Synthesis and Magnetic Properties of the Novel Dithiadiazolyl Radical, p-NCC6F4C6F4CNSSN

Abstract
:Introduction

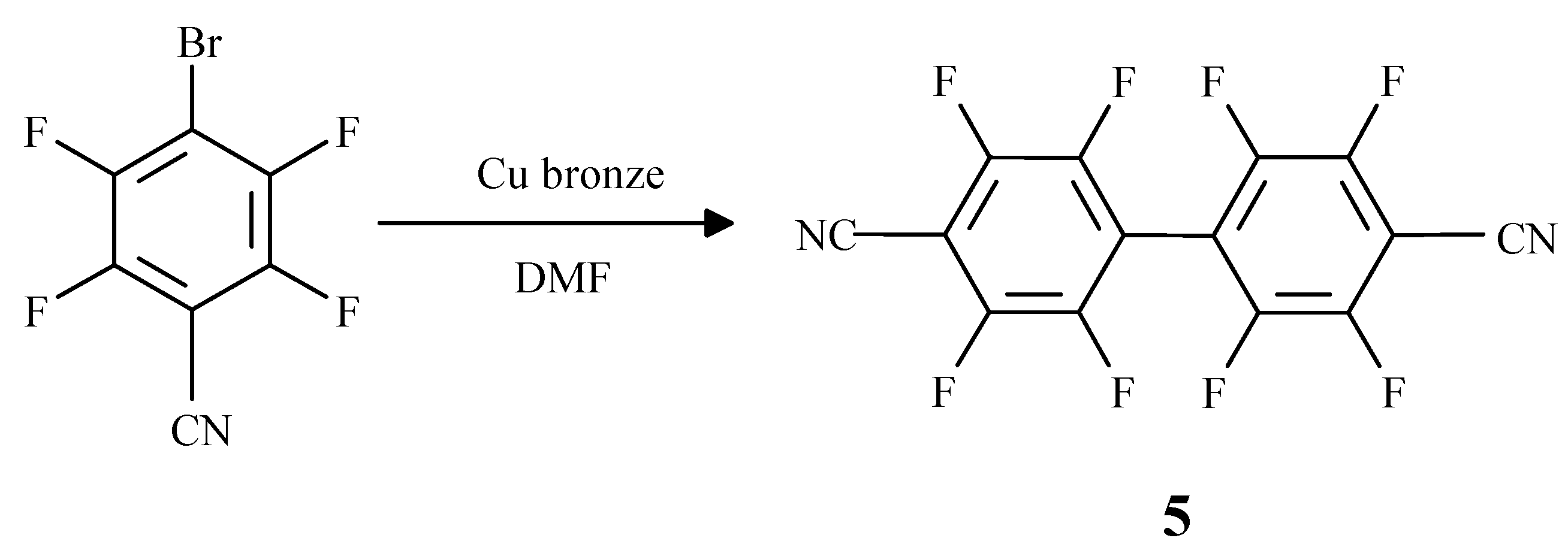
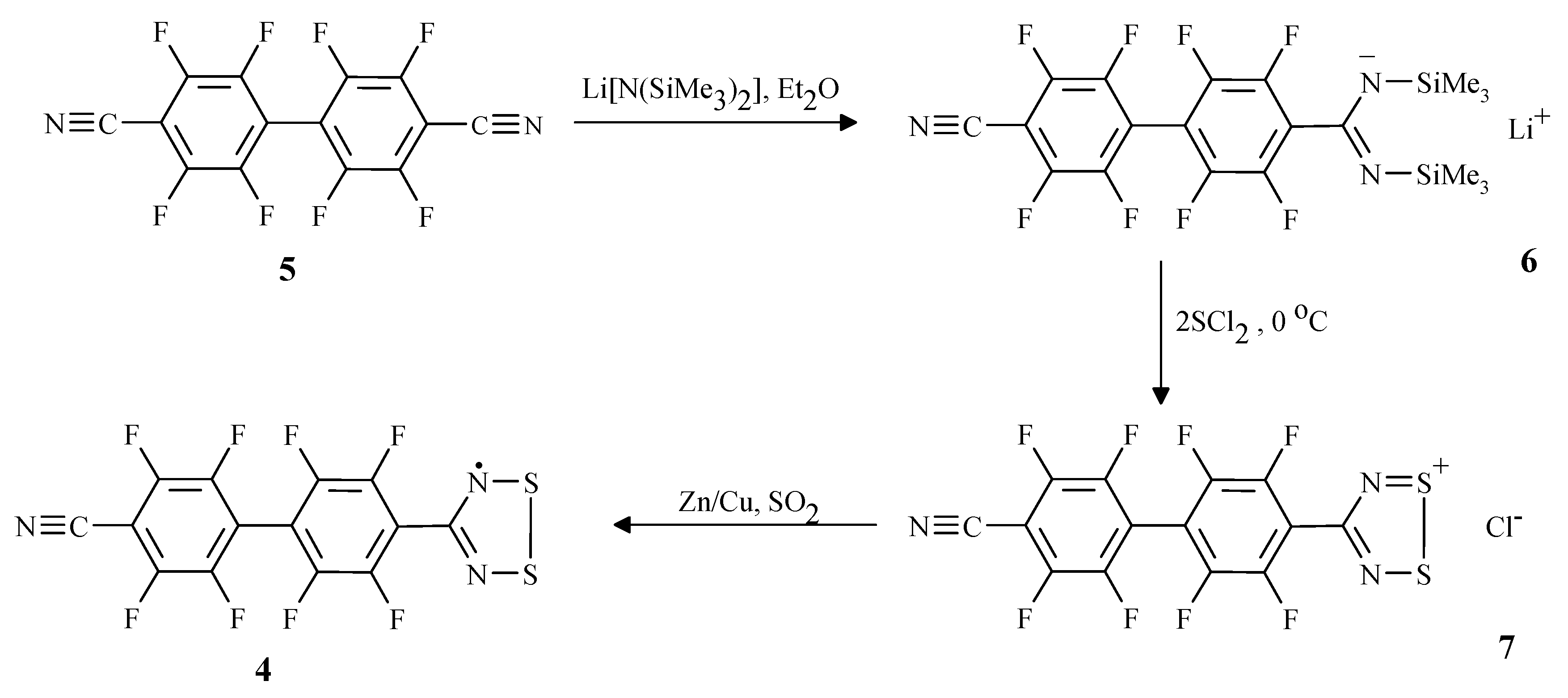
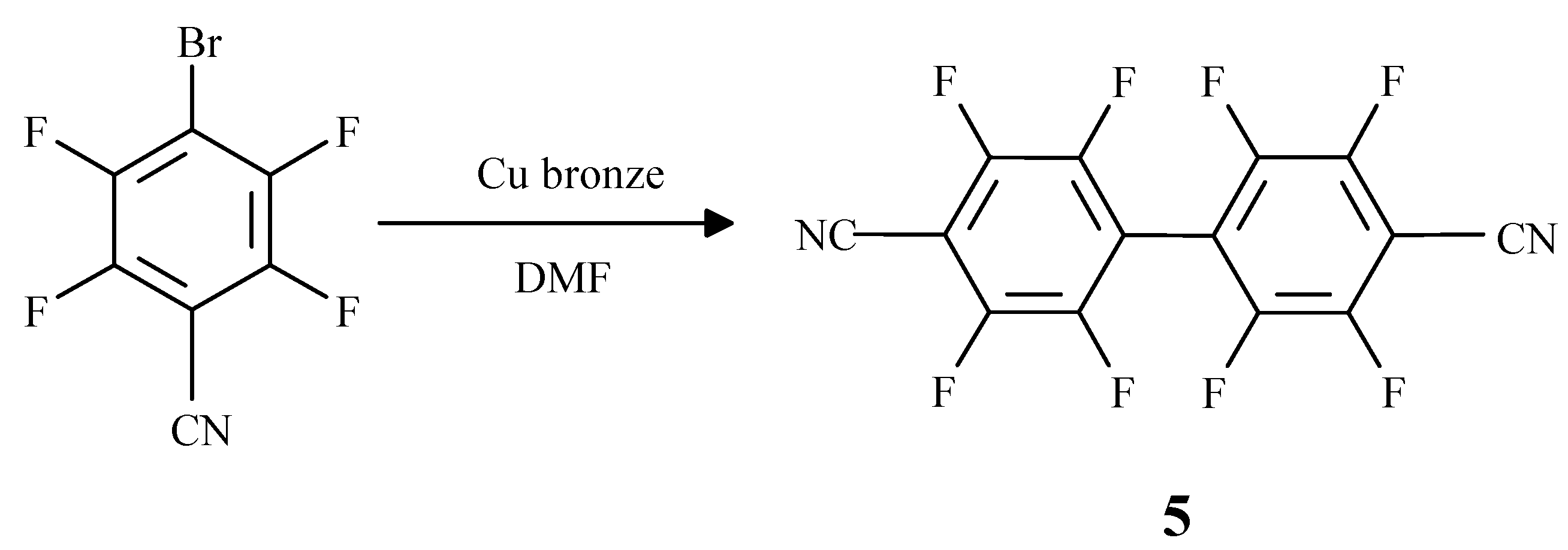
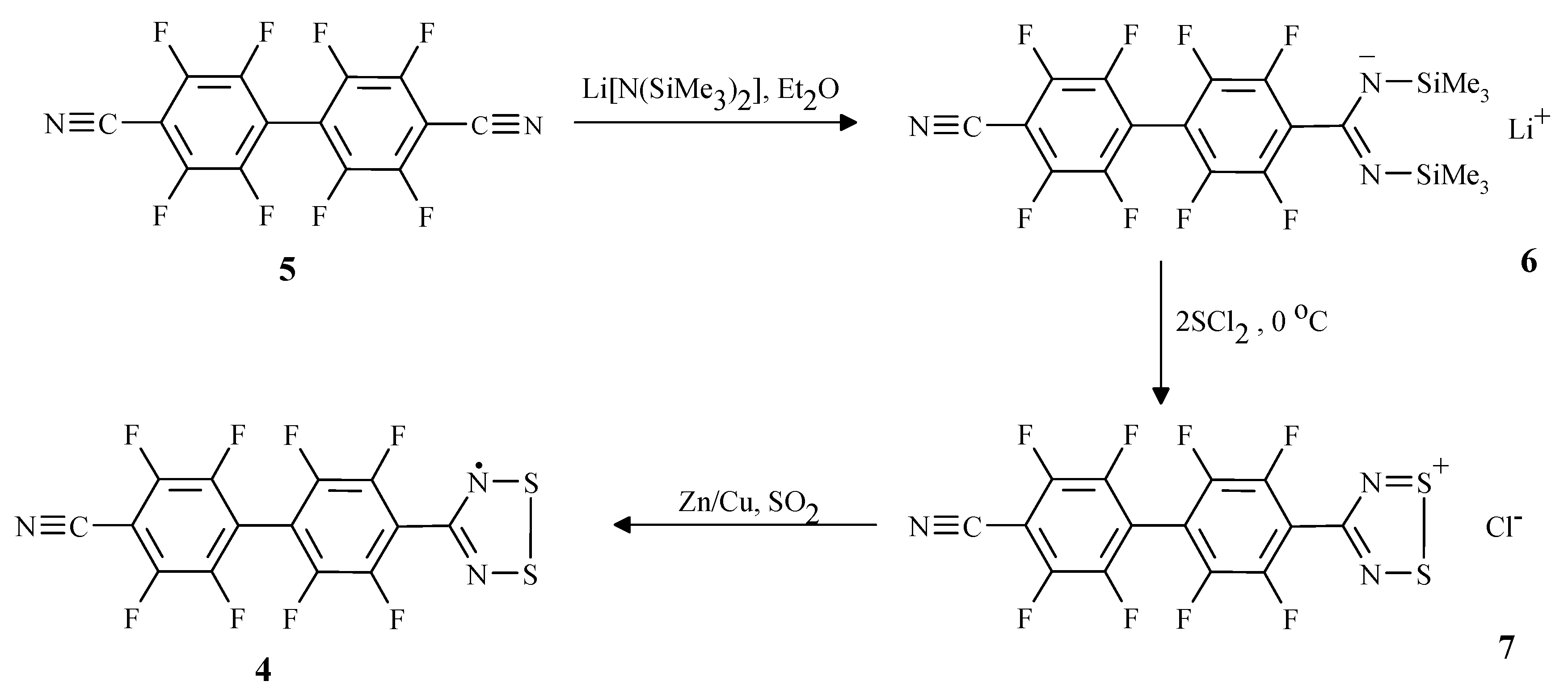
Results
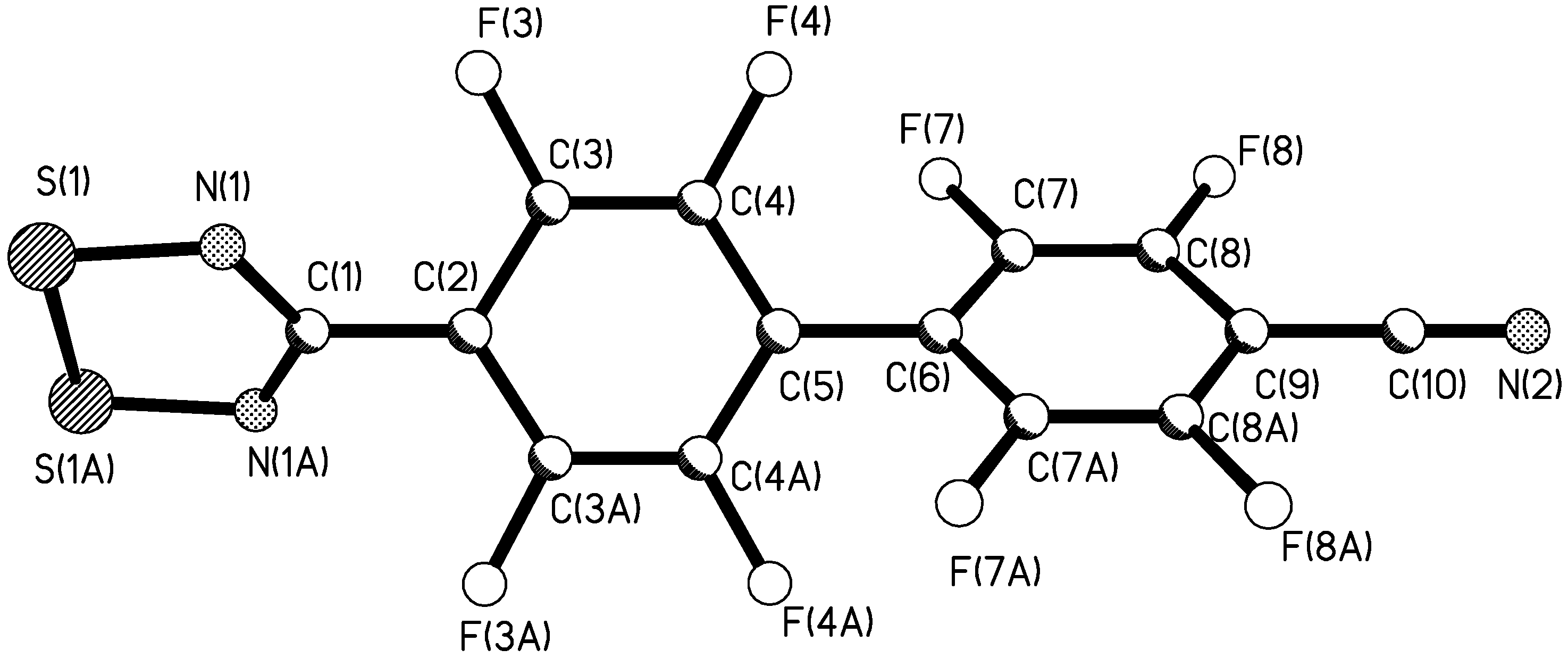
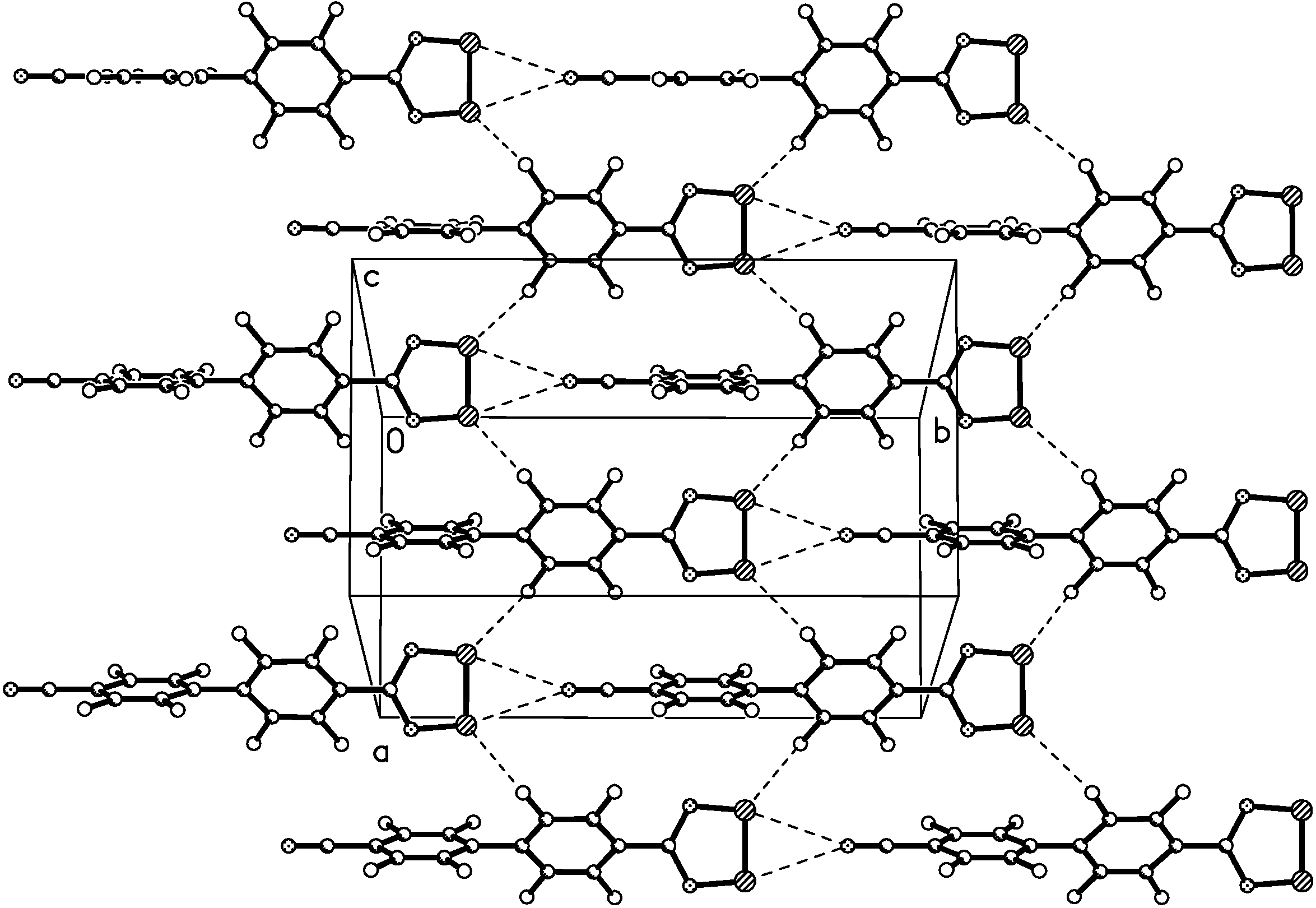
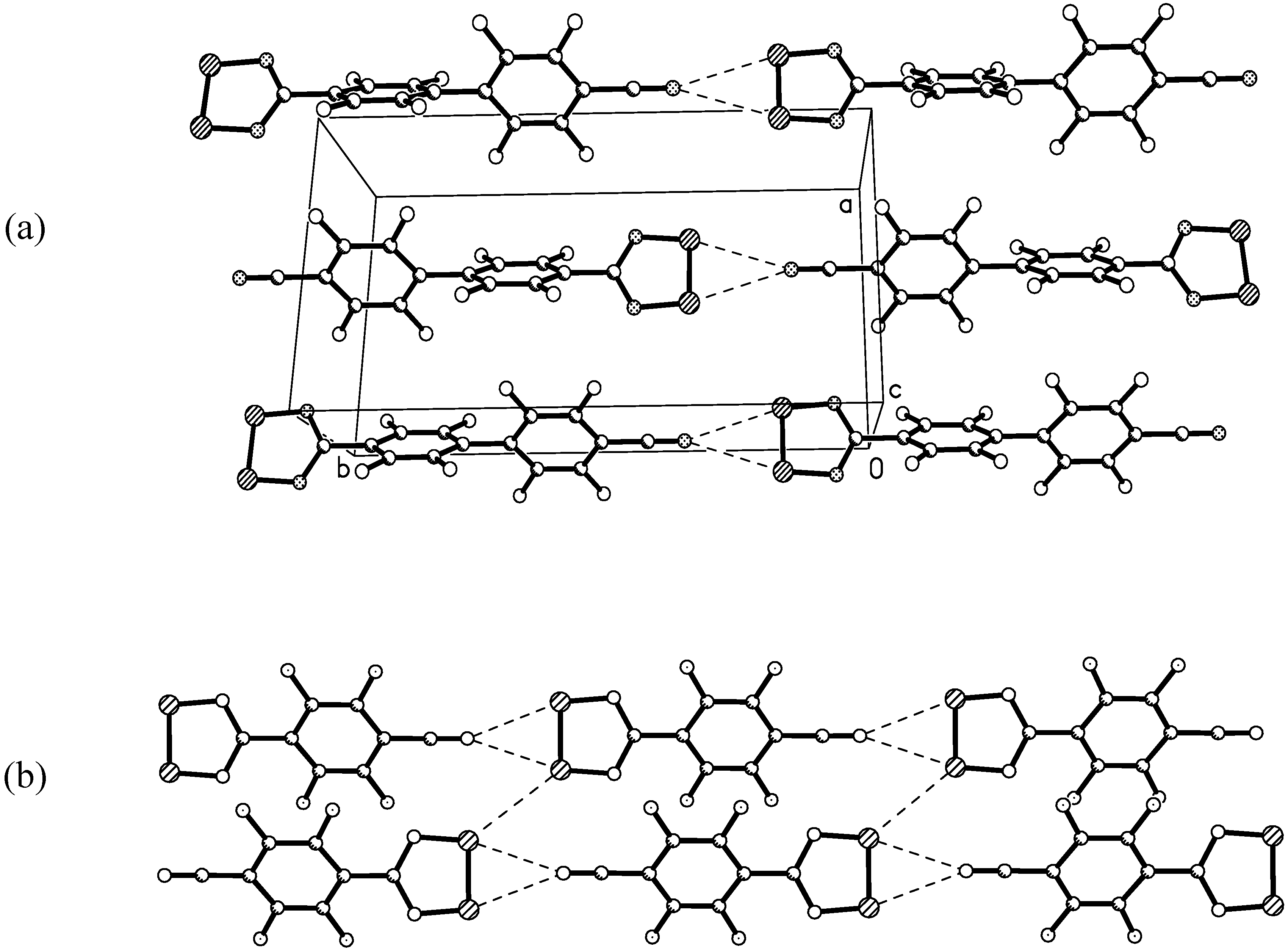
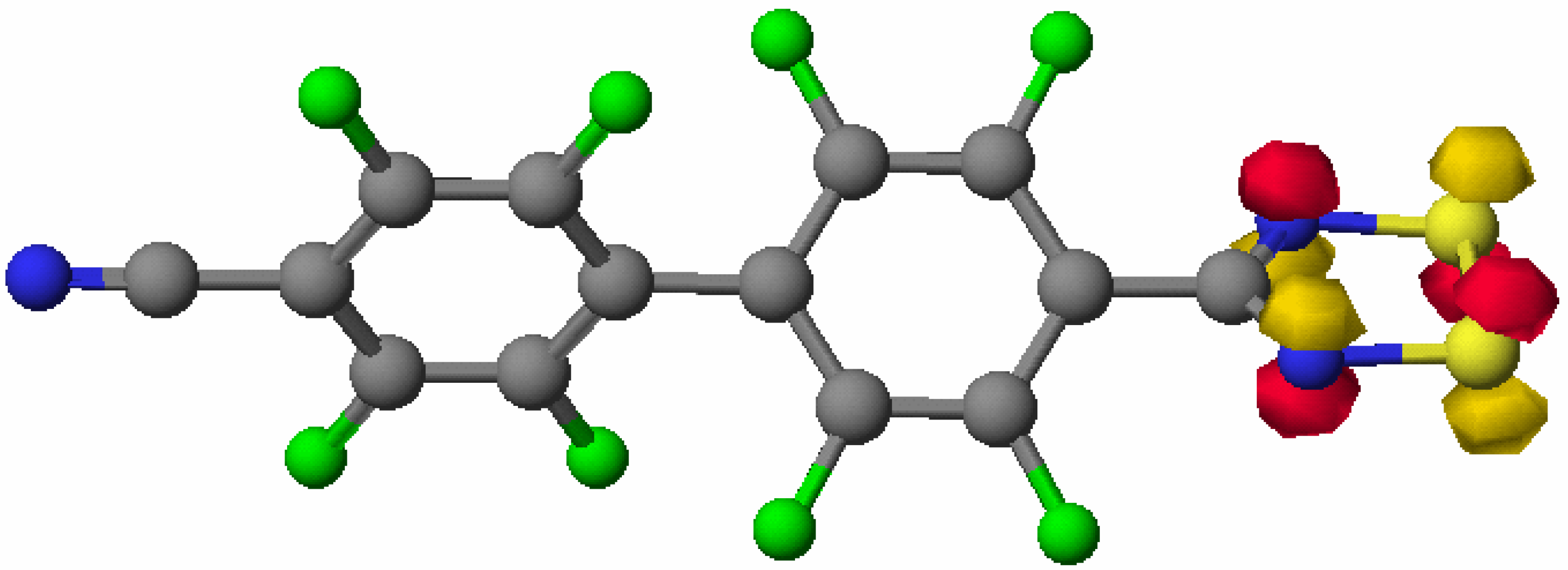
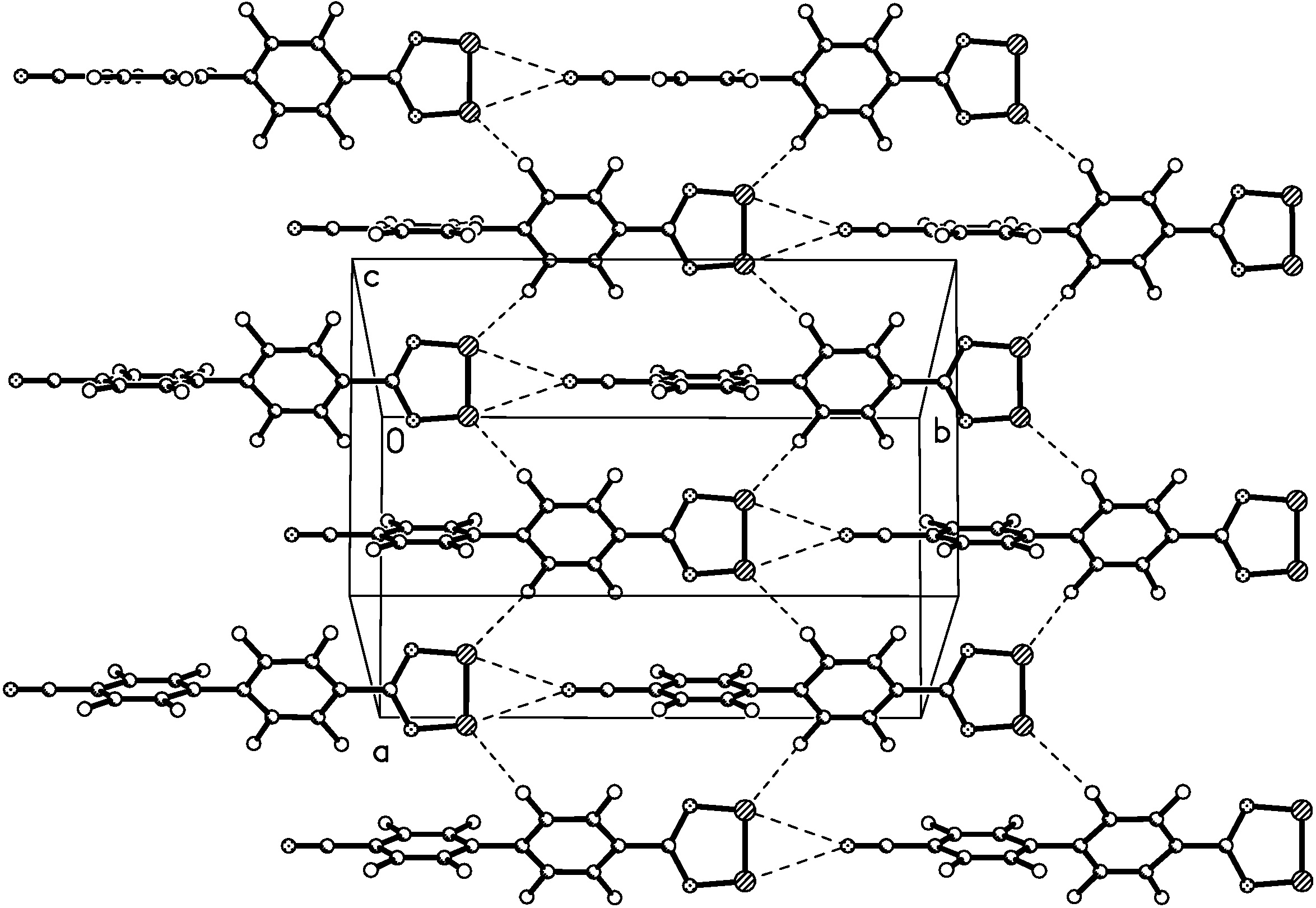
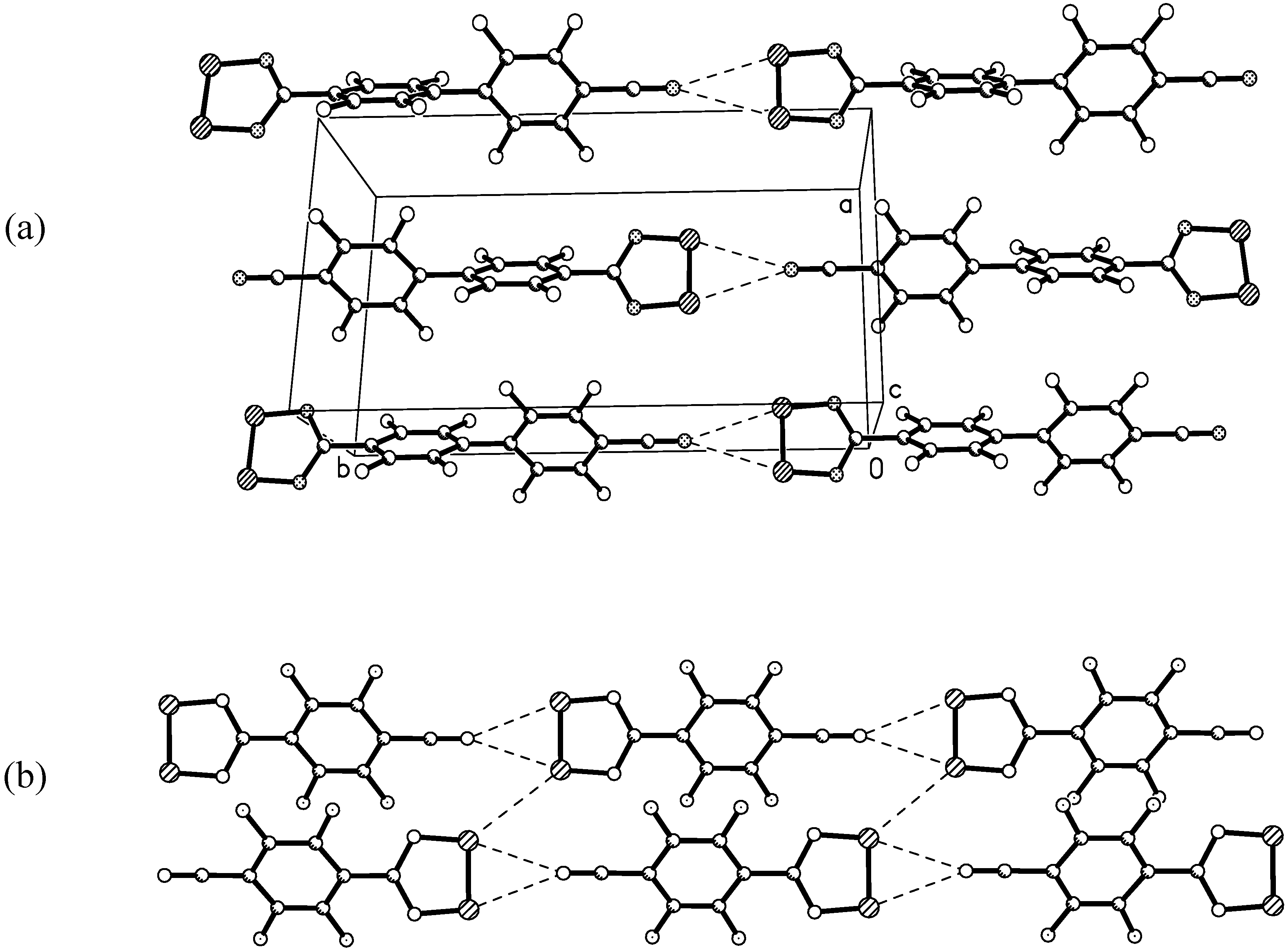
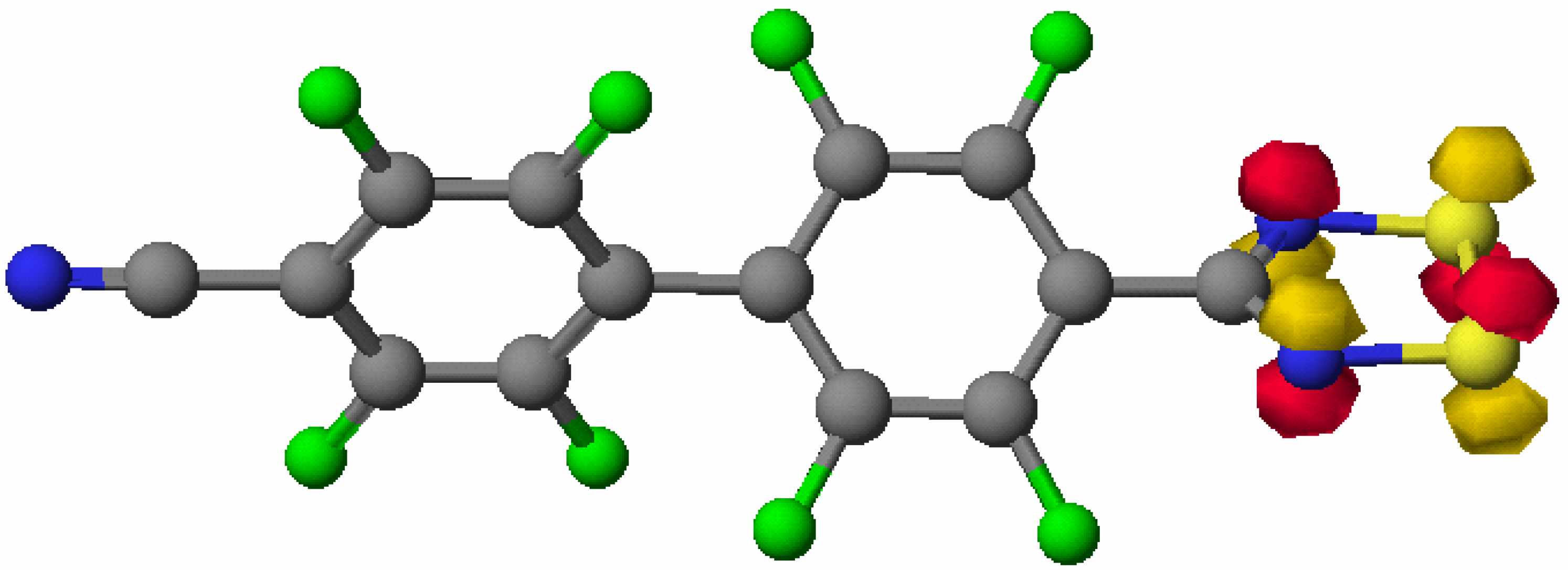
X-ray crystal structure of 4

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1α | 1β | 2 | 3 | 4 | |
|---|---|---|---|---|---|
| C-N/Å | 1.331(2) | 1.325 | 1.317(14) | 1.325(2) | 1.331(2) |
| 1.324(2) | 1.327(12) | ||||
| N-S/Å | 1.637(2) | 1.638 | 1.640(8) | 1.635(2) | 1.6340(19) |
| 1.638(2) | 1.624(9) | ||||
| S-S/Å | 2.0897(14) | 2.082 | 2.070(4) | 2.0815(13) | 2.0856(11) |
| NCN/o | 123.9(2) | 124.0 | 124.0(9) | 123.9(3) | 123.5(3) |
| CNS/o | 113.52(13) | 113.4 | 113.9(7) | 113.55(17) | 113.67(16) |
| 113.74(13) | 112.8(7) | ||||
| NSS/o | 94.49(8) | 94.53 | 93.7(4) | 94.51(7) | 94.57(6) |
| 94.37(8) | 95.5(3) | ||||
| θ/o | 32 | 58 | 51.8 | 58.1 | 42.9 |
| Reference | 5a | 5b | 6 | 7 | This work |
Theoretical Studies on 4
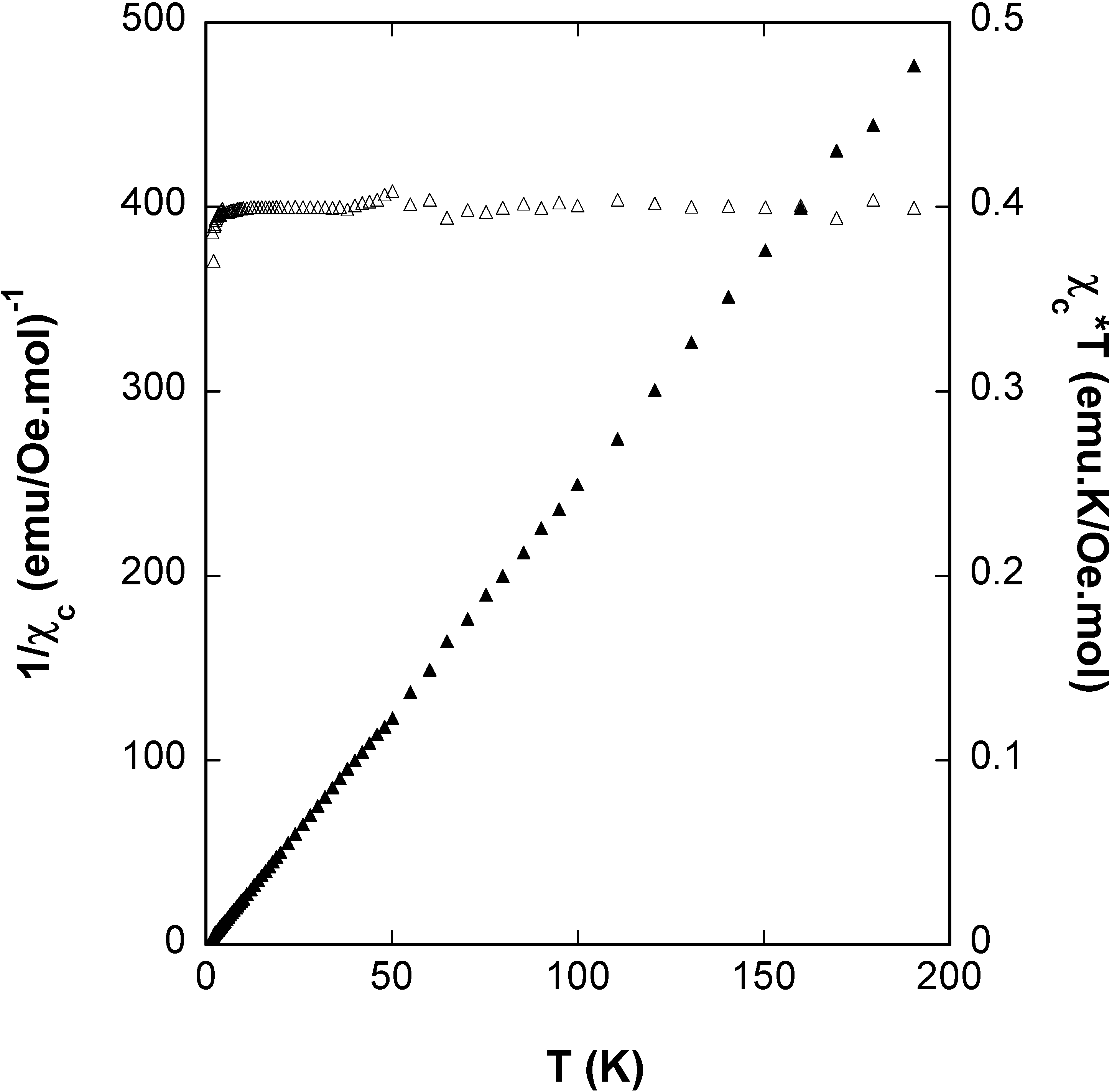
Magnetic Behaviour of 4

Discussion
Conclusions
Experimental
General
Synthesis of p-NCC6F4C6F4CN
Synthesis of p-NCC6F4C6F4CNSSN• (4)
X-ray crystallography
References
- Palacio, F. Molecular Magnetism: From Molecular Assemblies to the Devices; NATO-ASI, Series E; Vol. 321, Coronado, E., Delhaes, P., Gatteschi, D., Miller, J., Eds.; Kluwer Academic: Dordrect (The Netherlands), 1996. [Google Scholar]
- McConnell, H.M. J. Chem. Phys. 1963, 39, 1910.McConnell, H.M. Proc. Robert A. Welch Found. Conf. Chem. Res. 1967, 11, 144.
- For recent studies on crystal structure prediction see: Lommerse, J.P.M.; Motherwell, S.D.W.; Ammon, H.L.; Dunitz, J.D.; Gavezzotti, A.; Hofmann, D.; Leusen, F.J.J.; Mooij, W.T.M.; Price, S.L.; Scweitzer, B.; Schmidt, M.U.; van Eijck, B.P.; Verwer, P.; Williams, D.E. Acta. Crystallogr. 2000, B56, 697.Motherwell, S.D.W.; Ammon, H.L.; Dunitz, J.D.; Dzyabchenko, A.; Erk, P.; Gavezzotti, A.; Hofmann, D.W.M.; Leusen, F.J.J.; Lommerse, J.P.M.; Mooij, W.T.M.; Price, S.L.; Scherage, H.; Scweitzer, B.; Schmidt, M.U.; van Eijck, B.P.; Verwer, P.; Williams, D.E. Acta. Crystallogr. 2002, B58, 647.
- Rawson, J.M.; Banister, A.J.; Lavender, I. Adv. Heterocycl. Chem. 1995, 62, 137.
- Banister, A.J.; Bricklebank, N.; Clegg, W.; Elsegood, M.R.J.; Gregory, C.I.; Lavender, I.; Rawson, J.M.; Tanner, B.K. Chem. Commun. 1995, 679.Banister, A.J.; Bricklebank, N.; Lavender, I.; Rawson, J.M.; Gregory, C.I.; Tanner, B.K.; Clegg, W.; Elsegood, M.R.J.; Palacio, F. Angew. Chem. Int Ed. Engl. 1996, 35, 2533.
- Antorrena, G.; Davies, J.E.; Hartley, M.; Palacio, F.; Rawson, J.M.; Smith, J.N.B.; Steiner, A. Chem. Commun. 1999, 1393.
- Alberola, A.; Less, R.J.; Pask, C.M.; Rawson, J.M.; Palacio, F.; Oliete, P.; Paulsen, C.; Yamaguchi, A.; Farley, R.D.; Murphy, D.M. Angew. Chem. Int. Ed. Engl. 2003, 42, 4782.
- Antorrena, G.; Brownridge, S.; Cameron, T.S.; Palacio, F.; Parsons, S.; Passmore, J.; Thompson, L.K.; Zarlaida, F. Can. J. Chem. 2002, 80, 1568.Cordes, A.W.; Haddon, R.C.; Hicks, R.G.; Oakley, R.T.; Palstra, T.T.M. Inorg. Chem. 1992, 1802.Cordes, A.W.; Chamchoumis, C.M.; Hicks, R.G.; Oakley, R.T.; Young, K.M.; Haddon, R.C. Can. J. Chem. 1992, 919.
- Birchall, J.M.; Haszeldine, R.N.; Jones, M.E. J. Chem. Soc. 1971, 1343.
- Fanta, P.E. Synthesis 1974, 9.
- Nyburg, S.C.; Faerman, C.H. Acta Crystallogr. Sect. B. 1985, 41, 274.
- Alonso, P.J.; Antorrena, G.; Martinez, J.I.; Novoa, J.J.; Palacio, F.; Rawson, J.M.; Smith, J.N.B. Appl. Magn. Reson. 2001, 20, 231.Fairhurst, S.A.; Sutcliffe, L.H.; Preston, K.F.; Banister, A.J.; Partington, A.S.; Rawson, J.M.; Passmore, J.; Schriver, M.J. Magn. Reson. Chem. 1993, 31, 1027.
- Luzon, J.; Campo, J.; Palacio, F.; McIntyre, G.J.; Goeta, A.E.; Pask, C.M.; Rawson, J.M. Polyhedron 2003, 2301.
- Kahn, O. Molecular Magnetism; VCH: New York, 1993. [Google Scholar]
- Novoa, J.J.; Deumal, M. Struct. Bond. 2001, 100, 33.
- Langley, P.J.; Rawson, J.M.; Smith, J.N.B.; Schuler, M.; Bachmann, R.; Schweiger, A.; Palacio, F.; Antorrena, G.; Gescheidt, G.; Quintel, A.; Rechsteiner, P.; Hulliger, J. J. Mater. Chem. 1999, 9, 1431.
- Samples Availability: Available from the authors.
© 2004 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Alberola, A.; Less, R.J.; Palacio, F.; Pask, C.M.; Rawson, J.M. Synthesis and Magnetic Properties of the Novel Dithiadiazolyl Radical, p-NCC6F4C6F4CNSSN. Molecules 2004, 9, 771-781. https://doi.org/10.3390/90900771
Alberola A, Less RJ, Palacio F, Pask CM, Rawson JM. Synthesis and Magnetic Properties of the Novel Dithiadiazolyl Radical, p-NCC6F4C6F4CNSSN. Molecules. 2004; 9(9):771-781. https://doi.org/10.3390/90900771
Chicago/Turabian StyleAlberola, Antonio, Robert J. Less, Fernando Palacio, Christopher M. Pask, and Jeremy M. Rawson. 2004. "Synthesis and Magnetic Properties of the Novel Dithiadiazolyl Radical, p-NCC6F4C6F4CNSSN" Molecules 9, no. 9: 771-781. https://doi.org/10.3390/90900771