Crystal Structure-Based Exploration of Arginine-Containing Peptide Binding in the ADP-Ribosyltransferase Domain of the Type III Effector XopAI Protein

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
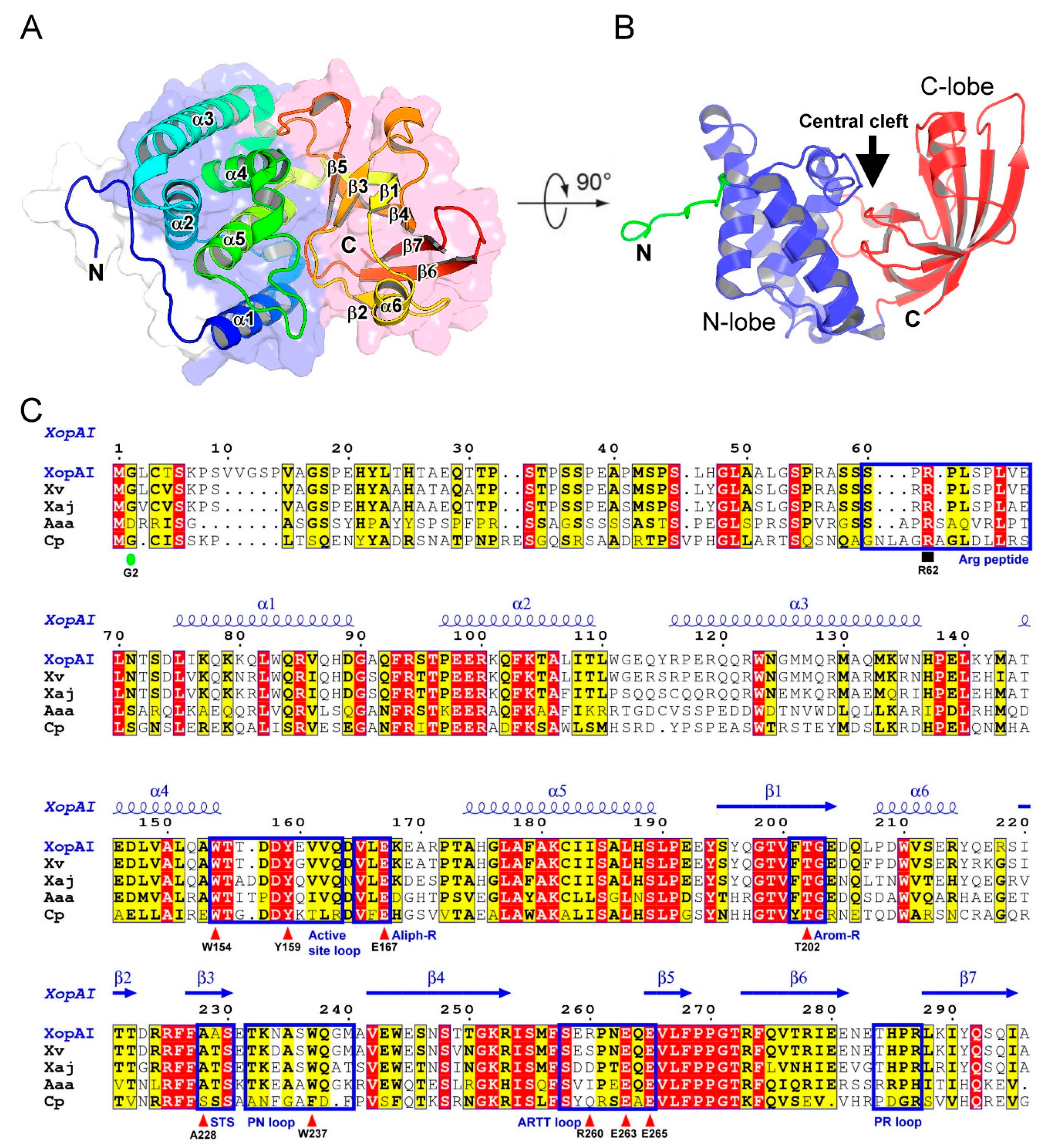
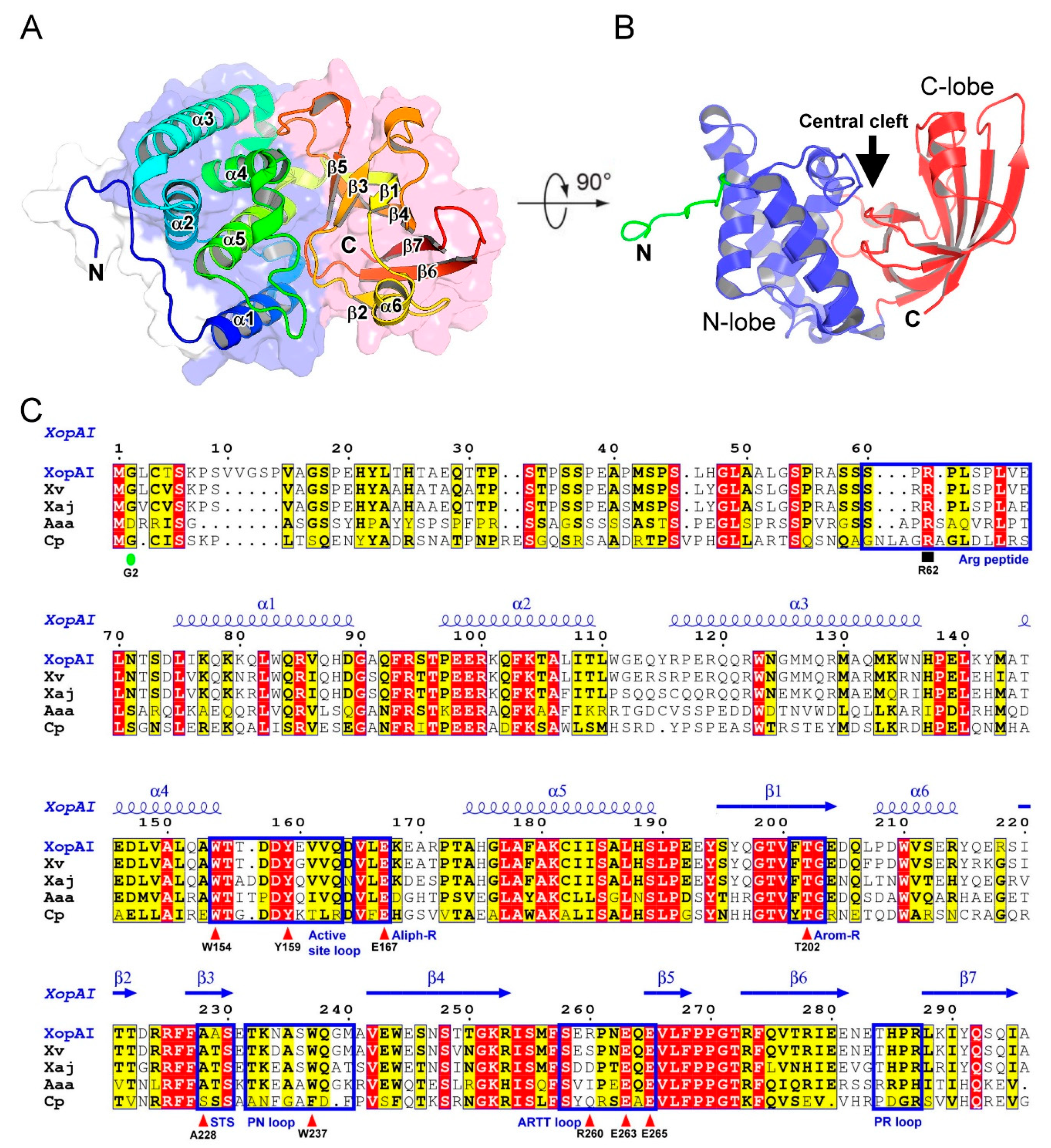
2.1. Structure of XopAI
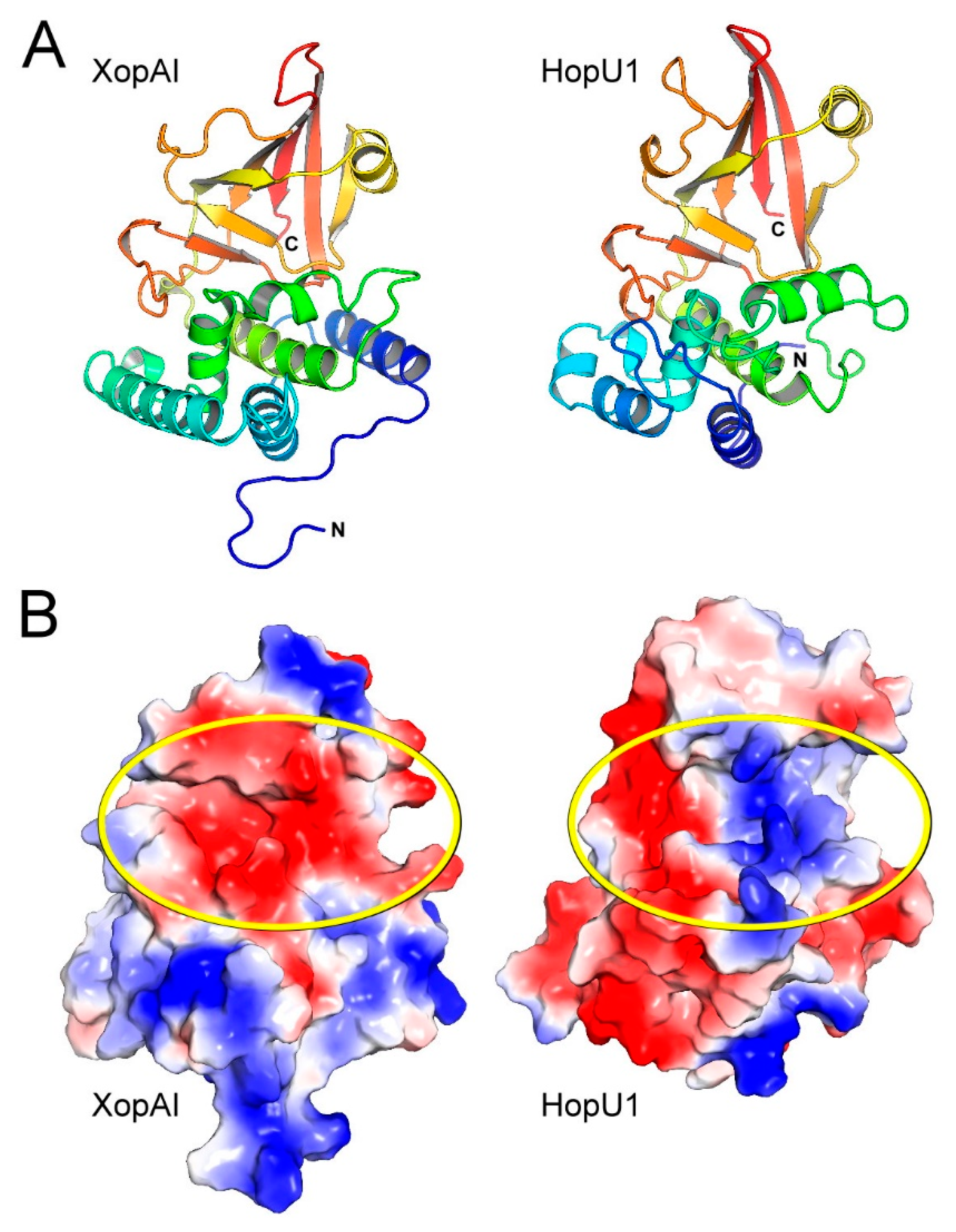
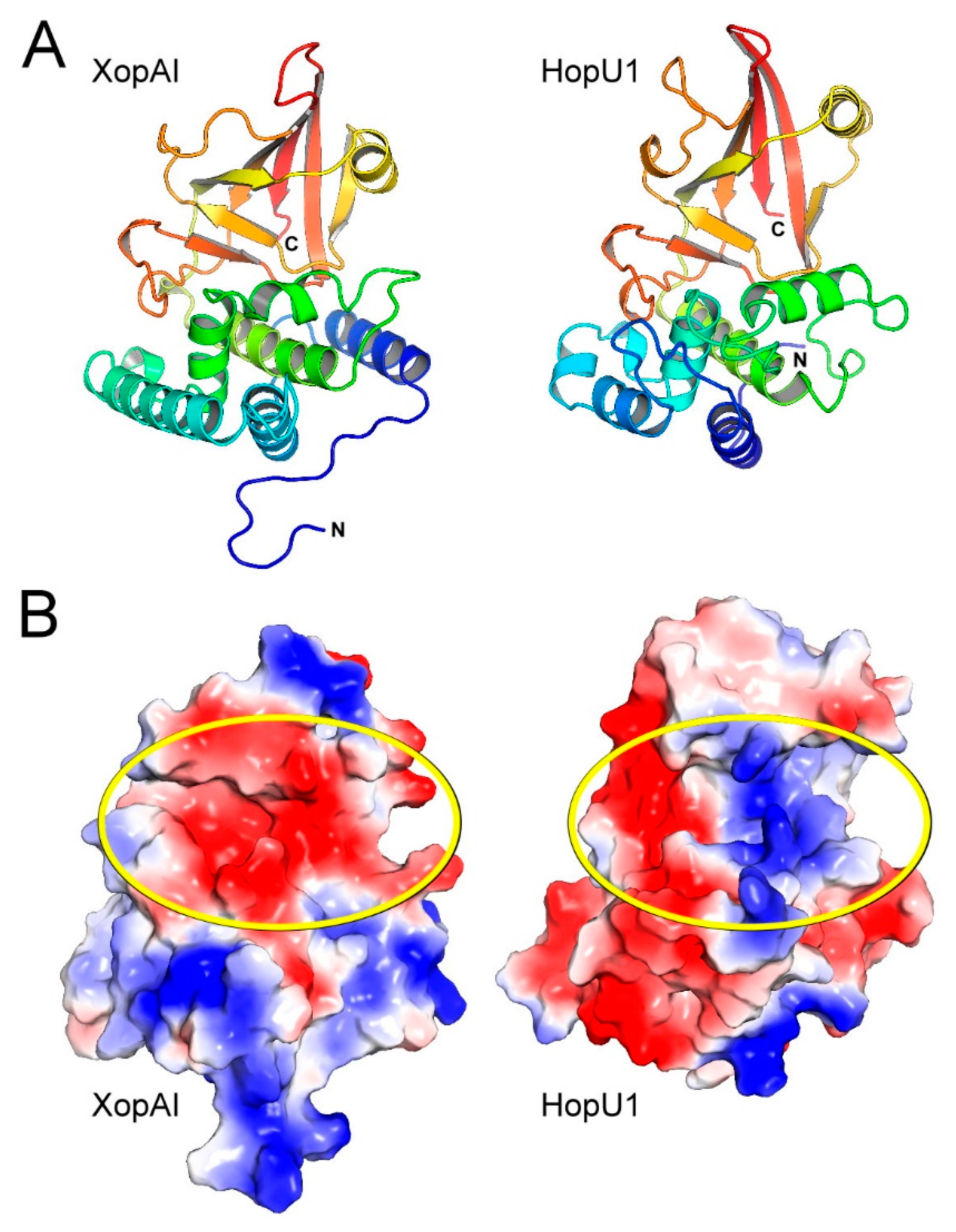
2.2. Structural Comparison of XopAI with mARTs
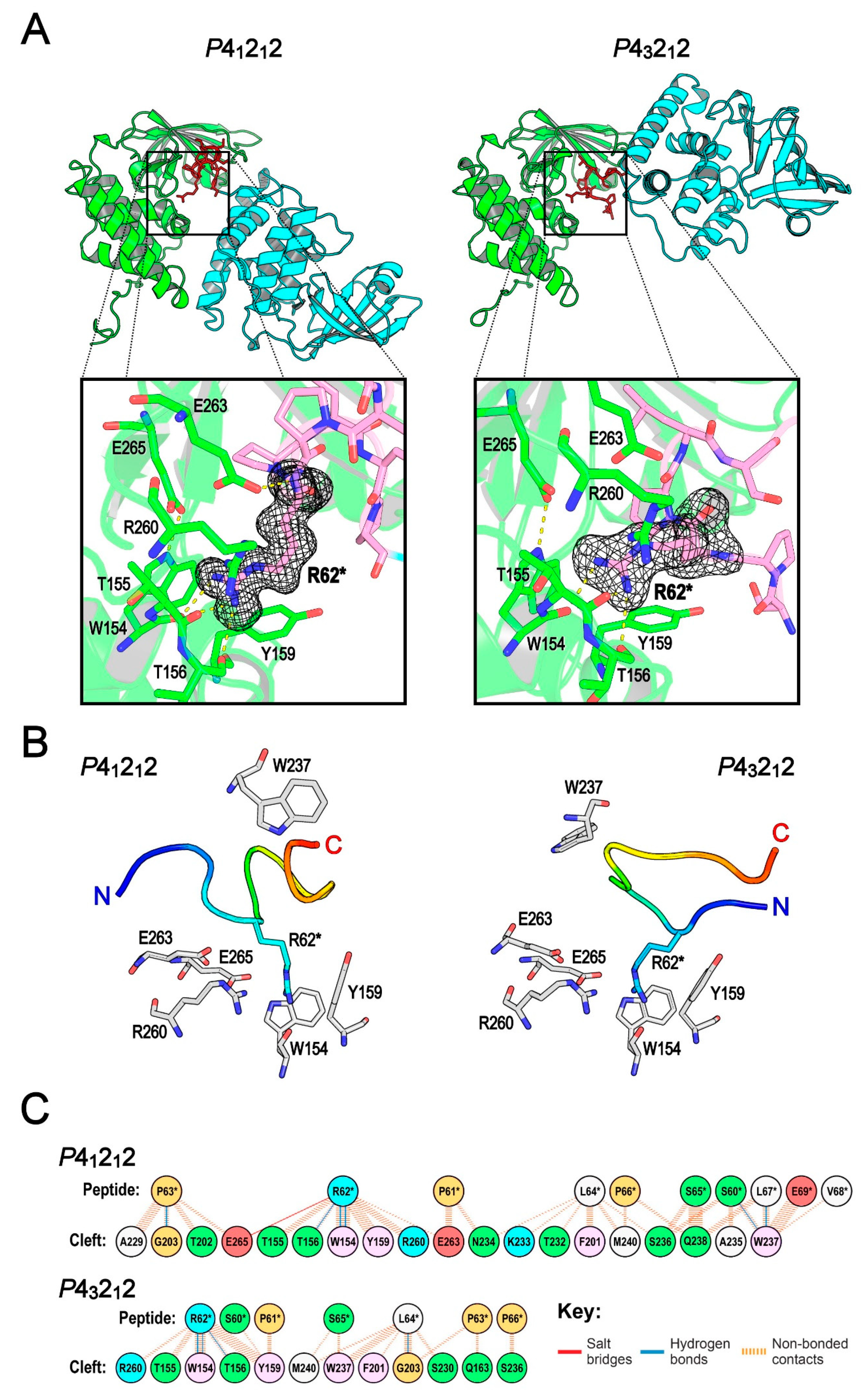
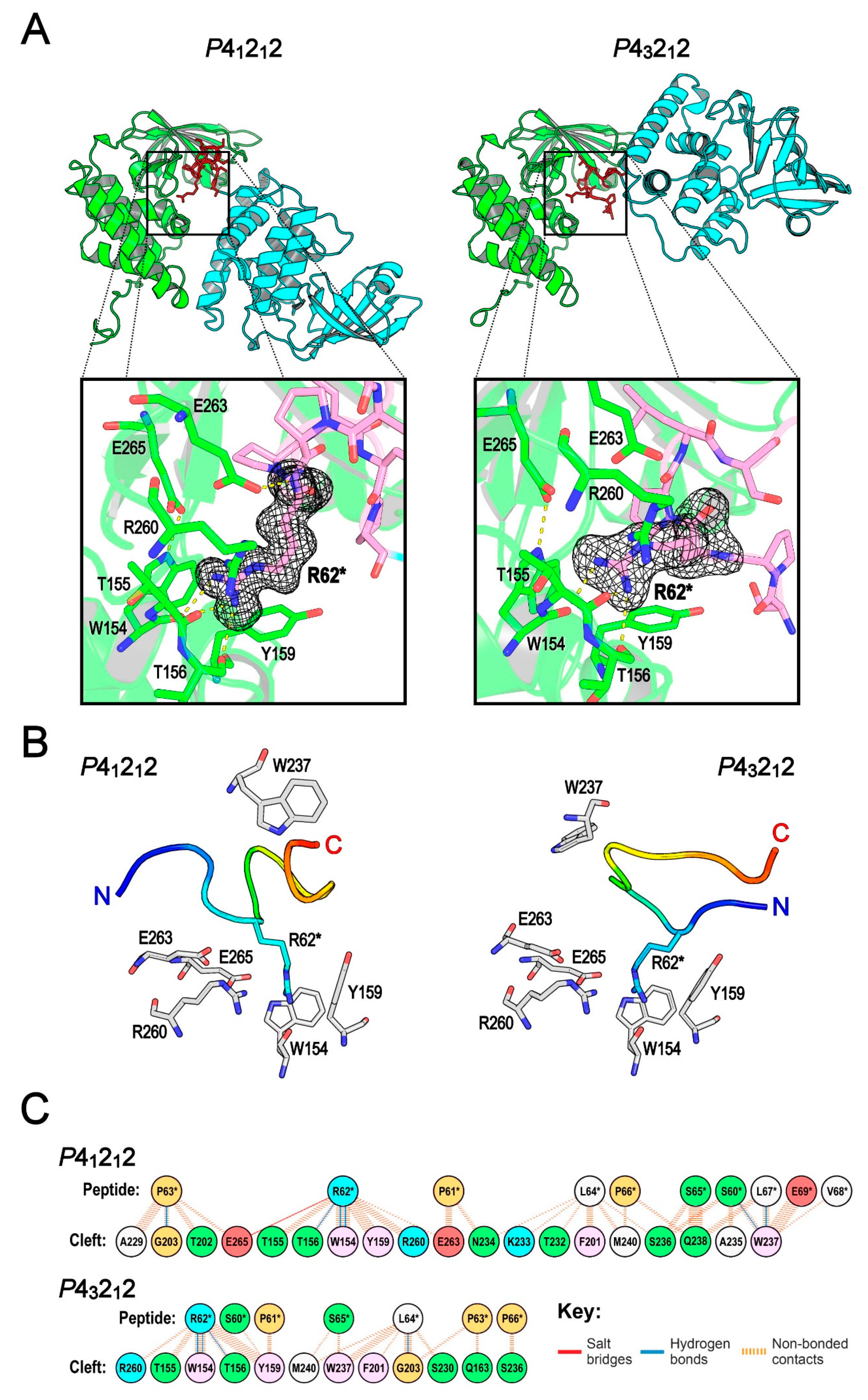
2.3. The Central Cleft of XopAI Has the Ability to Bind Peptides
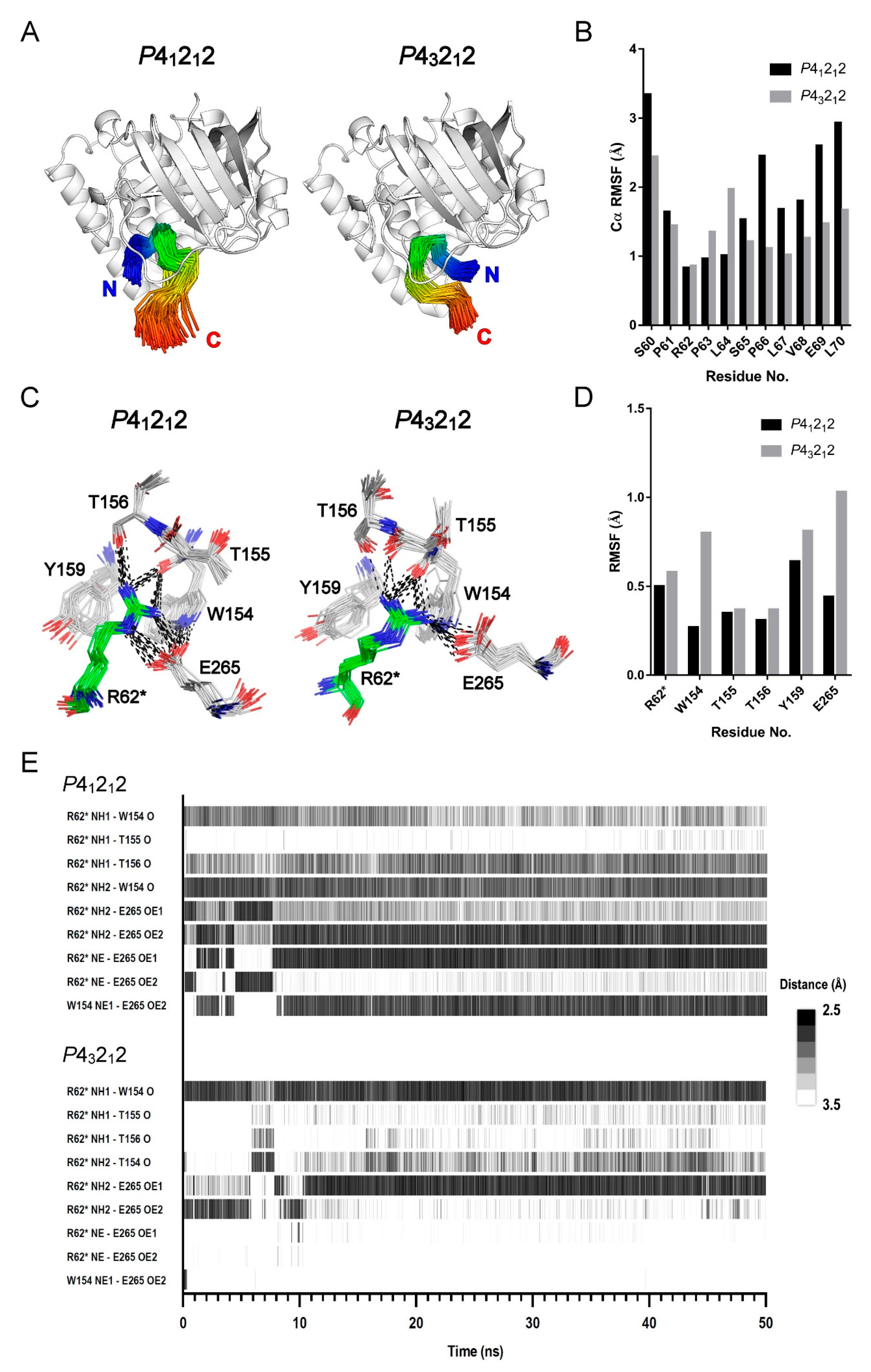
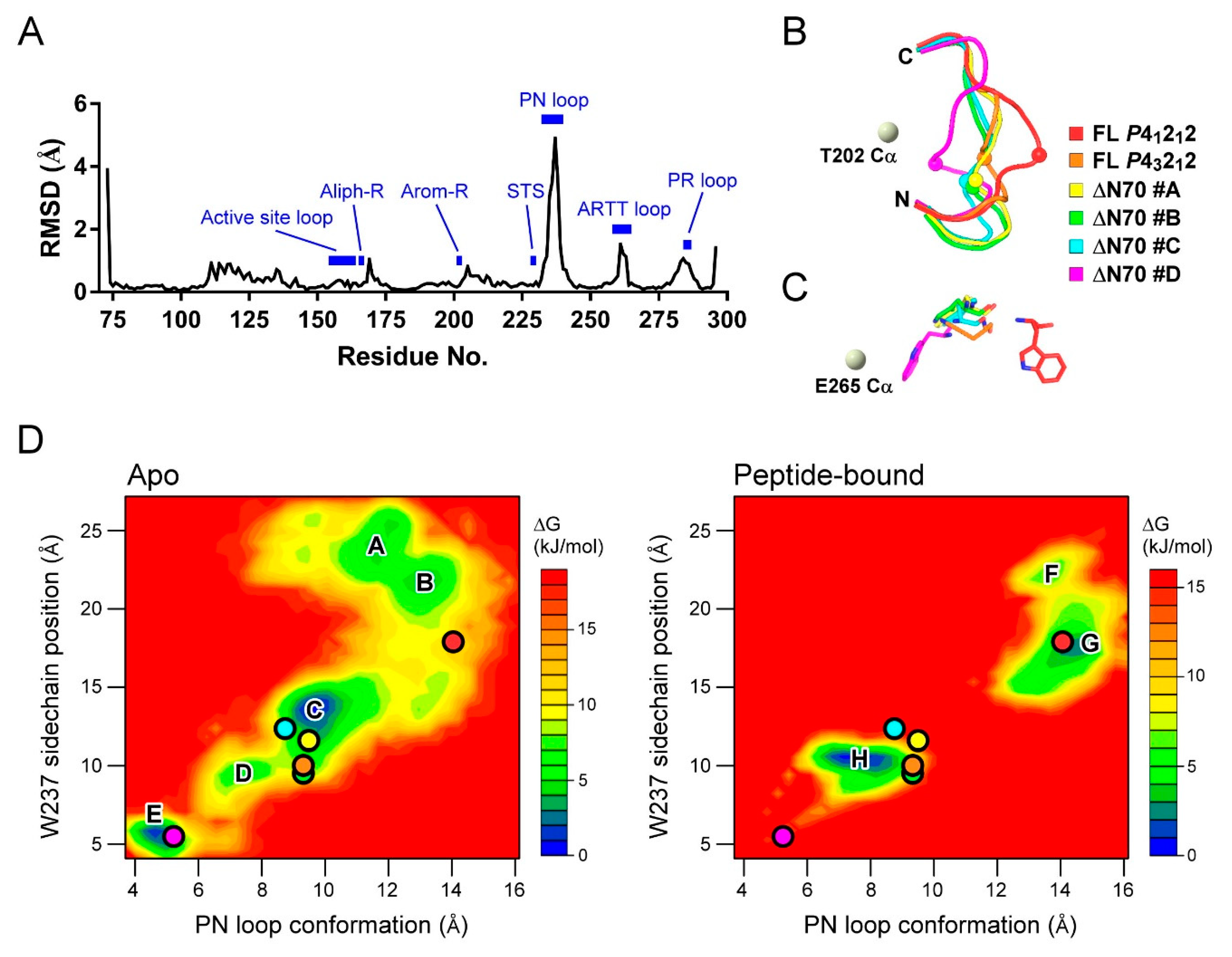
2.4. MD Simulation Study Supports the Protein–Peptide Interaction of XopAI
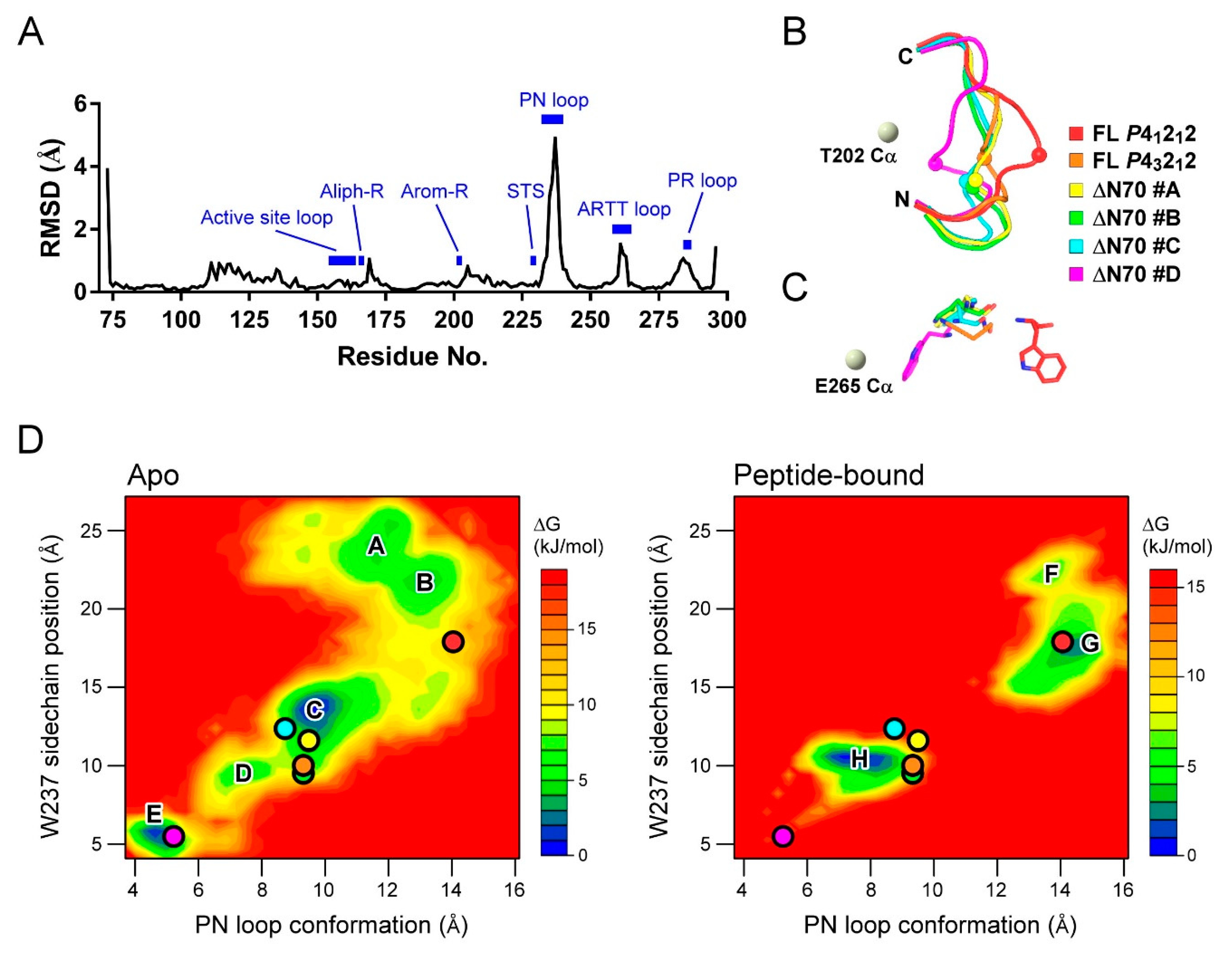
2.5. MD Study for the PN Loop Dynamics of XopAI in Response to the Arg Peptide Binding
3. Materials and Methods
3.1. Plasmid Constructions
3.2. Protein Expression and Purification
3.3. Protein Crystallization
3.4. Data Collection and Structure Determination
3.5. Sedimentation-Velocity Analytical Ultracentrifugation
3.6. Fluorescence Spectroscopic Assay
3.7. Bioinformatic Analyses
3.8. MD Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ARTT | ADP-ribosyl-turn-turn |
| Br-MAD | bromide multiple-wavelength anomalous diffraction |
| mART | mono-ADP-ribosyltransferase |
| MD | molecular dynamics |
| PN | phosphate-nicotinamide |
| RMSD | root-mean-square deviation |
| RMSF | root-mean-square fluctuation |
| Xac | Xanthomonas axonopodis pv. citri |
References
- Graham, J.H.; Gottwald, T.R.; Cubero, J.; Achor, D.S. Xanthomonas axonopodis pv. citri: Factors affecting successful eradication of citrus canker. Mol. Plant Pathol. 2004, 5, 1–15. [Google Scholar] [PubMed]
- Buttner, D.; He, S.Y. Type III protein secretion in plant pathogenic bacteria. Plant Physiol. 2009, 150, 1656–1664. [Google Scholar] [CrossRef] [PubMed]
- He, S.Y. Type III protein secretion systems in plant and animal pathogenic bacteria. Annu. Rev. Phytopathol. 1998, 36, 363–392. [Google Scholar] [CrossRef] [PubMed]
- Dunger, G.; Arabolaza, A.L.; Gottig, N.; Orellano, E.G.; Ottado, J. Participation of Xanthomonas axonopodis pv. citri hrp cluster in citrus canker and nonhost plant responses. Plant Pathol. 2005, 54, 781–788. [Google Scholar]
- Swarup, S.; Yang, Y.; Kingsley, M.T.; Gabriel, D.W. An Xanthomonas citri pathogenicity gene, pthA, pleiotropically encodes gratuitous avirulence on nonhosts. Mol. Plant Microbe Interact. 1992, 5, 204–213. [Google Scholar] [CrossRef] [PubMed]
- da Silva, R.A.; Pereira, L.M.; Silveira, M.C.; Jardim, R.; de Miranda, A.B. Mining of potential drug targets through the identification of essential and analogous enzymes in the genomes of pathogens of Glycine max, Zea mays and Solanum lycopersicum. PLoS ONE 2018, 13, e0197511. [Google Scholar] [CrossRef] [PubMed]
- Moreira, L.M.; Almeida, N.F., Jr.; Potnis, N.; Digiampietri, L.A.; Adi, S.S.; Bortolossi, J.C.; da Silva, A.C.; da Silva, A.M.; de Moraes, F.E.; de Oliveira, J.C.; et al. Novel insights into the genomic basis of citrus canker based on the genome sequences of two strains of Xanthomonas fuscans subsp. aurantifolii. BMC Genom. 2010, 11, 238. [Google Scholar] [CrossRef]
- Ferreira, R.M.; de Oliveira, A.C.; Moreira, L.M.; Belasque, J., Jr.; Gourbeyre, E.; Siguier, P.; Ferro, M.I.; Ferro, J.A.; Chandler, M.; Varani, A.M. A TALE of transposition: Tn3-like transposons play a major role in the spread of pathogenicity determinants of Xanthomonas citri and other xanthomonads. MBio 2015, 6, e02505-14. [Google Scholar] [CrossRef]
- White, F.F.; Potnis, N.; Jones, J.B.; Koebnik, R. The type III effectors of Xanthomonas. Mol. Plant Pathol. 2009, 10, 749–766. [Google Scholar] [CrossRef]
- Thieme, F.; Szczesny, R.; Urban, A.; Kirchner, O.; Hause, G.; Bonas, U. New type III effectors from Xanthomonas campestris pv. vesicatoria trigger plant reactions dependent on a conserved N-myristoylation motif. Mol. Plant Microbe Interact. 2007, 20, 1250–1261. [Google Scholar]
- da Silva, A.C.; Ferro, J.A.; Reinach, F.C.; Farah, C.S.; Furlan, L.R.; Quaggio, R.B.; Monteiro-Vitorello, C.B.; Van Sluys, M.A.; Almeida, N.F.; Alves, L.M.; et al. Comparison of the genomes of two Xanthomonas pathogens with differing host specificities. Nature 2002, 417, 459–463. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Barbieri, J.T. Molecular mechanisms of the cytotoxicity of ADP-ribosylating toxins. Annu. Rev. Microbiol. 2008, 62, 271–288. [Google Scholar] [CrossRef] [PubMed]
- Simon, N.C.; Aktories, K.; Barbieri, J.T. Novel bacterial ADP-ribosylating toxins: Structure and function. Nat. Rev. Microbiol. 2014, 12, 599–611. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.Q.; Guo, M.; Jeong, B.R.; Tian, F.; Elthon, T.E.; Cerny, R.L.; Staiger, D.; Alfano, J.R. A type III effector ADP-ribosylates RNA-binding proteins and quells plant immunity. Nature 2007, 447, 284–288. [Google Scholar] [CrossRef] [PubMed]
- Dauter, M.; Dauter, Z. Phase determination using halide ions. Methods Mol. Biol. 2007, 364, 149–158. [Google Scholar] [PubMed]
- Schaad, N.W.; Postnikova, E.; Sechler, A.; Claflin, L.E.; Vidaver, A.K.; Jones, J.B.; Agarkova, I.; Ignatov, A.; Dickstein, E.; Ramundo, B.A. Reclassification of subspecies of Acidovorax avenae as A. Avenae (Manns 1905) emend., A. cattleyae (Pavarino, 1911) comb. nov., A. citrulli Schaad et al., 1978) comb. nov., and proposal of A. oryzae sp. nov. Syst. Appl. Microbiol. 2008, 31, 434–446. [Google Scholar] [CrossRef]
- Hoppener-Ogawa, S.; de Boer, W.; Leveau, J.H.; van Veen, J.A.; de Brandt, E.; Vanlaere, E.; Sutton, H.; Dare, D.J.; Vandamme, P. Collimonas arenae sp. nov. and Collimonas pratensis sp. nov., isolated from (semi-)natural grassland soils. Int J. Syst. Evol. Microbiol. 2008, 58 (Pt 2), 414–419. [Google Scholar] [CrossRef]
- Jeong, B.R.; Lin, Y.; Joe, A.; Guo, M.; Korneli, C.; Yang, H.; Wang, P.; Yu, M.; Cerny, R.L.; Staiger, D.; et al. Structure function analysis of an ADP-ribosyltransferase type III effector and its RNA-binding target in plant immunity. J. Biol. Chem. 2011, 286, 43272–43281. [Google Scholar] [CrossRef]
- Ting, S.Y.; Bosch, D.E.; Mangiameli, S.M.; Radey, M.C.; Huang, S.; Park, Y.J.; Kelly, K.A.; Filip, S.K.; Goo, Y.A.; Eng, J.K.; et al. Bifunctional immunity proteins protect bacteria against FtsZ-targeting ADP-ribosylating toxins. Cell 2018, 175, 1380–1392. [Google Scholar] [CrossRef]
- Mueller-Dieckmann, C.; Ritter, H.; Haag, F.; Koch-Nolte, F.; Schulz, G.E. Structure of the ecto-ADP-ribosyl transferase ART2.2 from rat. J. Mol. Biol 2002, 322, 687–696. [Google Scholar] [CrossRef]
- Karlberg, T.; Hornyak, P.; Pinto, A.F.; Milanova, S.; Ebrahimi, M.; Lindberg, M.; Pullen, N.; Nordstrom, A.; Loverli, E.; Caraballo, R.; et al. 14-3-3 proteins activate Pseudomonas exotoxins-S and -T by chaperoning a hydrophobic surface. Nat. Commun. 2018, 9, 3785. [Google Scholar] [CrossRef] [PubMed]
- Hottiger, M.O.; Hassa, P.O.; Luscher, B.; Schuler, H.; Koch-Nolte, F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci. 2010, 35, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Fieldhouse, R.J.; Turgeon, Z.; White, D.; Merrill, A.R. Cholera- and anthrax-like toxins are among several new ADP-ribosyltransferases. PLoS Comput. Biol. 2010, 6, e1001029. [Google Scholar] [CrossRef] [PubMed]
- Holbourn, K.P.; Shone, C.C.; Acharya, K.R. A family of killer toxins. Exploring the mechanism of ADP-ribosylating toxins. FEBS 2006, 273, 4579–4593. [Google Scholar] [CrossRef] [PubMed]
- Lo Conte, L.; Chothia, C.; Janin, J. The atomic structure of protein–protein recognition sites. J. Mol. Biol 1999, 285, 2177–2198. [Google Scholar] [CrossRef]
- Petsalaki, E.; Russell, R.B. Peptide-mediated interactions in biological systems: New discoveries and applications. Curr. Opin. Biotechnol. 2008, 19, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Wilton, M.; Subramaniam, R.; Elmore, J.; Felsensteiner, C.; Coaker, G.; Desveaux, D. The type III effector HopF2Pto targets Arabidopsis RIN4 protein to promote Pseudomonas syringae virulence. Proc. Natl. Acad. Sci. USA 2010, 107, 2349–2354. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Jablonska, J.; Pravda, L.; Varekova, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef]
- Weiss, D.R.; Levitt, M. Can morphing methods predict intermediate structures? J. Mol. Biol. 2009, 385, 665–674. [Google Scholar] [CrossRef]
- Powell, H.R.; Battye, T.G.G.; Kontogiannis, L.; Johnson, O.; Leslie, A.G.W. Integrating macromolecular X-ray diffraction data with the graphical user interface iMosflm. Nat. Protoc. 2017, 12, 1310–1325. [Google Scholar] [CrossRef] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 (Pt 2), 213–221. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- DeLano, W.L. PyMOL: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Schuck, P.; Perugini, M.A.; Gonzales, N.R.; Howlett, G.J.; Schubert, D. Size-distribution analysis of proteins by analytical ultracentrifugation: Strategies and application to model systems. Biophys J. 2002, 82, 1096–1111. [Google Scholar] [CrossRef]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Ostell, J.; Pruitt, K.D.; Karsch-Mizrachi, I. GenBank. Nucleic Acids Res. 2019, 47, D94–D99. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinf 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Gouet, P.; Robert, X.; Courcelle, E. ESPript/ENDscript: Extracting and rendering sequence and 3D information from atomic structures of proteins. Nucleic Acids Res. 2003, 31, 3320–3323. [Google Scholar] [CrossRef]
- Ward, J.J.; McGuffin, L.J.; Bryson, K.; Buxton, B.F.; Jones, D.T. The DISOPRED server for the prediction of protein disorder. Bioinf 2004, 20, 2138–2139. [Google Scholar] [CrossRef]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef] [Green Version]
- Hanson, J.; Yang, Y.; Paliwal, K.; Zhou, Y. Improving protein disorder prediction by deep bidirectional long short-term memory recurrent neural networks. Bioinf 2017, 33, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.S.; Eramian, D.; Shen, M.Y.; Pieper, U.; Sali, A. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinf 2006, 5. [Google Scholar] [CrossRef]
- Holm, L.; Laakso, L.M. Dali server update. Nucleic Acids Res. 2016, 44. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, 344–350. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Delgado, J.; Radusky, L.G.; Cianferoni, D.; Serrano, L. FoldX 5.0: Working with RNA, small molecules and a new graphical interface. Bioinf 2019. [Google Scholar] [CrossRef]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed atlas of surface topography of proteins. Nucleic Acids Res. 2018, 46, 363–367. [Google Scholar] [CrossRef]
- Pronk, S.; Pall, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinf 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Open Source Drug Discovery, C.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Krebs, W.G.; Gerstein, M. The morph server: A standardized system for analyzing and visualizing macromolecular motions in a database framework. Nucleic Acids Res. 2000, 28, 1665–1675. [Google Scholar] [CrossRef] [PubMed]
- Gerlt, J.A.; Babbitt, P.C.; Rayment, I. Divergent evolution in the enolase superfamily: The interplay of mechanism and specificity. Arch. Biochem. Biophys. 2005, 433, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Nardini, M.; Dijkstra, B.W. Alpha/beta hydrolase fold enzymes: The family keeps growing. Curr. Opin. Struct. Biol. 1999, 9, 732–737. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Native | Br-MAD | ||
|---|---|---|---|---|
| λ1 (H rem) 1 | λ2 (infl) | λ3 (Peak) | ||
| Wavelength (Å) | 0.9762 | 0.9056 | 0.9193 | 0.9190 |
| Space group | P43212 | |||
| Resolution (Å) | 2.0 | 2.3 | 2.4 | 2.5 |
| Redundancy | 4.1 | 2.4 | 4.6 | 2.5 |
| Completeness (%) | 93.7 (85.3) 2 | 98.5 (99.7) | 98.4 (99.7) | 98.2 (99.7) |
| Average I/σ(I) | 11.1 (2.5) | 17.7 (3.5) | 19.5 (4.0) | 18.9 (3.8) |
| Rmerge (%) | 8.2 (52.5) | 11.2 (49.2) | 10.2 (44.7) | 10.3 (43.6) |
| Protein | XopAI | XopAI-ΔN70 | |
|---|---|---|---|
| Data collection | |||
| Wavelength (Å) | 0.9762 | 0.9198 | 0.9762 |
| Space group | P43212 | P41212 | P21 |
| Cell parameters (Å) | a = b = 73.05 Å, c = 114.06 Å | a = b = 52.98 Å, c = 212.09 Å | a = 62.78 Å, b = 98.76 Å, c = 77.45 Å, β = 91.21° |
| Resolution (Å) | 2.01 | 1.53 | 2.26 |
| Mosaicity (°) | 1.07 | 0.29 | 1.23 |
| Redundancy | 4.1 | 10.9 | 3.5 |
| Completeness (%) | 93.7 (85.3) 1 | 100.0 (100.0) | 99.4 (99.3) |
| Average I/σ(I) | 11.1 (2.5) | 9.6 (1.7) | 7.5 (1.8) |
| Rmerge (%) | 8.2 (52.5) | 13.1 (106.4) | 14.2 (78.3) |
| CC1/2 | 0.997 (0.712) | 0.997 (0.647) | 0.990 (0.609) |
| Refinement | |||
| Resolution limit (Å) | 26.56–2.01 | 36.89–1.53 | 31.53–2.26 |
| Rwork (%) | 16.6 | 16.1 | 18.9 |
| Rfree (%) | 19.5 | 17.1 | 23.2 |
| Number of non-H atoms | |||
| Protein | 1961 | 1973 | 7354 |
| Water | 236 | 374 | 558 |
| Ramachandran plot statistics | |||
| Favored regions (%) | 99.15 | 98.73 | 98.43 |
| Allowed regions (%) | 0.85 | 1.27 | 1.46 |
| Disallowed regions (%) | 0 | 0 | 0.11 |
| Average B factor (Å2) | 25.9 | 22.7 | 35.1 |
| R.m.s. deviation from ideality | |||
| Bond length (Å) | 0.004 | 0.010 | 0.004 |
| Bond angle (˚) | 0.655 | 1.024 | 0.561 |
| Protein Data Bank (PDB) code | 6KLY | 6K93 | 6K94 |
| Domain Name | Binding Energy (kcal/mol) | Number of Bound Residues | Interface Area (Å2) | PDB ID |
|---|---|---|---|---|
| XopAI (P41212) | −8.3 | 12 | 673 | 6K93 (this study) |
| XopAI (P43212) | −7.6 | 7 | 470 | 6KLY (this study) |
| PDZ | −3.7 ± 2.3 | 4~9 (6) 1 | 432 ± 92 | 1BE9, 1L6O, 1MFG, 1N7F, 1OBX, 1OBZ, 1Q3P, 2I0I, 2QT5, 3DIW, 3LNY |
| IRS | −4.9 ± 1.3 | 9 | 617 ± 20 | 1UEF, 3ML4 |
| VHS | −5.5 ± 3.2 | 5~7 (6) | 420 ± 45 | 1JUQ, 1UJK |
| 14-3-3 | −5.7 ± 1.7 | 5~10 (7) | 554 ± 122 | 1QJB, 2BTP, 2C74, 3MHR, 3UBW |
| WH1 | −6.6 ± 2.6 | 5~6 (6) | 315 ± 35 | 1DDV, 1EVH, 1QC6 |
| Skp1 | −6.9 ± 2.3 | 6~12 (10) | 548 ± 151 | 2AST, 2OVQ, 2P1Q |
| SH2 | −7.0 | 4 | 363 | 1FYR |
| TPR | −7.0 ± 1.1 | 5~8 (7) | 502 ± 29 | 1ELR, 1ELW |
| PID | −7.7 ± 2.7 | 9~10 (10) | 717 ± 28 | 1AQC, 1M7E, 1NTV |
| BIR | −8.3 ± 1.7 | 4~7 (6) | 420 ± 54 | 1JD5, 1SE0, 3D9T |
| WD40 | −8.4 ± 2.3 | 8~12 (9) | 567 ± 44 | 2CE8, 4ERY, 5IGO, 5IGQ |
| SH3 | −9.4 ± 0.6 | 9~10 (10) | 480 ± 24 | 1CKA, 1N5Z, 1W70 |
| SPRY | −10.0 ± 0.5 | 7 | 352 ± 2 | 2JK9, 3EMW |
| Complex | ΔEvdW 1 | ΔEelec | ΔGpolar | ΔGnonpolar | ΔGbinding |
|---|---|---|---|---|---|
| P41212 | −248.2 ± 25.2 | −582.9 ± 42.2 | 496.4 ± 42.8 | −33.8 ± 2.1 | −368.5 ± 40.3 |
| P43212 | −173.3 ± 22.1 | −539.2 ± 40.4 | 505.1 ± 38.9 | −27.7 ± 2.0 | −235.1 ± 30.9 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.-H.; Yang, J.-Y.; Hsu, D.-W.; Lai, Y.-H.; Li, Y.-P.; Tsai, Y.-R.; Hou, M.-H. Crystal Structure-Based Exploration of Arginine-Containing Peptide Binding in the ADP-Ribosyltransferase Domain of the Type III Effector XopAI Protein. Int. J. Mol. Sci. 2019, 20, 5085. https://doi.org/10.3390/ijms20205085
Liu J-H, Yang J-Y, Hsu D-W, Lai Y-H, Li Y-P, Tsai Y-R, Hou M-H. Crystal Structure-Based Exploration of Arginine-Containing Peptide Binding in the ADP-Ribosyltransferase Domain of the Type III Effector XopAI Protein. International Journal of Molecular Sciences. 2019; 20(20):5085. https://doi.org/10.3390/ijms20205085
Chicago/Turabian StyleLiu, Jyung-Hurng, Jun-Yi Yang, Duen-Wei Hsu, Yi-Hua Lai, Yun-Pei Li, Yi-Rung Tsai, and Ming-Hon Hou. 2019. "Crystal Structure-Based Exploration of Arginine-Containing Peptide Binding in the ADP-Ribosyltransferase Domain of the Type III Effector XopAI Protein" International Journal of Molecular Sciences 20, no. 20: 5085. https://doi.org/10.3390/ijms20205085