Systematic Approach to Developing Splice Modulating Antisense Oligonucleotides

 , ,
, ,  ,
,
Abstract
:1. Introduction
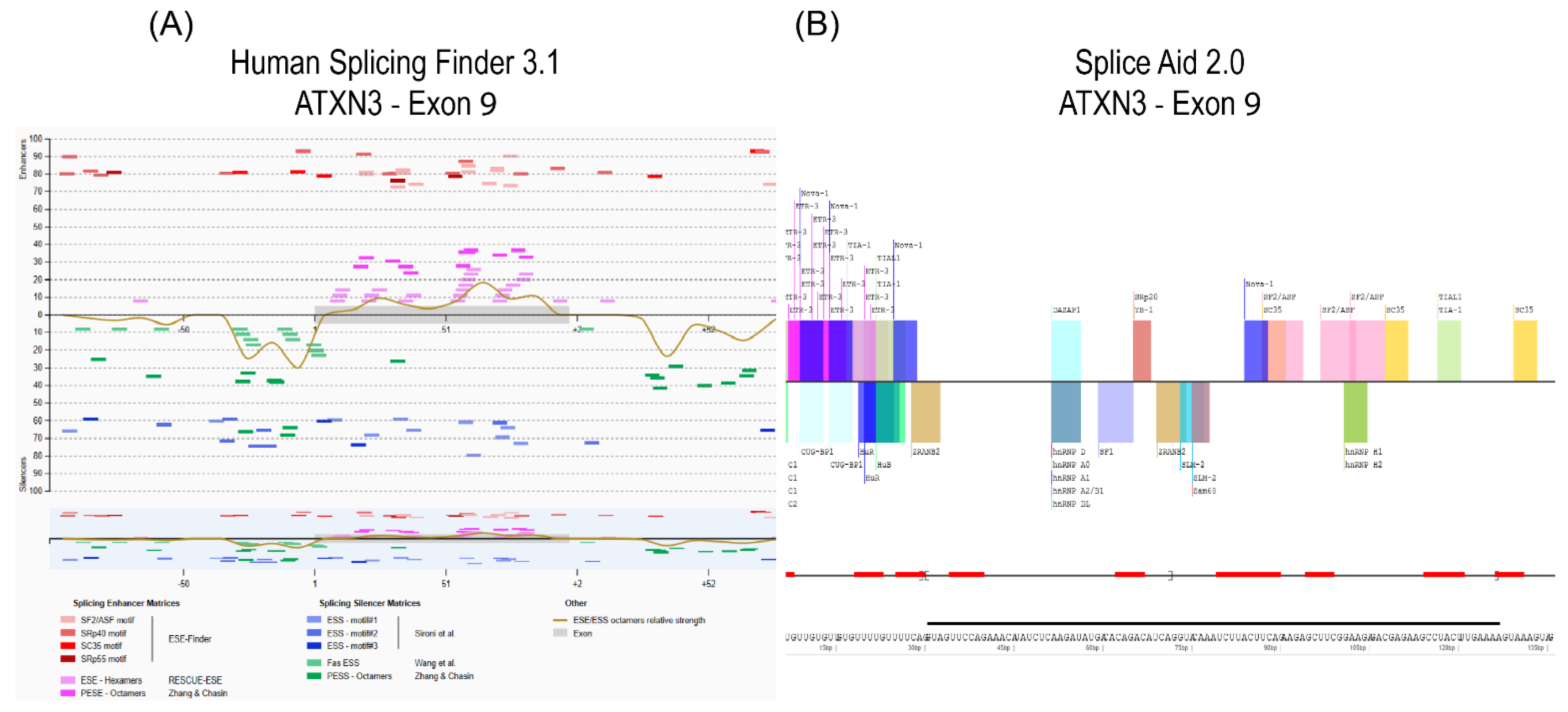
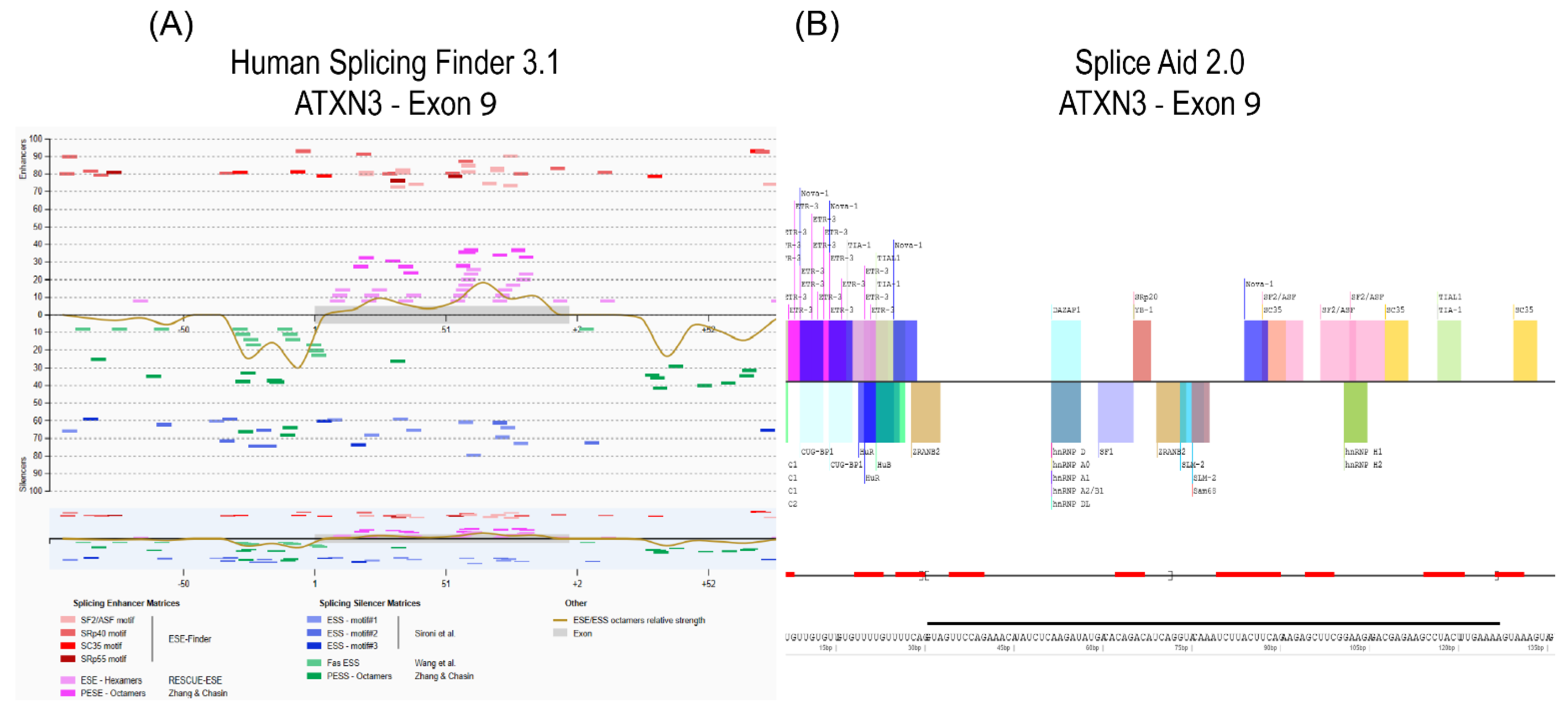
- The pre-mRNA sequence is interrogated by one or more in silico prediction programs to identify potential splice enhancer or silencer motifs.
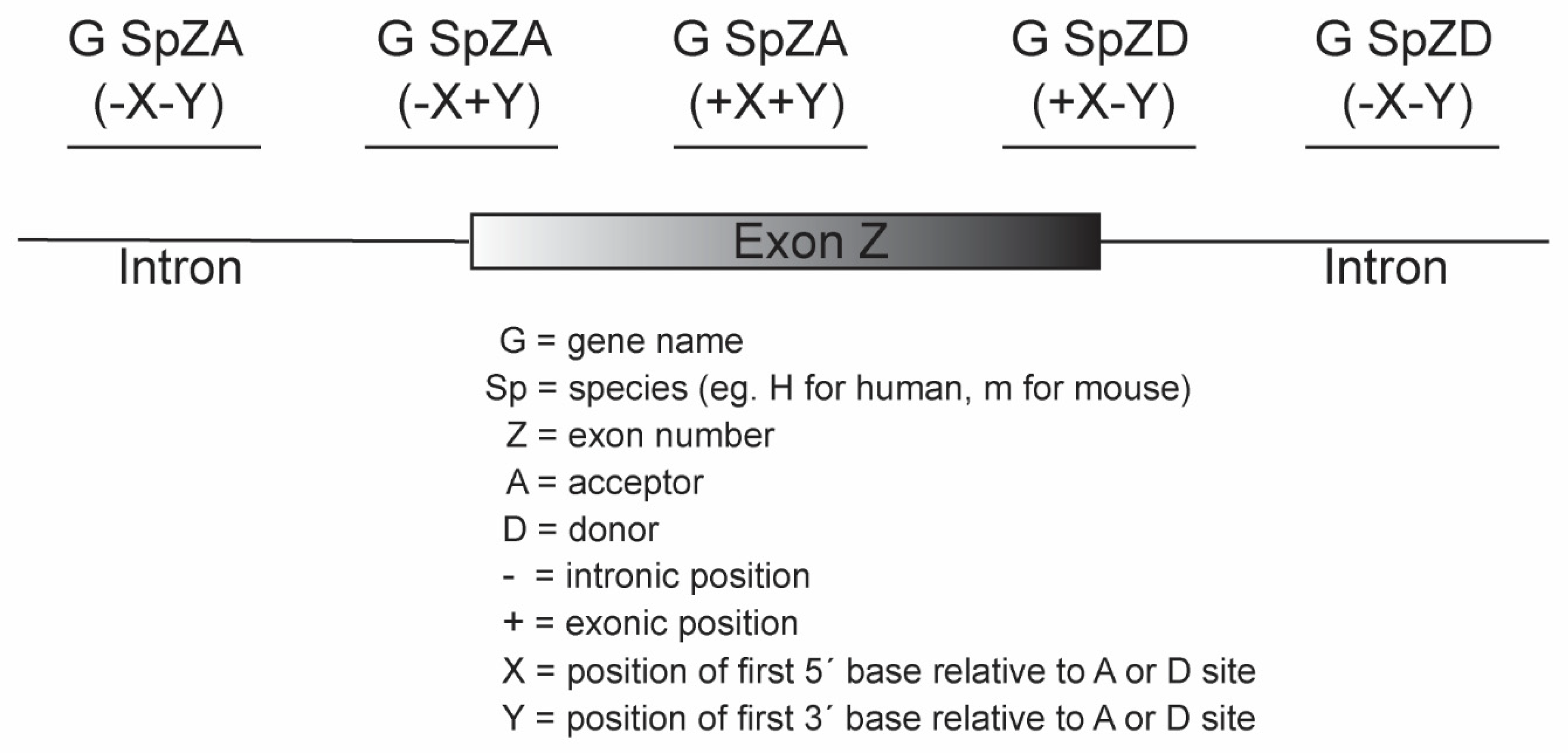
- Antisense oligonucleotides, typically 20 to 25 mers, are designed to anneal to the target motifs and synthesised as 2′-O-methyl (2-OMe) modified bases on a phosphorothioate (PS) backbone.
- The test compounds are complexed with cationic liposome preparations and transfected into cells.
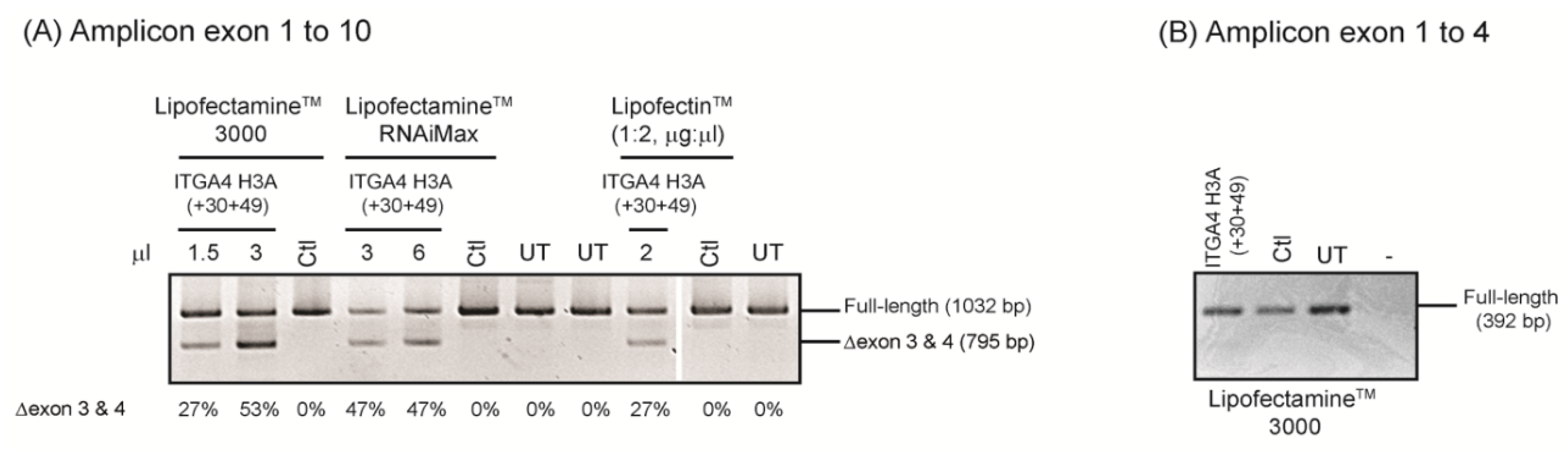
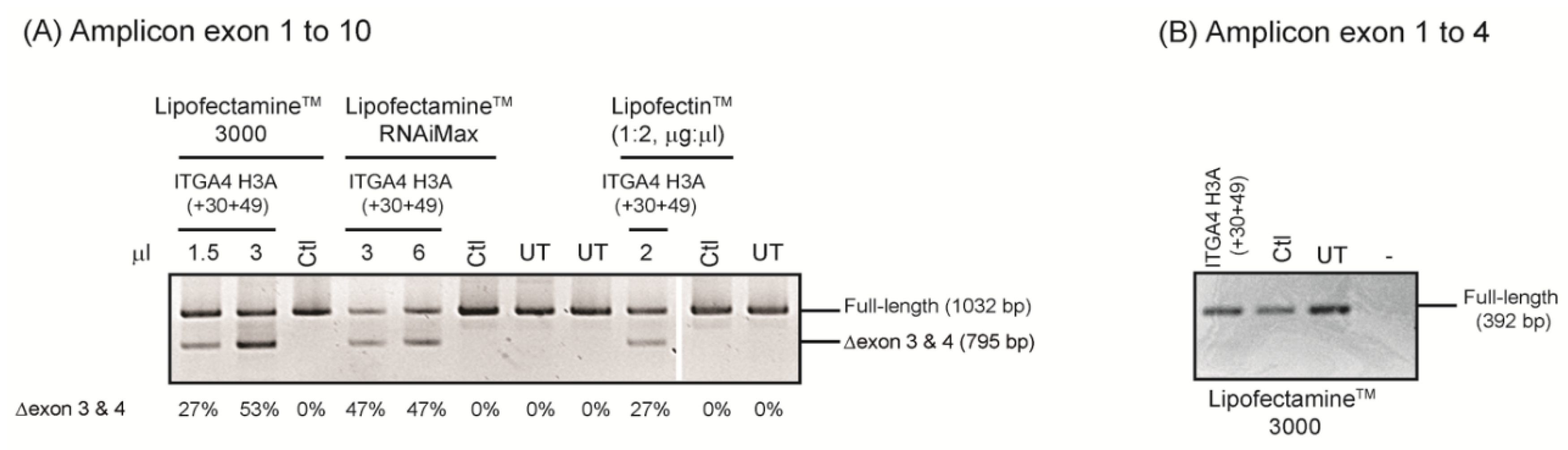
- After incubation, total RNA is extracted and the target transcript is amplified using RT-PCR to assess differences in pre-mRNA processing, with and without AO treatment.
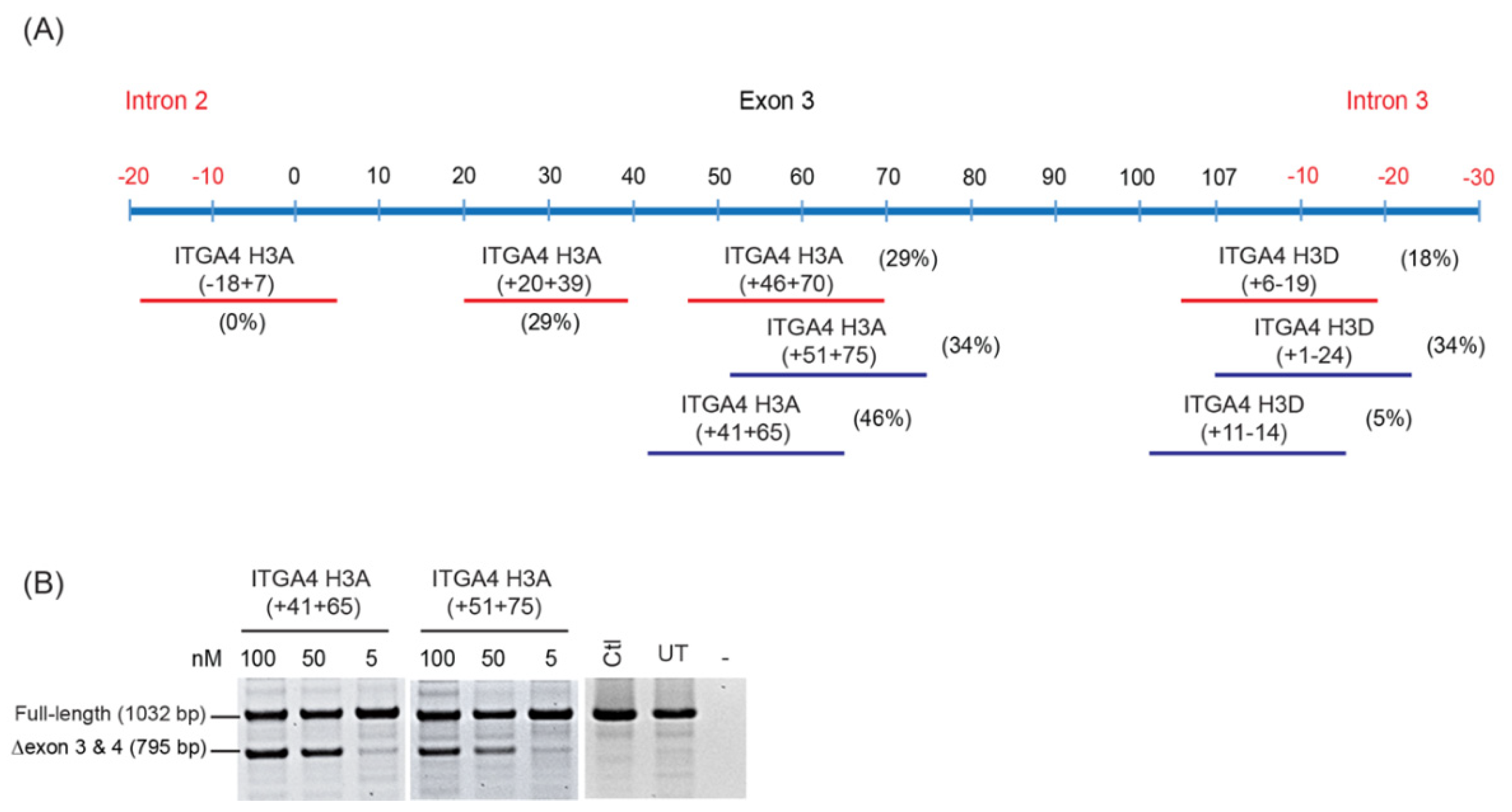
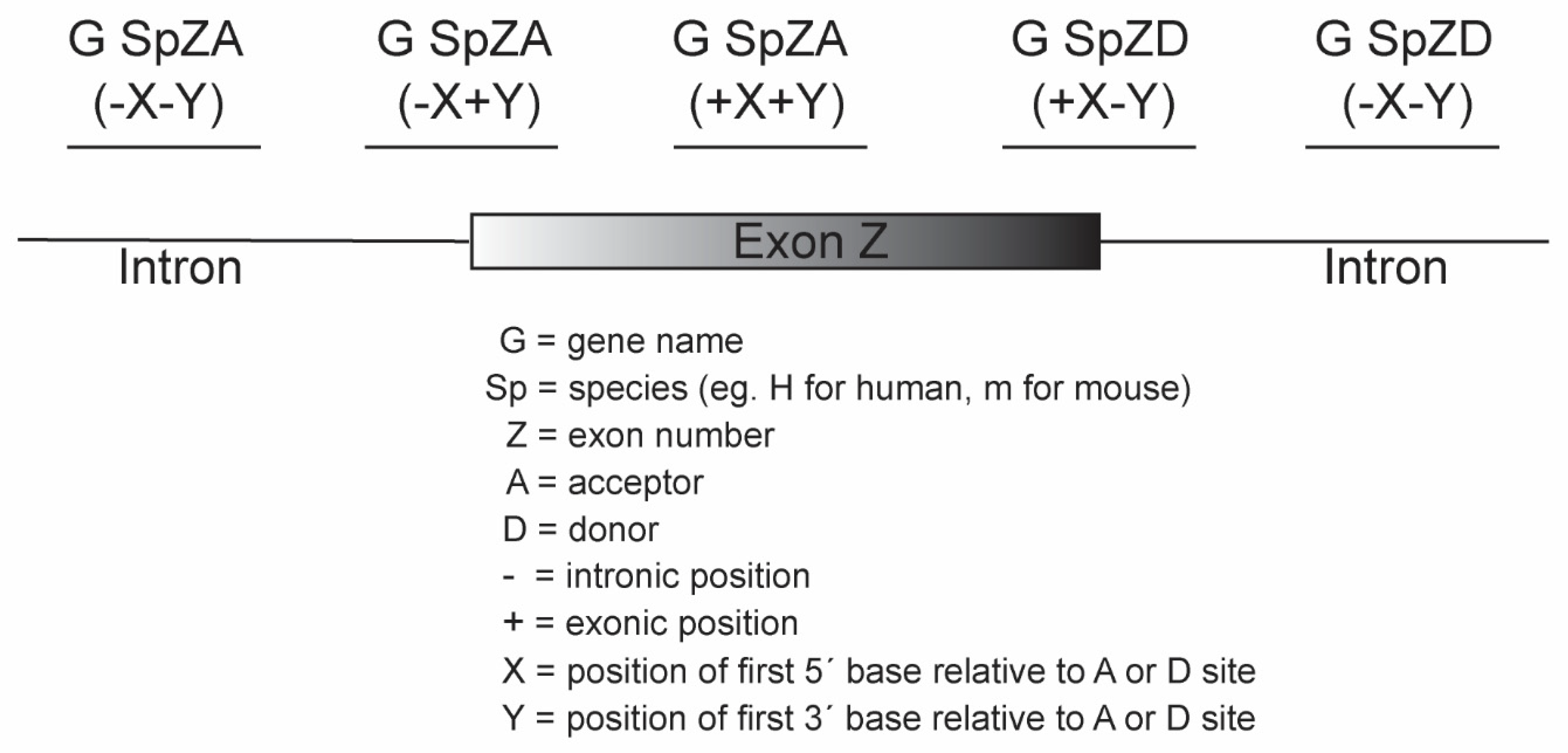
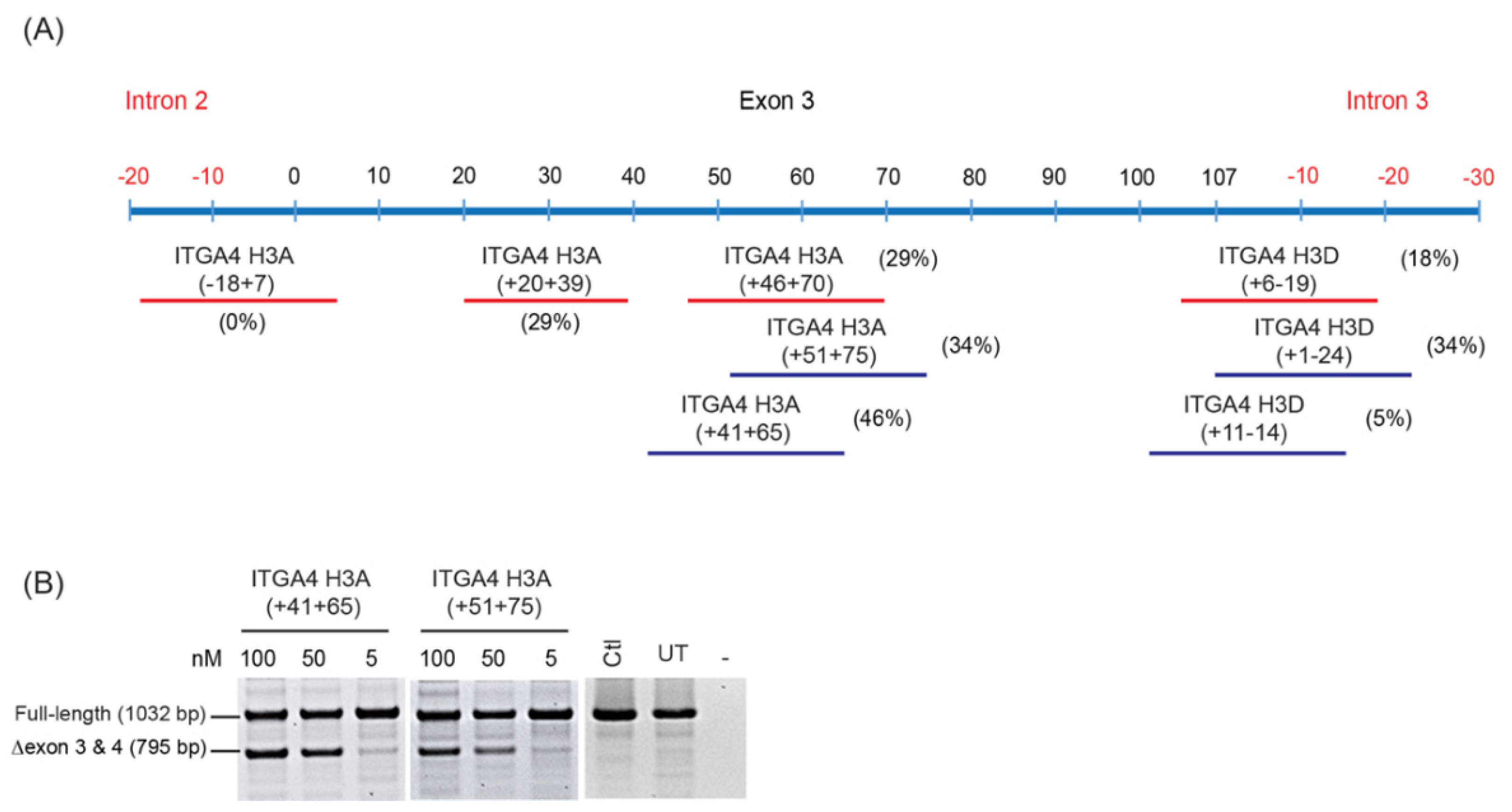
- Oligomers shown to induce the desired changes in pre-mRNA processing are further refined by micro-walking around the annealing site and/or altering AO length.
- Transfection studies over a range of concentrations are performed to identify compound(s) that modify splicing in a dose-dependent manner, and at the lowest concentration.
2. Results
Guidelines for Developing Splice Switching AOs
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Antisense Oligonucleotides (AOs)
4.3. 2′-O-Methyl Phosphorothioate AO Transfection
4.4. RT-PCR
Author Contributions
Funding
Conflicts of Interest
References
- Mendell, J.R.; Goemans, N.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Shao, J.; Kaye, E.M.; Mercuri, E.; Eteplirsen Study Group; Telethon Foundation DMD Italian Networkc; et al. Longitudinal effect of eteplirsen versus historical control on ambulation in D uchenne muscular dystrophy. Ann. Neurol. 2016, 79, 257–271. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Sahenk, Z.; Rodino-Klapac, L.R. Clinical trials of exon skipping in duchenne muscular dystrophy. Expert Opin. Orphan Drugs 2017, 5, 683–690. [Google Scholar] [CrossRef]
- Finkel, R.S.; Chiriboga, C.A.; Vajsar, J.; Day, J.W.; Montes, J.; De Vivo, D.C.; Yamashita, M.; Rigo, F.; Hung, G.; Schneider, E. Treatment of infantile-onset spinal muscular atrophy with nusinersen: A phase 2, open-label, dose-escalation study. Lancet 2017, 388, 3017–3026. [Google Scholar] [CrossRef]
- Rinaldi, C.; Wood, M.J. Antisense oligonucleotides: The next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 2018, 14, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Scoles, D.R.; Meera, P.; Schneider, M.D.; Paul, S.; Dansithong, W.; Figueroa, K.P.; Hung, G.; Rigo, F.; Bennett, C.F.; Otis, T.S. Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature 2017, 544, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.; Hansen, J.B.; Lai, J.; Wu, S.; Voskresenskiy, A.; H⊘g, A.; Worm, J.; Hedtjärn, M.; Souleimanian, N.; Miller, P. Efficient gene silencing by delivery of locked nucleic acid antisense oligonucleotides, unassisted by transfection reagents. Nucleic Acids Res. 2009, 38, e3. [Google Scholar] [CrossRef] [PubMed]
- Julien, T.; Frankel, B.; Longo, S.; Kyle, M.; Gibson, S.; Shillitoe, E.; Ryken, T. Antisense-mediated inhibition of the bcl-2 gene induces apoptosis in human malignant glioma. Surg. Neurol. 2000, 53, 360–369. [Google Scholar] [CrossRef]
- Mendell, J.R.; Rodino-Klapac, L.R.; Sahenk, Z.; Roush, K.; Bird, L.; Lowes, L.P.; Alfano, L.; Gomez, A.M.; Lewis, S.; Kota, J. Eteplirsen for the treatment of duchenne muscular dystrophy. Ann. Neurol. 2013, 74, 637–647. [Google Scholar] [CrossRef]
- Havens, M.A.; Hastings, M.L. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016, 44, 6549–6563. [Google Scholar] [CrossRef]
- Flynn, L.L.; Mitrpant, C.; Pitout, I.L.; Fletcher, S.; Wilton, S.D. Antisense oligonucleotide-mediated terminal intron retention of the smn2 transcript. Mol. Ther. Nucleic Acids 2018, 11, 91–102. [Google Scholar] [CrossRef]
- Piva, F.; Giulietti, M.; Burini, A.B.; Principato, G. Spliceaid 2: A database of human splicing factors expression data and rna target motifs. Hum. Mutat. 2012, 33, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Desmet, F.-O.; Hamroun, D.; Lalande, M.; Collod-Béroud, G.; Claustres, M.; Béroud, C. Human splicing finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-Y.; Chien, C.-H.; Jen, K.-H.; Huang, H.-D. Regrna: An integrated web server for identifying regulatory rna motifs and elements. Nucleic Acids Res. 2006, 34, W429–W434. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A. Overview on aon design. In Exon Skipping; Springer: Heidelberg, Germany, 2012; pp. 117–129. [Google Scholar]
- Pramono, Z.A.D.; Wee, K.B.; Wang, J.L.; Chen, Y.J.; Xiong, Q.B.; Lai, P.S.; Yee, W.C. A prospective study in the rational design of efficient antisense oligonucleotides for exon skipping in the dmd gene. Hum. Gene Ther. 2012, 23, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Van Vliet, L.; Hirschi, M.; Janson, A.A.; Heemskerk, H.; De Winter, C.L.; De Kimpe, S.; Van Deutekom, J.C.; Ac’t Hoen, P.; van Ommen, G.-J.B. Guidelines for antisense oligonucleotide design and insight into splice-modulating mechanisms. Mol. Ther. 2009, 17, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Gebski, B.L.; Mann, C.J.; Fletcher, S.; Wilton, S.D. Morpholino antisense oligonucleotide induced dystrophin exon 23 skipping in mdx mouse muscle. Hum. Mol. Genet. 2003, 12, 1801–1811. [Google Scholar] [CrossRef]
- Mitrpant, C.; Adams, A.M.; Meloni, P.L.; Muntoni, F.; Fletcher, S.; Wilton, S.D. Rational design of antisense oligomers to induce dystrophin exon skipping. Mol. Ther. 2009, 17, 1418–1426. [Google Scholar] [CrossRef]
- Greer, K.L.; Lochmüller, H.; Flanigan, K.; Fletcher, S.; Wilton, S.D. Targeted exon skipping to correct exon duplications in the dystrophin gene. Mol. Ther. Nucleic Acids 2014, 3, e155. [Google Scholar] [CrossRef]
- Summerton, J.E. Morpholino, sirna, and s-DNA compared: Impact of structure and mechanism of action on off-target effects and sequence specificity. Curr. Top. Med. Chem. 2007, 7, 651–660. [Google Scholar] [CrossRef]
- Zuhorn, I.S.; Engberts, J.B.; Hoekstra, D. Gene delivery by cationic lipid vectors: Overcoming cellular barriers. Eur. Biophys. J. 2007, 36, 349–362. [Google Scholar] [CrossRef]
- Aung-Htut, M.T.; McIntosh, C.S.; West, K.A.; Fletcher, S.; Wilton, S.D. In vitro validation of phosphorodiamidate morpholino oligomers. Molecules 2019, 24, 2922. [Google Scholar] [CrossRef] [PubMed]
- Mann, C.J.; Honeyman, K.; McClorey, G.; Fletcher, S.; Wilton, S.D. Improved antisense oligonucleotide induced exon skipping in the mdx mouse model of muscular dystrophy. J. Gene Med. Cross-Discip. J. Res. Sci. Gene Transf. Clin. Appl. 2002, 4, 644–654. [Google Scholar]
- Weizmann-Insitite. Gene Cards Human Gene Database. Available online: https://www.genecards.org/ (accessed on 14 March 2019).
- Murry, C.E.; Kay, M.A.; Bartosek, T.; Hauschka, S.D.; Schwartz, S.M. Muscle differentiation during repair of myocardial necrosis in rats via gene transfer with myod. J. Clin. Investig. 1996, 98, 2209–2217. [Google Scholar] [CrossRef] [PubMed]
- Lattanzi, L.; Salvatori, G.; Coletta, M.; Sonnino, C.; De Angelis, M.C.; Gioglio, L.; Murry, C.E.; Kelly, R.; Ferrari, G.; Molinaro, M. High efficiency myogenic conversion of human fibroblasts by adenoviral vector-mediated myod gene transfer. An alternative strategy for ex vivo gene therapy of primary myopathies. J. Clin. Investig. 1998, 101, 2119–2128. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Costa, M.; Mermelstein, C.; Chagas, C.; Holtzer, S.; Holtzer, H. Myod converts primary dermal fibroblasts, chondroblasts, smooth muscle, and retinal pigmented epithelial cells into striated mononucleated myoblasts and multinucleated myotubes. Proc. Natl. Acad. Sci. USA 1990, 87, 7988–7992. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S.; Adkin, C.F.; Meloni, P.; Wong, B.; Muntoni, F.; Kole, R.; Fragall, C.; Greer, K.; Johnsen, R.; Wilton, S.D. Targeted exon skipping to address “leaky” mutations in the dystrophin gene. Mol. Ther. Nucleic Acids 2012, 1, e48. [Google Scholar] [CrossRef] [PubMed]
- Adams, A.M.; Harding, P.L.; Iversen, P.L.; Coleman, C.; Fletcher, S.; Wilton, S.D. Antisense oligonucleotide induced exon skipping and the dystrophin gene transcript: Cocktails and chemistries. BMC Mol. Biol. 2007, 8, 57. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S.; Ly, T.; Duff, R.; Howell, J.M.; Wilton, S. Cryptic splicing involving the splice site mutation in the canine model of duchenne muscular dystrophy. Neuromuscul. Disord. 2001, 11, 239–243. [Google Scholar] [CrossRef]
- Harding, P.; Fall, A.; Honeyman, K.; Fletcher, S.; Wilton, S. The influence of antisense oligonucleotide length on dystrophin exon skipping. Mol. Ther. 2007, 15, 157–166. [Google Scholar] [CrossRef]
- Jing, N.; Li, Y.; Xiong, W.; Sha, W.; Jing, L.; Tweardy, D.J. G-quartet oligonucleotides: A new class of signal transducer and activator of transcription 3 inhibitors that suppresses growth of prostate and breast tumors through induction of apoptosis. Cancer Res. 2004, 64, 6603–6609. [Google Scholar] [CrossRef]
- Williamson, J.R. G-quartet structures in telomeric DNA. Annu. Rev. Biophys. Biomol. Struct. 1994, 23, 703–730. [Google Scholar] [CrossRef] [PubMed]
- Flanigan, K.M.; Voit, T.; Rosales, X.Q.; Servais, L.; Kraus, J.E.; Wardell, C.; Morgan, A.; Dorricott, S.; Nakielny, J.; Quarcoo, N. Pharmacokinetics and safety of single doses of drisapersen in non-ambulant subjects with duchenne muscular dystrophy: Results of a double-blind randomized clinical trial. Neuromuscul. Disord. 2014, 24, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Dias, N.; Stein, C. Antisense oligonucleotides: Basic concepts and mechanisms. Mol. Cancer Ther. 2002, 1, 347–355. [Google Scholar] [PubMed]
- Wong, E.; Goldberg, T. Mipomersen (kynamro): A novel antisense oligonucleotide inhibitor for the management of homozygous familial hypercholesterolemia. Pharm. Ther. 2014, 39, 119–122. [Google Scholar]
- Chi, X.; Gatti, P.; Papoian, T. Safety of antisense oligonucleotide and sirna-based therapeutics. Drug Discov. Today 2017, 22, 823–833. [Google Scholar] [CrossRef]
- Summerton, J.; Weller, D. Morpholino antisense oligomers: Design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 1997, 7, 187–195. [Google Scholar] [CrossRef]
- Wilton, S.D.; Fall, A.M.; Harding, P.L.; McClorey, G.; Coleman, C.; Fletcher, S. Antisense oligonucleotide-induced exon skipping across the human dystrophin gene transcript. Mol. Ther. 2007, 15, 1288–1296. [Google Scholar] [CrossRef]
- Bjourson, A.J.; Cooper, J.E. Band-stab pcr: A simple technique for the purification of individual pcr products. Nucleic Acids Res. 1992, 20, 4675. [Google Scholar] [CrossRef]
- Aung-Htut, M.T.; Comerford, I.; Johnsen, R.; Foyle, K.; Fletcher, S.; Wilton, S.D. Reduction of integrin alpha 4 activity through splice modulating antisense oligonucleotides. Sci. Rep. 2019, 9, 12994. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Lines | Transfection Reagents |
|---|---|
| Dermal fibroblasts | Lipofectin™, Lipofectamine™ 3000 |
| Myoblasts and myotubes | Lipofectamine™ 2000 |
| Lymphoblasts and lymphocytes | Nucleofection P3 Primary Cell Kit |
| Huh7 | Lipofectamine™ 3000, Lipofectamine™ RNAiMax |
| HEK293 | Lipofectamine™ 3000 |
| H2k mdx | Lipofectin™ |
| MO3.13 | Lipofectamine™ 3000 |
| iPSCs and neural stem cells | Lipofectamine™ Stem |
| Name | Sequence (5′ – 3′) |
|---|---|
| ITGA4 H3A(+30+49) | UCUCUCUCUUCCAAACAAGU |
| ITGA4 H3A(-18+7) | GGGCUACCUAUAGCAUGUGAAAAUA |
| ITGA4 H3A(+20+39) | CCAAACAAGUCUUUCCACAA |
| ITGA4 H3A(+46+70) | GUGACCCCCAACCACUGAUUGUCUC |
| ITGA4 H3A(+41+65) | CCCCAACCACUGAUUGUCUCUCUCU |
| ITGA4 H3A(+51+75) | AAAGUGUGACCCCCAACCACUGAUU |
| ITGA4 H3D(+6-19) | GACCAGUUCCAAUACCUACCACGAU |
| ITGA4 H3D(+11-14) | GUUCCAAUACCUACCACGAUGGAUC |
| ITGA4 H3D(+1-24) | CUGUGGACCAGUUCCAAUACCUACC |
| Ctl | GGAUGUCCUGAGUCUAGACCCUCCG |
| Name | Sequence (5′ – 3′) | Amplification Performed after Treatment with the Following AOs |
|---|---|---|
| ITGA4 ex1_F | gagagcgcgctgctttaccagg | All AOs |
| ITGA4 ex10_R | gccatcattgtcaatgtcgcca | |
| ITGA4 ex1_F | gagagcgcgctgctttaccagg | ITGA4 H3A(+30+49) |
| ITGA4 ex4_R | ggcactccatagcaaccacc |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aung-Htut, M.T.; McIntosh, C.S.; Ham, K.A.; Pitout, I.L.; Flynn, L.L.; Greer, K.; Fletcher, S.; Wilton, S.D. Systematic Approach to Developing Splice Modulating Antisense Oligonucleotides. Int. J. Mol. Sci. 2019, 20, 5030. https://doi.org/10.3390/ijms20205030
Aung-Htut MT, McIntosh CS, Ham KA, Pitout IL, Flynn LL, Greer K, Fletcher S, Wilton SD. Systematic Approach to Developing Splice Modulating Antisense Oligonucleotides. International Journal of Molecular Sciences. 2019; 20(20):5030. https://doi.org/10.3390/ijms20205030
Chicago/Turabian StyleAung-Htut, May T., Craig S. McIntosh, Kristin A. Ham, Ianthe L. Pitout, Loren L. Flynn, Kane Greer, Sue Fletcher, and Steve D. Wilton. 2019. "Systematic Approach to Developing Splice Modulating Antisense Oligonucleotides" International Journal of Molecular Sciences 20, no. 20: 5030. https://doi.org/10.3390/ijms20205030