
Oxoisoaporphines and Aporphines: Versatile Molecules with Anticancer Effects

 ,
,  ,
,  ,
,
Abstract
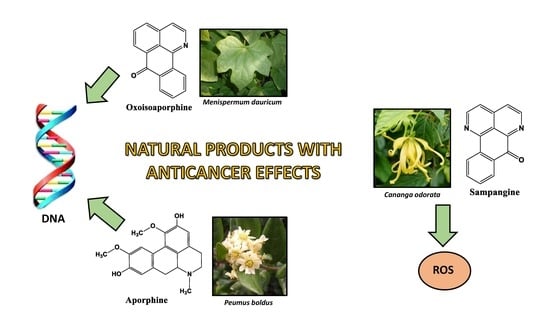
1. Introduction
2. Anticancer Natural Compounds: Oxoisoaporphines, Sampangines, and Boldine—Synthesis and Structural Description
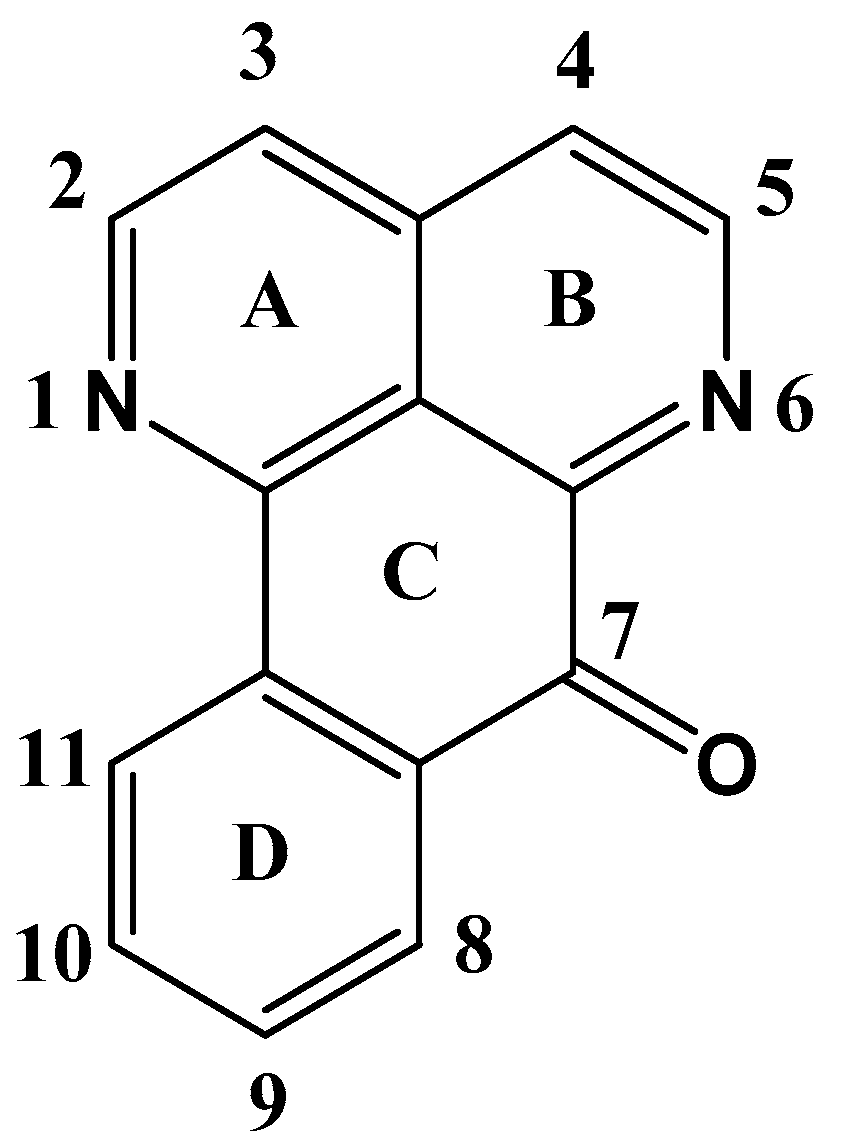
2.1. Oxoisoaporfines
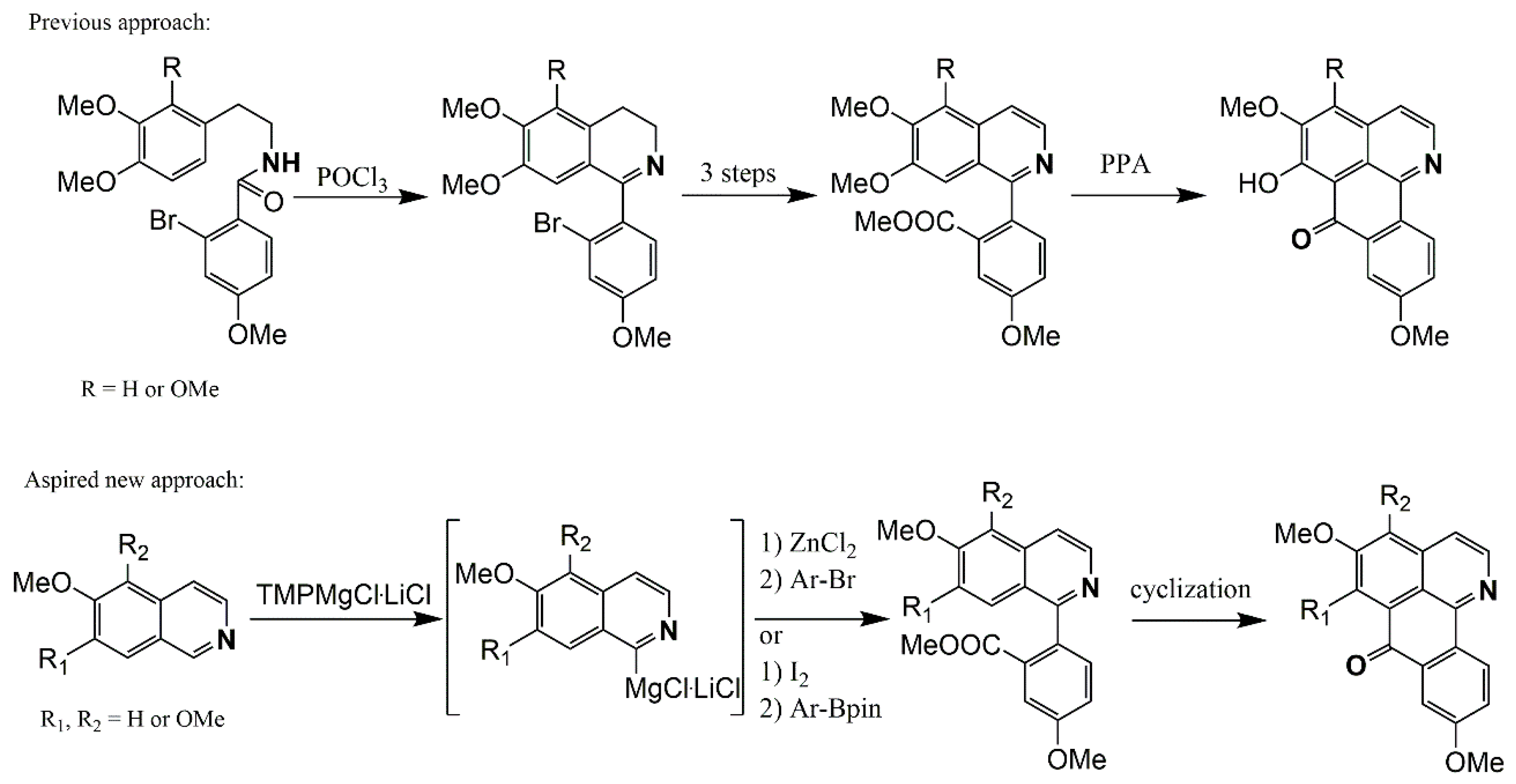
Synthesis
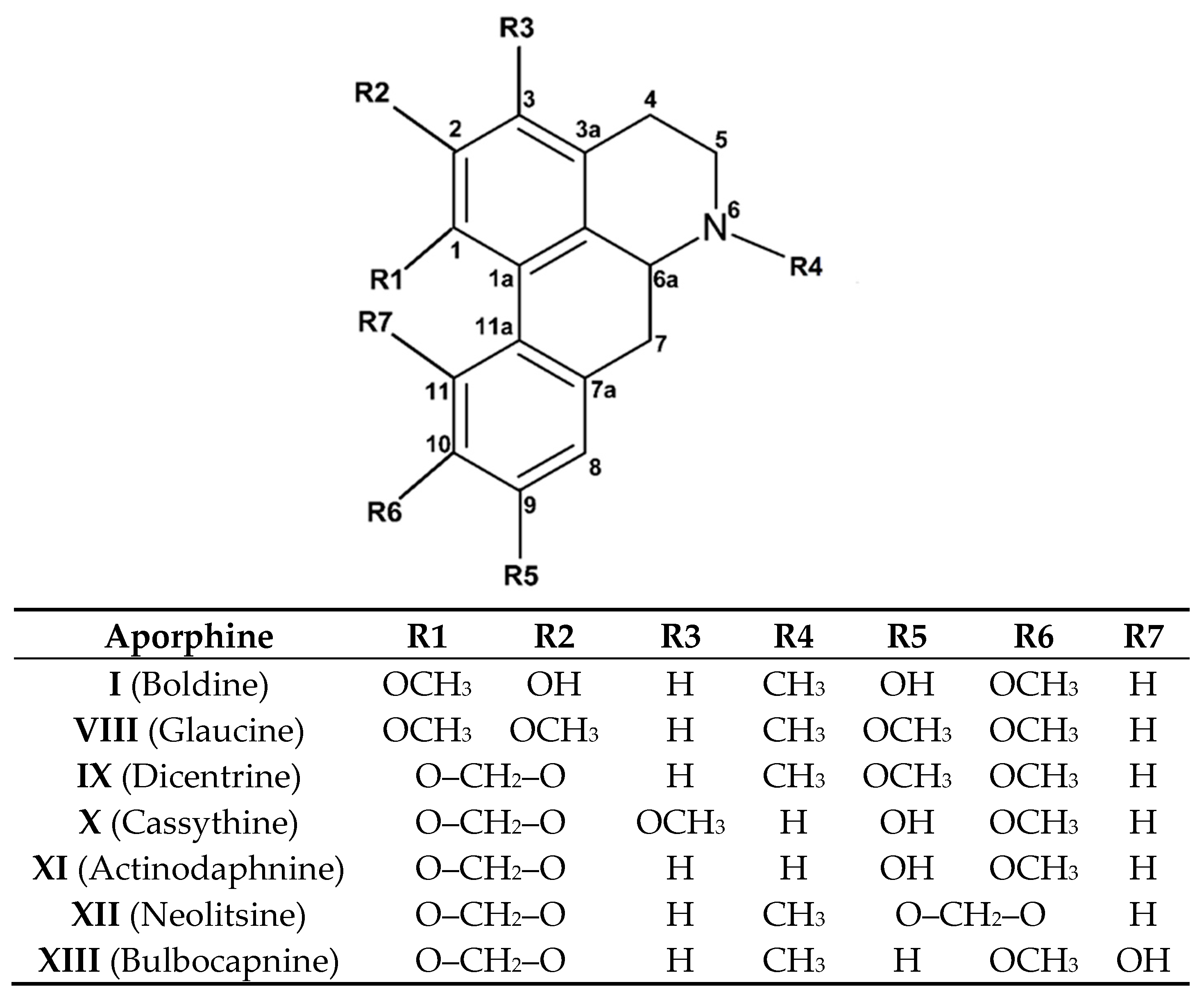
2.2. Aporphine Compounds: Boldine and Their Derivatives, Medicinal Application of Natural Aporphines
Extractive Methodology, Synthesis, and Purification
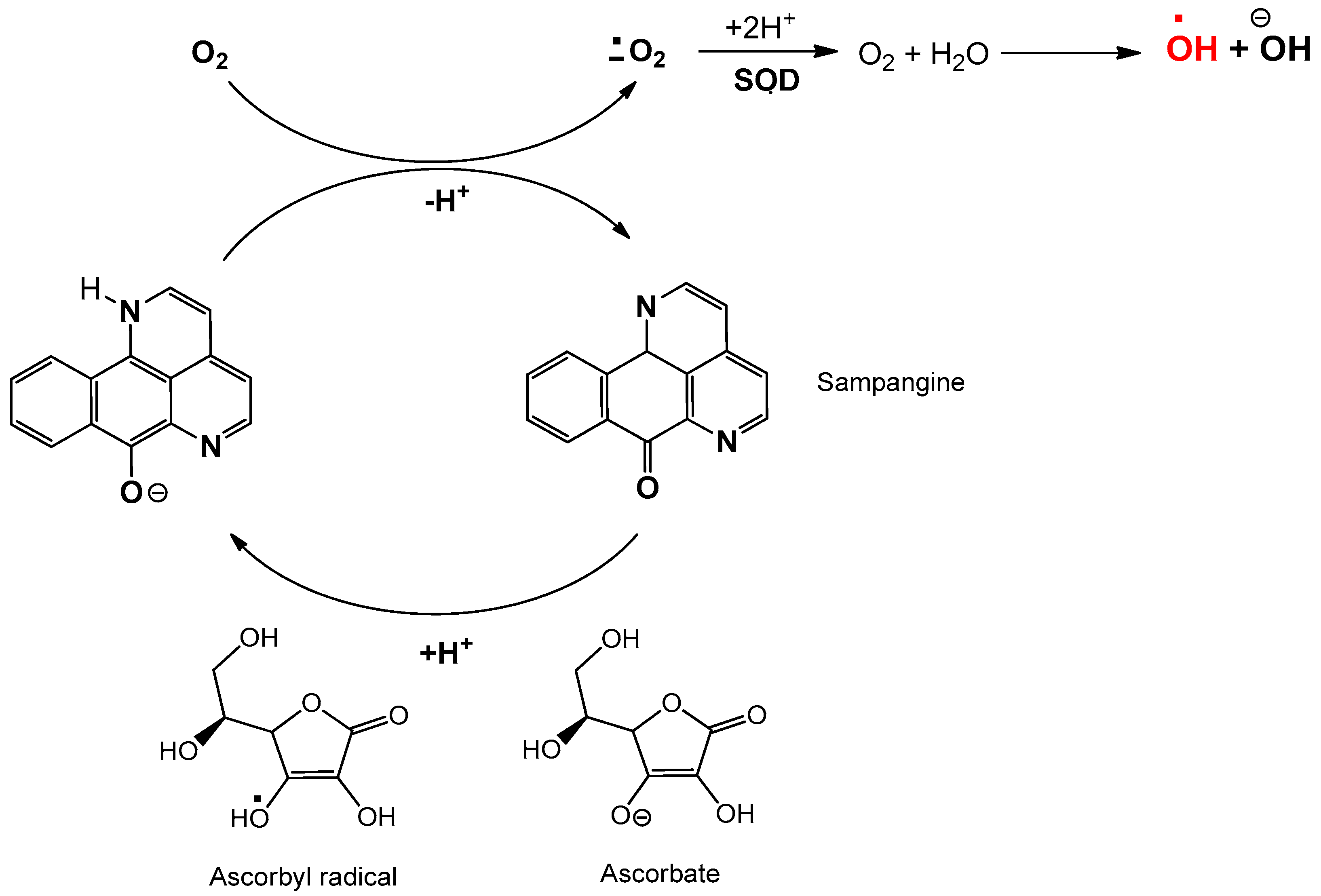
2.3. Sampangine
3. Oxoisoaporphines, Sampangines, Boldine: Structural and Cytotoxicity Correlation
3.1. Oxoisoaporphines as a Versatile Framework for Tuning Bioinorganic Properties
3.1.1. Docking Studies
3.1.2. Perspective
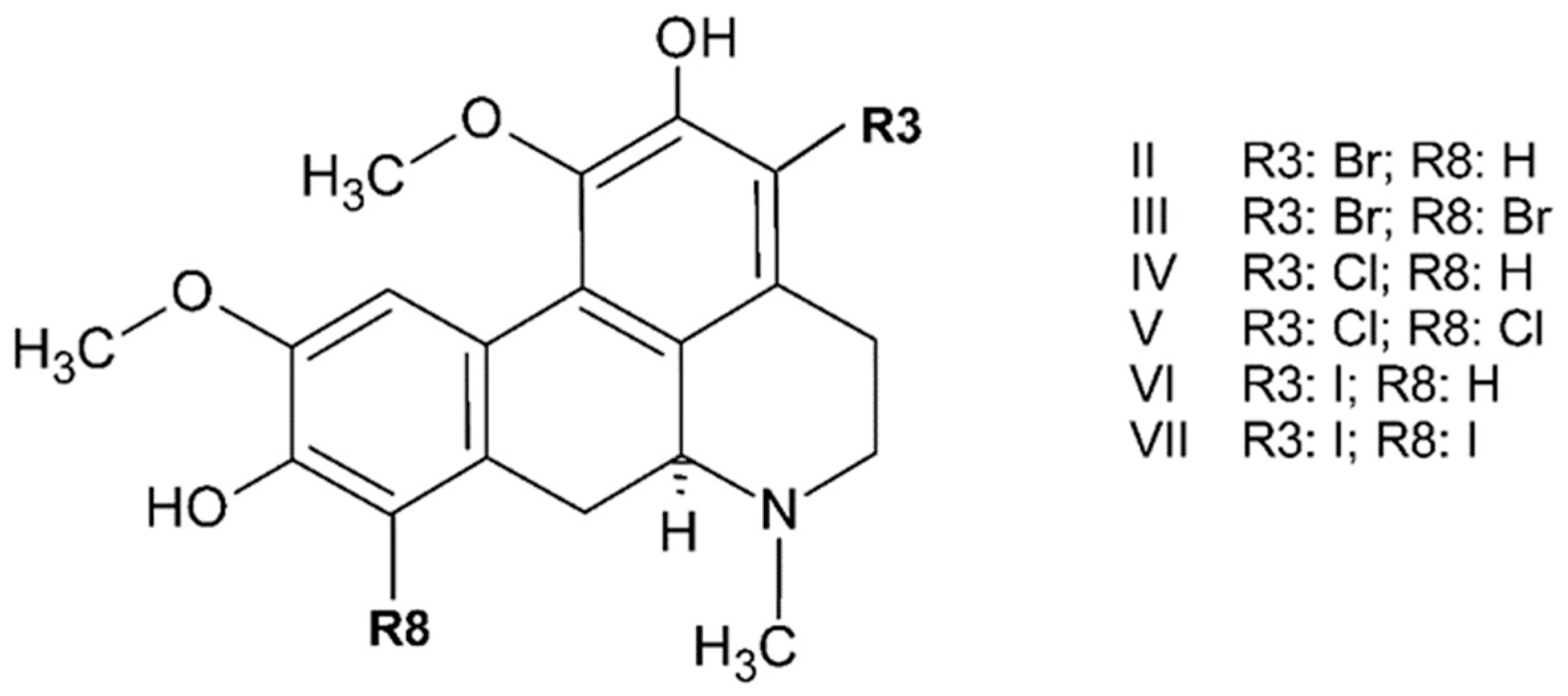
3.2. Boldine and Derivatives
3.2.1. Exceptional Family of Compounds in DNA Presence
3.2.2. Perspective
3.3. Sampangine: Anticancer Activity and Action Mechanism
3.3.1. Sampangine Derivatives
3.3.2. Perspective
4. Final Comments
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar]
- Goodman, L.S.; Wintrobe, M.M.; Dameshek, W.; Goodman, M.J.; Gilman, A.; McLennan, M.T. Nitrogen mustard therapy: Use of methyl-bis(beta-chloroethyl)amine hydrochloride and tris(beta-chloroethyl)amine hydrochloride for hodgkin’s disease, lymphosarcoma, leukemia and certain allied and miscellaneous disorders. J. Am. Med. Assoc. 1946, 132, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Neidle, S.; Thurston, D.E. Chemical approaches to the discovery and development of cancer therapies. Nat. Rev. Cancer 2005, 5, 285–296. [Google Scholar] [CrossRef] [PubMed]
- DeVita, V.T.; Chu, E. A history of cancer chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef]
- Avendaño, C.; Menéndez, J.C. General aspects of cancer chemotherapy. In Medicinal Chemistry of Anticancer Drugs, 2nd ed.; Avendaño, C., Menéndez, J.C., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 1–22. [Google Scholar]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar]
- Gerhardt, D.; Horn, A.P.; Gaelzer, M.M.; Frozza, R.L.; Delgado-Cañedo, A.; Pelegrini, A.L.; Henriques, A.T.; Lenz, G.; Salbego, C. Boldine: A potential new antiproliferative drug against glioma cell lines. Investig. New Drugs 2009, 27, 517–525. [Google Scholar] [CrossRef]
- Berdis, A.J. Inhibiting DNA polymerases as a therapeutic intervention against cancer. Front. Mol. Biosci. 2017, 4, 78. [Google Scholar] [CrossRef]
- Ubhi, T.; Brown, G.W. Exploiting DNA replication stress for cancer treatment. Cancer Res. 2019, 79, 1730–1739. [Google Scholar] [CrossRef]
- Yang, Y.; Karakhanova, S.; Werner, J.; Bazhin, A.V. Reactive oxygen species in cancer biology and anticancer therapy. Curr. Med. Chem. 2013, 20, 3677–3692. [Google Scholar] [CrossRef]
- Peng, X.; Gandhi, V. ROS-activated anticancer prodrugs: A new strategy for tumor-specific damage. Ther. Deliv. 2012, 3, 823–833. [Google Scholar]
- Bai, J.; Li, Y.; Zhang, G. Cell cycle regulation and anticancer drug discovery. Cancer Biol. Med. 2017, 14, 348–362. [Google Scholar] [PubMed]
- Shapiro, G.I.; Harper, J.W. Anticancer drug targets: Cell cycle and checkpoint control. J. Clin. Investig. 1999, 104, 1645–1653. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A target for anticancer therapy. Int. J. Mol. Sci 2018, 19, 448. [Google Scholar] [CrossRef] [PubMed]
- Foerster, F.; Braig, S.; Moser, C.; Kubisch, R.; Busse, J.; Wagner, E.; Schmoeckel, E.; Mayr, D.; Schmitt, S.; Huettel, S.; et al. Targeting the actin cytoskeleton: Selective antitumor action via trapping PKCε. Cell Death Dis. 2014, 5, e1398. [Google Scholar] [CrossRef] [PubMed]
- de Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- Greider, C.W. Telomeres. Curr. Opin. Cell Biol. 1991, 3, 444–451. [Google Scholar] [CrossRef]
- Veverka, P.; Janovič, T.; Hofr, C. Quantitative biology of human shelterin and telomerase: Searching for the weakest point. Int. J. Mol. Sci. 2019, 20, 3186. [Google Scholar] [CrossRef]
- Shay, J.W. Role of telomeres and telomerase in aging and cancer. Cancer Discov. 2016, 6, 584–593. [Google Scholar] [CrossRef]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific association of human telomerase activity with immortal cells and cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef]
- Shay, J.W.; Wright, W.E. Telomerase therapeutics for cancer: Challenges and new directions. Nat. Rev. Drug Discov. 2006, 5, 577–584. [Google Scholar] [CrossRef]
- Ganesan, K.; Xu, B. Telomerase inhibitors from natural products and their anticancer potential. Int. J. Mol. Sci. 2018, 19, 13. [Google Scholar] [CrossRef] [PubMed]
- Autexier, C.; Lue, N.F. The structure and function of telomerase reverse transcriptase. Annu. Rev. Biochem. 2006, 75, 493–517. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Tang, W.-J.; Shi, J.B.; Liu, M.M.; Liu, X.-H. Therapeutic strategies for targeting telomerase in cancer. In Medicinal Research Reviews; John Wiley and Sons: Hoboken, NJ, USA, 2019. [Google Scholar]
- Gillis, A.J.; Schuller, A.P.; Skordalakes, E. Structure of the Tribolium castaneum telomerase catalytic subunit TERT. Nature 2008, 455, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Gavory, G.; Symmons, M.F.; Krishnan Ghosh, Y.; Klenerman, D.; Balasubramanian, S. Structural analysis of the catalytic core of human telomerase RNA by FRET and molecular modeling. Biochemistry 2006, 45, 13304–13311. [Google Scholar] [CrossRef]
- Sengupta, A.; Ganguly, A.; Chowdhury, S. Promise of G-quadruplex structure binding ligands as epigenetic modifiers with anti-cancer effects. Molecules 2019, 24, 582. [Google Scholar] [CrossRef]
- Fernando, H.; Reszka, A.P.; Huppert, J.; Ladame, S.; Rankin, S.; Venkitaraman, A.R.; Neidle, S.; Balasubramanian, S. A conserved quadruplex motif located in a transcription activation site of the human c-kit oncogene. Biochemistry 2006, 45, 7854–7860. [Google Scholar] [CrossRef]
- Siddiqui-Jain, A.; Grand, C.L.; Bearss, D.J.; Hurley, L.H. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYCranscription. Proc. Natl. Acad. Sci. USA 2002, 99, 11593–11598. [Google Scholar] [CrossRef]
- Sun, D.; Thompson, B.; Cathers, B.E.; Salazar, M.; Kerwin, S.M.; Trent, J.O.; Jenkins, T.C.; Neidle, S.; Hurley, L.H. Inhibition of human telomerase by a G-quadruplex-interactive compound. J. Med. Chem. 1997, 40, 2113–2116. [Google Scholar] [CrossRef]
- Cao, Q.; Li, Y.; Freisinger, E.; Qin, P.Z.; Sigel, R.K.O.; Mao, Z.-W. G-quadruplex DNA targeted metal complexes acting as potential anticancer drugs. Inorg. Chem. Front. 2017, 4, 10–32. [Google Scholar] [CrossRef]
- Nasiri, H.R.; Hohmann, K.; Hatemler, M.G.; Plodek, A.; Bracher, F.; Schwalbe, H. In vitro production of reactive oxygen species (ROS) by sampangine. Med. Chem. Res. 2017, 26, 1170–1175. [Google Scholar] [CrossRef]
- Chio, I.I.C.; Tuveson, D.A. ROS in cancer: The burning question. Trends Mol. Med. 2017, 23, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2008, 417, 1–13. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Toyokuni, S.; Okamoto, K.; Yodoi, J.; Hiai, H. Persistent oxidative stress in cancer. FEBS Lett. 1995, 358, 1–3. [Google Scholar] [CrossRef]
- Oberley, L.W.; Buettner, G.R. Role of superoxide dismutase in cancer: A review. Cancer Res. 1979, 39, 1141–1149. [Google Scholar] [PubMed]
- Ozben, T. Oxidative stress and apoptosis: Impact on cancer therapy. J. Pharm. Sci. 2007, 96, 2181–2196. [Google Scholar] [CrossRef]
- Takahashi, A.; Ohtani, N.; Yamakoshi, K.; Iida, S.-i.; Tahara, H.; Nakayama, K.; Nakayama, K.I.; Ide, T.; Saya, H.; Hara, E. Mitogenic signalling and the p16INK4a–Rb pathway cooperate to enforce irreversible cellular senescence. Nat. Cell Biol. 2006, 8, 1291–1297. [Google Scholar] [CrossRef]
- Yang, H.; Villani, R.M.; Wang, H.; Simpson, M.J.; Roberts, M.S.; Tang, M.; Liang, X. The role of cellular reactive oxygen species in cancer chemotherapy. J. Exp. Clin. Cancer Res. 2018, 37, 266. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Tang, H.; Wang, X.-D.; Wei, Y.-B.; Huang, S.-L.; Huang, Z.-S.; Tan, J.-H.; An, L.-K.; Wu, J.-Y.; Chan, A.S.-C.; Gu, L.-Q. Oxoisoaporphine alkaloid derivatives: Synthesis, DNA binding affinity and cytotoxicity. Eur. J. Med. Chem. 2008, 43, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Wei, Y.-B.; Zhang, C.; Ning, F.-X.; Qiao, W.; Huang, S.-L.; Ma, L.; Huang, Z.-S.; Gu, L.-Q. Synthesis, biological evaluation and molecular modeling of oxoisoaporphine and oxoaporphine derivatives as new dual inhibitors of acetylcholinesterase/butyrylcholinesterase. Eur. J. Med. Chem. 2009, 44, 2523–2532. [Google Scholar] [CrossRef] [PubMed]
- Castro-Castillo, V.; Suárez-Rozas, C.; Pabón, A.; Pérez, E.G.; Cassels, B.K.; Blair, S. Synthesis and antiplasmodial activity of some 1-azabenzanthrone derivatives. Bioorg. Med. Chem. Lett. 2013, 23, 327–329. [Google Scholar] [CrossRef]
- Wei, Z.-Z.; Qin, Q.-P.; Chen, J.-N.; Chen, Z.-F. Oxoisoaporphine as potent telomerase inhibitor. Molecules 2016, 21, 1534. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, L.; Sun, J. Oxoisoaporphine alkaloids: Prospective anti-Alzheimer’s disease, anticancer, and antidepressant agents. ChemMedChem 2018, 13, 1262–1274. [Google Scholar] [CrossRef]
- Huang, L.; Luo, Y.; Pu, Z.; Kong, X.; Fu, X.; Xing, H.; Wei, S.; Chen, W.; Tang, H. Oxoisoaporphine alkaloid derivative 8-1 reduces Aβ1-42 secretion and toxicity in human cell and Caenorhabditis elegans models of Alzheimer’s disease. Neurochem. Int. 2017, 108, 157–168. [Google Scholar] [CrossRef]
- Nabavi, S.; Kessels, H.W.; Alfonso, S.; Aow, J.; Fox, R.; Malinow, R. Metabotropic NMDA receptor function is required for NMDA receptor-dependent long-term depression. Proc. Natl. Acad. Sci. USA 2013, 110, 4027–4032. [Google Scholar] [CrossRef]
- Sobarzo-Sánchez, E.; Soto, P.G.; Valdés Rivera, C.; Sánchez, G.; Hidalgo, M.E. Applied biological and physicochemical activity of isoquinoline alkaloids: Oxoisoaporphine and boldine. Molecules 2012, 17, 10958–10970. [Google Scholar] [CrossRef]
- Yu, B.-W.; Meng, L.-H.; Chen, J.-Y.; Zhou, T.-X.; Cheng, K.-F.; Ding, J.; Qin, G.-W. Cytotoxic oxoisoaporphine alkaloids from Menispermum dauricum. J. Nat. Prod. 2001, 64, 968–970. [Google Scholar] [CrossRef]
- Zhang, X.; Ye, W.; Zhao, S.; Che, C.-T. Isoquinoline and isoindole alkaloids from menispermum dauricum. Phytochemistry 2004, 65, 929–932. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, Y.; Babiker, H.A.A.; Inanaga, S.; Kato, M.; Isogai, A. Oxoisoaporphines from menispermum dauricum. Phytochemistry 1999, 52, 1431–1435. [Google Scholar] [CrossRef]
- Hu, S.-M.; Xu, S.-X.; Yao, X.-S.; Cui, C.-B.; Tezuka, Y.; Kikuchi, T. Dauricoside, a new glycosidal alkaloid having an inhibitory activity against blood-platelet aggregation. Chem. Pharm. Bull. 1993, 41, 1866–1868. [Google Scholar] [CrossRef] [PubMed]
- Kunitomo, J.; Kaede, S.; Satoh, M. The structure of 2, 3-dihydromenisporphine and the synthesis of dauriporphine, oxoisoaporphine alkaloids from menispermum dauricum DC. Chem. Pharm. Bull. 1985, 33, 2778–2782. [Google Scholar] [CrossRef]
- Kunitomo, J.; Satoh, M.; Shingu, T. Structure and synthesis of menisporphine, a new type of isoquinoline alkaloid: Alkaloids of menispermum dauricum DC. Tetrahedron 1983, 39, 3261–3265. [Google Scholar] [CrossRef]
- Hou, C.; Xue, H. Studies on the chemical constituents of menispermum dauricum DC. Acta Pharm. Sin. 1985, 20, 112–117. [Google Scholar]
- Takani, M.; Takasu, Y.; Takahashi, K. Studies on constituents of medicinal plants. XXIII. Constituents of the vines of menispermum dauricum DC. Chem. Pharm. Bull. 1983, 31, 3091–3093. [Google Scholar] [CrossRef]
- Jia-Qing, Q. Cardiovascular pharmacological effects of bisbenzylisoquinoline alkaloid derivatives. Acta Pharmacol. Sin. 2016, 23, 1086–1092. [Google Scholar]
- Wang, F.; Qu, L.; Lv, Q.; Guo, L.J. Effect of phenolic alkaloids from menispermum dauricum on myocardial-cerebral ischemia-reperfusion injury in rabbits. Acta Pharm. Sin. 2001, 22, 1130–1134. [Google Scholar]
- Schmeda-Hirschmann, G.; Rodriguez, J.A.; Theoduloz, C.; Astudillo, S.L.; Feresin, G.E.; Tapia, A. Free-radical scavengers and antioxidants from peumus boldus mol. (“Boldo”). Free Radic. Res. 2003, 37, 447–452. [Google Scholar] [CrossRef]
- Kunitomo, J.; Satoh, M. Structure of menisporphine: A new type of isoquinoline alkaloid. Chem. Pharm. Bull. 1982, 30, 2659–2660. [Google Scholar] [CrossRef]
- Sobarzo-Sánchez, E.; Cassels, B.K.; Jullian, C.; Castedo, L. Complete structural and spectral assignment of oxoisoaporphines by HMQC and HMBC experiments. Mag. Reson. Chem. 2003, 41, 296–300. [Google Scholar] [CrossRef]
- Iwashima, S.; Ueda, T.; Honda, H.; Tsujioka, T.; Ohno, M.; Aoki, J.; Kan, T. Synthesis and physical properties of azapolycyclic hydrocarbons. Part 1. Preparation of 1-azabenzanthrone and its condensation products and their structural determination. J. Chem. Soc. Perkin Trans. 1984, 2177–2187. [Google Scholar] [CrossRef]
- Castro-Castillo, V.; Rebolledo-Fuentes, M.; Theoduloz, C.; Cassels, B.K. Synthesis of lakshminine and antiproliferative Testing of related oxoisoaporphines. J. Nat. Prod. 2010, 73, 1951–1953. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-F.; Qin, Q.-P.; Qin, J.-L.; Liu, Y.-C.; Huang, K.-B.; Li, Y.-L.; Meng, T.; Zhang, G.-H.; Peng, Y.; Luo, X.-J.; et al. Stabilization of G-Quadruplex DNA, inhibition of telomerase activity, and tumor cell apoptosis by organoplatinum(II) complexes with oxoisoaporphine. J. Med. Chem. 2015, 58, 2159–2179. [Google Scholar] [CrossRef]
- Achari, K.M.M.; Karthick, M.; Ramanathan, C.R. Metal free synthesis of functionalized 1-aryl isoquinolines via iodine mediated oxidative dehydrogenation and ring opening of lactam in isoindoloisoquinolinones. J. Chem. Sci. 2017, 129, 679–690. [Google Scholar] [CrossRef]
- Walker, G.N.; Kempton, R.J. Aromatic demethoxylation in the cyclization of 3-(.beta.-dialkoxyarylethylamino)phthalides to 2, 3-dihydro-7H-dibenzo[da,h]quinolines. J. Org. Chem. 1971, 36, 1413–1416. [Google Scholar] [CrossRef]
- Fodor, G.; Nagubandi, S. Correlation of the von Braun, Ritter, Bischler-Napieralski, Beckmann and Schmidt reactions via nitrilium salt intermediates. Tetrahedron 1980, 36, 1279–1300. [Google Scholar] [CrossRef]
- Kupchan, S.M.; O’Brien, P.F. Novel oxidative photochemical aporphine synthesis. Total synthesis of corunnine and nandazurine. J. Chem. Soc. Chem. Commun. 1973, 915–916. [Google Scholar] [CrossRef]
- Kupchan, S.M.; Moniot, J.L.; Kanojia, R.M.; O’Brien, J.B. Photochemical synthesis of aporphines. J. Org. Chem. 1971, 36, 2413–2418. [Google Scholar] [CrossRef]
- Castedo, L.; Suau, R.; Villaverde, C.; Saá, J.M. On the structure of glauvine: Synthesis of oxolirioferine, norlirioferine and N,O-diacetylnorlirioferine. Heterocycles 1980, 14, 1131–1134. [Google Scholar] [CrossRef]
- Kessar, S.V.; Gupta, Y.P.; Yadav, V.S.; Narula, M.; Mohammad, T. Synthetic photochemistry. synthesis of (±)oliveroline and (±)ushinsunine. Tetrahedron Lett. 1980, 21, 3307–3308. [Google Scholar] [CrossRef]
- Melzer, B.; Bracher, F. A divergent approach to benzylisoquinoline-type and oxoaporphine alkaloids via regioselective direct ring metalation of alkoxy isoquinolines. Org. Biomol. Chem. 2015, 13, 7664–7672. [Google Scholar] [CrossRef] [PubMed]
- Orito, K.; Uchiito, S.; Satoh, Y.; Tatsuzawa, T.; Harada, R.; Tokuda, M. Aryl radical cyclizations of 1-(2‘-Bromobenzyl)isoquinolines with AIBN−Bu3SnH: Formation of aporphines and indolo[2,1-a]isoquinolines. Org. Lett. 2000, 2, 307–310. [Google Scholar] [CrossRef]
- Cuny, G.D. Synthesis of (+/-)-aporphine utilizing Pictet-Spengler and intramolecular phenol ortho-arylation reactions. Tetrahedron Lett. 2004, 45, 5167–5170. [Google Scholar] [CrossRef]
- Chaudhary, S.; Pecic, S.; Legendre, O.; Harding, W.W. Microwave-Assisted direct biaryl coupling: First Application to the synthesis of aporphines. Tetrahedron Lett. 2009, 50, 2437–2439. [Google Scholar] [CrossRef]
- Jia, X.; Yang, D.; Zhang, S.; Cheng, J. Chelation-assisted palladium-catalyzed direct cyanation of 2-arylpyridine C−H bonds. Org. Lett. 2009, 11, 4716–4719. [Google Scholar] [CrossRef]
- Chaitanya, M.; Yadagiri, D.; Anbarasan, P. Rhodium catalyzed cyanation of chelation assisted C–H bonds. Org. Lett. 2013, 15, 4960–4963. [Google Scholar] [CrossRef]
- Krasovskiy, A.; Krasovskaya, V.; Knochel, P. Mixed Mg/Li amides of the type R2NMgCl⋅LiCl as highly efficient bases for the regioselective generation of functionalized aryl and heteroaryl magnesium compounds. Angew. Chem. Int. Ed. 2006, 45, 2958–2961. [Google Scholar] [CrossRef]
- Metzger, A.; Schade, M.A.; Knochel, P. LiCl-mediated preparation of highly functionalized benzylic zinc chlorides. Org. Lett. 2008, 10, 1107–1110. [Google Scholar] [CrossRef]
- Melzer, B.C.; Bracher, F. A novel approach to oxoisoaporphine alkaloids via regioselective metalation of alkoxy isoquinolines. Beilstein J. Org. Chem. 2017, 13, 1564–1571. [Google Scholar] [CrossRef] [PubMed]
- Leboeuf, M.; Cavé, A.; Bhaumik, P.K.; Mukherjee, B.; Mukherjee, R. The phytochemistry of the annonaceae. Phytochemistry 1980, 21, 2783–2813. [Google Scholar] [CrossRef]
- Bannach, R.; Valenzuela, A.; Cassels, B.K.; Núnez-Vergara, L.J.; Speisky, H. Cytoprotective and antioxidant effects of boldine on tert-butyl hydroperoxide—Induced damage to isolated hepatocytes. Cell Biol. Toxicol. 1996, 12, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Speisky, H.; Cassels, B.K. Boldo and boldine: An emerging case of natural drug development. Pharmacol. Res. 1994, 29, 1–12. [Google Scholar] [CrossRef]
- Kazemi Noureini, S.; Kheirabadi, M.; Masoumi, F.; Khosrogerdi, F.; Zarei, Y.; Suárez-Rozas, C.; Salas-Norambuena, J.; Kennedy Cassels, B. Telomerase Inhibition by a new synthetic derivative of the aporphine alkaloid boldine. Int. J. Mol. Sci. 2018, 19, 1239. [Google Scholar] [CrossRef]
- Kazemi Noureini, S.; Tanavar, F. Boldine, a natural aporphine alkaloid, inhibits telomerase at non-toxic concentrations. Chem. Biol. Interact. 2015, 231, 27–34. [Google Scholar] [CrossRef]
- Paydar, M.; Kamalidehghan, B.; Wong, Y.L.; Wong, W.F.; Looi, C.Y.; Mustafa, M.R. Evaluation of cytotoxic and chemotherapeutic properties of boldine in breast cancer using in vitro and in vivo models. Drug Des. Dev. Ther. 2014, 8, 719–733. [Google Scholar]
- Reveco, P.G.; Asencio, M.; Sanguinetti, M.E.; Thomet, F.A. The Synthesis of methoxymethyl derivatives of boldine: Versatile protected precursors of substituted boldine derivatives. Synth. Commun. 2005, 35, 341–347. [Google Scholar] [CrossRef]
- Hughes, D.W.; Genest, K.; Skakum, W. Alkaloids of Peumus boldus. Isolation of (+) reticuline and isoboldine. J. Pharm. Sci. 1968, 57, 1023–1025. [Google Scholar] [CrossRef]
- Cámara, C.I.; Bornancini, C.A.; Cabrera, J.L.; Ortega, M.G.; Yudi, L.M. Quantitative analysis of boldine alkaloid in natural extracts by cyclic voltammetry at a liquid–liquid interface and validation of the method by comparison with high performance liquid chromatography. Talanta 2010, 83, 623–630. [Google Scholar] [CrossRef]
- Pharmacopoeia, E. Boldo leaf. In European Pharmacopoeia, 8th ed.; Council of Europe: Strasbourg, French, 2013; pp. 1188–1990. [Google Scholar]
- Fuentes-Barros, G.; Castro-Saavedra, S.; Liberona, L.; Acevedo-Fuentes, W.; Tirapegui, C.; Mattar, C.; Cassels, B.K. Variation of the alkaloid content of Peumus boldus (boldo). Fitoterapia 2018, 127, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Gafner, S.; Dietz, B.M.; McPhail, K.L.; Scott, I.M.; Glinski, J.A.; Russell, F.E.; McCollom, M.M.; Budzinski, J.W.; Foster, B.C.; Bergeron, C.; et al. Alkaloids from eschscholzia californica and their capacity to inhibit binding of [3H]8-Hydroxy-2-(di-N-propylamino)tetralin to 5-HT1A receptors in vitro. J. Nat. Prod. 2006, 69, 432–435. [Google Scholar] [CrossRef] [PubMed]
- Cassels, B.K.; Asencio, M.; Conget, P.; Speisky, H.; Videla, L.A.; Lissi, E.A. Structure-antioxidative activity relationships in benzylisoquinoline alkaloids. Pharmacol. Res. 1995, 31, 103–107. [Google Scholar] [CrossRef]
- Sobarzo-Sánchez, E.M.; Arbaoui, J.; Protais, P.; Cassels, B.K. Halogenated boldine derivatives with enhanced monoamine receptor selectivity. J. Nat. Prod. 2000, 63, 480–484. [Google Scholar] [CrossRef]
- Rao, J.U.M.; Giri, G.S.; Hanumaiah, T.; Rao, K.V.J. Sampangine, a new alkaloid from cananga odorata. J. Nat. Prod. 1986, 49, 346–347. [Google Scholar] [CrossRef]
- Kluza, J.; Mazinghien, R.; Degardin, K.; Lansiaux, A.; Bailly, C. Induction of apoptosis by the plant alkaloid sampangine in human HL-60 leukemia cells is mediated by reactive oxygen species. Eur. J. Pharmacol. 2005, 525, 32–40. [Google Scholar] [CrossRef]
- Mahdi, F.; Morgan, J.B.; Liu, W.; Agarwal, A.K.; Jekabsons, M.B.; Liu, Y.; Zhou, Y.-D.; Nagle, D.G. Sampangine (a copyrine alkaloid) exerts biological activities through cellular redox cycling of Its quinone and semiquinone intermediates. J. Nat. Prod. 2015, 78, 3018–3023. [Google Scholar] [CrossRef]
- Zjawiony, J.K.; Srivastava, A.R.; Hufford, C.D.; Clark, A.M. Chemistry of sampangines. Heterocycles 1994, 39, 779–800. [Google Scholar]
- Sharma, V.; Sharma, P.C.; Kumar, V. A mini review on pyridoacridines: Prospective lead compounds in medicinal chemistry. J. Adv. Res. 2015, 6, 63–71. [Google Scholar] [CrossRef]
- Eder, C.; Schupp, P.; Proksch, P.; Wray, V.; Steube, K.; Müller, C.E.; Frobenius, W.; Herderich, M.; van Soest, R.W.M. Bioactive pyridoacridine alkaloids from the Micronesian Sponge Oceanapia sp. J. Nat. Prod. 1998, 61, 301–305. [Google Scholar] [CrossRef]
- Rezler, E.M.; Bearss, D.J.; Hurley, L.H. Telomeres and telomerases as drug targets. Curr. Opin. Pharmacol. 2002, 2, 415–423. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Hurley, L.H.; Neidle, S. Targeting G-quadruplexes in gene promoters: A novel anticancer strategy? Nat. Rev. Drug Discov. 2011, 10, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y. Chemistry in human telomere biology: Structure, function and targeting of telomere DNA/RNA. Chem. Soc. Rev. 2011, 40, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Sekaran, V.; Soares, J.; Jarstfer, M.B. Telomere maintenance as a target for drug discovery. J. Med. Chem. 2014, 57, 521–538. [Google Scholar] [CrossRef]
- Mergny, J.-L.; Riou, J.-F.; Mailliet, P.; Teulade-Fichou, M.-P.; Gilson, E. Natural and pharmacological regulation of telomerase. Nucleic Acids Res. 2002, 30, 839–865. [Google Scholar] [CrossRef]
- Phan, A.T.; Patel, D.J. Two-repeat human telomeric d(TAGGGTTAGGGT) sequence forms interconverting parallel and antiparallel G-quadruplexes in solution: Distinct topologies, thermodynamic properties, and folding/unfolding kinetics. J. Am. Chem. Soc. 2003, 125, 15021–15027. [Google Scholar] [CrossRef]
- Ying, L.; Green, J.J.; Li, H.; Klenerman, D.; Balasubramanian, S. Studies on the structure and dynamics of the human telomeric G quadruplex by single-molecule fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. USA 2003, 100, 14629–14634. [Google Scholar] [CrossRef]
- Parkinson, G.N.; Lee, M.P.H.; Neidle, S. Crystal structure of parallel quadruplexes from human telomeric DNA. Nature 2002, 417, 876–880. [Google Scholar] [CrossRef]
- Neidle, S.; Parkinson, G.N. The structure of telomeric DNA. Curr. Opin. Struct. Biol. 2003, 13, 275–283. [Google Scholar] [CrossRef]
- Qin, Q.-P.; Qin, J.-L.; Meng, T.; Yang, G.-A.; Wei, Z.-Z.; Liu, Y.-C.; Liang, H.; Chen, Z.-F. Preparation of 6/8/11-amino/chloro-oxoisoaporphine and group-10 metal complexes and evaluation of their in vitro and in vivo antitumor activity. Sci. Rep. 2016, 6, 37644. [Google Scholar] [CrossRef]
- Cheng, J.-J.; Tsai, T.-H.; Lin, L.-C. New alkaloids and cytotoxic principles from Sinomenium acutum. Planta Med. 2012, 78, 1873–1877. [Google Scholar] [CrossRef] [PubMed]
- Andrews, L.G.; Tollefsbol, T.O. Methods of Telomerase Inhibition; Humana Press: Totowa, NJ, USA, 2007; Volume 405, pp. 1–8. [Google Scholar]
- Zhang, Q.; Kim, N.-K.; Feigon, J. Architecture of human telomerase RNA. Proc. Natl. Acad. Sci. USA 2011, 108, 20325–20332. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Brown, A.F.; Wu, J.; Xue, J.; Bley, C.J.; Rand, D.P.; Wu, L.; Zhang, R.; Chen, J.J.L.; Lei, M. Structural basis for protein-RNA recognition in telomerase. Nat. Struct. Mol. Biol. 2014, 21, 507. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Liu, N.; Dong, G.; Jiang, Y.; Liu, Y.; He, X.; Huang, Y.; He, S.; Chen, W.; Li, Z.; et al. Scaffold hopping of sampangine: Discovery of potent antifungal lead compound against Aspergillus fumigatus and Cryptococcus neoformans. Bioorg. Med. Chem. Lett. 2014, 24, 4090–4094. [Google Scholar] [CrossRef]
- Bryan, C.; Rice, C.; Hoffman, H.; Harkisheimer, M.; Sweeney, M.; Skordalakes, E. Structural basis of telomerase inhibition by the highly specific BIBR1532. Structure 2015, 23, 1934–1942. [Google Scholar] [CrossRef]
- Gehm, B.D.; McAndrews, J.M.; Chien, P.Y.; Jameson, J.L. Resveratrol, a polyphenolic compound found in grapes and wine, is an agonist for the estrogen receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 14138–14143. [Google Scholar] [CrossRef]
- Tan, T.-W.; Tsai, H.-R.; Lu, H.-F.; Lin, H.-L.; Tsou, M.-F.; Lin, Y.-T.; Tsai, H.-Y.; Chen, Y.-F.; Chung, J.-G. Curcumin-induced cell cycle arrest and apoptosis in human acute promyelocytic leukemia HL-60 cells via MMP changes and caspase-3 activation. Anticancer Res. 2006, 26, 4361–4371. [Google Scholar]
- Hoet, S.; Stévigny, C.; Block, S.; Opperdoes, F.; Colson, P.; Baldeyrou, B.; Lansiaux, A.; Bailly, C.; Quetin-Leclercq, J. Alkaloids from cassytha filiformis and related aporphines: Antitrypanosomal activity, cytotoxicity, and interaction with DNA and topoisomerases. Planta Med. 2004, 70, 407–413. [Google Scholar]
- Stévigny, C.; Block, S.; De Pauw-Gillet, M.C.; de Hoffmann, E.; Llabrès, G.; Adjakidjé, V.; Quetin-Leclercq, J. Cytotoxic aporphine alkaloids from cassytha filiformis. Planta Med. 2002, 68, 1042–1044. [Google Scholar] [CrossRef]
- Maung, K.; Palau, V.; Lightner, J.; Brannon, M.; Krishnan, K. Gamma-tocotrienol and simvastatin synergistically induce cytotoxicity in leukemia cell lines, K-562 and HL-60. Blood 2013, 122, 4927. [Google Scholar] [CrossRef]
- Kim, N.-Y.; Pae, H.-O.; Oh, G.-S.; Kang, T.-H.; Kim, Y.-C.; Rhew, H.-Y.; Chung, H.-T. Butein, a plant polyphenol, induces apoptosis concomitant with increased caspase-3 activity, decreased Bcl-2 expression and increased Bax expression in HL-60 cells. Pharmacol. Toxicol. 2001, 88, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Effenberger-Neidnicht, K.; Schobert, R. Combinatorial effects of thymoquinone on the anti-cancer activity of doxorubicin. Cancer Chemother. Pharmacol. 2011, 67, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Kluza, J.; Clark, A.M.; Bailly, C. Apoptosis induced by the alkaloid sampangine in HL-60 leukemia cells. Ann. N. Y. Acad. Sci. 2003, 1010, 331–334. [Google Scholar] [CrossRef] [PubMed]
- Joshi, B.; Li, L.; Taffe, B.G.; Zhu, Z.; Wahl, S.; Tian, H.; Ben-Josef, E.; Taylor, J.D.; Porter, A.T.; Tang, D.G. Apoptosis induction by a novel anti-prostate cancer compound, BMD188 (a fatty acid-containing hydroxamic acid), requires the mitochondrial respiratory chain. Cancer Res. 1999, 59, 4343–4355. [Google Scholar]
- Dassonneville, L.; Wattez, N.; Baldeyrou, B.; Mahieu, C.; Lansiaux, A.; Banaigs, B.; Bonnard, I.; Bailly, C. Inhibition of topoisomerase II by the marine alkaloid ascididemin and induction of apoptosis in leukemia cells. Biochem. Pharmacol. 2000, 60, 527–537. [Google Scholar] [CrossRef]
- Matsumoto, S.S.; Biggs, J.; Copp, B.R.; Holden, J.A.; Barrows, L.R. Mechanism of ascididemin-induced cytotoxicity. Chem. Res. Toxicol. 2003, 16, 113–122. [Google Scholar] [CrossRef]
- Salvioli, S.; Ardizzoni, A.; Franceschi, C.; Cossarizza, A. JC-1, but not DiOC6(3) or rhodamine 123, is a reliable fluorescent probe to assess ΔΨ changes in intact cells: Implications for studies on mitochondrial functionality during apoptosis. FEBS Lett. 1997, 411, 77–82. [Google Scholar] [CrossRef]
- Costantini, P.; Belzacq, A.-S.; Vieira, H.L.A.; Larochette, N.; de Pablo, M.A.; Zamzami, N.; Susin, S.A.; Brenner, C.; Kroemer, G. Oxidation of a critical thiol residue of the adenine nucleotide translocator enforces Bcl-2-independent permeability transition pore opening and apoptosis. Oncogene 2000, 19, 307. [Google Scholar] [CrossRef]
- Huang, Z.; Chen, K.; Xu, T.; Zhang, J.; Li, Y.; Li, W.; Agarwal, A.K.; Clark, A.M.; Phillips, J.D.; Pan, X. Sampangine inhibits heme biosynthesis in both yeast and human. Eukaryot. Cell 2011, 10, 1536–1544. [Google Scholar] [CrossRef]
- Li, Y.; Zhu, T.; Zhao, J.; Xu, B. Interactive enhancements of ascorbic acid and iron in hydroxyl radical generation in quinone redox cycling. Environ. Sci. Technol. 2012, 46, 10302–10309. [Google Scholar] [CrossRef]
- Beck, R.; Dejeans, N.; Glorieux, C.; Pedrosa, R.C.; Vásquez, D.; Valderrama, J.A.; Calderon, P.B.; Verrax, J. Molecular chaperone Hsp90 as a target for oxidant-based anticancer therapies. Curr. Med. Chem. 2011, 18, 2816–2825. [Google Scholar] [CrossRef] [PubMed]
- Felipe, K.B.; Benites, J.; Glorieux, C.; Sid, B.; Valenzuela, M.; Kviecinski, M.R.; Pedrosa, R.C.; Valderrama, J.A.; Levêque, P.; Gallez, B.; et al. Antiproliferative effects of phenylaminonaphthoquinones are increased by ascorbate and associated with the appearance of a senescent phenotype in human bladder cancer cells. Biochem. Biophys. Res. Commun. 2013, 433, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Kviecinski, M.R.; Pedrosa, R.C.; Felipe, K.B.; Farias, M.S.; Glorieux, C.; Valenzuela, M.; Sid, B.; Benites, J.; Valderrama, J.A.; Verrax, J.; et al. Inhibition of cell proliferation and migration by oxidative stress from ascorbate-driven juglone redox cycling in human bladder-derived T24 cells. Biochem. Biophys. Res. Commun. 2012, 421, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Vásquez, D.R.; Verrax, J.; Valderrama, J.A.; Calderon, P.B. Aminopyrimidoisoquinolinequinone (APIQ) redox cycling is potentiated by ascorbate and induces oxidative stress leading to necrotic-like cancer cell death. Investig. New Drugs 2012, 30, 1003–1011. [Google Scholar] [CrossRef]
- Semenza, G.L. Oxygen homeostasis. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 336–361. [Google Scholar] [CrossRef]
- Semenza, G.L. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. Mech. Dis. 2014, 9, 47–71. [Google Scholar] [CrossRef]
- Hodges, T.W.; Hossain, C.F.; Kim, Y.-P.; Zhou, Y.-D.; Nagle, D.G. Molecular-targeted antitumor agents: the saururus cernuus dineolignans manassantin b and 4-O-demethylmanassantin B are potent inhibitors of Hypoxia-activated HIF-1. J. Nat. Prod. 2004, 67, 767–771. [Google Scholar] [CrossRef]
- Lin, X.; David, C.A.; Donnelly, J.B.; Michaelides, M.; Chandel, N.S.; Huang, X.; Warrior, U.; Weinberg, F.; Tormos, K.V.; Fesik, S.W.; et al. A chemical genomics screen highlights the essential role of mitochondria in HIF-1 regulation. Proc. Natl. Acad. Sci. USA 2008, 105, 174–179. [Google Scholar] [CrossRef]
- Du, L.; Mahdi, F.; Datta, S.; Jekabsons, M.B.; Zhou, Y.-D.; Nagle, D.G. Structures and Mechanisms of antitumor agents: Xestoquinones uncouple cellular respiration and disrupt HIF signaling in human breast tumor cells. J. Nat. Prod. 2012, 75, 1553–1559. [Google Scholar] [CrossRef]
- Du, L.; Mahdi, F.; Jekabsons, M.B.; Nagle, D.G.; Zhou, Y.-D. Mammea E/BB, an isoprenylated dihydroxycoumarin protonophore that potently uncouples mitochondrial electron transport, disrupts Hypoxic signaling in tumor cells. J. Nat. Prod. 2010, 73, 1868–1872. [Google Scholar] [CrossRef]
- Jiang, Z.; Liu, N.; Hu, D.; Dong, G.; Miao, Z.; Yao, J.; He, H.; Jiang, Y.; Zhang, W.; Wang, Y.; et al. The discovery of novel antifungal scaffolds by structural simplification of the natural product sampangine. Chem. Commun. 2015, 51, 14648–14651. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.K.; Xu, T.; Jacob, M.R.; Feng, Q.; Lorenz, M.C.; Walker, L.A.; Clark, A.M. Role of heme in the antifungal activity of the azaoxoaporphine alkaloid sampangine. Eukaryot. Cell 2008, 7, 387–400. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, B.; Pedrajas, J.R.; Padilla, C.A.; Bárcena, J.A. Thiol redox sensitivity of two key enzymes of heme biosynthesis and pentose phosphate pathways: Uroporphyrinogen decarboxylase and transketolase. Oxid. Med. Cell. Longev. 2013, 2013, 932472. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Lim, C.K.; De Matteis, F. Peroxylated and hydroxylated uroporphyrins: A study of their production in vitro in enzymic and chemical model systems. Biomed. Chromatogr. 1996, 10, 213–220. [Google Scholar] [CrossRef]
- Miller, D.M.; Woods, J.S. Redox activities of mercury-thiol complexes: Implications for mercury-induced porphyria and toxicity. Chem.Biol. Interact. 1993, 88, 23–35. [Google Scholar] [CrossRef]
- Woods, J.S.; Calas, C.A. Iron stimulation of free radical-mediated porphyrinogen oxidation by hepatic and renal mitochondria. Biochem. Biophys. Res. Commun. 1989, 160, 101–108. [Google Scholar] [CrossRef]
- Peterson, J.; Zjawiony, J.; Clark, A.; Hufford, C.; Graves, D.; Walker, L. Abstract, Interscience Conference on Antimicrobial Agents and Chemoterapy; Publons: Chicago, IL, USA, 1991. [Google Scholar]
- Bracher, F. Polycyclische aromatische Alkaloide, I. Synthese von Cleistopholin und Sampangin. Lieb. Ann. Chem. 1989, 1989, 87–88. [Google Scholar] [CrossRef]
- Peterson, J.R.; Zjawiony, J.K.; Liu, S.; Hufford, C.D.; Clark, A.M.; Rogers, R.D. Copyrine alkaloids: Synthesis, spectroscopic characterization, and antimycotic/antimycobacterial activity of A- and B-ring-functionalized sampangines. J. Med. Chem. 1992, 35, 4069–4077. [Google Scholar] [CrossRef]
- Orabi, K.Y. Microbial models of mammalian metabolism. Sampangines. In Studies in Natural Products Chemstry; Attaur, R., Ed.; Elsevier: Amsterdam, The Netherlands, 2000; Volume 23, pp. 3–49. [Google Scholar]
- Liu, S.C.; Oguntimein, B.; Hufford, C.D.; Clark, A.M. 3-Methoxysampangine, a novel antifungal copyrine alkaloid from Cleistopholis patens. Antimicrob. Agents Chemother. 1990, 34, 529–533. [Google Scholar] [CrossRef]
- Zjawiony, J.K.; Kbalil, A.A.; Clark, A.M.; Hufford, C.D.; Buolamwini, J.K. Studies on methoxylation in the 7H-naphtho[1,2,3-I,j][2,7]naphthyridin-7-one system. J. Heterocycl. Chem. 1997, 34, 1233–1237. [Google Scholar] [CrossRef]
- Isamu, K.; Zjawiony Jordan, K. Theoretical and experimental aspects of bromination of sampangine. Chem. Lett. 2000, 29, 568–569. [Google Scholar]
- Zjawiony, J.; Katsuyama, I.; Khalil, A.; Ren, J. The identification, mechanism, and improved synthesis of a new and unique heterocyclic system with a fused imidazole ring. Heterocycles 2001, 54, 721–726. [Google Scholar] [CrossRef]
- Claes, P.; Cappoen, D.; Mbala, B.M.; Jacobs, J.; Mertens, B.; Mathys, V.; Verschaeve, L.; Huygen, K.; De Kimpe, N. Synthesis and antimycobacterial activity of analogues of the bioactive natural products sampangine and cleistopholine. Eur. J. Med. Chem. 2013, 67, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.-Z.; Qin, Q.-P.; Meng, T.; Deng, C.-X.; Liang, H.; Chen, Z.-F. 5-Bromo-oxoisoaporphine platinum(II) complexes exhibit tumor cell cytotoxcicity via inhibition of telomerase activity and disruption of c-myc G-quadruplex DNA and mitochondrial functions. Eur. J. Med. Chem. 2018, 145, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Mink, K.; Bracher, F. Hetero Analogues of the antimicrobial alkaloids cleistopholine and sampangine. Arch. Pharm. 2007, 340, 429–433. [Google Scholar] [CrossRef]
- Shrivastava, S.; Jeengar, M.K.; Reddy, V.S.; Reddy, G.B.; Naidu, V.G.M. Anticancer effect of celastrol on human triple negative breast cancer: Possible involvement of oxidative stress, mitochondrial dysfunction, apoptosis and PI3K/Akt pathways. Exp. Mol. Pathol. 2015, 98, 313–327. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | IC50 (μM) | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 | 24 | |
| T-24 | 38.4 | 35 | 16.8 | |||||||||||||||||||||
| Hep-G2 | 20.5 | 18 | 17.4 | 19.7 | 8.14 | 9.7 | 6.4 | 12.1 | 21.02 | 3.9 | 8.2 | 7.7 | 12.9 | 29.8 | 128.5 | 141.2 | 108.3 | 60.6 | 127.3 | |||||
| SK-OV-3 | >100 | 40 | >100 | 130 | 141.9 | 143.6 | 154.1 | 55.9 | 90.2 | |||||||||||||||
| SK-OV-3/DDP | 13.9 | 17 | 36.8 | 85 | 145.1 | |||||||||||||||||||
| BEL-7402 | 54.7 | 50 | 36.7 | 95 | 69.0 | |||||||||||||||||||
| NCI-H460 | 66.9 | 57 | >100 | 35.5 | 2.1 | 9.2 | 4.2 | 5.0 | 30.3 | 2.4 | 16.7 | 6.7 | 9.1 | 19.3 | 95.3 | 94.7 | 91.0 | 88.1 | 72.4 | |||||
| HCT-8 | 60.2 | 56 | >100 | 80 | 112.5 | 135.8 | 167.1 | 42.7 | 52.3 | |||||||||||||||
| HL-7702 | 64.5 | 75 | >100 | 100 | 98.1 | 101.2 | 100.1 | 132.2 | 99.7 | |||||||||||||||
| A549 | >50 | 8.8 | 108.4 | 86.1 | 115.2 | 72.2 | ||||||||||||||||||
| HL-60 | >50 | >50 | ||||||||||||||||||||||
| MCF-7 | 6.2 | 3.0 | 20.4 | 11.8 | 17.5 | 5.1 | 12.7 | 28.1 | 4.3 | 8.0 | 9.1 | 27.4 | 11.5 | 33.1 | >100 | 0.39 | 122.9 | |||||||
| P-388 | 9.6 | 30.5 | ||||||||||||||||||||||
| H460 | 45.2 | >100 | 3.18 | |||||||||||||||||||||
| HT-29 | >100 | >100 | >100 | |||||||||||||||||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez-Arce, E.; Cancino, P.; Arias-Calderón, M.; Silva-Matus, P.; Saldías, M. Oxoisoaporphines and Aporphines: Versatile Molecules with Anticancer Effects. Molecules 2020, 25, 108. https://doi.org/10.3390/molecules25010108
Rodríguez-Arce E, Cancino P, Arias-Calderón M, Silva-Matus P, Saldías M. Oxoisoaporphines and Aporphines: Versatile Molecules with Anticancer Effects. Molecules. 2020; 25(1):108. https://doi.org/10.3390/molecules25010108
Chicago/Turabian StyleRodríguez-Arce, Esteban, Patricio Cancino, Manuel Arias-Calderón, Paul Silva-Matus, and Marianela Saldías. 2020. "Oxoisoaporphines and Aporphines: Versatile Molecules with Anticancer Effects" Molecules 25, no. 1: 108. https://doi.org/10.3390/molecules25010108
APA StyleRodríguez-Arce, E., Cancino, P., Arias-Calderón, M., Silva-Matus, P., & Saldías, M. (2020). Oxoisoaporphines and Aporphines: Versatile Molecules with Anticancer Effects. Molecules, 25(1), 108. https://doi.org/10.3390/molecules25010108