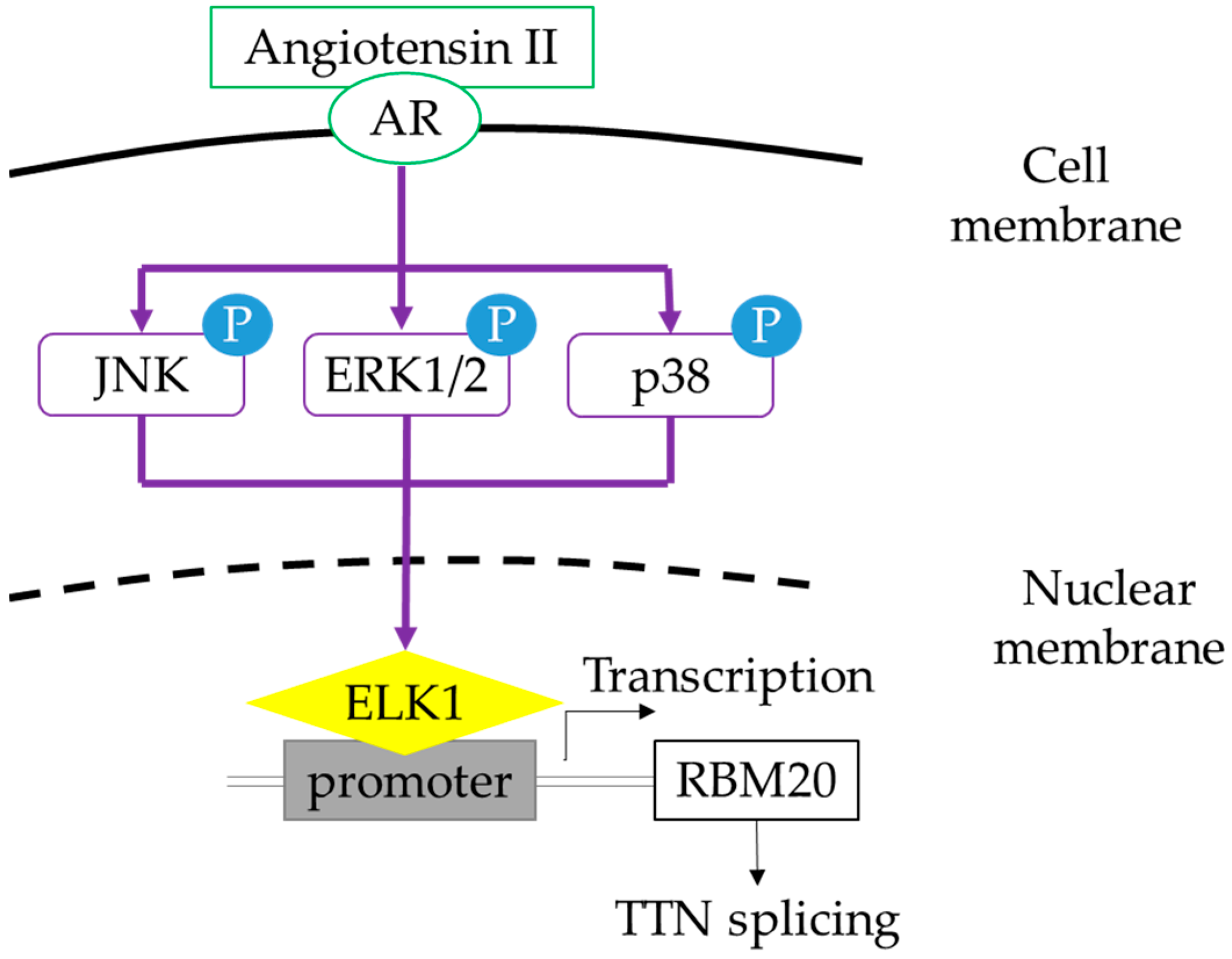
Angiotensin II Influences Pre-mRNA Splicing Regulation by Enhancing RBM20 Transcription Through Activation of the MAPK/ELK1 Signaling Pathway

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
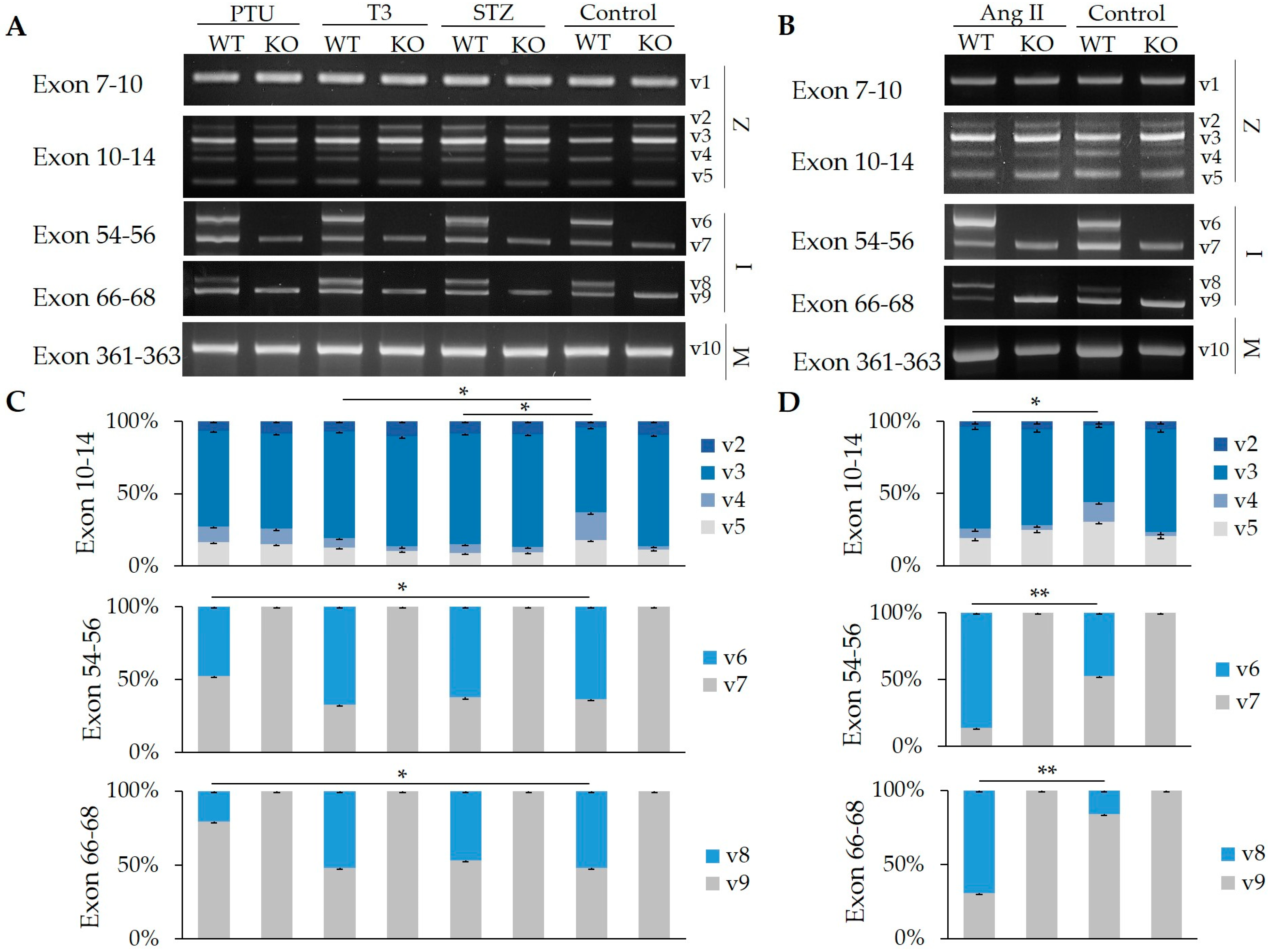
2.1. Splicing Regulation of RBM20 in Z-, I-, and M-Band Titin at Different Ages
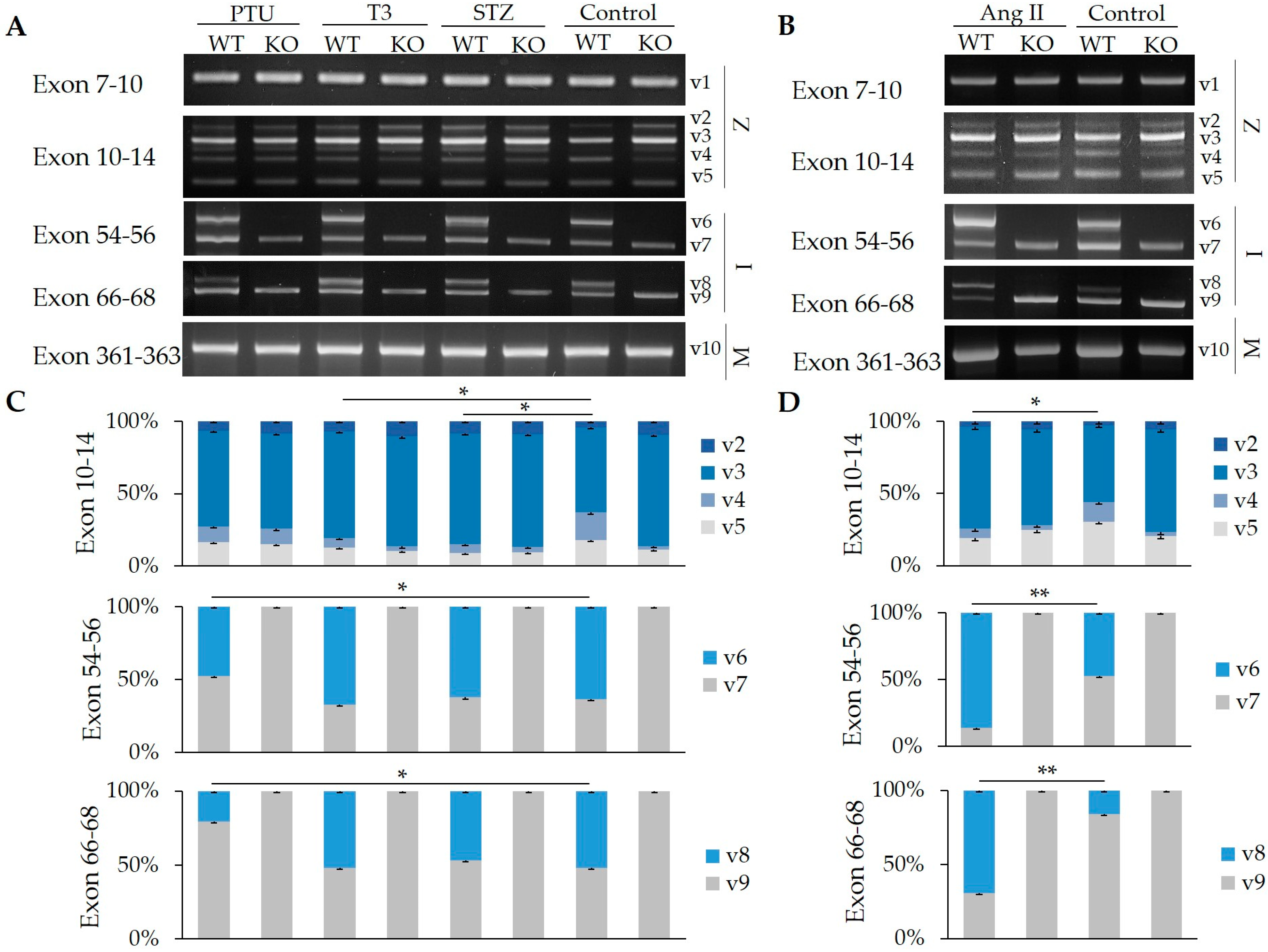
2.2. Impact of Hormones on the Regulation of Z-, I-, and M-Band Titin Splicing with and without RBM20
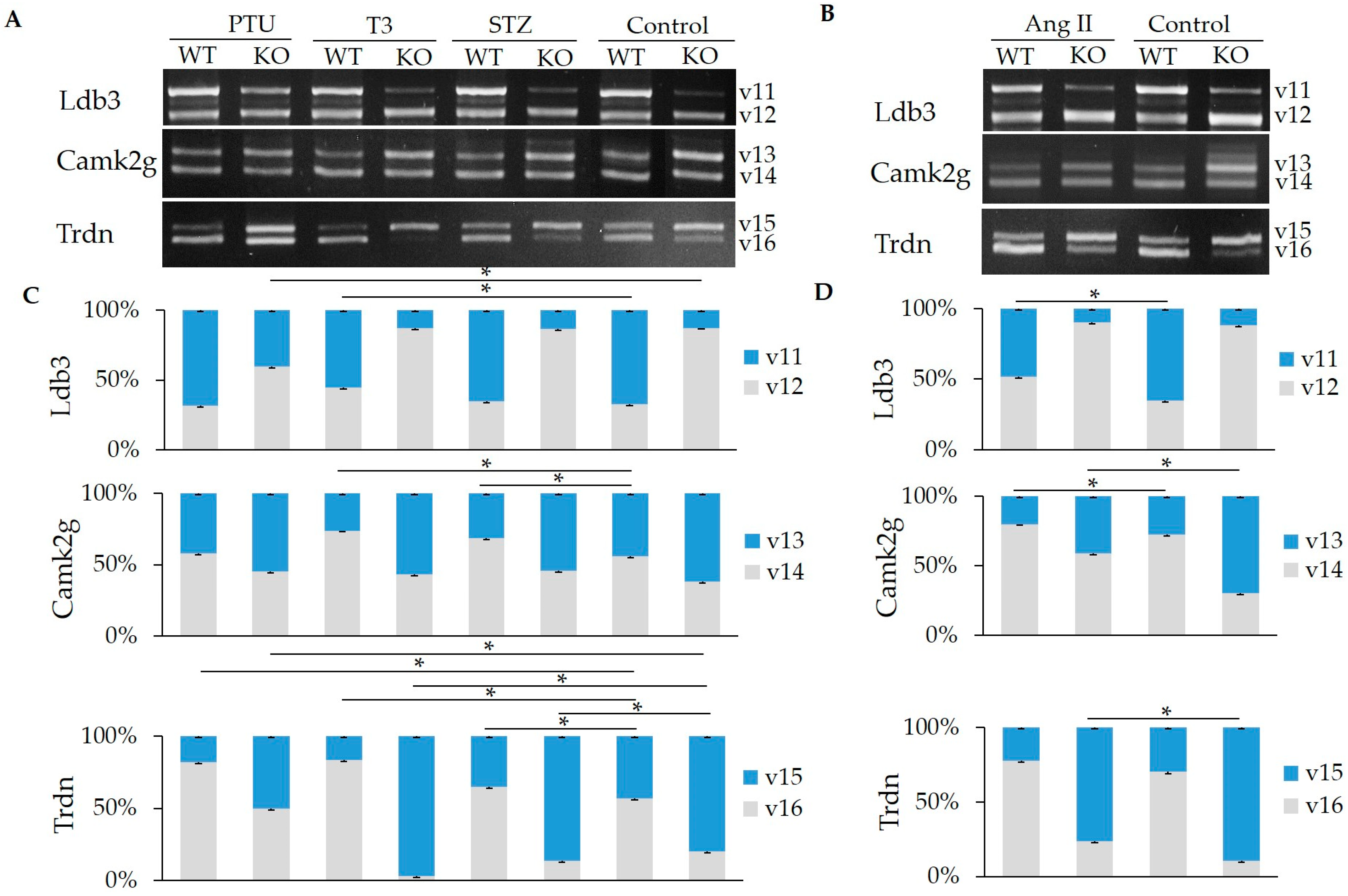
2.3. Hormone Impact on the Splicing of Other Substrates
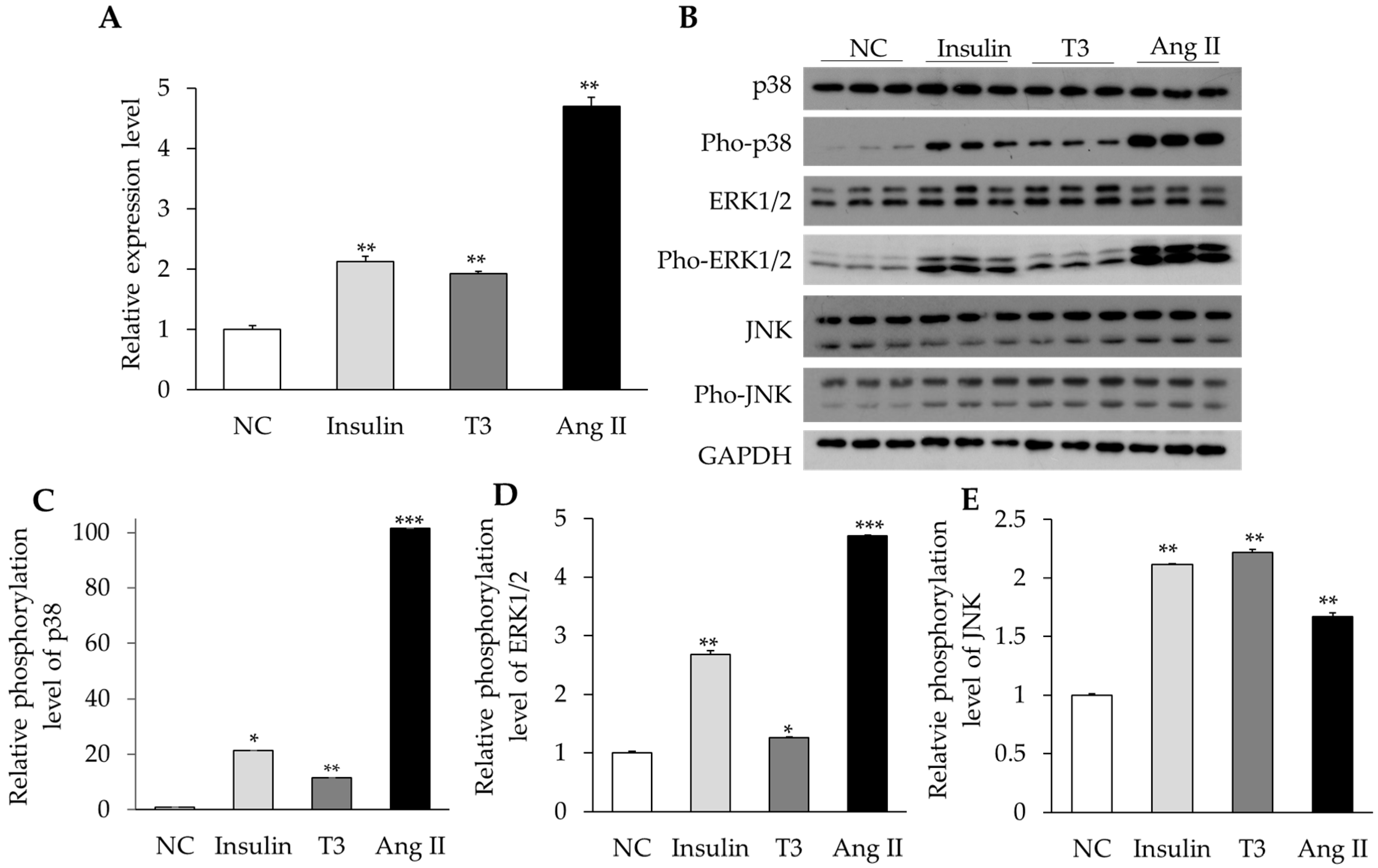
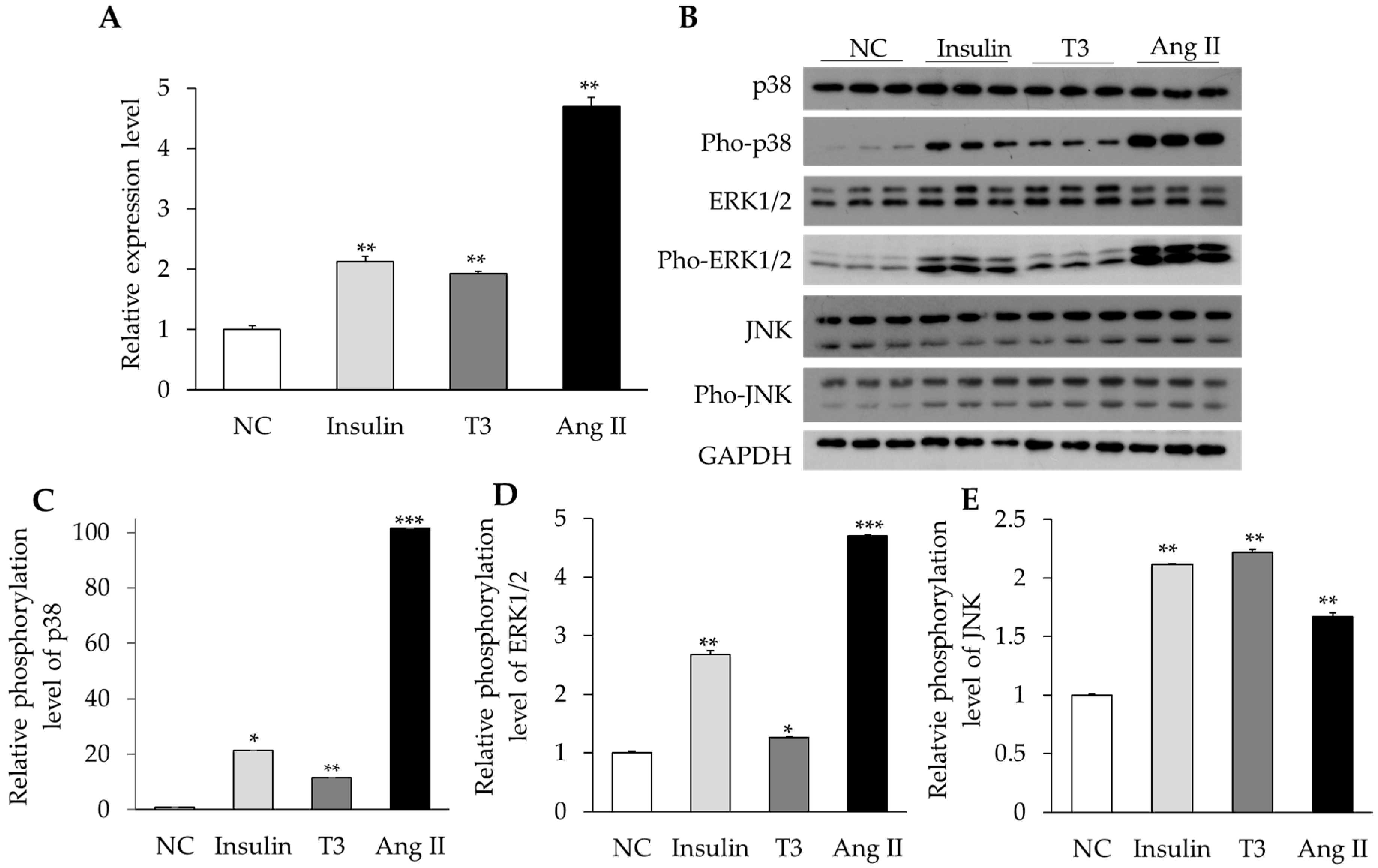
2.4. Activation of MAPK Signaling by Insulin, Thyroid Hormone, and Ang II in Cultured Neonatal Cardiomyocytes
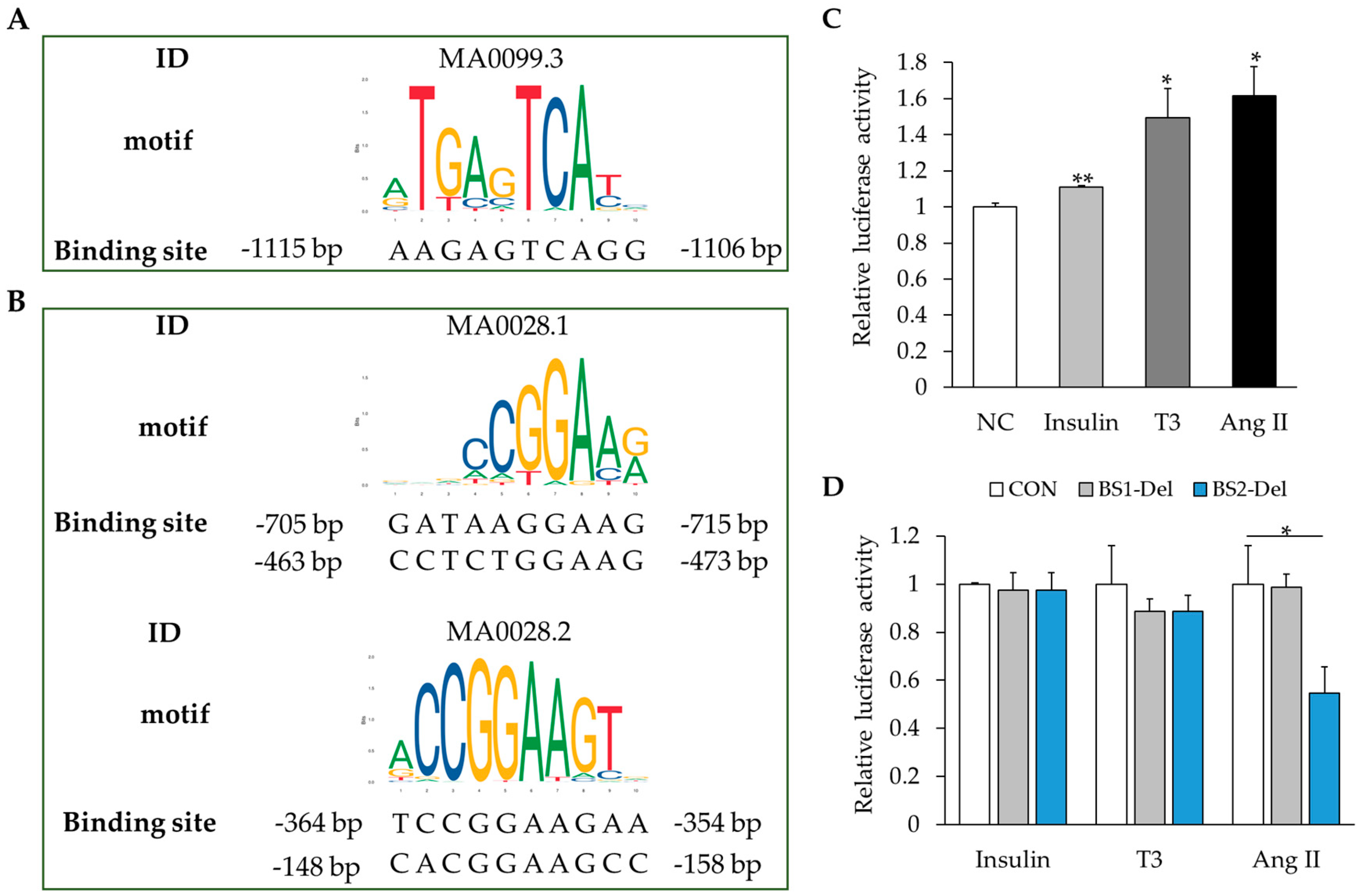
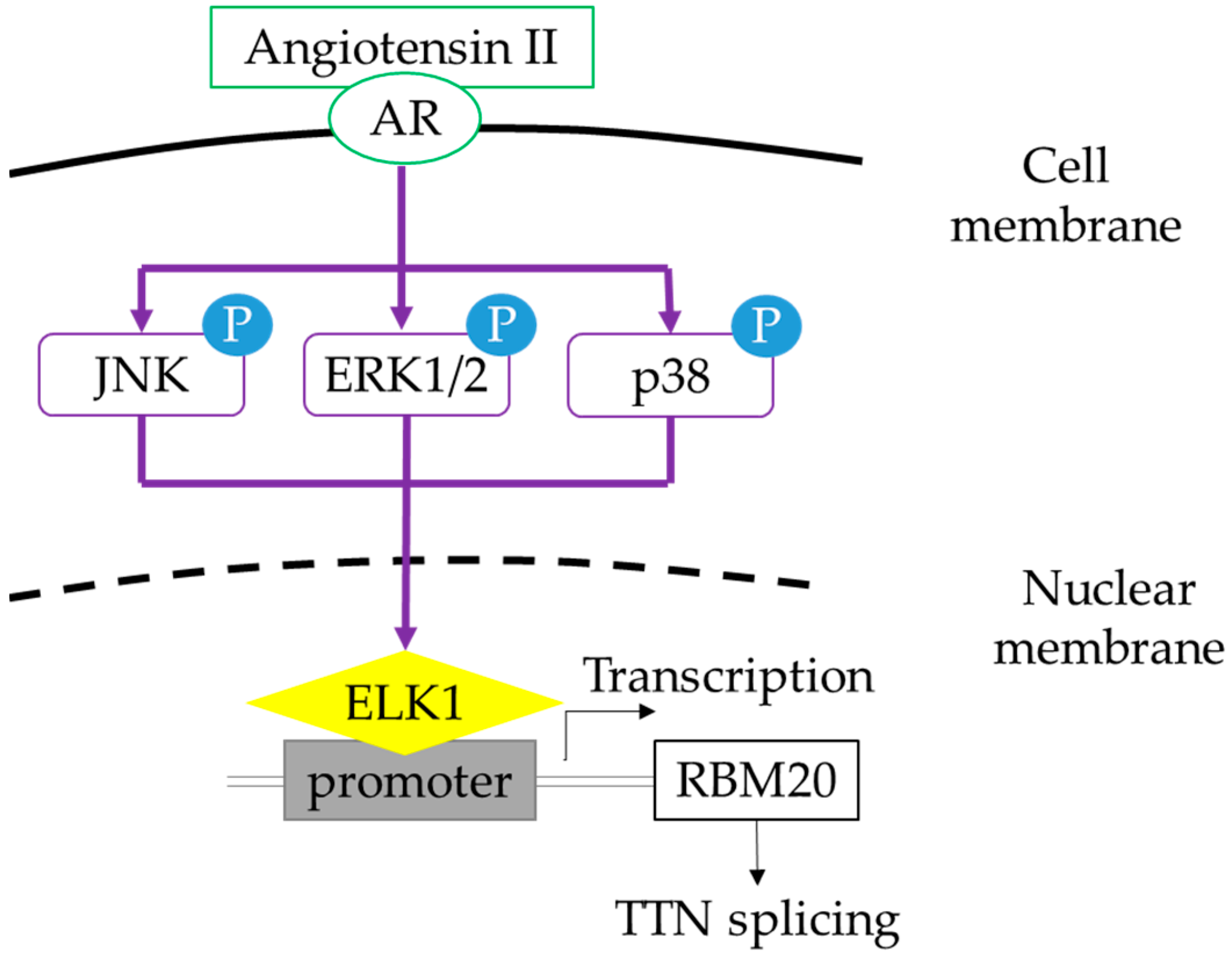
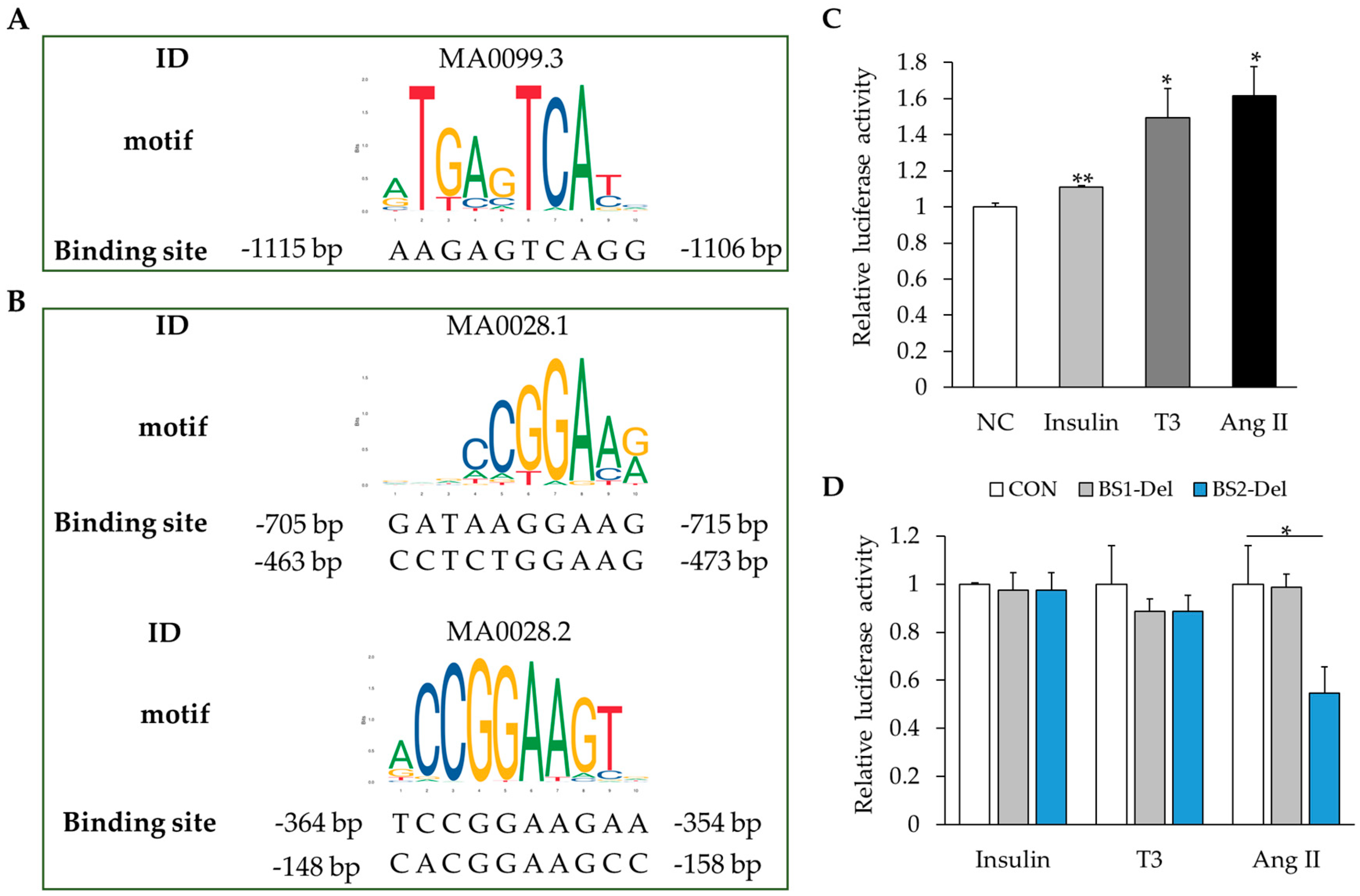
2.5. MAPK Signaling Activation Enhances RBM20 Transcription in Primary Neonatal Cardiomyocyte Cultures
3. Discussion
4. Materials and Methods
4.1. Experimental Animals and Tissue Samples
4.2. Neonatal Rat Cardiomyocytes (NRCMs) Isolation and Treatment
4.3. Reverse Transcriptional PCR (RT-PCR) and Quantitative Real-Time PCR (qPCR) Analysis
4.4. Western Blot Analysis
4.5. Bioinformatics Analysis
4.6. Dual-Luciferase Activity Assay
4.7. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| T3 | Triiodothyronine |
| Ang II | Angiotensin II |
| WT | Wild type |
| KO | RBM20 knockout |
| STZ | Streptozotocin |
| RBM20 | RNA binding motif 20 |
| SR | Serine-arginine |
| PTU | Propylthiouracil |
| Camk2g | Calcium/calmodulin dependent protein kinase II gamma |
| Ldb3 | LIM domain binding 3 |
| Trdn | Triadin |
| MAPK | Mitogen-Activated Protein Kinase |
| SD | Sprague Dawley |
| BN | Brown Norway |
| GAPDH | Glyceraldehyde-3-phosphate |
| SE | Standard error |
References
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Burgess, D.J. Alternative splicing: Proteomic rewiring through transcriptomic diversity. Nat. Rev. Genet. 2012, 13, 518. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.J.; Cooper, T.A. The pathobiology of splicing. J. Pathol. 2010, 220, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Kelemen, O.; Convertini, P.; Zhang, Z.; Wen, Y.; Shen, M.; Falaleeva, M.; Stamm, S. Function of alternative splicing. Gene 2013, 514, 1–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidalgo, C.; Granzier, H. Tuning the molecular giant titin through phosphorylation: Role in health and disease. Trends Cardiovas. Med. 2013, 23, 165–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Bharmal, S.J.; Esbona, K.; Greaser, M.L. Titin diversity-alternative splicing gone wild. J. Biomed. Biotechnol. 2010, 2010, 753675. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Kimura, A.; Kuroyanagi, H. Alternative splicing regulator RBM20 and cardiomyopathy. Front. Mol. Biosci. 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Gigli, M.; Begay, R.L.; Morea, G.; Graw, S.L.; Sinagra, G.; Taylor, M.R.; Granzier, H.; Mestroni, L. A review of the giant protein titin in clinical molecular diagnostics of cardiomyopathies. Front. Cardiovasc. Med. 2016, 3, 21–29. [Google Scholar] [CrossRef]
- Guo, W.; Schafer, S.; Greaser, M.L.; Radke, M.H.; Liss, M.; Govindarajan, T.; Maatz, H.; Schulz, H.; Li, S.; Parrish, A.M.; et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat. Med. 2012, 18, 766–773. [Google Scholar] [CrossRef]
- Zhu, C.; Chen, Z.; Guo, W. Pre-mRNA mis-splicing of sarcomeric genes in heart failure. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2056–2063. [Google Scholar] [CrossRef]
- Guo, W.; Sun, M. RBM20, a potential target for treatment of cardiomyopathy via titin isoform switching. Biophys. Rev. 2018, 10, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Guo, W.; Dewey, C.N.; Greaser, M.L. Rbm20 regulates titin alternative splicing as a splicing repressor. Nucleic. Acids. Res. 2013, 41, 2659–2672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Song, J.; Chen, L.; Zhu, C.; Cai, H.; Sun, M.; Sterm, A.; Mozdziak, P.; Ge, Y.; Mean, W.J.; et al. Characterization of TTN Novex Splicing Variants across Species and the Role of RBM20 in Novex-Specific Exon Splicing. Genes 2018, 9, 86. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Maimaiti, R.; Zhu, C.; Cai, H.; Stern, A.; Mozdziak, P.; Ge, Y.; Ford, S.P.; Nathanielsz, P.W.; Guo, W. Z-band and M-band titin splicing and regulation by RNA binding motif 20 in striated muscles. J. Cell. Biochem. 2018, 119, 9986–9996. [Google Scholar] [CrossRef] [PubMed]
- Maatz, H.; Jens, M.; Liss, M.; Schafer, S.; Heinig, M.; Kirchner, M.; Adami, E.; Rintisch, C.; Dauksite, V.; Radke, M.H.; et al. RNA-binding protein RBM20 represses splicing to orchestrate cardiac pre-mRNA processing. J. Clin. Investig. 2014, 124, 3419–3430. [Google Scholar] [CrossRef] [PubMed]
- Krüger, M.; Babicz, K.; von Frieling-Salewsky, M.; Linke, W.A. Insulin signaling regulates cardiac titin properties in heart development and diabetic cardiomyopathy. J. Mol. Cell. Cardiol. 2010, 48, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Krüger, M.; Sachse, C.; Zimmermann, W.H.; Eschenhagen, T.; Klede, S.; Linke, W.A. Thyroid hormone regulates developmental titin isoform transitions via the phosphatidylinositol-3-kinase/AKT pathway. Circ. Res. 2008, 102, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Yin, Z.; Ren, J.; McCormick, R.J.; Ford, S.P.; Guo, W. RBM20 is an essential factor for thyroid hormone-regulated titin isoform transition. J. Mol. Cell. Biol. 2015, 7, 88–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Yin, Z.; Tan, B.; Guo, W. Insulin regulates titin pre-mRNA splicing through the PI3K-Akt-mTOR kinase axis in a RBM20-dependent manner. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2363–2371. [Google Scholar] [CrossRef]
- Warren, C.M.; Krzesinski, P.R.; Greaser, M.L. Vertical agarose gel electrophoresis and electroblotting of high-molecular-weight proteins. Electrophoresis 2003, 24, 1695–1702. [Google Scholar] [CrossRef]
- Warren, C.M.; Krzesinski, P.R.; Campbell, K.S.; Moss, R.L.; Greaser, M.L. Titin isoform changes in rat myocardium during development. Mech. Dev. 2004, 121, 1301–1312. [Google Scholar] [CrossRef] [PubMed]
- Greaser, M.L.; Warren, C.M.; Esbona, K.; Guo, W.; Duan, Y.; Parrish, A.M.; Krzesinski, P.R.; Norman, H.S.; Dunning, S.; Fitzsimons, D.P.; et al. Mutation that dramatically alters rat titin isoform expression and cardiomyocyte passive tension. J. Mol. Cell. Cardiol. 2008, 44, 983–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rexiati, M.; Sun, M.; Guo, W. Muscle-Specific Mis-Splicing and Heart Disease Exemplified by RBM20. Genes 2018, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Laviola, L.; Belsanti, G.; Davalli, A.M.; Napoli, R.; Perrini, S.; Weir, G.C.; Giorgino, R.; Giorgino, F. Effects of streptozocin diabetes and diabetes treatment by islet transplantation on in vivo insulin signaling in rat heart. Diabetes 2001, 50, 2709–2720. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.G.; Yu, Y.; Zhang, Z.H.; Weiss, R.M.; Felder, R.B. Mitogen-activated protein kinases mediate upregulation of hypothalamic angiotensin II type 1 receptors in heart failure rats. Hypertension 2008, 52, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Mariash, C.N.; Bhargava, M.; Wattenberg, E.V.; Ingbar, D.H. T3 increases Na-K-ATPase activity via a MAPK/ERK1/2-dependent pathway in rat adult alveolar epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L749–L754. [Google Scholar] [CrossRef] [Green Version]
- Lara-Pezzi, E.; Gómez-Salinero, J.; Gatto, A.; García-Pavía, P. The alternative heart: Impact of alternative splicing in heart disease. J. Cardiovasc. Trans. Res. 2013, 6, 945–955. [Google Scholar] [CrossRef]
- Noyes, A.M.; Zhou, A.; Gao, G.; Gu, L.; Day, S.; Wasserstrom, J.A.; Dudley, S.C. Abnormal sodium channel mRNA splicing in hypertrophic cardiomyopathy. Int. J. Cardiol. 2017, 249, 282–286. [Google Scholar] [CrossRef]
- Van den Hoogenhof, M.M.; Pinto, Y.M.; Creemers, E.E. RNA splicing: Regulation and dysregulation in the heart. Circ. Res. 2016, 118, 454–468. [Google Scholar] [CrossRef]
- Yin, Z.; Ren, J.; Guo, W. Sarcomeric protein isoform transitions incardiac muscle: A journey to heart failure. Biochim. Biophys. Acta Mol. Basis Dis. 2015, 1852, 47–52. [Google Scholar] [CrossRef]
- Opitz, C.A.; Leake, M.C.; Makarenko, I.; Benes, V.; Linke, W.A. Developmentally regulated switching of titin size alters myofibrillar stiffness in the perinatal heart. Circ. Res. 2004, 94, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Chauveau, C.; Rowell, J.; Ferreiro, A. A rising titan: TTN review and mutation update. Hum. Mutat. 2014, 35, 1046–1059. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Pleitner, J.M.; Saupe, K.W.; Greaser, M.L. Pathophysiological defects and transcriptional profiling in the RBM20-/-rat model. PLoS ONE 2013, 8, e84281. [Google Scholar] [CrossRef] [PubMed]
- Seger, R.; Krebs, E.G. The MAPK signaling cascade. FASEB J. 1995, 9, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Riehle, C.; Abel, E.D. Insulin signaling and heart failure. Circ. Res. 2016, 118, 1151–1169. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Ott, U.M.; Ascheim, D.D. Thyroid hormone and heart failure. Curr. Heart Fail. Rep. 2006, 3, 114–119. [Google Scholar] [CrossRef]
- Dai, D.F.; Johnson, S.C.; Villarin, J.J.; Chin, M.T.; Nieves-Cintrón, M.; Chen, T.; Marcinek, D.J.; Dornll, G.W.; Kang, J.; Prolla, T.A.; et al. Mitochondrial oxidative stress mediates angiotensin II–induced cardiac hypertrophy and Gαq overexpression–induced heart failure. Circ. Res. 2011, 108, 837–846. [Google Scholar] [CrossRef]
- Naro, C.; Sette, C. Phosphorylation-mediated regulation of alternative splicing in cancer. Int. J. Cell. Biol. 2013, 2013, 151839. [Google Scholar] [CrossRef]
- Azzout-Marniche, D.; Bécard, D.; Guichard, C.; Foretz, M.; Ferré, P.; Foufelle, F. Insulin effects on sterol regulatory-element-binding protein-1c (SREBP-1c) transcriptional activity in rat hepatocytes. Biochem. J. 2000, 350, 389–393. [Google Scholar] [CrossRef]
- Liang, F.; Webb, P.; Marimuthu, A.; Zhang, S.; Gardner, D.G. Triiodothyronine increases brain natriuretic peptide (BNP) gene transcription and amplifies endothelin-dependent BNP gene transcription and hypertrophy in neonatal rat ventricular myocytes. J. Biol. Chem. 2003, 278, 15073–15083. [Google Scholar] [CrossRef]
- Han, Y.; Runge, M.S.; Brasier, A.R. Angiotensin II induces interleukin-6 transcription in vascular smooth muscle cells through pleiotropic activation of nuclear factor-κB transcription factors. Circ. Res. 1999, 84, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Duan, R.; Xie, W.; Burghardt, R.C.; Safe, S. Estrogen receptor-mediated activation of the serum response element in MCF-7 cells through MAPK-dependent phosphorylation of Elk-1. J. Biol. Chem. 2001, 276, 11590–11598. [Google Scholar] [CrossRef] [PubMed]
- Hodge, C.; Liao, J.; Stofega, M.; Guan, K.; Carter-Su, C.; Schwartz, J. Growth Hormone Stimulates Phosphorylation and Activation of Elk-1 and Expression of c-fos, egr-1, andjunB through Activation of Extracellular Signal-regulated Kinases 1 and 2. J. Biol. Chem. 1998, 273, 31327–31336. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, B.A.; Harvey, B.J.; Healy, V. 17β-Estradiol rapidly stimulates c-fos expression via the MAPK pathway in T84 cells. Mol. Cell. Endocrinol. 2005, 229, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Fornes, O.; Stigliani, A.; Gheorghe, M.; Castro-Mondragon, J.A.; van der Lee, R.; Bessy, A.; Jeanne, C.; Shubhada, R.K.; Ge, T.; et al. JASPAR 2018: Update of the open-access database of transcription factor binding profiles and its web framework. Nucl. Acids Res. 2017, 46, D260–D266. [Google Scholar] [CrossRef] [PubMed]
- Farré, D.; Roset, R.; Huerta, M.; Adsuara, J.E.; Roselló, L.; Albà, M.M.; Messeguer, X. Identification of patterns in biological sequences at the ALGGEN server: PROMO and MALGEN. Nucl. Acids Res. 2003, 31, 3651–3653. [Google Scholar] [CrossRef] [Green Version]
- Messeguer, X.; Escudero, R.; Farré, D.; Núñez, O.; Martínez, J.; Albà, M.M. PROMO: Detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 2002, 18, 333–334. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, H.; Zhu, C.; Chen, Z.; Maimaiti, R.; Sun, M.; McCormick, R.J.; Lan, X.; Chen, H.; Guo, W. Angiotensin II Influences Pre-mRNA Splicing Regulation by Enhancing RBM20 Transcription Through Activation of the MAPK/ELK1 Signaling Pathway. Int. J. Mol. Sci. 2019, 20, 5059. https://doi.org/10.3390/ijms20205059
Cai H, Zhu C, Chen Z, Maimaiti R, Sun M, McCormick RJ, Lan X, Chen H, Guo W. Angiotensin II Influences Pre-mRNA Splicing Regulation by Enhancing RBM20 Transcription Through Activation of the MAPK/ELK1 Signaling Pathway. International Journal of Molecular Sciences. 2019; 20(20):5059. https://doi.org/10.3390/ijms20205059
Chicago/Turabian StyleCai, Hanfang, Chaoqun Zhu, Zhilong Chen, Rexiati Maimaiti, Mingming Sun, Richard J. McCormick, Xianyong Lan, Hong Chen, and Wei Guo. 2019. "Angiotensin II Influences Pre-mRNA Splicing Regulation by Enhancing RBM20 Transcription Through Activation of the MAPK/ELK1 Signaling Pathway" International Journal of Molecular Sciences 20, no. 20: 5059. https://doi.org/10.3390/ijms20205059