
Copper(I)-Catalyzed Cross-Coupling of 1-Bromoalkynes with N-Heterocyclic Organozinc Reagents

Abstract
:1. Introduction
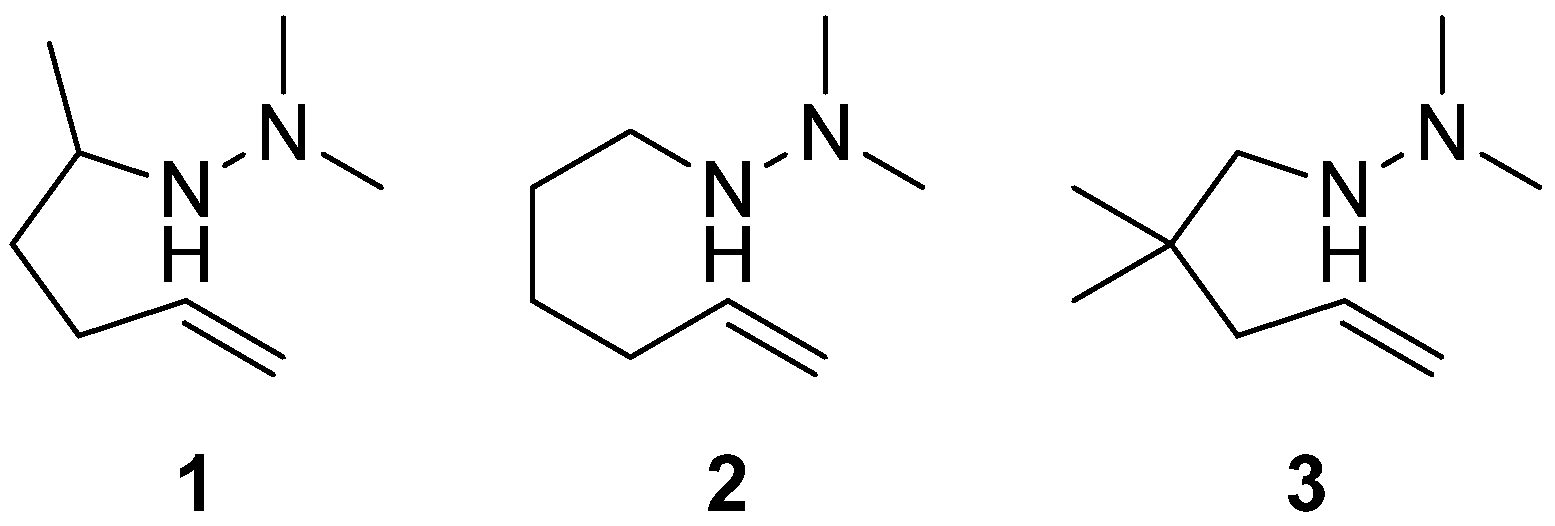
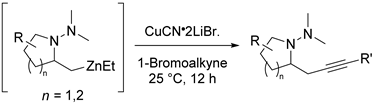
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Experimental Procedures
3.2.1. General Procedure for the Synthesis of Alkynyl N,N-Dimethylhydrazinoalkenes
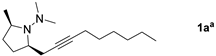
3.2.2. Procedure for Preparative Scale Synthesis of 3a
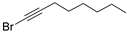
3.2.3. General Procedure for the Synthesis of 1-Bromoalkynes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Meanwell, N.A. Improving Drug Candidates by Design: A Focus on Physicochemical Properties as a Means of Improving Compound Disposition and Safety. Chem. Res. Toxicol. 2011, 24, 1420–1456. [Google Scholar] [CrossRef] [PubMed]
- Welsch, M.E.; Snyder, S.A.; Stockwell, B.R. Privileged scaffolds for library design and drug discovery. Curr. Opin. Chem. Biol. 2010, 14, 347–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roughley, S.D.; Jordan, A.M. The Medicinal Chemist’s Toolbox: An Analysis of Reactions Used in the Pursuit of Drug Candidates. J. Med. Chem. 2011, 54, 3451–3479. [Google Scholar] [CrossRef] [PubMed]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef] [PubMed]
- de la Torre, B.G.; Albericio, F. The Pharmaceutical Industry in 2021. An Analysis of FDA Drug Approvals from the Perspective of Molecules. Molecules 2022, 27, 1075. [Google Scholar] [CrossRef] [PubMed]
- de la Torre, B.G.; Albericio, F. The Pharmaceutical Industry in 2020. An Analysis of FDA Drug Approvals from the Perspective of Molecules. Molecules 2021, 26, 627. [Google Scholar] [CrossRef]
- Jones, L.; Bunnage, M. Applications of chemogenomic library screening in drug discovery. Nat. Rev. Drug. Discov. 2017, 16, 285–296. [Google Scholar] [CrossRef]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational Methods in Drug Discovery. Pharmacol. Rev. 2012, 66, 334–395. [Google Scholar] [CrossRef] [Green Version]
- Mickelsen, K.; Zabawa, S.; Livinghouse, T. Diethylzinc-Mediated Metalloamination–Alkylation of N,N-Dimethylhydrazinoalkenes Catalysis of C–Zn Alkylation Using Simple Cu(I) Salts. Synlett 2018, 29, 181–184. [Google Scholar] [CrossRef]
- Sunsdahl, B.; Mickelsen, K.; Zabawa, S.; Anderson, B.K.; Livinghouse, T. 1,2-Disubstituted Alkenes as Migratory Insertion Participants in Zn(II)-Promoted Metalloamination/Cyclization of N,N-Dimethylhydrazinoalkenes. J. Org. Chem. 2016, 81, 10160–10164. [Google Scholar] [CrossRef]
- Sunsdahl, B.; Smith, A.R.; Livinghouse, T. Intramolecular Metalloamination of N,N-Dimethylhydrazinoalkenes: A Versatile Method to Access Functionalized Piperidines and Pyrrolidines. Angew. Chem. Int. Ed. 2014, 53, 14352–14356. [Google Scholar] [CrossRef] [PubMed]
- Babij, N.R.; Jordan, R.B.; McKenna, G.M.; Fornwald, R.M.; Wolfe, J.P. Stereocontrolled Synthesis of Bicyclic Ureas and Sulfamides via Pd-Catalyzed Alkene Carboamination Reactions. Tetrahedron 2019, 75, 4228–4243. [Google Scholar] [CrossRef] [PubMed]
- Hinds, E.J.; Wolfe, J.P. A Cross-Metathesis/Aza-Michael Reaction Strategy for the Synthesis of Cyclic and Bicyclic Ureas. J. Org. Chem. 2018, 83, 10668–10676. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, J.P. Palladium-Catalyzed Carboetherification and Carboamination Reactions of γ-Hydroxy and γ-Aminoalkenes for the Synthesis of Tetrahydrofurans and Pyrrolidines. Eur. J. Org. Chem. 2007, 4, 571–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolai, S.; Orcel, U.; Muriel, B.; Greenwood, P.D.G.; Buzzetti, L.; Purins, M.; Waser, J. Palladium-Catalyzed Functionalization of olefins and Alkynes: From Oxyalkynylation to Tethered Dynaimc Kinetic Asymmetric Transforamtions (DYKAT). Synlett 2021, 32, 472–487. [Google Scholar] [CrossRef]
- Nicolai, S.; Sedigh-Zadeh, R.; Waser, J. Pd(0)-Catalyzed Alkene Oxy- and Aminoalkynylation with Aliphatic Bromoacetylenes. J. Org. Chem. 2013, 78, 3783–3801. [Google Scholar] [CrossRef] [Green Version]
- Waser, J.; Nicolai, S. Pd(0)-Catalyzed Oxy- and Aminoalkynylation of Olefins for the Synthesis of Tetrahydrofurans and Pyrrolidines. Org. Lett. 2011, 13, 6324–6327. [Google Scholar]
- Shikora, J.M.; Um, C.; Khoder, A.M.; Chemler, S.R. Saturated oxygen and nitrogen heterocycles via oxidative coupling of alkylfluroborates with alkenols, alkenoic acids and protected alkenylamines. Chem. Sci. 2019, 40, 9265–9269. [Google Scholar] [CrossRef]
- Chemler, S.R.; Karyakarte, S.D.; Khoder, Z.M. Stereoselective and Regioselective Synthesis of hetereocycles via Copper-Catalyzed Additions of Amine Derivatives and Alcohols to Alkenes. J. Org. Chem. 2017, 82, 11311–11325. [Google Scholar] [CrossRef]
- Wdowik, T.; Chemler, S. R Direct Synthesis of 2-Formylpyrrolidines, 2-Pyrrolidinones and 2-Dihydrofuranones via Aerobic Copper-Catalyzed Aminooxygenation and deoxygenation of 4-Pentenylsulonaminds and 4-Pentenylalcohols. J. Am. Chem. Soc. 2017, 139, 9515–9518. [Google Scholar] [CrossRef]
- Yeh, M.C.P.; Knochel, P. The Reactivity of Highly Functionalized Copper, Zinc Reagents RCu(CN)ZnI Toward 1-Haloalkynes and Acetylenic Esters. Tetrahedron Lett. 1989, 30, 4799–4802. [Google Scholar] [CrossRef] [Green Version]
- Lipshutz, B.H.; Sharma, S.; Ellsworth, E.L. Higher order cyanocuprates R2Cu(CN)Li2: Discrete reagents or lower order lithium cyanide (LiCN) modified Gilman cuprates? J. Am. Chem. Soc. 1990, 112, 4032–4034. [Google Scholar] [CrossRef]
- Knochel, P.; Millot, N.; Rodriguez, A.L.; Tucker, C.E. Preparation and Applications of Functionalized Organozinc Compounds. In Organic Reactions; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2004. [Google Scholar]
- Thapa, S.; Shrestha, B.; Gurung, S.K.; Giri, R. Copper-catalysed cross-coupling: An untapped potential. Org. Biomol. Chem. 2015, 13, 4816–4827. [Google Scholar] [CrossRef] [PubMed]
- Klapars, A.; Antilla, J.C.; Huang, X.H.; Buchwald, S.L. A general and efficient copper catalyst for the amidation of aryl halides and the N-arylation of nitrogen heterocycles. J. Am. Chem. Soc. 2001, 123, 7727–7729. [Google Scholar] [CrossRef] [PubMed]
- Surry, D.S.; Buchwald, S.L. Diamine ligands in copper-catalyzed reactions. Chem. Sci. 2010, 1, 13–31. [Google Scholar] [CrossRef] [Green Version]
- Frabitore, C.; Lépeule, J.; Towey, B.; Livinghouse, T.; Robinson, W.C. Efficient reductions of dimethylhydrazones using preformed primary amine boranes. Syn. Comm. 2022, 52, 185–189. [Google Scholar] [CrossRef]
- Gao, Y.; Wu, G.; Zhou, Q.; Wang, J. Palladium-Catalyzed Oxygenative Cross-Coupling of Ynamide and Benzyl Bromides by Carbene Migratory Insertion. Angew. Chem. Int. Ed. 2018, 57, 2716–2720. [Google Scholar] [CrossRef]
- He, L.-Y.; Shulz-Senft, M.; Thiedemann, B.; Linshoeft, J.; Gates, P.G.; Staubitz, A. Nucleophile-Selective Cross-Coupling Reactions with Vinyl and Alkynyl Bromides on a Dinucleophilic Aromatic Substrate. Eur. J. Org. Chem. 2015, 11, 2498–2502. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| 1-Bromoalkyne | Product | Yield [%] |
|---|---|---|
 |  | 77 b |
| | 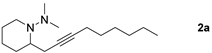 | 81 b 72 c |
| | 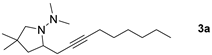 | 58 b 76 d |
 | 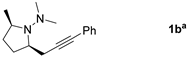 | 66 b |
| |  | 72 b 25 c |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frabitore, C.; Lépeule, J.; Livinghouse, T. Copper(I)-Catalyzed Cross-Coupling of 1-Bromoalkynes with N-Heterocyclic Organozinc Reagents. Molecules 2022, 27, 4561. https://doi.org/10.3390/molecules27144561
Frabitore C, Lépeule J, Livinghouse T. Copper(I)-Catalyzed Cross-Coupling of 1-Bromoalkynes with N-Heterocyclic Organozinc Reagents. Molecules. 2022; 27(14):4561. https://doi.org/10.3390/molecules27144561
Chicago/Turabian StyleFrabitore, Christian, Jérome Lépeule, and Tom Livinghouse. 2022. "Copper(I)-Catalyzed Cross-Coupling of 1-Bromoalkynes with N-Heterocyclic Organozinc Reagents" Molecules 27, no. 14: 4561. https://doi.org/10.3390/molecules27144561
APA StyleFrabitore, C., Lépeule, J., & Livinghouse, T. (2022). Copper(I)-Catalyzed Cross-Coupling of 1-Bromoalkynes with N-Heterocyclic Organozinc Reagents. Molecules, 27(14), 4561. https://doi.org/10.3390/molecules27144561