
The Synthesis of Biphasic Metabolites of Carfentanil

Abstract
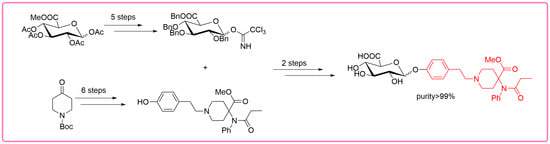
:
1. Introduction
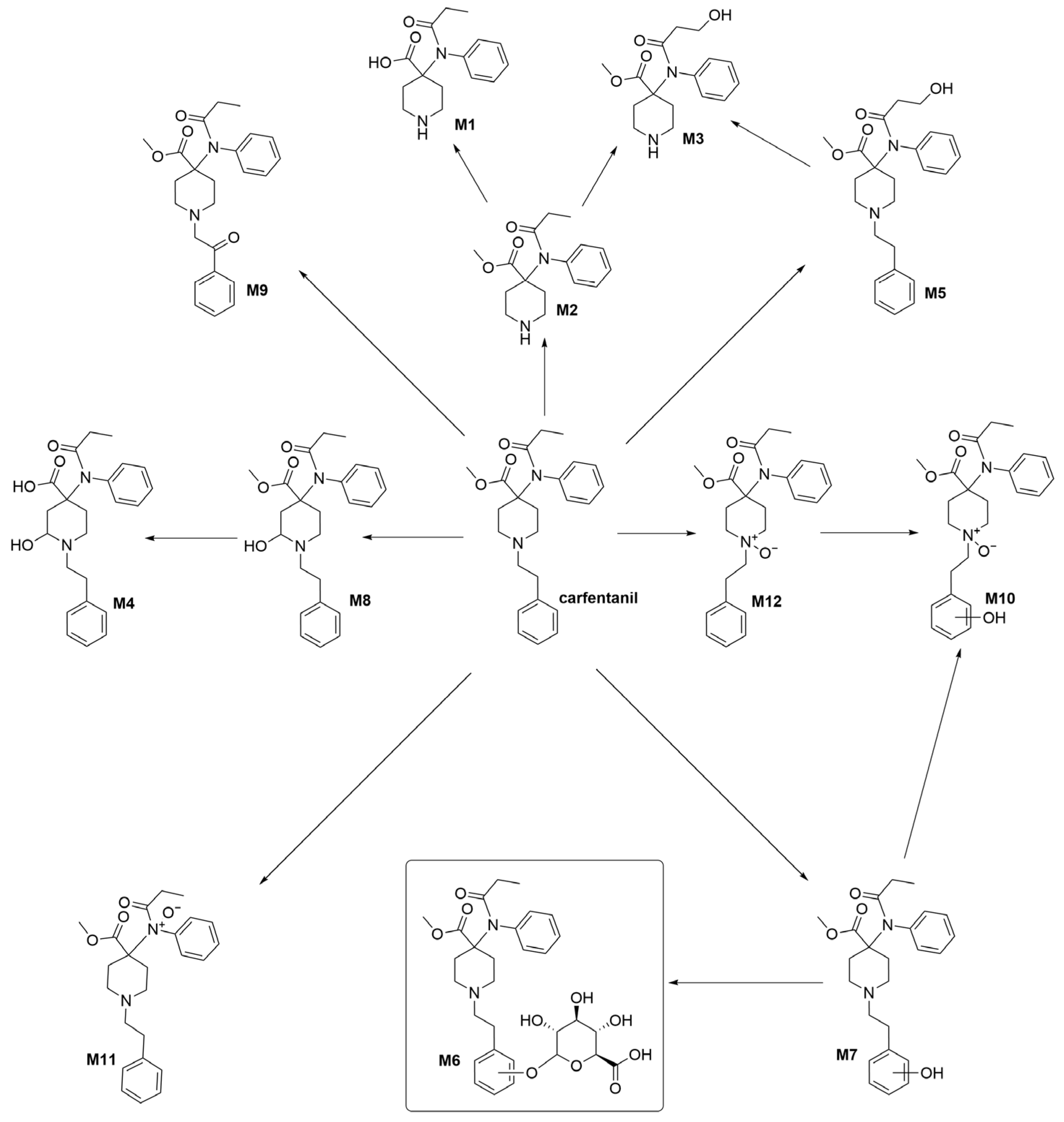
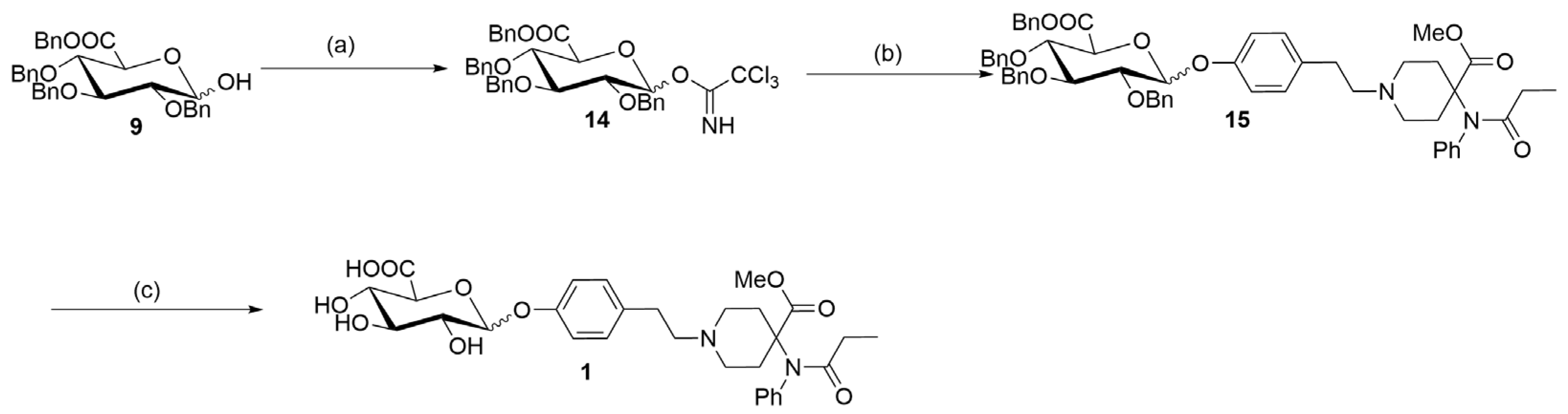
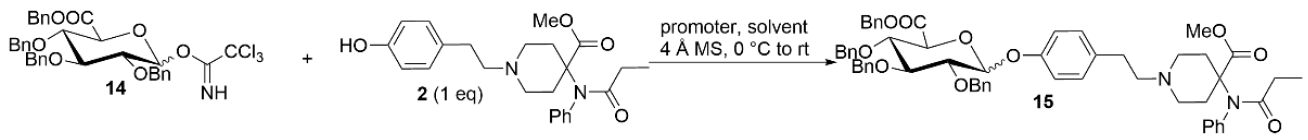
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. Experimental Section
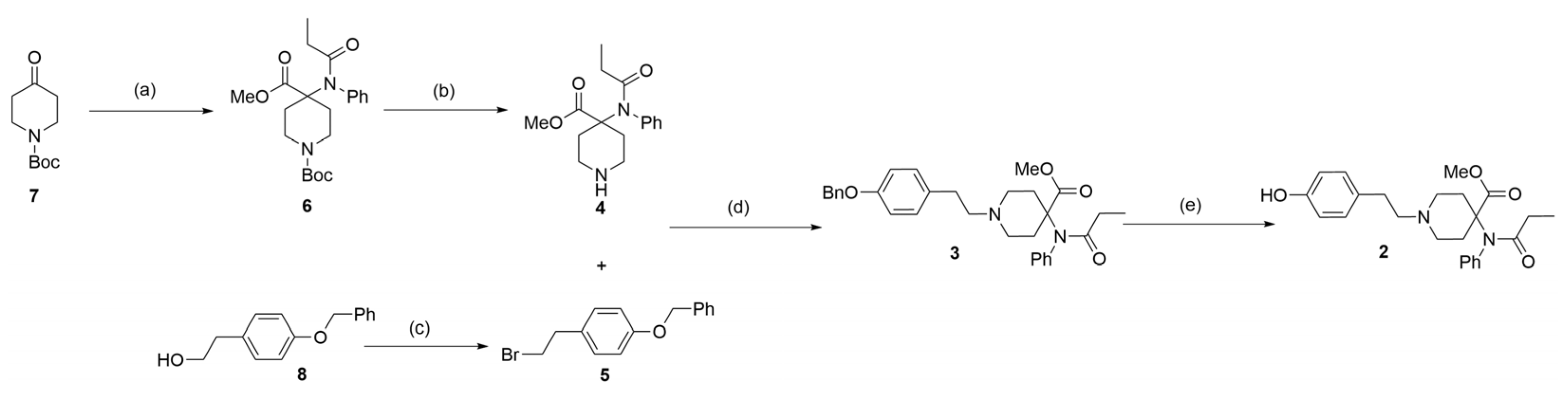
3.2.1. Synthesis of 1-(1,1-Dimethylethyl) 4-Methyl 4-[(1-Oxopropyl)phenylamino]-1,4-piperidinedicarboxylate (6)
3.2.2. Synthesis of Methyl 4-(N-Phenylpropionamido)piperidine-4-carboxylate (4)
3.2.3. Synthesis of 1-(2-Bromoethyl)-4-(phenylmethoxy)benzene (5)
3.2.4. Synthesis of Methyl 1-(4-(Benzyloxy)phenethyl)-4-(N-phenylpropionamido)-piperidine-4-carboxylate (3)
3.2.5. Synthesis of Methyl 1-[2-(4-Hydroxyphenyl)ethyl]-4-[(1-oxopropyl)phenylamino]-4-piperidinecarboxylate (2)
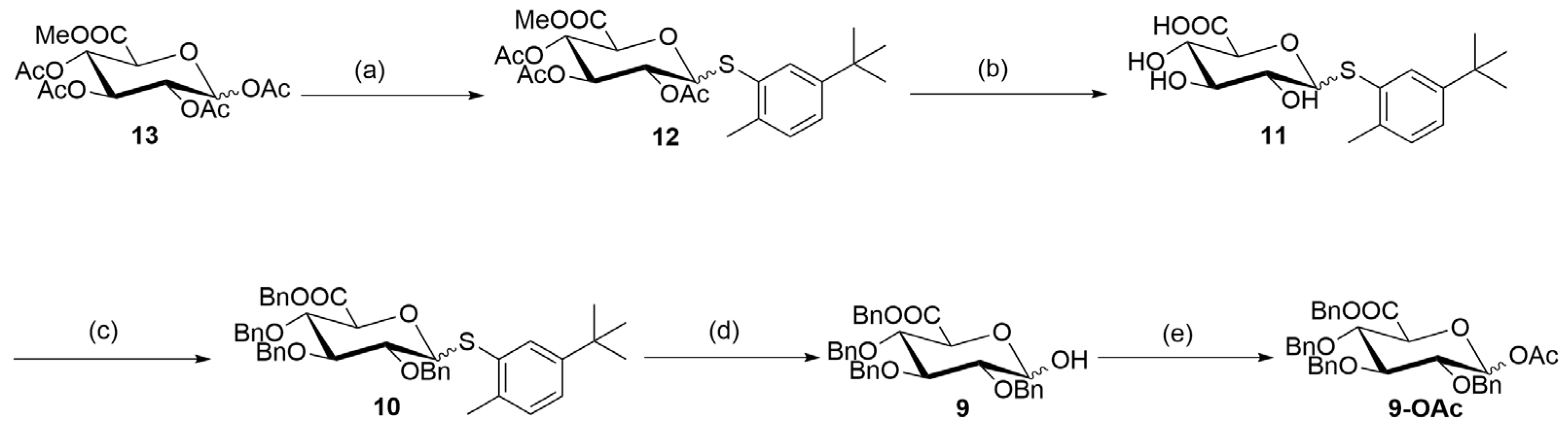
3.2.6. Synthesis of (3R,4S,5S,6S)-2-((5-(tert-Butyl)-2-methylphenyl)thio)-6-(methoxycarbonyl)-tetrahydro-2H-pyran-3,4,5-triyl Triacetate (12)
3.2.7. Synthesis of Benzyl (2S,3S,4S,5R)-3,4,5-tris(Benzyloxy)-6-((5-(tert-butyl)-2-methylphenyl)thio)tetrahydro-2H-pyran-2-carboxylate (10)
3.2.8. Synthesis of 2,3,4-Tri-O-benzyl-d-glucopyranuronic Acid Benzyl Ester (9)
3.2.9. Synthesis of 1-O-Acetyl-2,3,4-tri-O-benzyl-d-glucopyranuronic Acid Benzyl Ester (9-OAc)
3.2.10. Synthesis of Methyl 4-(N-Phenylpropionamido)-1-(4-(((3R,4S,5S,6S)-3,4,5-tris(benzyloxy)-6-((benzyloxy)carbonyl)tetrahydro-2H-pyran-2-yl)oxy)phenethyl)piperidine-4-carboxylate (15)
3.2.11. Synthesis of (2S,3S,4S,5R)-3,4,5-Trihydroxy-6-(4-(2-(4-(methoxycarbonyl)-4-(N-phenylpropionamido)-piperidin-1-yl)ethyl)phenoxy)tetrahydro-2H-pyran-2-carboxylic Acid (1)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cole, A.; Mutlow, A.; Isaza, R.; Carpenter, J.W.; Koch, D.E.; Hunter, R.P.; Dresser, B.L. Pharmacokinetics and pharmacodynamics of carfentanil and naltrexone in female common eland (Taurotragus oryx). J. Zoo Wildl. Med. 2006, 37, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Van Bever, W.F.M.; Niemegeers, C.J.E.; Schellekens, K.H.L.; Janssen, P.A.J. N-4- Substituted 1-(2-arylethyl)-4-piperidinyl-N-phenylpropanamides, a novel series of extremely potent analgesics with unusually high safety margin. Arzneimittelforschung 1976, 26, 1548–1551. [Google Scholar] [PubMed]
- Subramanian, G.; Paterlini, M.G.; Portoghese, P.S.; Ferguson, D.M. Molecular docking reveals a novel binding site model for fentanyl at the mu-opioid receptor. J. Med. Chem. 2000, 43, 381–391. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.; Gladden, R.M.; Mattson, C.L.; Kariisa, M. Overdose Deaths with Carfentanil and Other Fentanyl Analogs Detected-10 States, July 2016–June 2017. MMWR-Morb. Mortal. Wkly. Rep. 2018, 67, 767–768. [Google Scholar] [CrossRef] [PubMed]
- Riches, J.R.; Read, R.W.; Black, R.M.; Cooper, N.J.; Timperley, C.M. Analysis of Clothing and Urine from Moscow Theatre Siege Casualties Reveals Carfentanil and Remifentanil Use. J. Anal. Toxicol. 2012, 36, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, O.; Antoni, G. C-11 Carfentanil Binds Preferentially to mu-Opioid Receptor Subtype 1 Compared to Subtype 2. Mol. Imaging 2015, 14, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Raffa, R.B.; Pergolizzi, J.V., Jr.; LeQuang, J.A.; Taylor, R., Jr.; Colucci, S.; Annabi, M.H. The fentanyl family: A distinguished medical history tainted by abuse. J. Clin. Pharm. Ther. 2018, 43, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Ling, G.S.F.; Spiegel, K.; Nishimura, S.L.; Pasternak, G.W. Dissociation of morphine’s analgesic and respiratory depressant actions. Eur. J. Pharmacol. 1983, 86, 487–488. [Google Scholar] [CrossRef] [PubMed]
- Shook, J.E.; Watkins, W.D.; Camporesi, E.M. Differential roles of opioid receptors in respiration, respiratory disease, and opiate—Induced respiratory depression. Am. Rev. Respir. Dis. 1990, 142, 895–909. [Google Scholar] [CrossRef] [PubMed]
- Minkowski, C.P.; Epstein, D.; Frost, J.J.; Gorelick, D.A. Differential response to IV carfentanil in chronic cocaine users and healthy controls. Addict. Biol. 2012, 17, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Dannals, R.F.; Ravert, H.T.; Frost, J.J.; Wilson, A.A.; Burns, D.; Wagner, H.N., Jr. Radiosynthesis of an opiate receptor binding radiotracer: [11C]carfentanil. Int. J. Appl. Radiat. Isot. 1985, 36, 303–306. [Google Scholar] [CrossRef] [PubMed]
- Vuckovic, S.; Prostran, M.; Ivanovic, M.; Dosen-Micovic, L.; Todorovic, Z.; Nesic, Z.; Stojanovic, R.; Divac, N.; Mikovic, Z. Fentanyl analogs: Structure-activity-relationship study. Curr. Med. Chem. 2009, 16, 2468–2474. [Google Scholar] [CrossRef] [PubMed]
- Feasel, M.G.; Wohlfarth, A.; Nilles, J.M.; Pang, S.K.; Kristovich, R.L.; Huestis, M.A. Metabolism of Carfentanil, an Ultra-Potent Opioid, in Human Liver Microsomes and Human Hepatocytes by High-Resolution Mass Spectrometry. AAPS J. 2016, 18, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Butcher, K.J.; Hurst, J. Corrigendum to “Aromatic amines as nucleophiles in the Bargellini reaction”. Tetrahedron Lett. 2009, 50, 2497–2500. [Google Scholar] [CrossRef]
- Van Daele, P.G.H.; De Bruyn, M.F.L.; Boey, J.M.; Sanczuk, S.; Agten, J.T.M.; Janssen, P.A.J. Synthetic analgesics: N-(1-[2-arylethyl]-4-substituted 4-piperidinyl) N-arylalkanamides. Arzneim.-Forsch 1976, 26, 1521–1531. [Google Scholar]
- Feldman, P.L.; Brackeen, M.F. A novel route to the 4-anilido-4-(methoxycarbonyl)piperidine class of analgetics. J. Org. Chem. 1990, 55, 4207–4209. [Google Scholar] [CrossRef]
- Wagner Henry, N., Jr. Brain Imaging: The Chemistry of Mental Activity; Springer: Berlin/Heidelberg, Germany, 2009; 157p. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Entry | Compound 14 (Eq.) | Promoter (Eq.) | Solvent | Yield a |
| 1 | 1 | TMSOTf (0.3) b | CH2Cl2 | 8% |
| 2 | 1 | TBSOTf (0.3) c | CH2Cl2 | 13% |
| 3 | 1 | BF3.Et2O (0.3) | CH2Cl2 | 22% |
| 4 | 1 | BF3.Et2O (0.6) | CH2Cl2 | 32% |
| 5 | 1 | BF3.Et2O (1) | CH2Cl2 | 51% |
| 6 | 1 | BF3.Et2O (1) | toluene | 58% |
| 7 | 2 | BF3.Et2O (2) | toluene | 72% |
| 8 | 3 | BF3.Et2O (3) | toluene | 81% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Hu, J.; Liao, P.; Xue, S.; He, S.; Chen, R.; Zhao, X.; Liu, W. The Synthesis of Biphasic Metabolites of Carfentanil. Molecules 2023, 28, 7625. https://doi.org/10.3390/molecules28227625
Wang J, Hu J, Liao P, Xue S, He S, Chen R, Zhao X, Liu W. The Synthesis of Biphasic Metabolites of Carfentanil. Molecules. 2023; 28(22):7625. https://doi.org/10.3390/molecules28227625
Chicago/Turabian StyleWang, Junchang, Jianwen Hu, Pingyong Liao, Shang Xue, Shan He, Ruijia Chen, Xuejun Zhao, and Wenbin Liu. 2023. "The Synthesis of Biphasic Metabolites of Carfentanil" Molecules 28, no. 22: 7625. https://doi.org/10.3390/molecules28227625
APA StyleWang, J., Hu, J., Liao, P., Xue, S., He, S., Chen, R., Zhao, X., & Liu, W. (2023). The Synthesis of Biphasic Metabolites of Carfentanil. Molecules, 28(22), 7625. https://doi.org/10.3390/molecules28227625