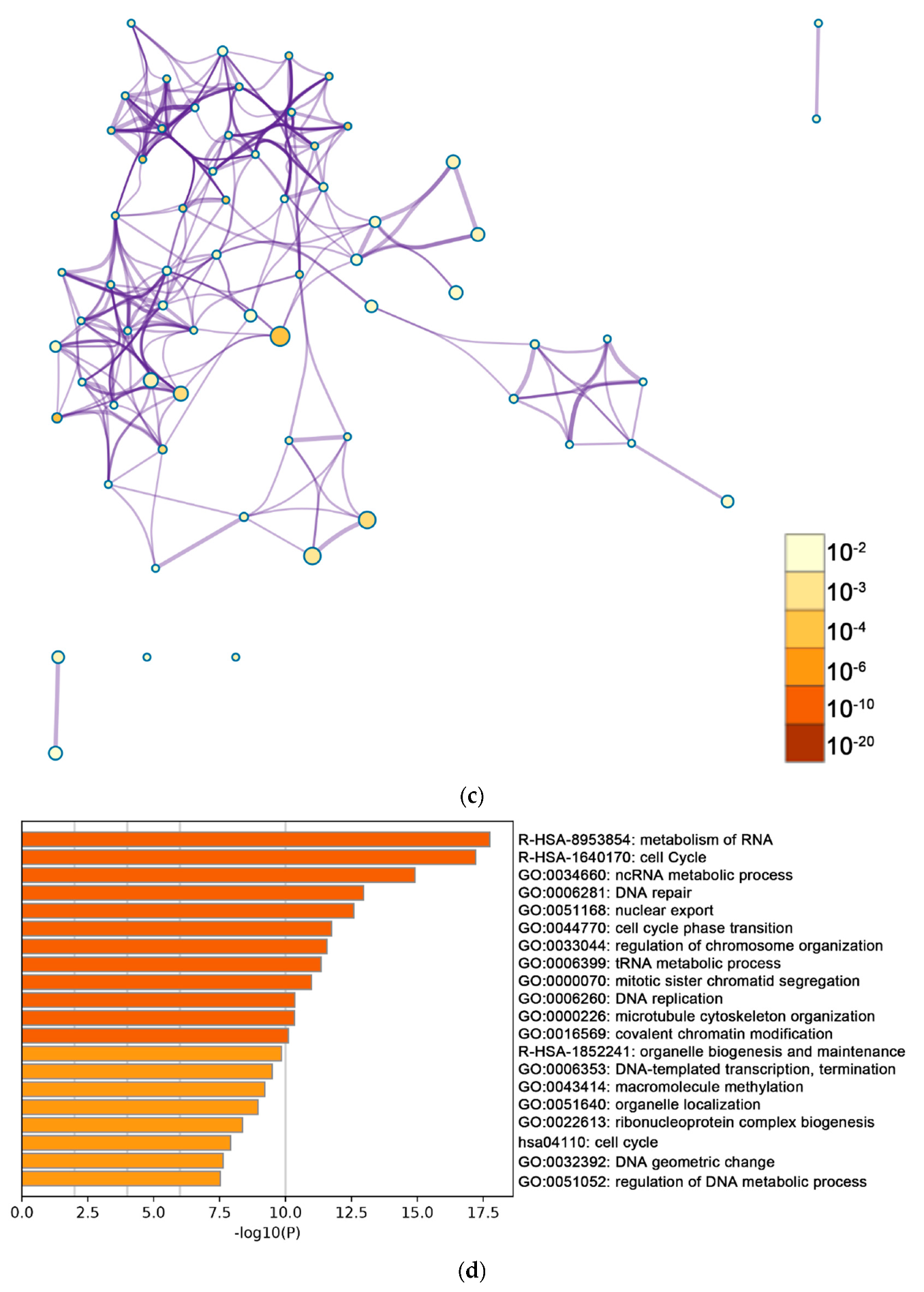
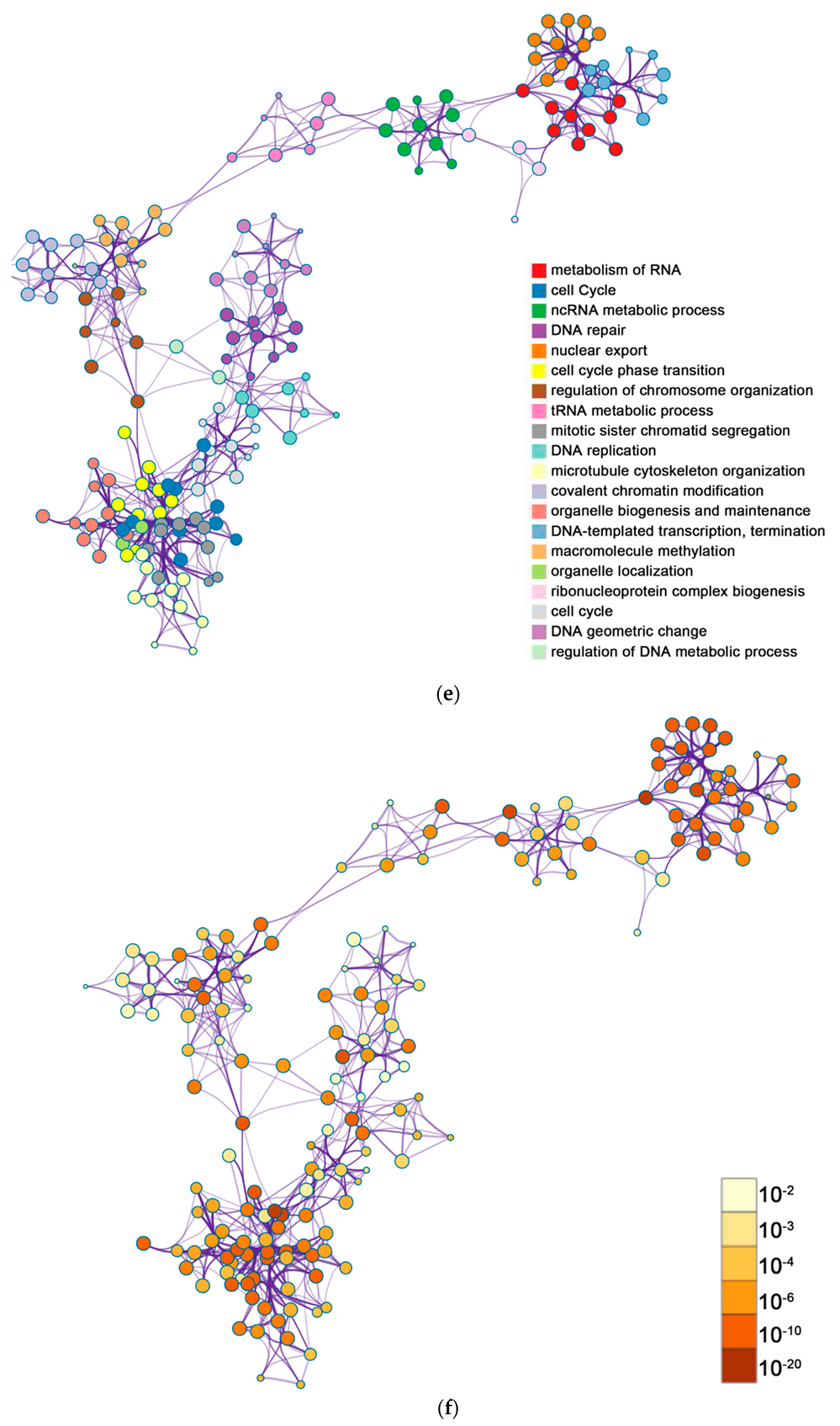
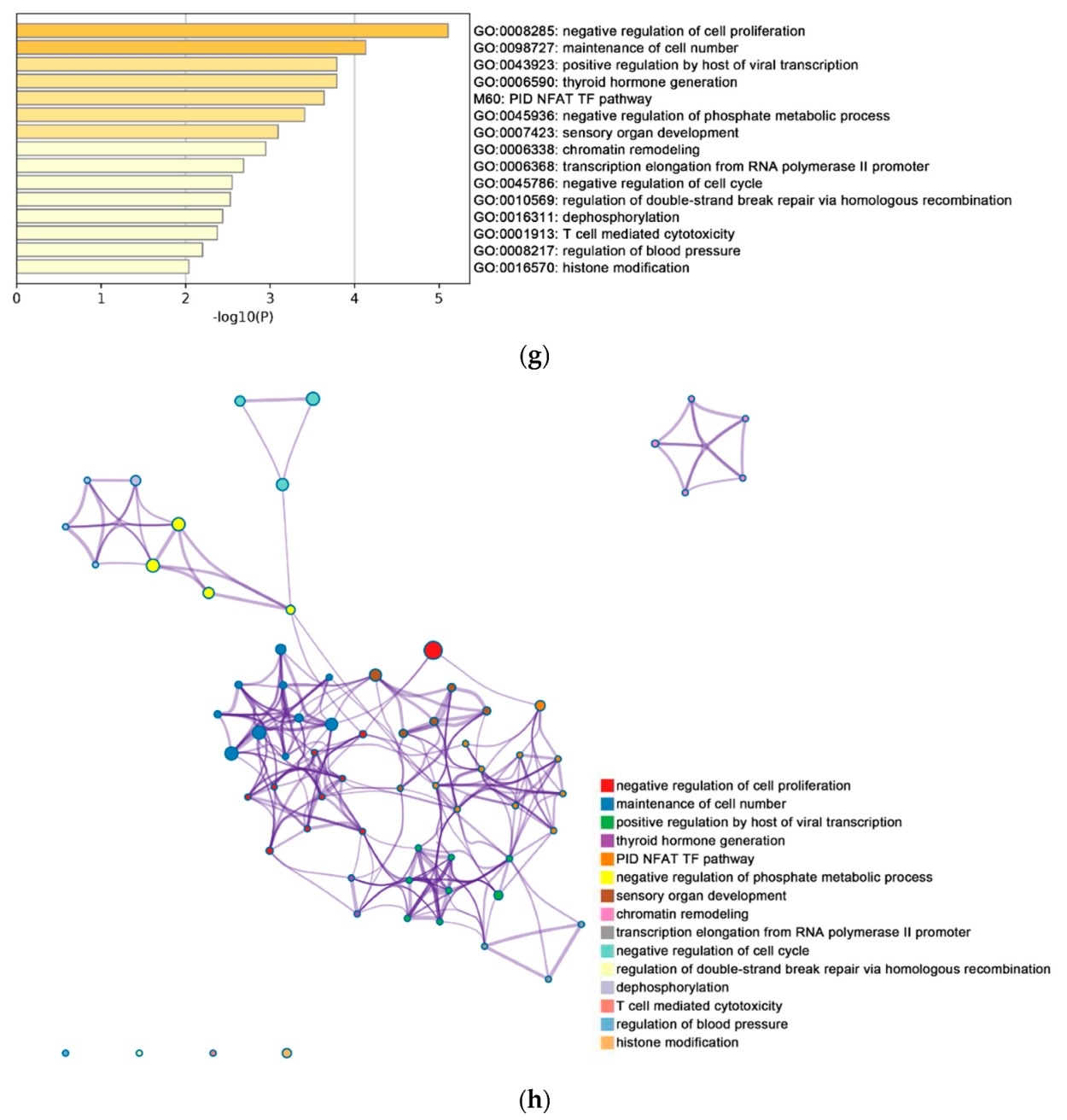
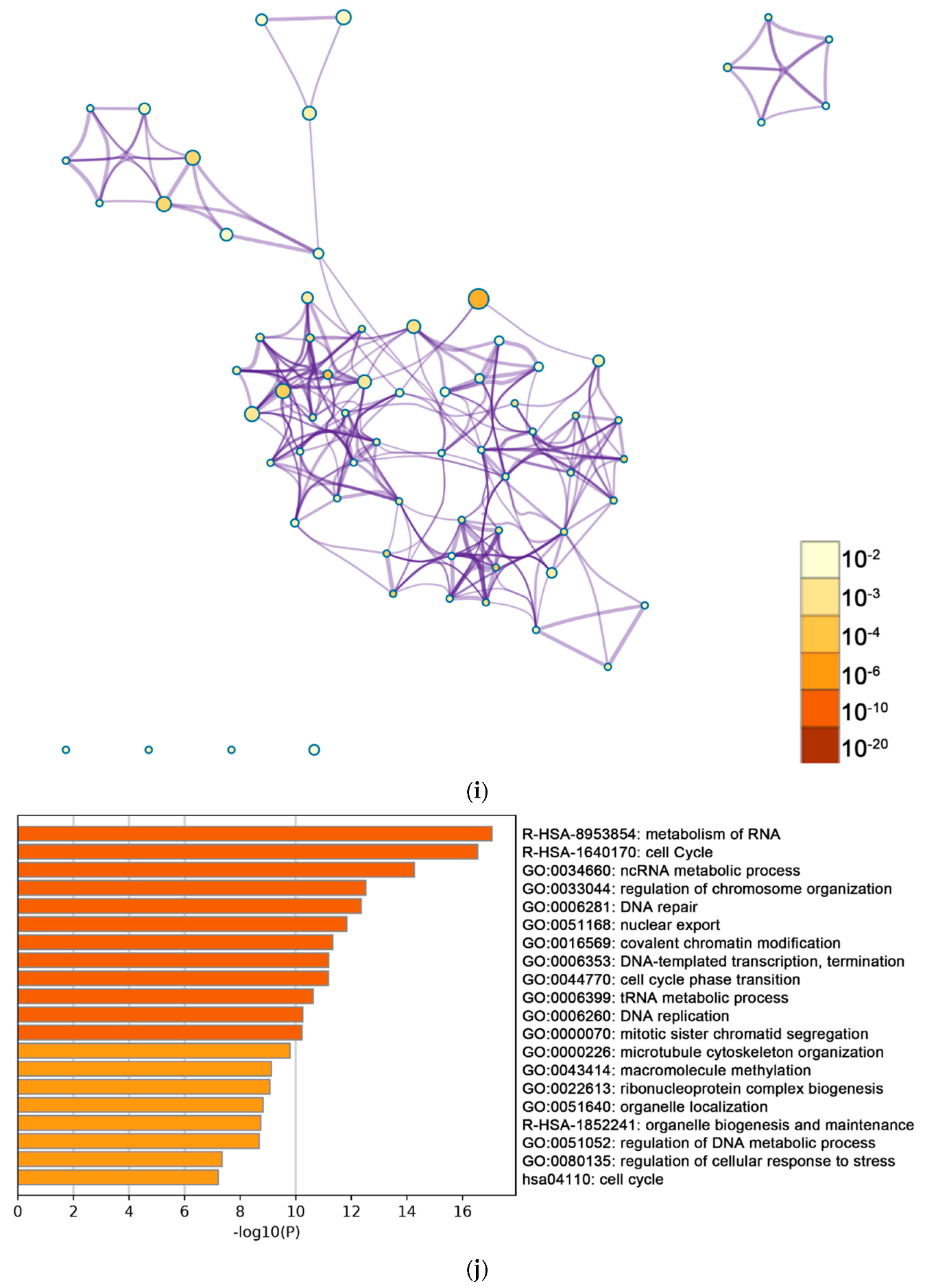
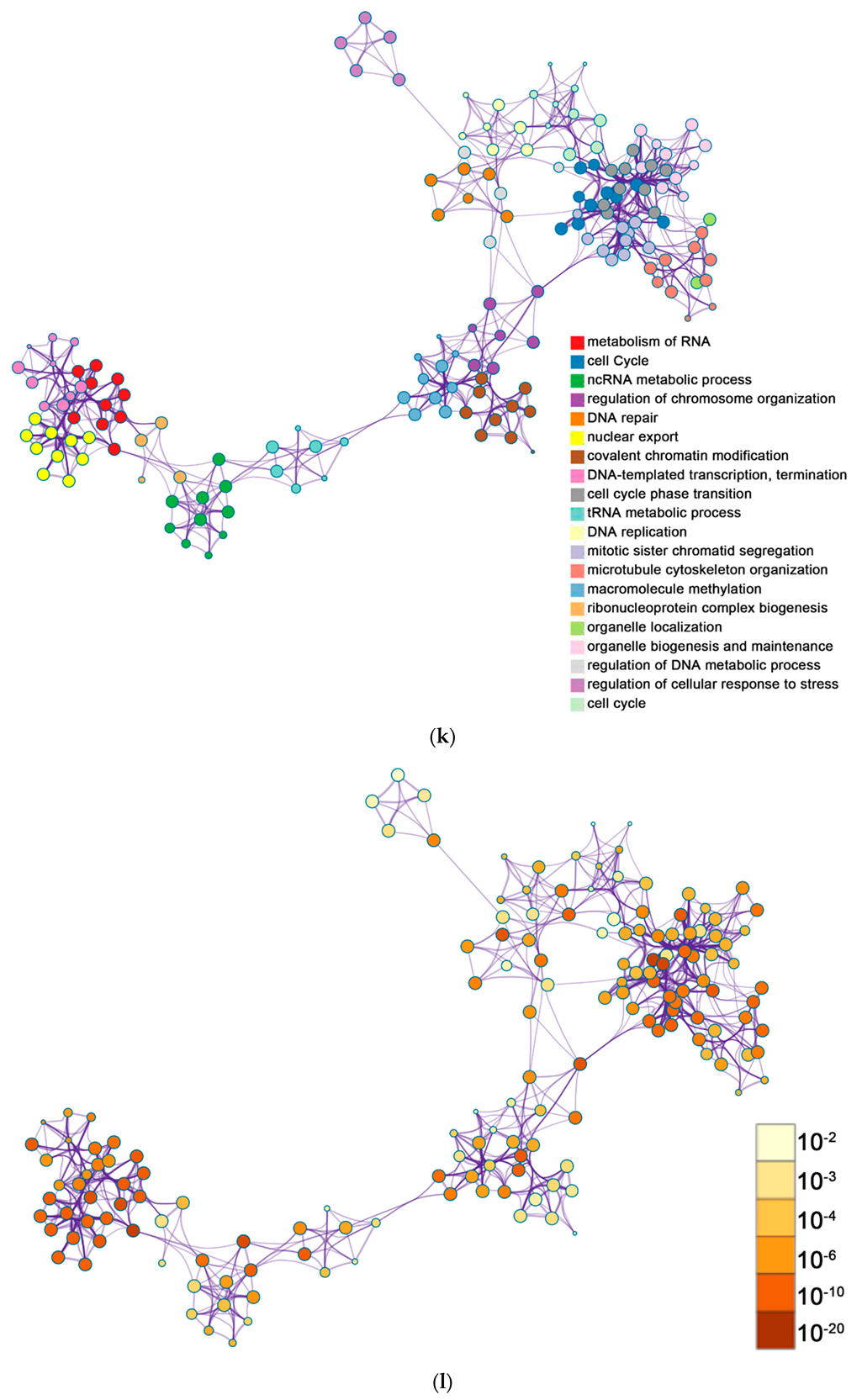
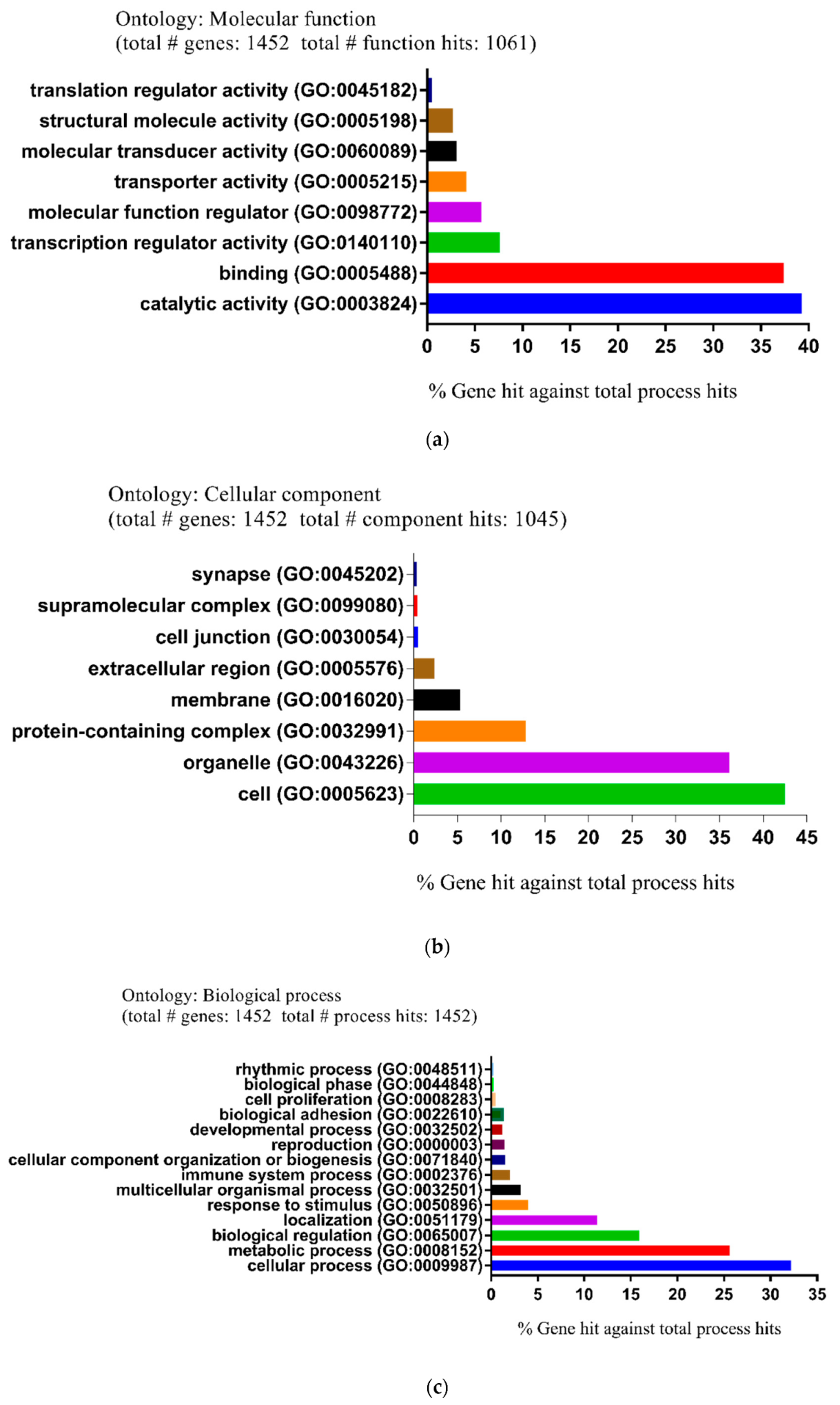
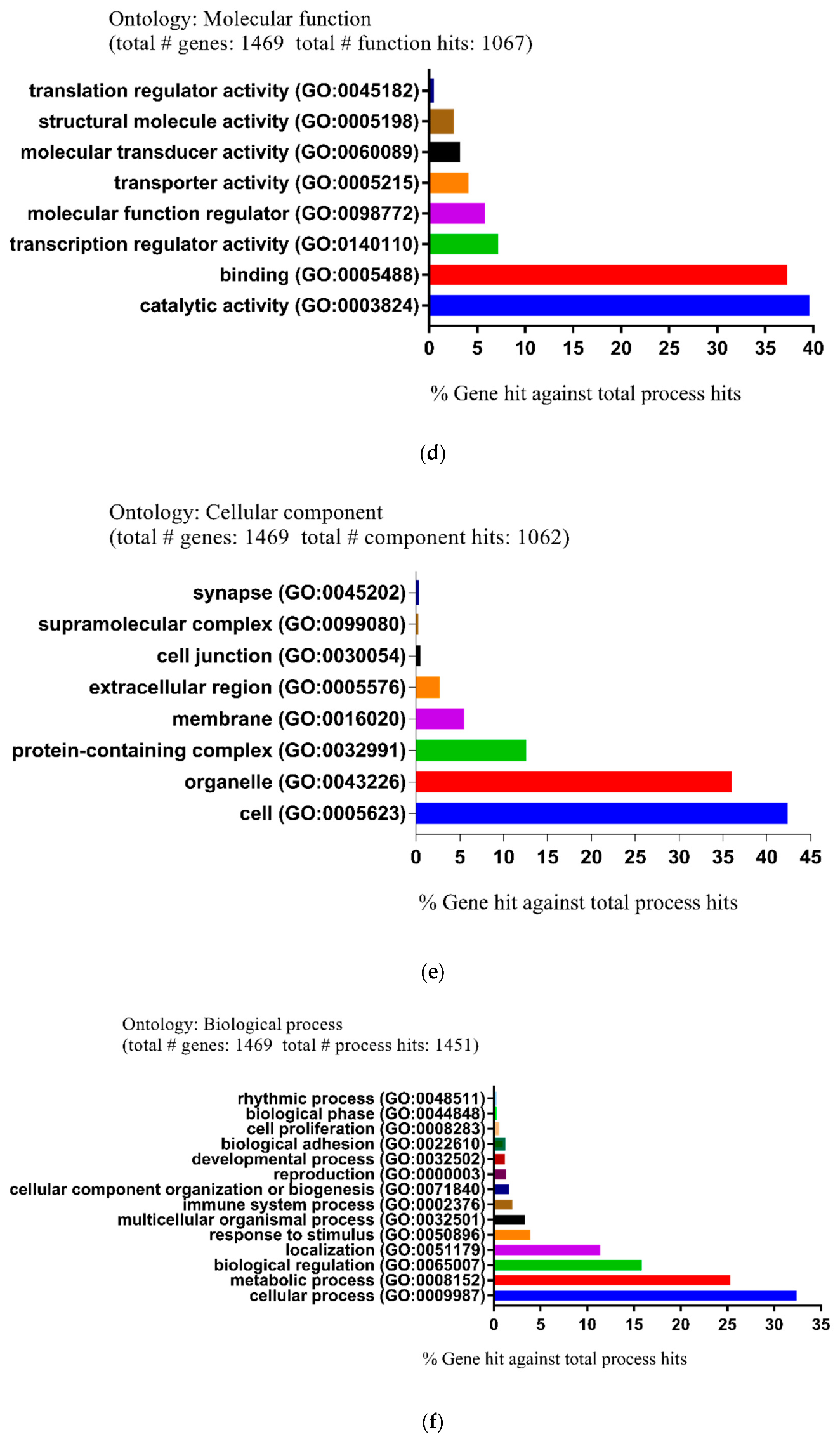
2.2. Functional and Pathway Enrichment Analysis of Identified Modules Associated with DEGs
2.3. Module Screening from the PPI Network
2.4. Sub-Localization Expression Analysis of Hub Genes
2.5. Mining Genetic Alterations Connected with Hub Genes by cBioportal
2.6. The Eight Hub Genes Expressed in Leukemia by Using Oncomine
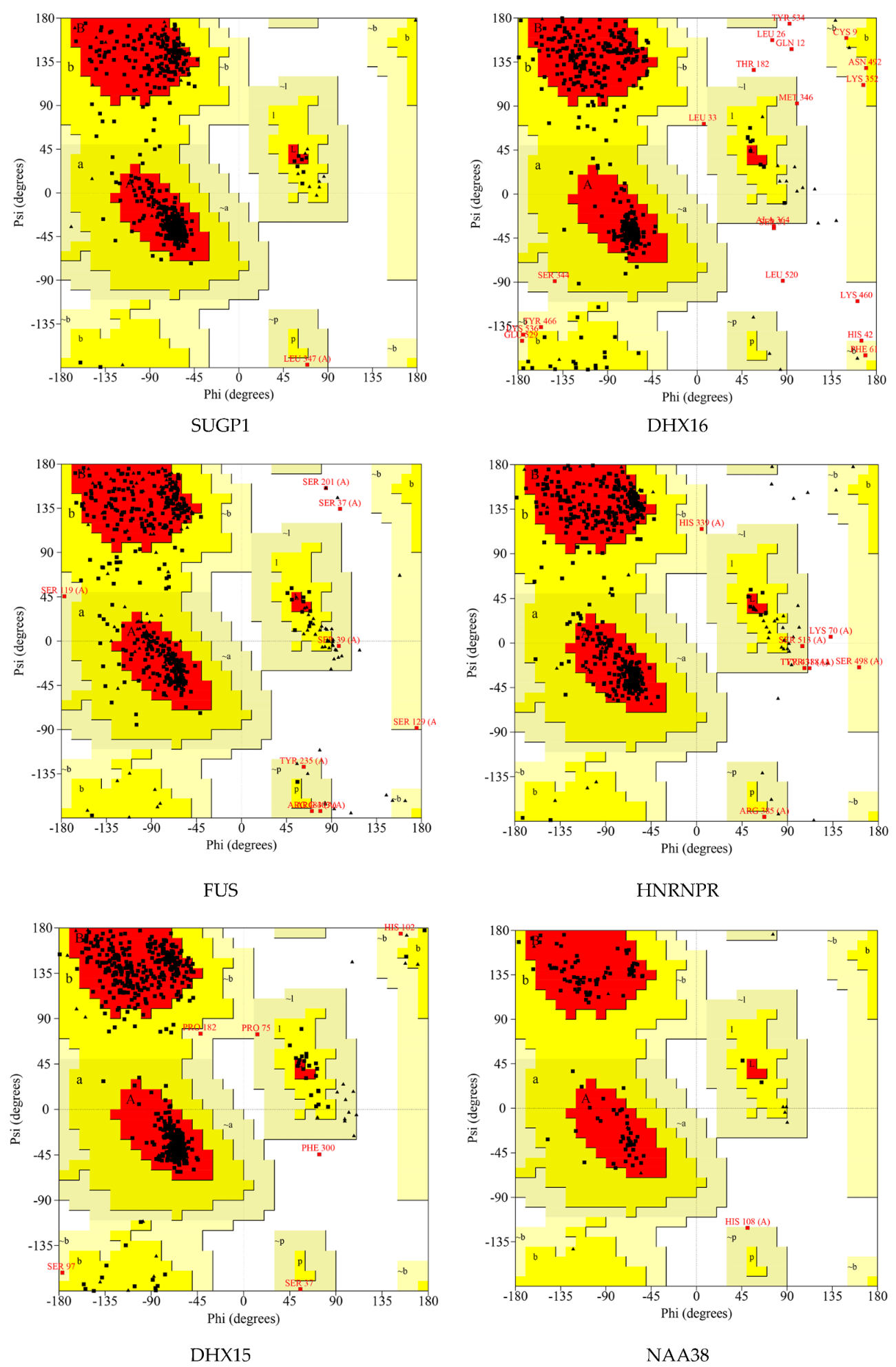
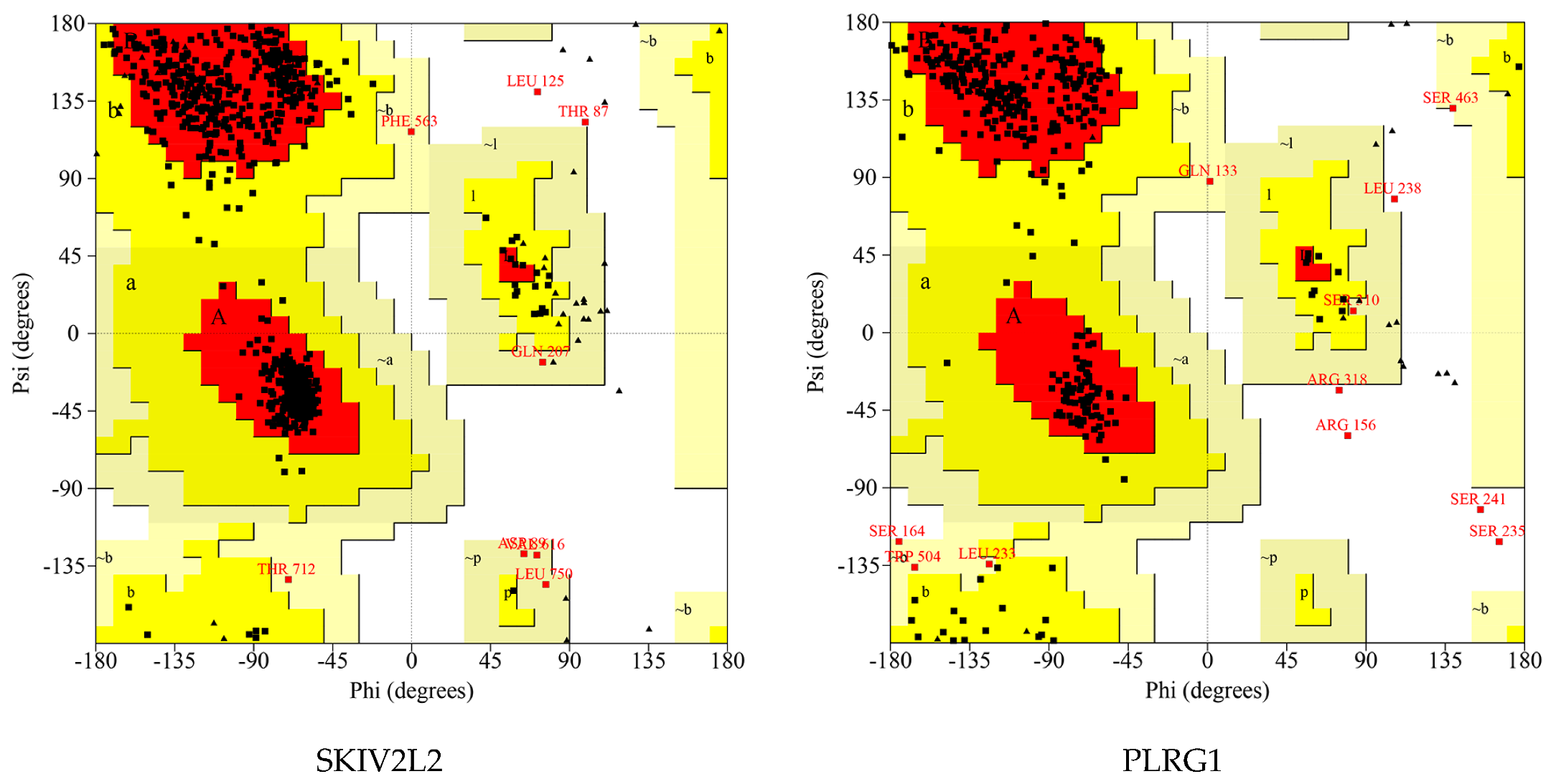
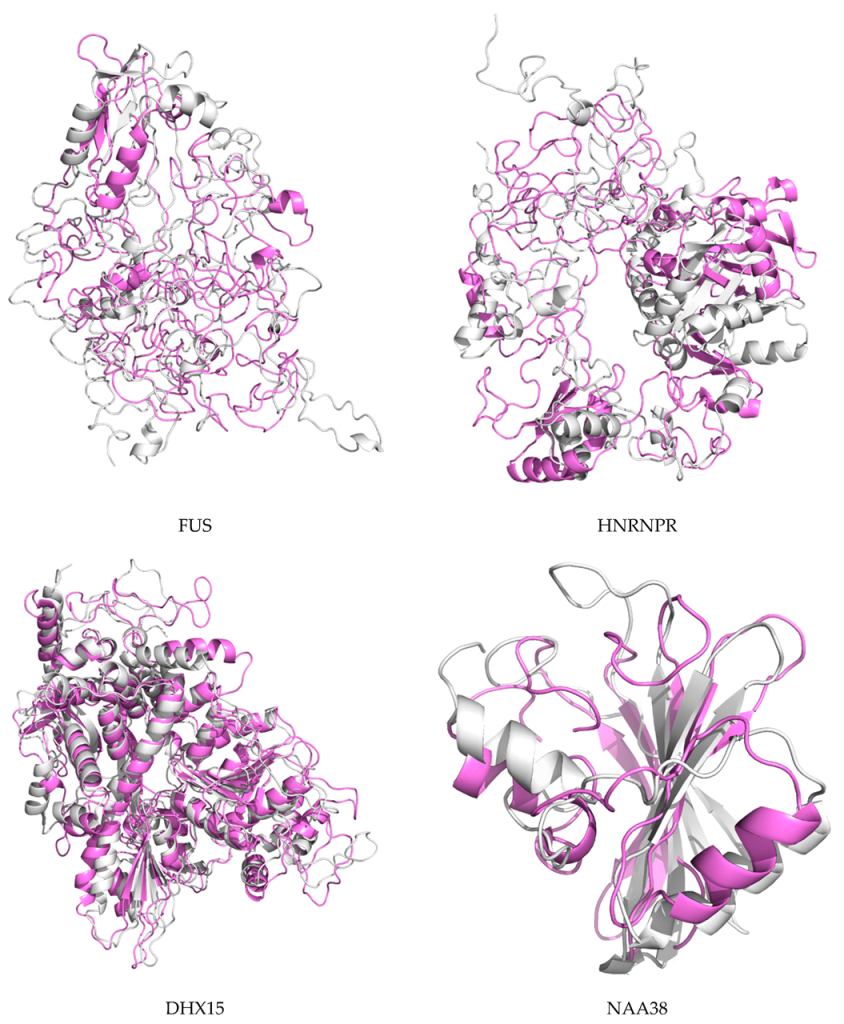
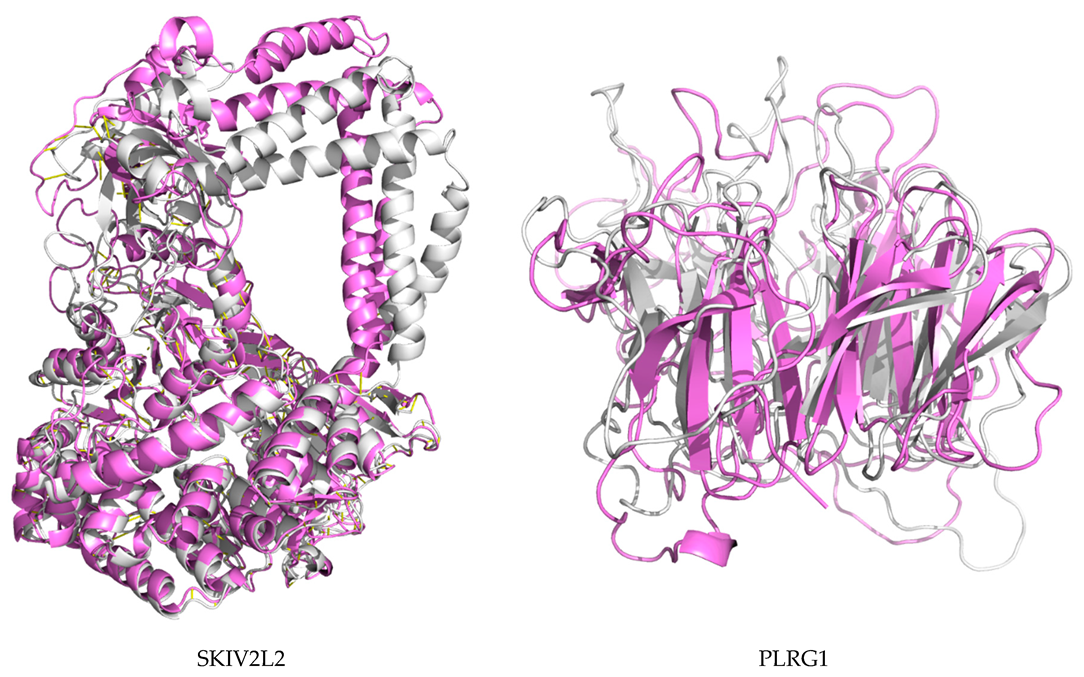
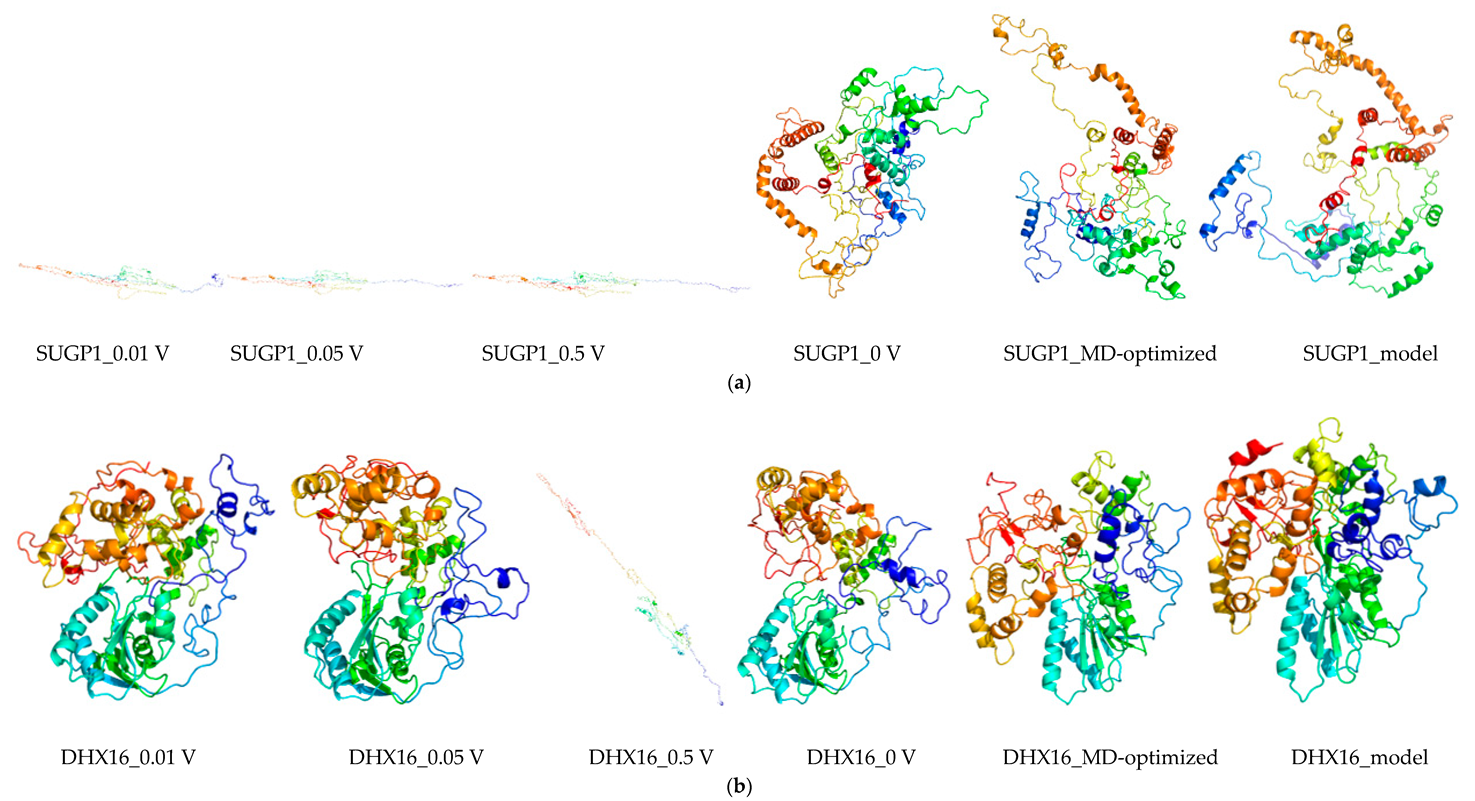
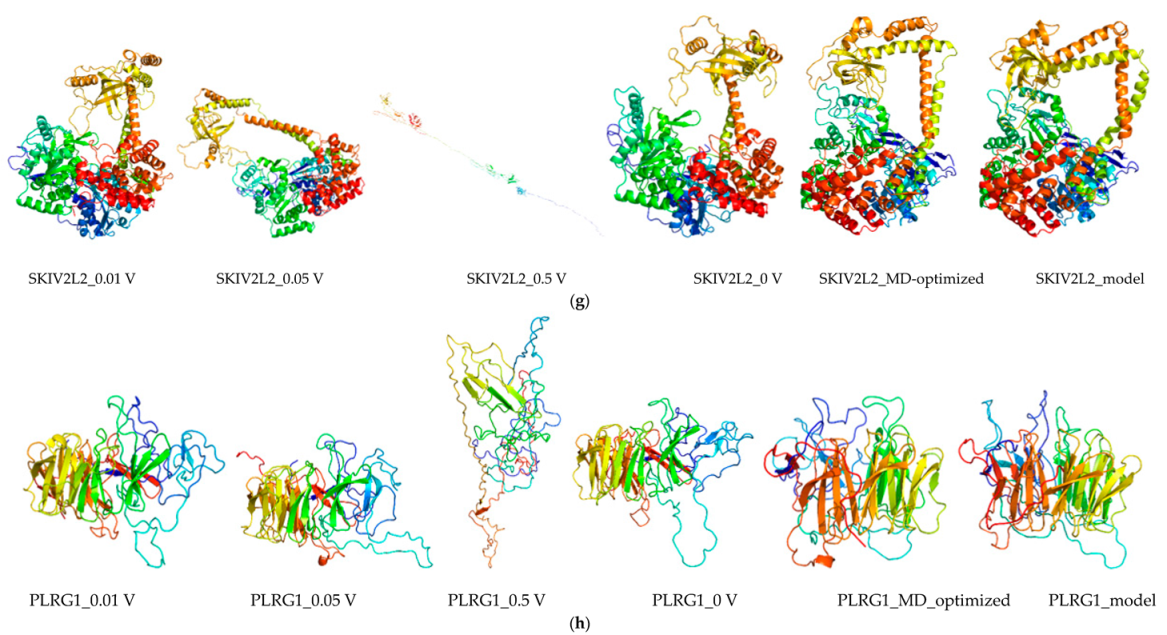
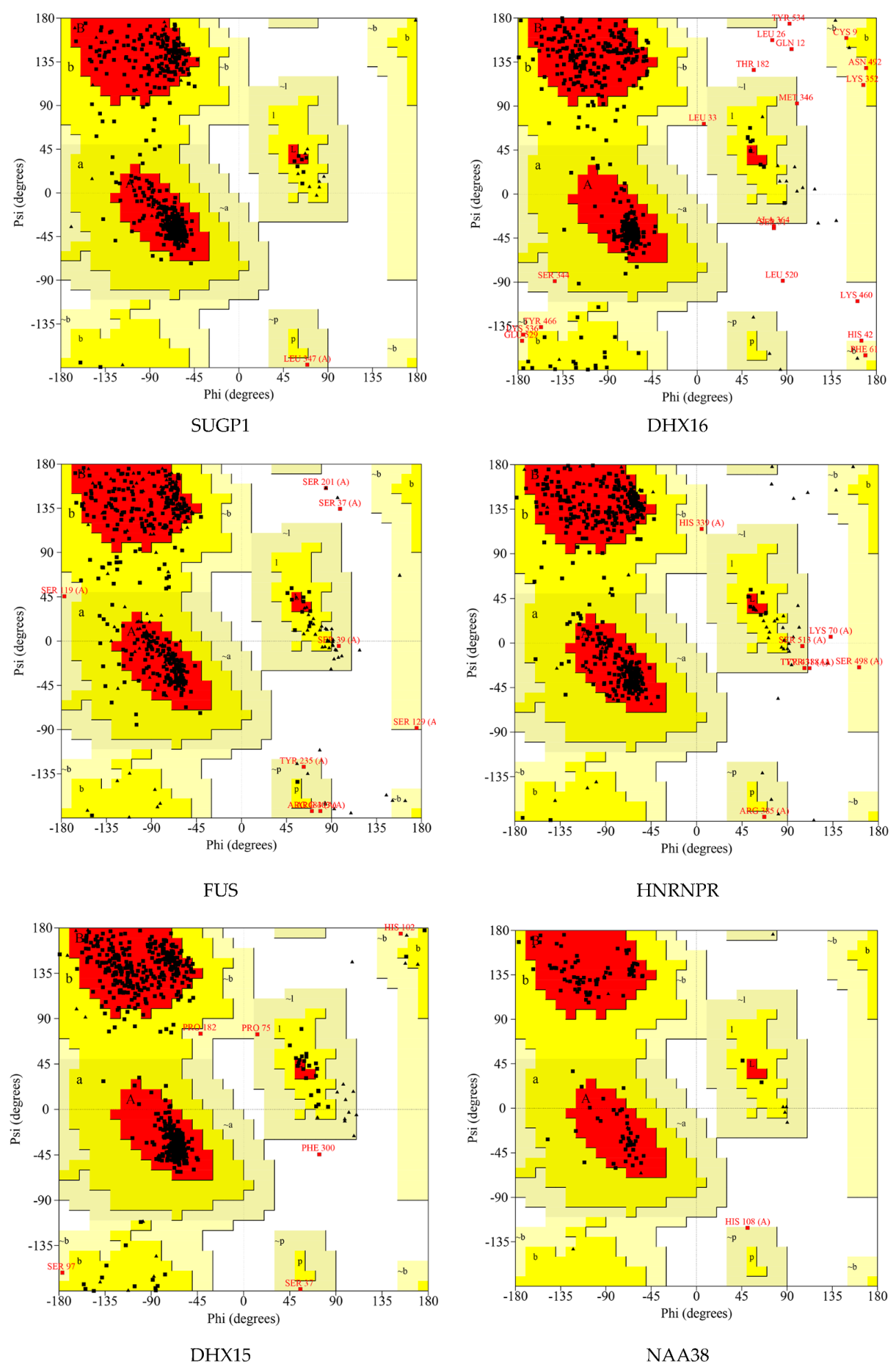
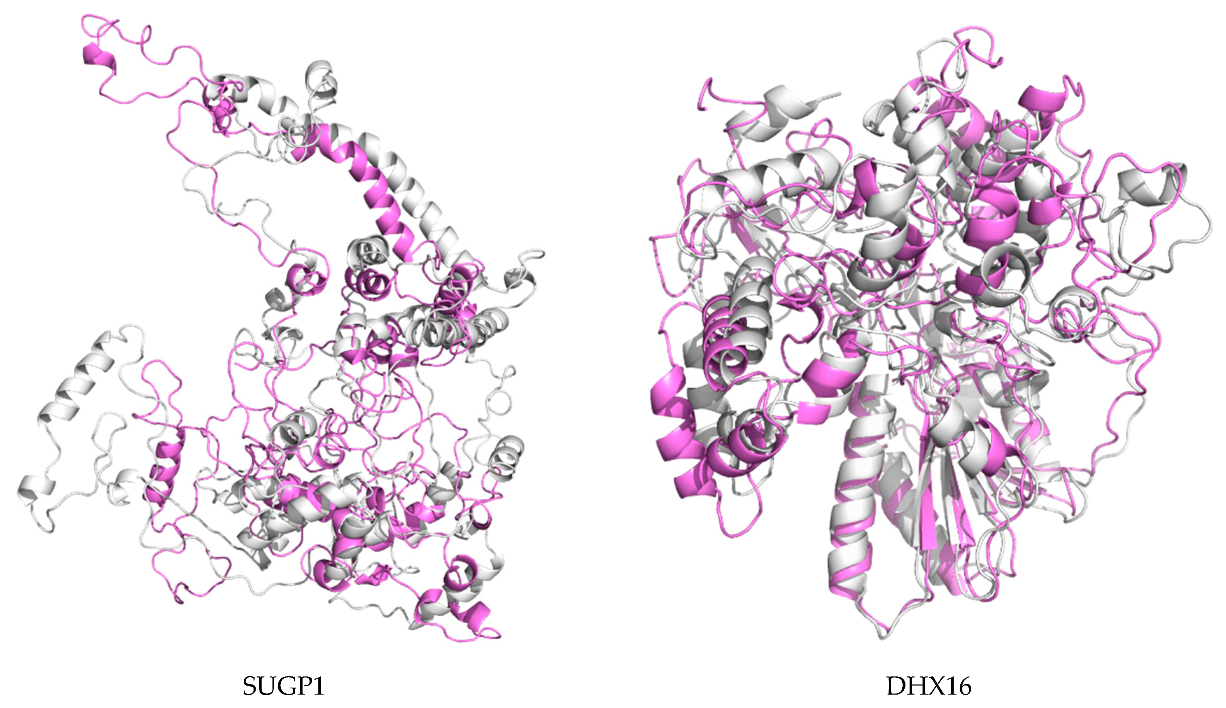
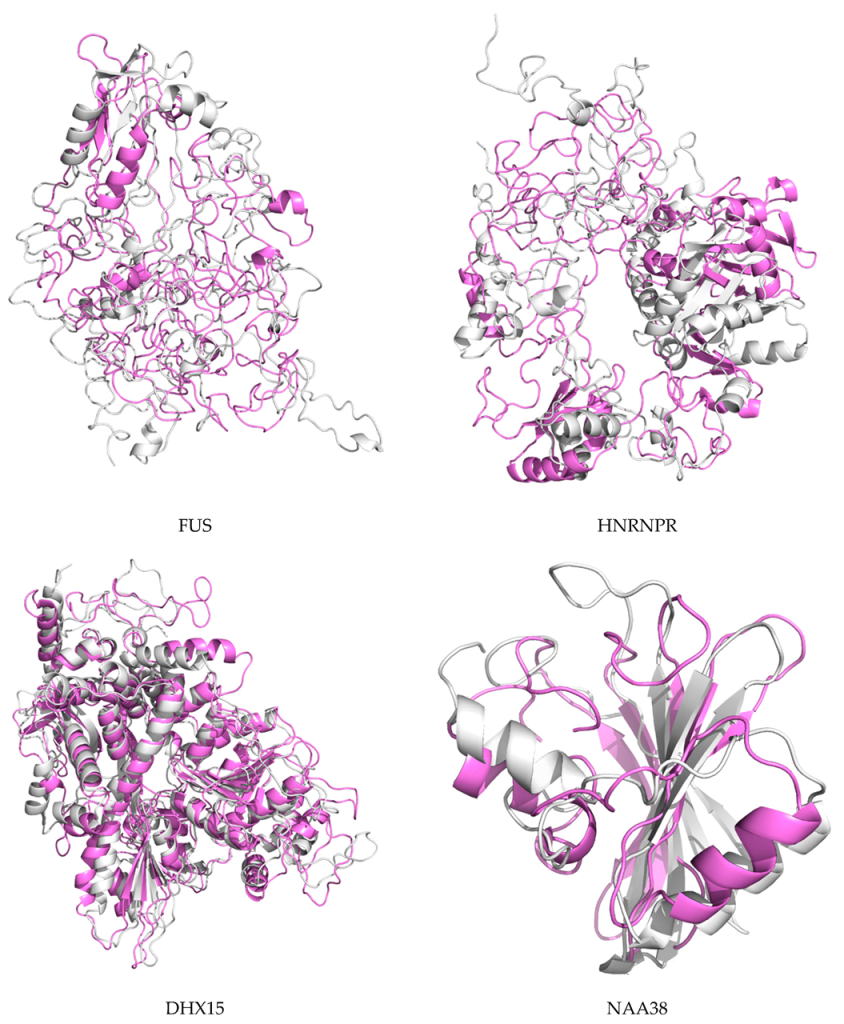
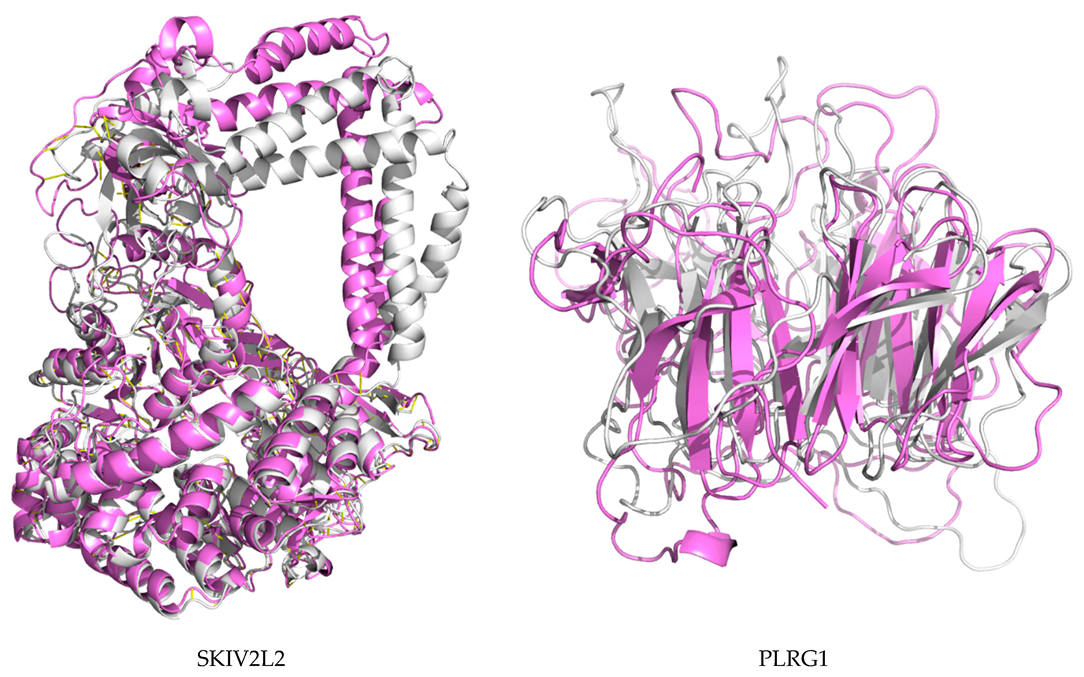
2.7. Protein Modeling
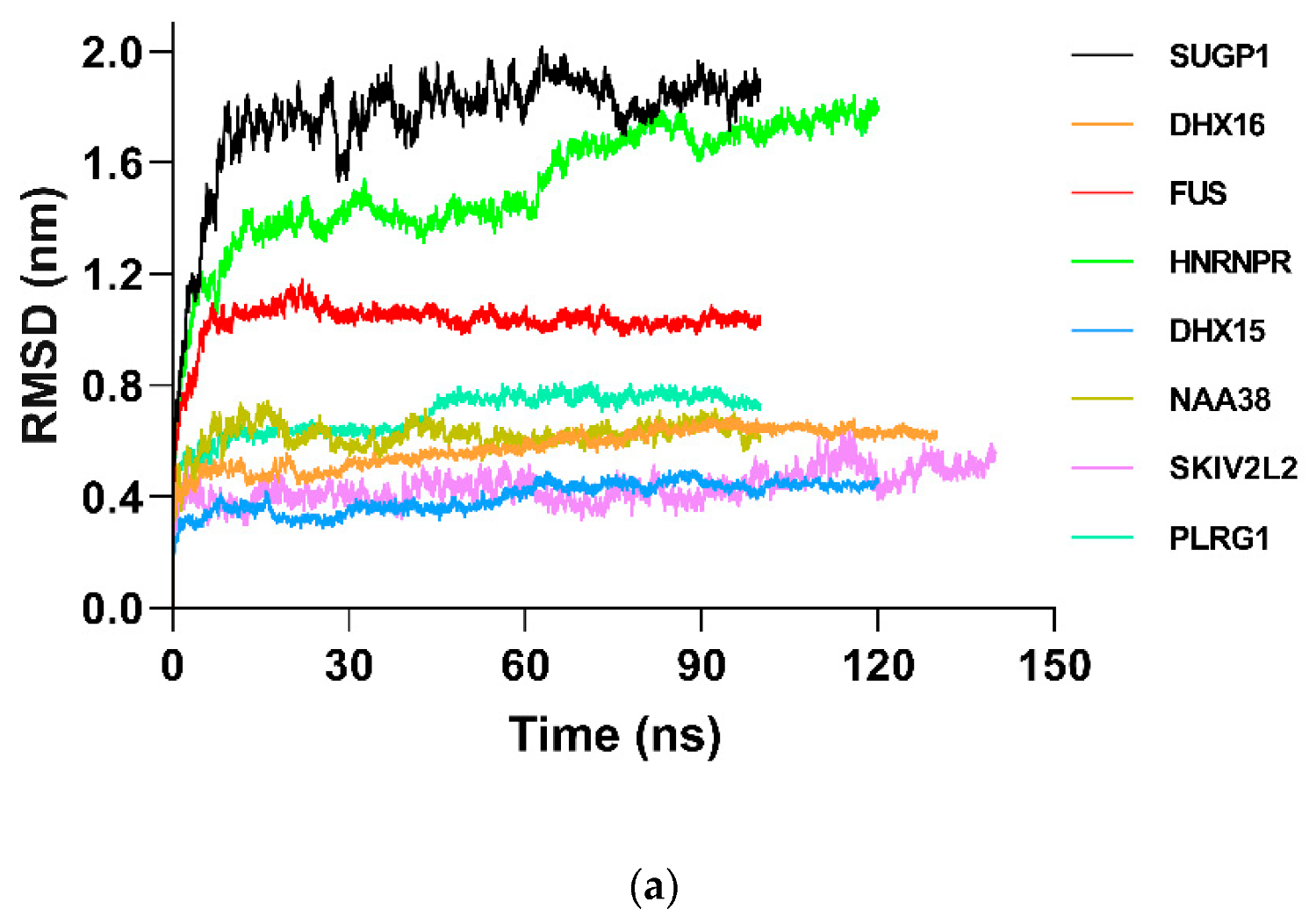
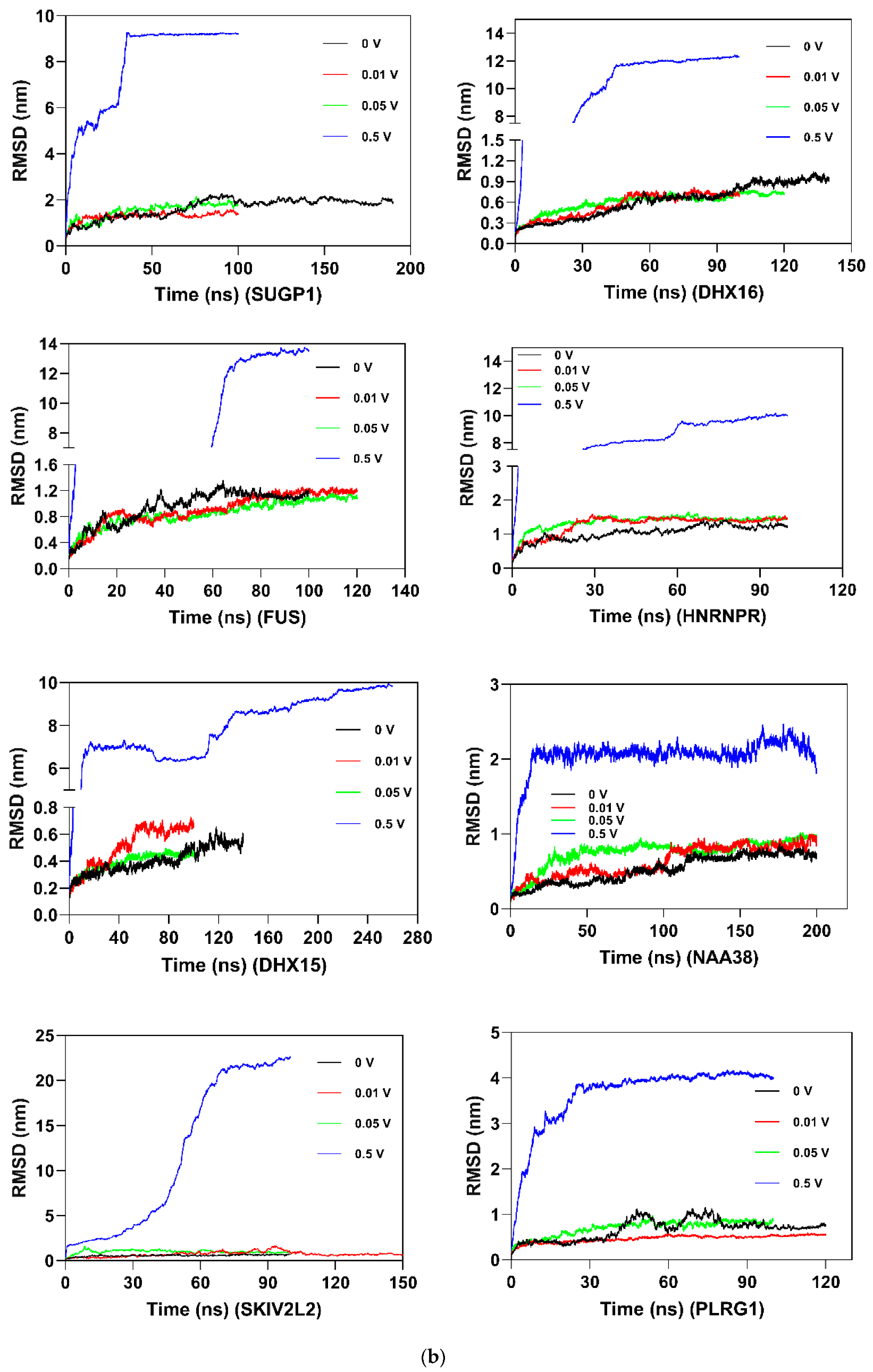
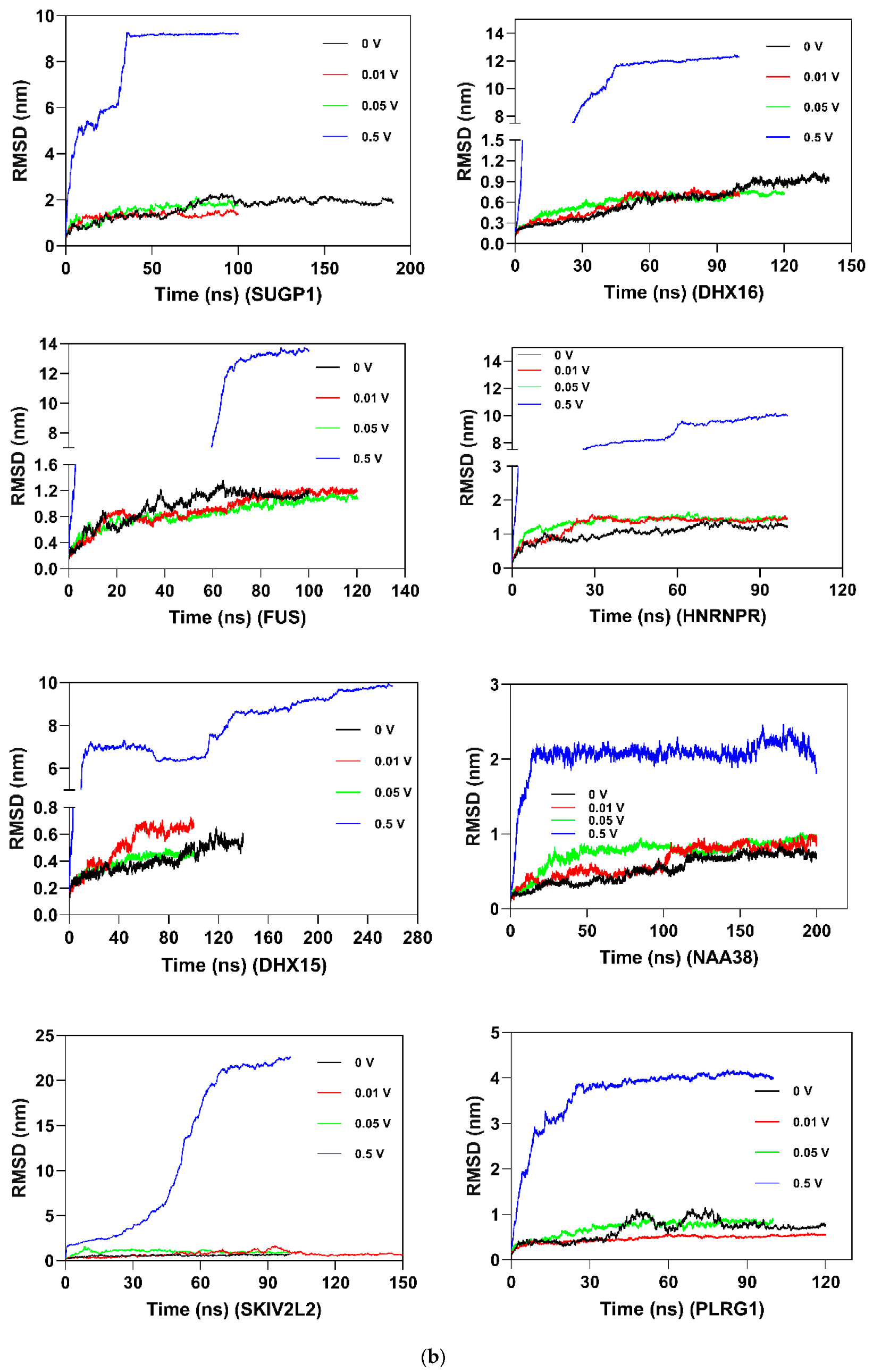
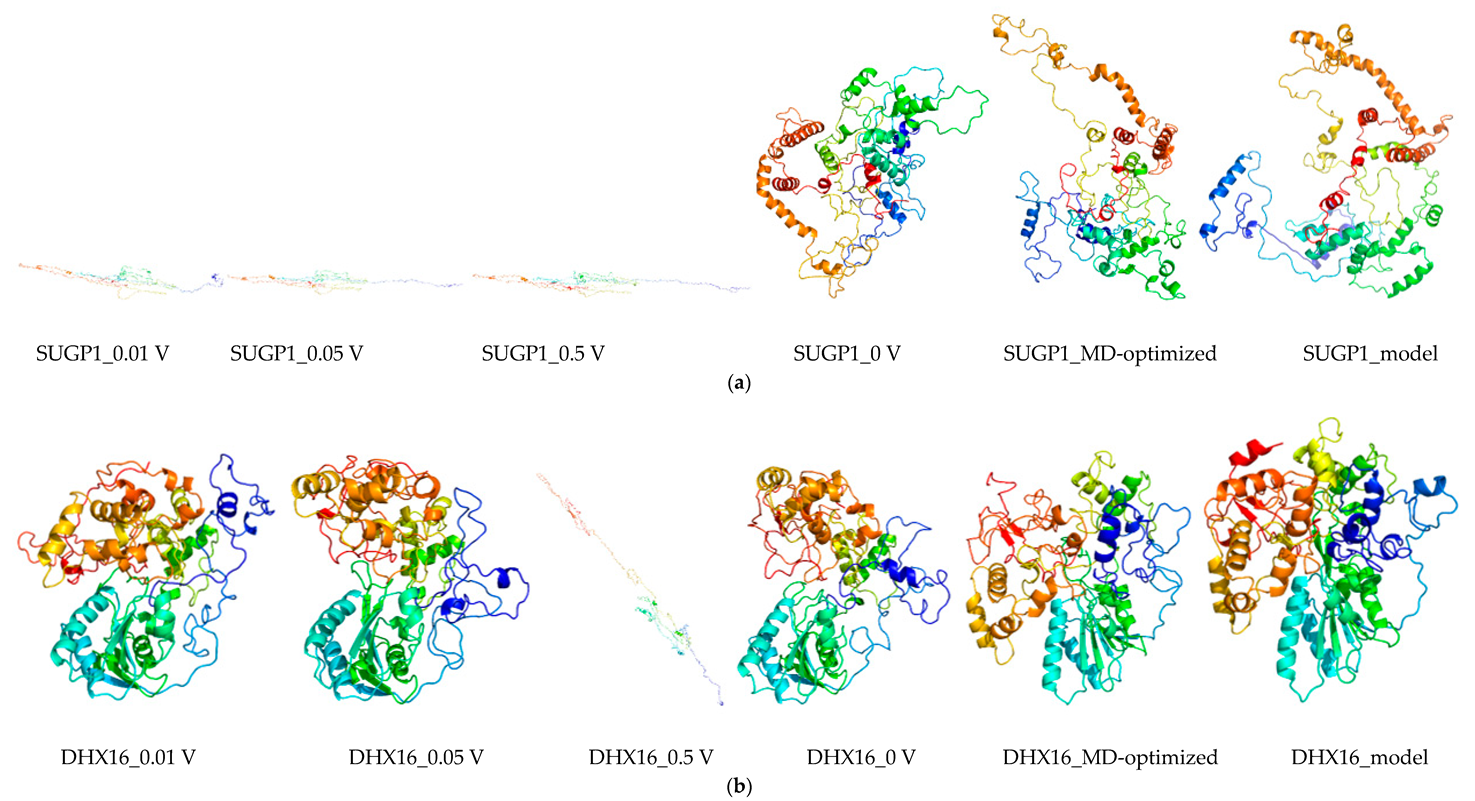
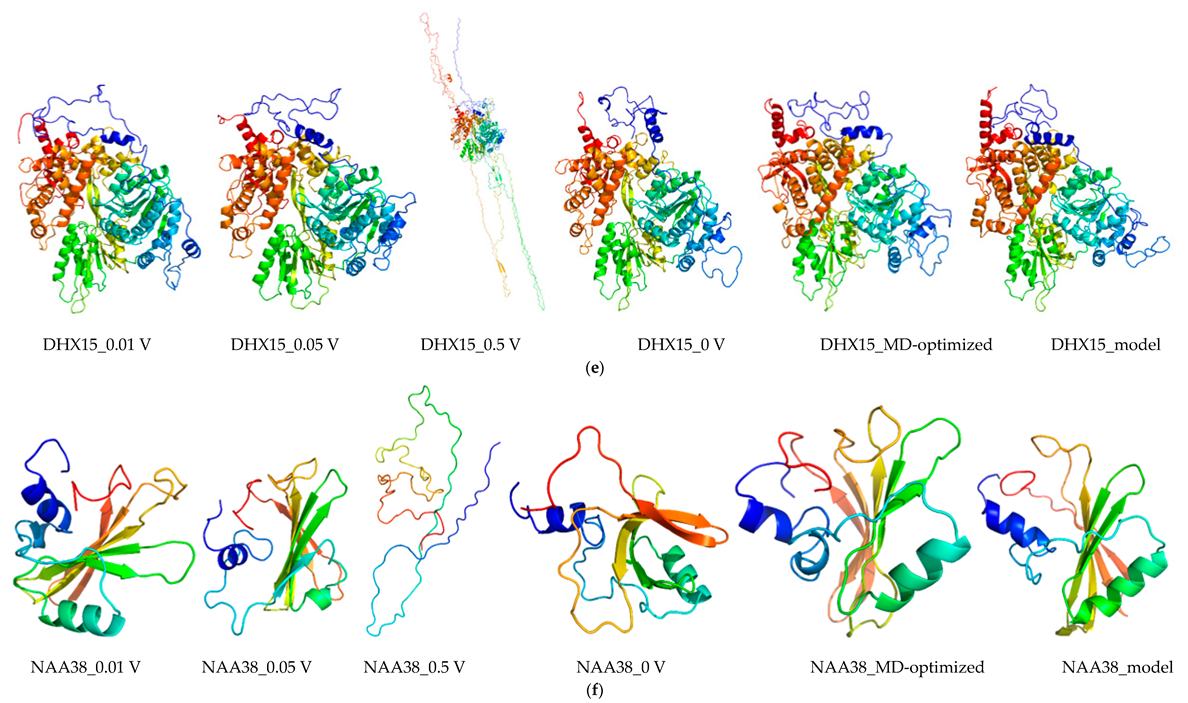
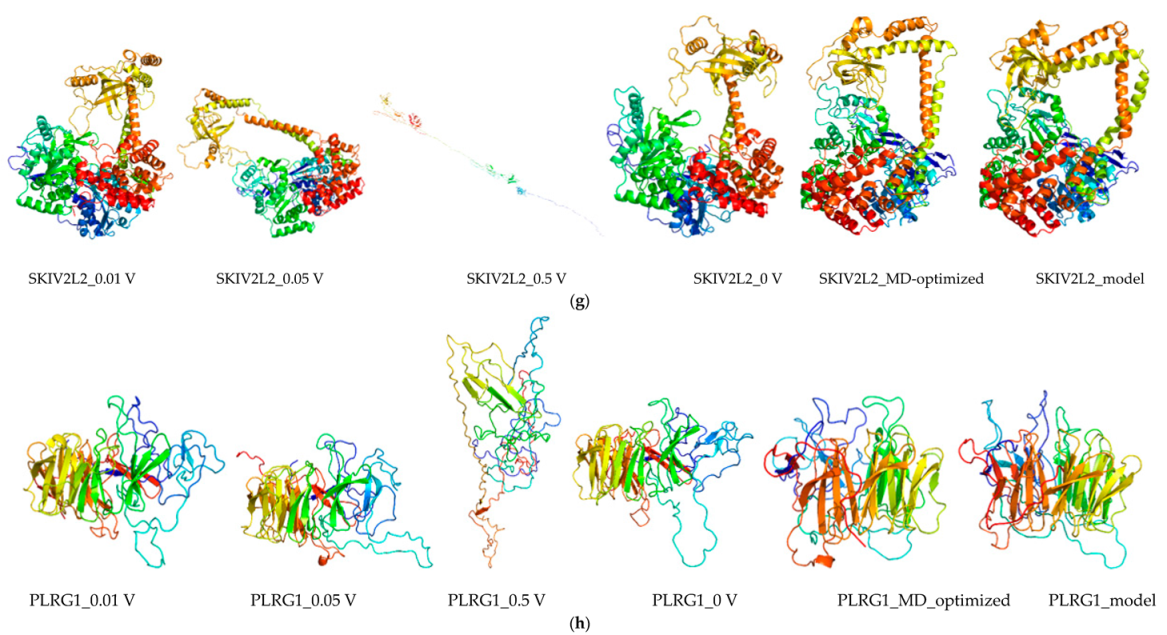
2.8. Molecular Dynamics and Simulation
3. Discussion
3.1. Electric Field: Jurkat Cells
3.2. Electric Field: Jurkat Cell Signal Pathway Change
3.3. Electric Field: Hub Gene Analysis
3.4. Electric Field: Jurkat Cell Gene Expression Change
3.5. Electric Field: The Effects of nsPEF on Cancer Cells
3.6. The Limitations of This Study
4. Materials and Methods
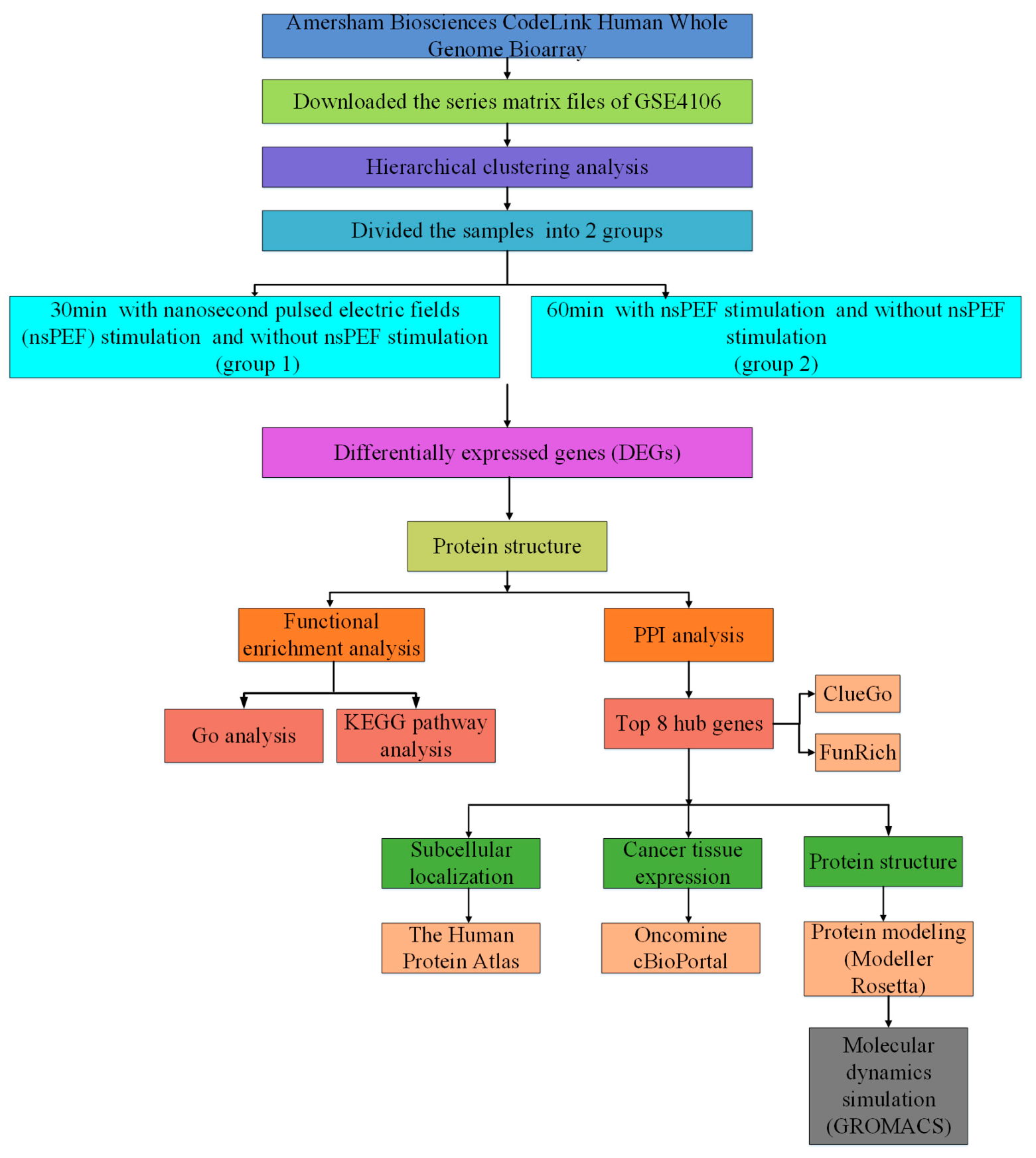
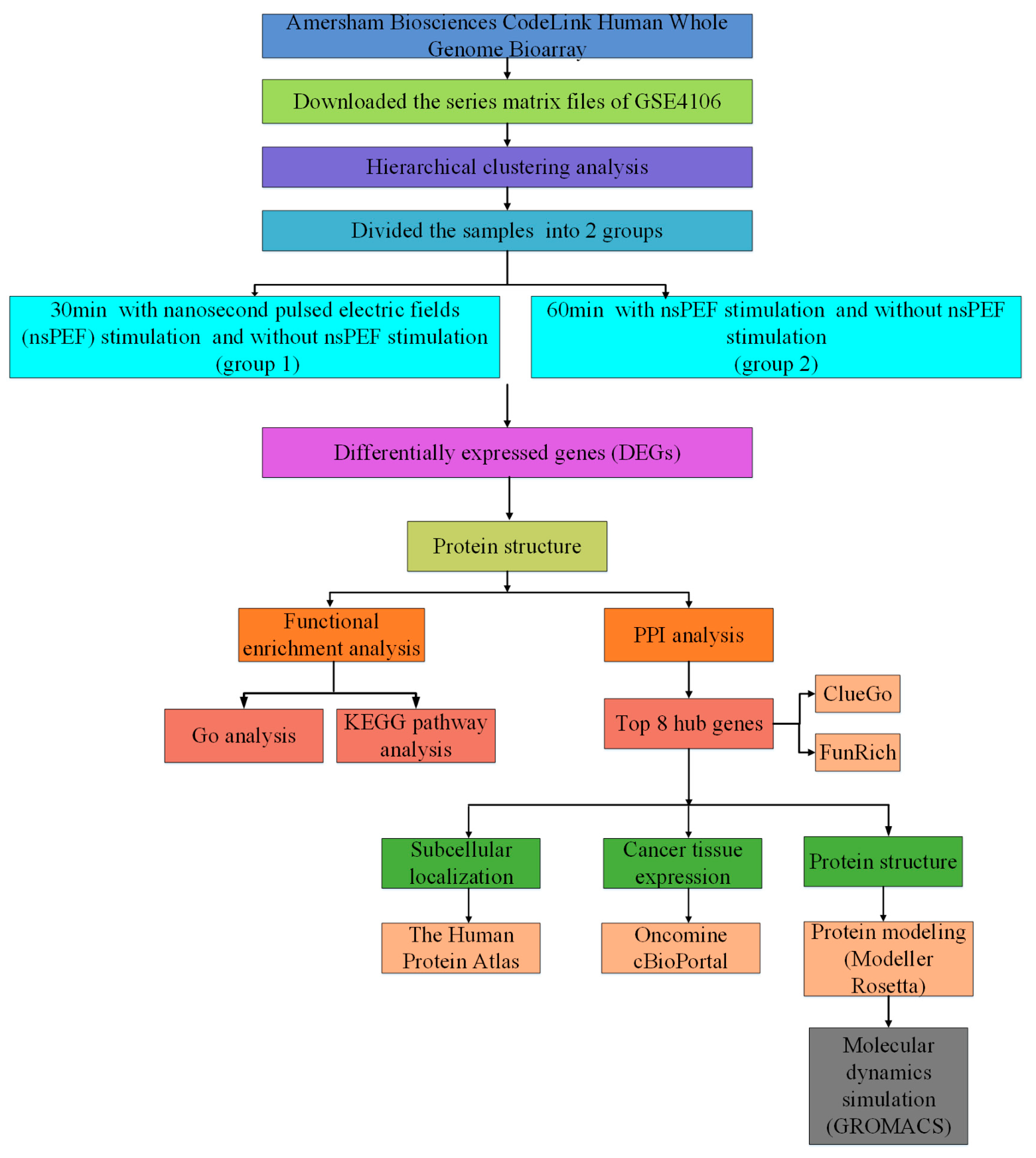
4.1. Main Steps
4.2. Microarray Data
4.3. Identification of DEGs
4.4. Functional Enrichment Analysis for DEGs
4.5. Protein–Protein Interaction (PPI) Network Construction and Module Analysis
4.6. Exploring Sub-Localization Expression of Hub Genes by Human Protein Atlas
4.7. Exploring Jurkat Genomics Data by cBio Cancer Genomics Portal
4.8. The Hub Genes Analysis by Using the Oncomine Database
4.9. Protein Modeling
4.10. De Novo Modeling of Proteins
4.11. Molecular Dynamics Simulations
4.11.1. Molecular Dynamic Simulation: Protein in Water
4.11.2. Molecular Dynamic Simulation: Protein under nsPEFs
4.11.3. Molecular Dynamic Simulation Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BP | biological process |
| BRD4/MYC | bromodomain-containing protein 4/MYC proto-oncogene |
| cBioPortal | cBio Cancer Genomics Portal |
| CC | cell component |
| DEGs | differentially expressed genes |
| FC | fold control |
| GO | gene ontology |
| IL7R | interleukin-7 receptor |
| KEGG | Kyoto Encyclopedia of Genes and Genomes pathway |
| MCODE | molecular complex detection |
| MF | molecular function |
| NOTCH1 | notch receptor 1 |
| nsPEF | nanosecond pulsed electric field |
| PPI | protein–protein interaction |
| PTEN | phosphatase and tensin homolog |
| RMSD | molecular dynamic analysis including root mean square deviation |
| RMSF | root mean square fluctuation |
| SINE | selective inhibitor of nuclear export |
| nsPEF | nanosecond pulsed electric field |
References
- Hijiya, N.; Suttorp, M. How I treat chronic myeloid leukemia in children and adolescents. Blood 2019, 133, 2374–2384. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Yin, S.; Chen, X.; Hu, C.; Zhang, X.; Hu, Z.; Yu, J.; Feng, X.; Jiang, K.; Ye, S.; Shen, K.; et al. Nanosecond pulsed electric field (nsPEF) treatment for hepatocellular carcinoma: A novel locoregional ablation decreasing lung metastasis. Cancer Lett. 2014, 346, 285–291. [Google Scholar] [CrossRef]
- Ferret, E.; Evrard, C.; Foucal, A.; Gervais, P. Volume changes of isolated human K562 leukemia cells induced by electric field pulses. Biotechnol. Bioeng. 2000, 67, 520–528. [Google Scholar] [CrossRef]
- Kennedy, S.M.; Aiken, E.J.; Beres, K.A.; Hahn, A.R.; Kamin, S.J.; Hagness, S.C.; Booske, J.H.; Murphy, W.L. Cationic peptide exposure enhances pulsed-electric-field-mediated membrane disruption. PLoS ONE 2014, 9, e92528. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Narita, M.; Ayres, F.; Satoh, N.; Abe, T.; Yanao, T.; Furukawa, T.; Toba, K.; Hirohashi, T.; Aizawa, Y. Cytoplasmic expression of EGFP in dendritic cells transfected with in vitro transcribed mRNA or cellular total RNA extracted from EGFP expressing leukemia cells. Med. Oncol. 2003, 20, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Sano, M.B.; Caldwell, J.L.; Davalos, R.V. Modeling and development of a low frequency contactless dielectrophoresis (cDEP) platform to sort cancer cells from dilute whole blood samples. Biosens. Bioelectron. 2011, 30, 13–20. [Google Scholar] [CrossRef]
- Kennedy, S.M.; Ji, Z.; Hedstrom, J.C.; Booske, J.H.; Hagness, S.C. Quantification of electroporative uptake kinetics and electric field heterogeneity effects in cells. Biophys. J. 2008, 94, 5018–5027. [Google Scholar] [CrossRef]
- Chen, X.; Cai, S.; Li, B.; Zhang, X.; Li, W.; Linag, H.; Cao, X. Identification of key genes and pathways for esophageal squamous cell carcinoma by bioinformatics analysis. Exp. Ther. Med. 2018, 16, 1121–1130. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Z.; Zhang, X.; Gong, T.; Yao, D. Examination of the expression and prognostic significance of DLGAPs in gastric cancer using the TCGA database and bioinformatic analysis. Mol. Med. Rep. 2018, 18, 5621–5629. [Google Scholar] [CrossRef]
- Persson, A.; Hober, S.; Uhlen, M. A human protein atlas based on antibody proteomics. Curr. Opin. Mol. 2006, 8, 185–190. [Google Scholar]
- Valk, P.J.; Verhaak, R.G.; Beijen, M.A.; Erpelinck, C.A.; Barjesteh van Waalwijk van Doorn-Khosrovani, S.; Boer, J.M.; Beverloo, H.B.; Moorhouse, M.J.; van der Spek, P.J.; Lowenberg, B.; et al. Prognostically useful gene-expression profiles in acute myeloid leukemia. N. Engl. J. Med. 2004, 350, 1617–1628. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.T.; Weiss, A. Jurkat T cells and development of the T-cell receptor signalling paradigm. Nat. Rev. Immunol. 2004, 4, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Morotomi-Yano, K.; Akiyama, H.; Yano, K. Different involvement of extracellular calcium in two modes of cell death induced by nanosecond pulsed electric fields. Arch. Biochem. Biophys. 2014, 555–556, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Asmar, A.; Stacey, M. Effects of nanosecond pulse electric fields on cellular elasticity. Micron 2015, 72, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Palmer, X.L.; Asmar, A.; Stacey, M.; Qian, S. Nanosecond pulsed electric field induced changes in cell surface charge density. Micron 2017, 100, 45–49. [Google Scholar] [CrossRef]
- Dutta, D.; Palmer, X.L.; Kim, J.; Qian, S.; Stacey, M. Energy dissipation mapping of cancer cells. Micron 2018, 105, 24–29. [Google Scholar] [CrossRef]
- Sozer, E.B.; Wu, Y.H.; Romeo, S.; Vernier, P.T. Nanometer-Scale Permeabilization and Osmotic Swelling Induced by 5-ns Pulsed Electric Fields. J. Membr. Biol. 2017, 250, 21–30. [Google Scholar] [CrossRef]
- Martelli, A.M.; Paganelli, F.; Fazio, A.; Bazzichetto, C.; Conciatori, F.; McCubrey, J.A. The Key Roles of PTEN in T-Cell Acute Lymphoblastic Leukemia Development, Progression, and Therapeutic Response. Cancers 2019, 11, 629. [Google Scholar] [CrossRef]
- Guo, W.; Schubbert, S.; Chen, J.Y.; Valamehr, B.; Mosessian, S.; Shi, H.; Dang, N.H.; Garcia, C.; Theodoro, M.F.; Varella-Garcia, M.; et al. Suppression of leukemia development caused by PTEN loss. Proc. Natl. Acad. Sci. USA 2011, 108, 1409–1414. [Google Scholar] [CrossRef]
- Zhou, J.; Nie, D.; Li, J.; Du, X.; Lu, Y.; Li, Y.; Liu, C.; Dai, W.; Wang, Y.; Jin, Y.; et al. PTEN Is Fundamental for Elimination of Leukemia Stem Cells Mediated by GSK126 Targeting EZH2 in Chronic Myelogenous Leukemia. Clin. Cancer Res. 2018, 24, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Yu, C.Y.; Theusch, E.; Naidoo, D.; Stevens, K.; Kuang, Y.L.; Schuetz, E.; Chaudhry, A.S.; Medina, M.W. SUGP1 is a novel regulator of cholesterol metabolism. Hum. Mol. Genet. 2016, 25, 3106–3116. [Google Scholar] [CrossRef] [PubMed]
- Gencheva, M.; Kato, M.; Newo, A.N.; Lin, R.J. Contribution of DEAH-box protein DHX16 in human pre-mRNA splicing. Biochem. J. 2010, 429, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Gencheva, M.; Lin, T.Y.; Wu, X.; Yang, L.; Richard, C.; Jones, M.; Lin, S.B.; Lin, R.J. Nuclear retention of unspliced pre-mRNAs by mutant DHX16/hPRP2, a spliceosomal DEAH-box protein. J. Biol. Chem. 2010, 285, 35624–35632. [Google Scholar] [CrossRef] [PubMed]
- Tyzack, G.E.; Luisier, R.; Taha, D.M.; Neeves, J.; Modic, M.; Mitchell, J.S.; Meyer, I.; Greensmith, L.; Newcombe, J.; Ule, J.; et al. Widespread FUS mislocalization is a molecular hallmark of amyotrophic lateral sclerosis. Brain 2019, 142, 2572–2580. [Google Scholar] [CrossRef] [PubMed]
- Reches, A.; Nachmani, D.; Berhani, O.; Duev-Cohen, A.; Shreibman, D.; Ophir, Y.; Seliger, B.; Mandelboim, O. HNRNPR Regulates the Expression of Classical and Nonclassical MHC Class I Proteins. J. Immunol. 2016, 196, 4967–4976. [Google Scholar] [CrossRef]
- Duijkers, F.A.; McDonald, A.; Janssens, G.E.; Lezzerini, M.; Jongejan, A.; van Koningsbruggen, S.; Leeuwenburgh-Pronk, W.G.; Wlodarski, M.W.; Moutton, S.; Tran-Mau-Them, F.; et al. HNRNPR Variants that Impair Homeobox Gene Expression Drive Developmental Disorders in Humans. Am. J. Hum. Genet. 2019, 104, 1040–1059. [Google Scholar] [CrossRef]
- Xu, Y.; Song, Q.; Pascal, L.E.; Zhong, M.; Zhou, Y.; Zhou, J.; Deng, F.M.; Huang, J.; Wang, Z. DHX15 is up-regulated in castration-resistant prostate cancer and required for androgen receptor sensitivity to low DHT concentrations. Prostate 2019, 79, 657–666. [Google Scholar] [CrossRef]
- Li, J.; Kang, F.; Gong, X.; Bai, Y.; Dai, J.; Zhao, C.; Dou, C.; Cao, Z.; Liang, M.; Dong, R.; et al. Ceria nanoparticles enhance endochondral ossification-based critical-sized bone defect regeneration by promoting the hypertrophic differentiation of BMSCs via DHX15 activation. Faseb J. 2019, 33, 6378–6389. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- Onderak, A.M.; Anderson, J.T. Loss of the RNA helicase SKIV2L2 impairs mitotic progression and replication-dependent histone mRNA turnover in murine cell lines. RNA 2017, 23, 910–926. [Google Scholar] [CrossRef] [PubMed]
- Lleres, D.; Denegri, M.; Biggiogera, M.; Ajuh, P.; Lamond, A.I. Direct interaction between hnRNP-M and CDC5L/PLRG1 proteins affects alternative splice site choice. Embo Rep. 2010, 11, 445–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleinridders, A.; Pogoda, H.M.; Irlenbusch, S.; Smyth, N.; Koncz, C.; Hammerschmidt, M.; Bruning, J.C. PLRG1 is an essential regulator of cell proliferation and apoptosis during vertebrate development and tissue homeostasis. Mol. Cell. Biol. 2009, 29, 3173–3185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martens, S.L.; Roth, C.C.; Ibey, B.L. Nanosecond pulsed electric field exposure does not induce the unfolded protein response in adult human dermal fibroblasts. Bioelectromagnetics 2018, 39, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.S.; Bai, L.; Smith, B.Y.; Patel, S.S.; Wang, M.D. Single-molecule studies reveal dynamics of DNA unwinding by the ring-shaped T7 helicase. Cell 2007, 129, 1299–1309. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Mao, Z.; Wang, B.; Zhang, J.; Lu, N.; Hong, R.; Dong, S.; Yao, C.; Liu, Q.H. Enhanced Antitumor Efficacy Achieved through Combination of nsPEFs and Low-Dosage Paclitaxel. IEEE Trans. Biomed. Eng. 2019, 66, 3129–3135. [Google Scholar] [CrossRef]
- Soueid, M.; Dobbelaar, M.C.F.; Bentouati, S.; Bardet, S.M.; O’Connor, R.P.; Bessieres, D.; Paillol, J.; Leveque, P.; Arnaud-Cormos, D. Delivery devices for exposure of biological cells to nanosecond pulsed electric fields. Med. Biol. Eng. Comput. 2018, 56, 85–97. [Google Scholar] [CrossRef]
- Muratori, C.; Pakhomov, A.G.; Heller, L.; Casciola, M.; Gianulis, E.; Grigoryev, S.; Xiao, S.; Pakhomova, O.N. Electrosensitization Increases Antitumor Effectiveness of Nanosecond Pulsed Electric Fields In Vivo. Technol. Cancer Res. Treat. 2017, 16, 987–996. [Google Scholar] [CrossRef]
- Chen, X.; Chen, Y.; Jiang, J.; Wu, L.; Yin, S.; Miao, X.; Swanson, R.J.; Zheng, S. Nano-pulse stimulation (NPS) ablate tumors and inhibit lung metastasis on both canine spontaneous osteosarcoma and murine transplanted hepatocellular carcinoma with high metastatic potential. Oncotarget 2017, 8, 44032–44039. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E.J.S. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3. [Google Scholar] [CrossRef] [PubMed]
- Matteucci, C.; Argaw-Denboba, A.; Balestrieri, E.; Giovinazzo, A.; Miele, M.; D’Agostini, C.; Pica, F.; Grelli, S.; Paci, M.; Mastino, A.; et al. Deciphering cellular biological processes to clinical application: A new perspective for Talpha1 treatment targeting multiple diseases. Expert Opin. Biol. 2018, 18, 23–31. [Google Scholar] [CrossRef]
- Ahn, J.H.; Hwang, S.H.; Cho, H.S.; Lee, M. Differential Gene Expression Common to Acquired and Intrinsic Resistance to BRAF Inhibitor Revealed by RNA-Seq Analysis. Biomol. Ther. 2019, 27, 302–310. [Google Scholar] [CrossRef]
- Jeanquartier, F.; Jean-Quartier, C.; Holzinger, A. Integrated web visualizations for protein-protein interaction databases. BMC Bioinform. 2015, 16, 195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Luo, B.; Wang, B.; Wu, Q.; Liang, Y.; He, Y. Identification of Potential Gene Interactions in Heart Failure Caused by Idiopathic Dilated Cardiomyopathy. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2018, 24, 7697–7709. [Google Scholar] [CrossRef]
- Li, P.; Wu, M.; Lin, Q.; Wang, S.; Chen, T.; Jiang, H. Key genes and integrated modules in hematopoietic differentiation of human embryonic stem cells: A comprehensive bioinformatic analysis. Stem Cell Res. Ther. 2018, 9, 301. [Google Scholar] [CrossRef]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. CytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. 4), S11. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Wang, P.; Yu, Y.; Huang, B.; Zhang, X.; Xu, C.; Zhao, X.; Yin, Z.; He, Z.; Jin, M.; et al. Tumor-preventing activity of aspirin in multiple cancers based on bioinformatic analyses. PeerJ 2018, 6, e5667. [Google Scholar] [CrossRef] [Green Version]
- Gong, L.; Zhang, D.; Dong, Y.; Lei, Y.; Qian, Y.; Tan, X.; Han, S.; Wang, J. Integrated Bioinformatics Analysis for Identificating the Therapeutic Targets of Aspirin in Small Cell Lung Cancer. J. Biomed. Inf. 2018, 88, 20–28. [Google Scholar] [CrossRef]
- Wang, L.L.; Chen, Z.S.; Zhou, W.D.; Shu, J.; Wang, X.H.; Jin, R.; Zhuang, L.L.; Hoda, M.A.; Zhang, H.; Zhou, G.P. Down-regulated GATA-1 up-regulates interferon regulatory factor 3 in lung adenocarcinoma. Sci. Rep. 2017, 7, 2551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [PubMed] [Green Version]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Leaver-Fay, A.; Tyka, M.; Lewis, S.M.; Lange, O.F.; Thompson, J.; Jacak, R.; Kaufman, K.; Renfrew, P.D.; Smith, C.A.; Sheffler, W.; et al. ROSETTA3: An object-oriented software suite for the simulation and design of macromolecules. Methods Enzym. 2011, 487, 545–574. [Google Scholar] [CrossRef] [Green Version]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway Type (Up-Regulated) | Up-Regulation (Group 1) | Up-Regulation (Group 2) | Pathway Type (Down-Regulated) | Down-Regulation (Group 1) | Down-Regulation (Group 2) |
|---|---|---|---|---|---|
| General (biological process—BP) | negative regulation of cell proliferation | negative regulation of cell proliferation | General (BP) | metabolism of RNA | metabolism of RNA |
| Specific (BP) | PID NFAT TF pathway | PID NFAT TF pathway | General (BP) | cell cycle | cell cycle |
| General (molecular function—MF) | regulation of double-strand break repair via homologous recombination | regulation of double-strand break repair via homologous recombination | General (MF) | ncRNA metabolic process | ncRNA metabolic process |
| General (MF) | negative regulation of phosphate metabolic process | negative regulation of phosphate metabolic process | General (BP) | DNA repair | DNA repair |
| General (BP) | chromatin remodeling | chromatin remodeling | General (BP) | nuclear export | nuclear export |
| General (MF) | maintenance of cell number | maintenance of cell number | General (MF) | cell cycle phase transition | cell cycle phase transition |
| Specific (BP) | O-glycan processing | T cell mediated cytotoxicity | General (MF) | regulation of chromosome organization | regulation of chromosome organization |
| General (BP) | mRNA processing | regulation of blood pressure | General (MF) | tRNA metabolic process | tRNA metabolic process |
| Specific (BP) | regulation of PTEN gene transcription | histone modification | General (BP) | mitotic sister chromatid segregation | mitotic sister chromatid segregation |
| General (MF) | glucose homeostasis | dephosphorylation | General (BP) | DNA replication | DNA replication |
| General (MF) | immune response-regulating cell surface receptor signaling pathway | transcription elongation from RNA polymerase II promoter | General (cellular component—CC) | microtubule cytoskeleton organization | microtubule cytoskeleton organization |
| negative regulation of cell cycle | General (BP) | covalent chromatin modification | covalent chromatin modification | ||
| General (BP) | organelle biogenesis and maintenance | organelle biogenesis and maintenance | |||
| General (MF) | DNA-templated transcription, termination | DNA-templated transcription, termination | |||
| General (CC) | macromolecule methylation | macromolecule methylation | |||
| General (BP) | organelle localization | organelle localization | |||
| General (BP) | ribonucleoprotein complex biogenesis | ribonucleoprotein complex biogenesis | |||
| General (BP) | cell cycle | cell cycle | |||
| General (BP) | regulation of DNA metabolic process | regulation of DNA metabolic process | |||
| General (BP) | DNA geometric change | regulation of cellular response to stress |
| Groups | Expression | Pathway ID | Name | Gene Count | % | Genes |
|---|---|---|---|---|---|---|
| Group 1 | Down-regulated | hsa01100 | Metabolic pathways | 117 | 9.04 | NAMPT, INMT, CHKA, GPAT4, CTPS1, DGKA, DLAT, DLD, DNMT1, EPRS, ALAS1, EXT1, ACSL3, FASN, FECH, SEPHS2, AKR1B1, GANAB, ACSL6, MLYCD, ETHE1, SHPK, PISD, KDSR, GART, GCNT1, GLDC, GLO1, GMDS, GOT2, GPI, PIGW, RIMKLA, GSTM3, HADHA, HK1, HMGCR, IDI1, INPP4A, ACADVL, STT3A, LSS, LTA4H, MAN2A2, MDH2, MGAT5, MPI, ASNS, NDUFA10, ATIC, NDUFB4, NDUFS1, OAT, ODC1, ACO2, OXCT1, PCCA, SEPSECS, PDE8A, INPP5K, PFKP, PGM1, ATP6V1B2, PIK3CD, PI4KA, PI4KB, PLCG1, PLCG2, PGM2, PPT1, ETNK1, SMPD4, CNDP2, AGK, CMAS, INPP5E, AGPAT4, QARS, BCAT1, RPN1, RPN2, SCD, GALNT11, RBKS, SGSH, SHMT1, SORD, TK2, UCK2, ALDH5A1, GALNT12, ALG9, SCD5, PGAP1, UXS1, PCBD2, ACSS1, DGKZ, DGKD, ALG2, AGPS, B4GALT4, CBS, CDS2, SUCLG2, GMPS, ALDH1A2, ST3GAL5, MTMR2, PAPSS1, CERS5, MARS2, PIGS, PIGB, PGS1, ENTPD4, FIG4 |
| hsa05200 | Pathways in cancer | 37 | 2.86 | CDK2, RASGRP2, CHUK, CRKL, AKT1, MTOR, GSTM3, MSH6, HDAC2, HSP90AB1, FAS, IKBKB, IL3RA, IL13, JAK1, LAMA5, LAMB1, SMAD2, MLH1, PIK3CD, PLCG1, PLCG2, PRKCA, MAPK9, MAP2K1, STAT5B, TGFBR1, TPR, TRAF1, TRAF3, ZBTB17, CXCR4, AXIN1, RASSF5, CASP9, CUL1, CCNE2 | ||
| hsa03013 | RNA transport | 27 | 2.09 | NXF1, RPP30, POP1, EIF2B1, EIF4EBP2, NUP210, GEMIN5, CYFIP2, KPNB1, NUP88, NUP98, NUP133, NUP107, UPF1, ELAC2, TPR, UBE2I, NUP85, FXR1, THOC5, EIF3C, PABPC4, NUP155, EIF5B, TGS1, NUP93, THOC1 | ||
| Up-regulated | hsa01100 | Metabolic pathways | 8 | 5.80 | AK2, LCLAT1, PGM2L1, IDS, G6PC2, GALNT14, NT5C1A, H6PD | |
| hsa04010 | MAPK signaling pathway | 5 | 3.62 | DUSP1, DUSP2, FOS, JUN, STK4, STK4 | ||
| has05200 | Pathways in cancer | 5 | 3.62 | FOS, DLL1, IL12A, JUN, STK4 | ||
| hsa05202 | Transcriptional misregulation in cancer | 4 | 2.90 | FUS, PBX1, SS18, CCNT1 | ||
| Group 2 | Down-regulated | hsa01100 | Metabolic pathways | 114 | 8.74 | NAMPT, INMT, CHKA, GPAT4, CTPS1, DGKA, DCTD, DLAT, DNMT1, EPRS, ALAS1, EXT1, ACSL3, FASN, FECH, SEPHS2, AKR1B1, GANAB, ACSL6, MLYCD, ETHE1, SHPK, PISD, KDSR, GART, GCNT1, GLDC, GLO1, GMDS, GOT2, GPI, PIGW, RIMKLA, GSTM3, HADHA, HK1, HMGCR, IDI1, INPP4A, ACADVL, STT3A, LSS, LTA4H, MAN2A2, MDH2, MPI, ASNS, NDUFA10, ATIC, NDUFB4, NDUFS1, OAT, ODC1, ACO2, OXCT1, PCCA, SEPSECS, PDE8A, PFKP, PGM1, ATP6V1B2, PIK3CD, PI4KB, PLCG1, PLCG2, PGM2, PPT1, SMPD4, CNDP2, AGK, CMAS, INPP5E, AGPAT4, QARS, BCAT1, RPN1, RPN2, SCD, GALNT11, RBKS, SGSH, SHMT1, SORD, TK2, UCK2, ALDH5A1, GALNT12, ALG9, SCD5, DGLUCY, PGAP1, UXS1, PCBD2, ACSS1, DGKZ, DGKD, ALG2, AGPS, B4GALT4, CBS, CDS2, SUCLG2, GMPS, ALDH1A2, ST3GAL5, MTMR2, PAPSS1, CERS5, MARS2, PIGS, PIGB, PGS1, ENTPD4, FIG4 |
| hsa05200 | Pathways in cancer | 37 | 2.84 | CDK2, RASGRP2, CRKL, AKT1, MTOR, GSTM3, MSH6, HDAC2, HSP90AB1, FAS, IKBKB, IL3RA, IL13, JAK1, LAMA5, LAMB1, SMAD2, MLH1, PIK3CD, PLCG1, PLCG2, PRKCA, PRKCB, MAPK9, MAP2K1, STAT5B, TGFBR1, TPR, TRAF1, TRAF3, ZBTB17, CXCR4, AXIN1, RASSF5, CASP9, CUL1, CCNE2 | ||
| hsa03013 | RNA transport | 27 | 2.07 | PRMT5, NXF1, RPP30, POP1, EIF2B1, EIF4EBP2, NUP210, GEMIN5, CYFIP2, KPNB1, NUP88, NUP98, NUP133, NUP107, UPF1, ELAC2, TPR, UBE2I, NUP85, FXR1, THOC5, EIF3C, PABPC4, NUP155, EIF5B, TGS1, NUP93 | ||
| Up-regulated | hsa01100 | Metabolic pathways | 9 | 6.21 | CYP2C9, AK2, LCLAT1, PGM2L1, IDS, G6PC2, GALNT14, NT5C1A, H6PD | |
| hsa05200 | Pathways in cancer | 6 | 4.14 | FOS, DLL1, IL12A, JUN, STK4, CCNA1 | ||
| hsa04010 | MAPK signaling pathway | 5 | 3.45 | DUSP1, DUSP2, FOS, JUN, STK4 | ||
| hsa05166 | Human T-cell leukemia virus 1 infection | 5 | 3.45 | EGR1, FOS, JUN, VAC14, CCNA1 |
| Gene Name | Cell Lines | Main Location |
|---|---|---|
| SUGP1 | A-431, U-2OS, U-251MG | nucleoplasm |
| DHX16 | HeLa, MCF7, U-2 OS | nucleoplasm |
| FUS | A-431, U-2 OS, U-251MG | nucleoplasm |
| HNRNPR | A-431, U-2 OS, U-251MG | nucleoplasm |
| DHX15 | A-431, HEK 293, U-2 OS | nuclear speckles |
| NAA38 | HEK 293, PC-3, U-2 OS | nucleus |
| SKIV2L2 | A-431, U-2 OS, U-251MG | nucleus |
| PLRG1 | A-431, U-2 OS, U-251MG | nuclear speckles, nuclear membrane |
| Proteins | Species | Protein Length (aa) | Model Templates (Query Cover, Identify) |
|---|---|---|---|
| SUGP1 | Homo sapiens | 645 | de novo |
| DHX16 | Homo sapiens | 560 | 5Z58_XX (94%, 99%) 5MQF_QQ (90%, 56%) 6FA9_A (89%, 55%) |
| FUS | Homo sapiens | 522 | de novo |
| HNRNPR | Homo sapiens | 535 | de novo |
| DHX15 | Homo sapiens | 795 | 5XDR_A (86%, 99%) 3KX2_B (84%, 66%) 2XAU_A (84%, 66%) |
| NAA38 | Homo sapiens | 125 | de novo |
| SKIV2L2 | Homo sapiens | 1042 | 6D6Q_M (100%, 100%) 6C90_A (70%, 100%) 2XGJ_A (89%, 56%) |
| PLRG1 | Homo sapiens | 514 | 6FF4_D (100%, 100%) 5MQF_D (100%, 99%) 4YVD_A (72%, 100%) |
| Proteins | Number of Residues in Favored Region | Number of Residues in Allowed Region | Number of Residues in Disallowed Region |
|---|---|---|---|
| SUGP1 | 448 (93.3%) | 32 (6.7%) | 0 (0.0%) |
| DHX16 | 423 (84.9%) | 65 (13.0%) | 10 (2.0%) |
| FUS | 284 (82.3%) | 59 (17.1%) | 2 (0.6%) |
| HNRNPR | 379 (90.2%) | 39 (9.3%) | 2 (0.5%) |
| DHX15 | 658 (92.4%) | 53 (7.4%) | 1 (0.1%) |
| NAA38 | 94 (89.5%) | 11 (10.5%) | 0 (0.0%) |
| SKIV2L2 | 870 (93.2%) | 61 (6.5%) | 2 (0.2%) |
| PLRG1 | 305 (84.3%) | 52 (14.4%) | 5 (1.4%) |
| Name and Ensembl ID | Species Gene Type | Location Length | Function | Refs |
|---|---|---|---|---|
| SUGP1 (SURP and G-patch domain containing 1) (ENSG00000105705) | Homo sapiens Protein coding | Chr 19 (2566 bp) | A novel modulator in cholesterol metabolism | [22] |
| DHX16 (DEAH-box helicase 16) (ENSG00000204560) | Homo sapiens Protein coding | Chr 6 (3406 bp) | Involved in the human pre-mRNA splicing | [23,24] |
| FUS (FUS RNA binding protein) (ENSG00000089280) | Homo sapiens Protein coding | Chr 16 (5119 bp) | A key player in neuronal cell-related diseases | [25] |
| HNRNPR (Heterogeneous nuclear ribonucleoprotein R) (ENSG00000125944) | Homo sapiens Protein coding | Chr 1 (7751 bp) | Is involved in processing the pre-mRNA in cell nucleus identified and is considered as a general positive modulator of MHC class I expression | [26,27] |
| DHX15 (DEAH-box helicase 15) (ENSG00000109606) | Homo sapiens Protein coding | Chr 4 (2998 bp) | Is involved in the regulation of tumor cell growth, such as prostate cancer progression and bone defect regeneration | [28,29] |
| NAA38 (N(alpha)-acetyltransferase 38, NatC auxiliary subunit) (ENSG00000183011) | Homo sapiens Protein coding | Chr 17 (999 bp) | Is related to the pathways of Golgi-to-ER retrograde transport and vesicle-induced transport | [30] |
| SKIV2L2 (Ski2-like RNA helicase 2) (ENSG00000204351) | Homo sapiens Protein coding | Chr 6 (3795 bp) | Regulates the cell proliferation | [31] |
| PLRG1 (Pleiotropic regulator 1) (ENSG00000171566) | Homo sapiens Protein coding | Chr 4 (3317 bp) | Regulates the cell proliferation | [32,33] |
| Gene | GO Analysis [30] |
|---|---|
| SUGP1 | MF: nucleic acid binding; RNA binding; protein binding |
| BP: mRNA splicing, via spliceosome; RNA processing; mRNA processing; RNA splicing | |
| CC: nucleus; nucleoplasm; spliceosomal complex | |
| DHX16 | MF: nucleic acid binding; RNA binding; RNA helicase activity; helicase activity; protein binding |
| BP: mRNA splicing, via spliceosome; mRNA processing; RNA splicing | |
| CC: nucleus; nucleoplasm; spliceosomal complex; U2-type precatalytic spliceosome | |
| FUS | MF: nucleic acid binding; DNA binding; chromatin binding; transcription coactivator activity; RNA binding |
| BP: mRNA splicing, via spliceosome; regulation of transcription, DNA-templated; regulation of transcription by RNA polymerase II; RNA splicing; regulation of RNA splicing | |
| CC: nucleus; nucleoplasm; cytoplasm; polysome; dendrite | |
| HNRNPR | MF: nucleic acid binding; RNA binding; mRNA binding; mRNA 3’-UTR binding; protein binding |
| BP: nucleus; nucleoplasm; spliceosomal complex; NOT nucleolus; cytoplasm | |
| CC: mRNA splicing, via spliceosome; mRNA processing; circadian rhythm; RNA splicing; RNA metabolic process | |
| DHX15 | MF: nucleic acid binding; RNA binding; RNA helicase activity; double-stranded RNA binding; helicase activity |
| BP: mRNA splicing, via spliceosome; mRNA processing; RNA splicing; response to toxic substance; response to alkaloid | |
| CC: nucleus; nucleoplasm; U12-type spliceosomal complex; nucleolus; nuclear speck | |
| NAA38 | MF: protein binding |
| BP: negative regulation of apoptotic process | |
| CC: nucleus; cytoplasm; colocalizes_with polysome; NatC complex | |
| SKIV2L2 | MF: nucleic acid binding; RNA binding; RNA helicase activity; ATP-dependent RNA helicase activity; helicase activity |
| BP: RNA catabolic process; exonucleolytic nuclear-transcribed mRNA catabolic process involved in deadenylation-dependent decay; nuclear-transcribed mRNA catabolic process, 3’-5’ exonucleolytic nonsense-mediated decay | |
| CC: nucleus; cytoplasm; cytosol; Ski complex | |
| PLRG1 | MF: protein binding |
| BP: mRNA splicing, via spliceosome; mRNA processing; RNA splicing; protein localization to nucleus; positive regulation of G1/S transition of mitotic cell cycle | |
| CC: Prp19 complex; fibrillar center; nucleus; nucleoplasm; colocalizes_with DNA replication factor A complex |
| Group | Experimental Grouping |
|---|---|
| Group 1 | Control 30 min: Jurkat cells were cultured for 30 min (without nsPEF) Experiment 30 min: Jurkat cells were cultured for 30 min (exposed to nsPEF) |
| Group 2 | Control 60 min: Jurkat cells were cultured for 60 min (without nsPEF) Experiment 60 min: Jurkat cells were cultured for 60 min (exposed to nsPEF) |
| Proteins | Species | Protein Length (aa) | Model Templates (Query Cover, Identify) |
|---|---|---|---|
| SUGP1 | Homo sapiens | 645 | de novo |
| DHX16 | Homo sapiens | 560 | 5Z58_XX (94%, 99%) 5MQF_QQ (90%, 56%) 6FA9_A (89%, 55%) |
| FUS | Homo sapiens | 522 | de novo |
| HNRNPR | Homo sapiens | 535 | de novo |
| DHX15 | Homo sapiens | 795 | 5XDR_A (86%, 99%) 3KX2_B (84%, 66%) 2XAU_A (84%, 66%) |
| NAA38 | Homo sapiens | 125 | de novo |
| SKIV2L2 | Homo sapiens | 1042 | 6D6Q_M (100%, 100%) 6C90_A (70%, 100%) 2XGJ_A (89%, 56%) |
| PLRG1 | Homo sapiens | 514 | 6FF4_D (100%, 100%) 5MQF_D (100%, 99%) 4YVD_A (72%, 100%) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Liu, S.; Yang, X.; Du, Y.; Luo, J.; Tan, J.; Sun, Y. Cellular Processes Involved in Jurkat Cells Exposed to Nanosecond Pulsed Electric Field. Int. J. Mol. Sci. 2019, 20, 5847. https://doi.org/10.3390/ijms20235847
Li H, Liu S, Yang X, Du Y, Luo J, Tan J, Sun Y. Cellular Processes Involved in Jurkat Cells Exposed to Nanosecond Pulsed Electric Field. International Journal of Molecular Sciences. 2019; 20(23):5847. https://doi.org/10.3390/ijms20235847
Chicago/Turabian StyleLi, Huijuan, Shibin Liu, Xue Yang, Yongqian Du, Jiezhang Luo, Jie Tan, and Yulong Sun. 2019. "Cellular Processes Involved in Jurkat Cells Exposed to Nanosecond Pulsed Electric Field" International Journal of Molecular Sciences 20, no. 23: 5847. https://doi.org/10.3390/ijms20235847
APA StyleLi, H., Liu, S., Yang, X., Du, Y., Luo, J., Tan, J., & Sun, Y. (2019). Cellular Processes Involved in Jurkat Cells Exposed to Nanosecond Pulsed Electric Field. International Journal of Molecular Sciences, 20(23), 5847. https://doi.org/10.3390/ijms20235847