Synthesis and Structural Determination of New Brassinosteroid 24-Nor-5α-Cholane Type Analogs

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
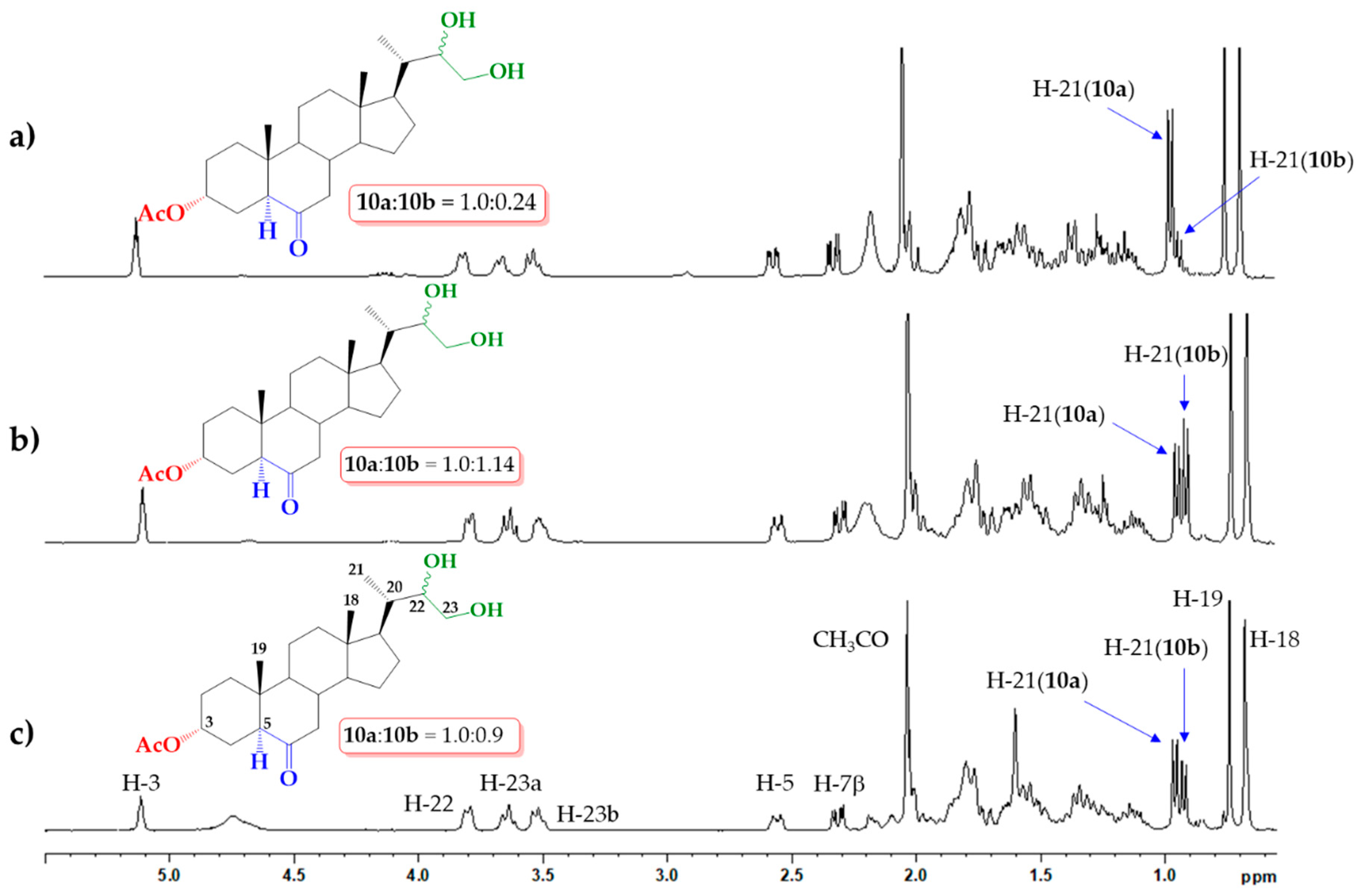
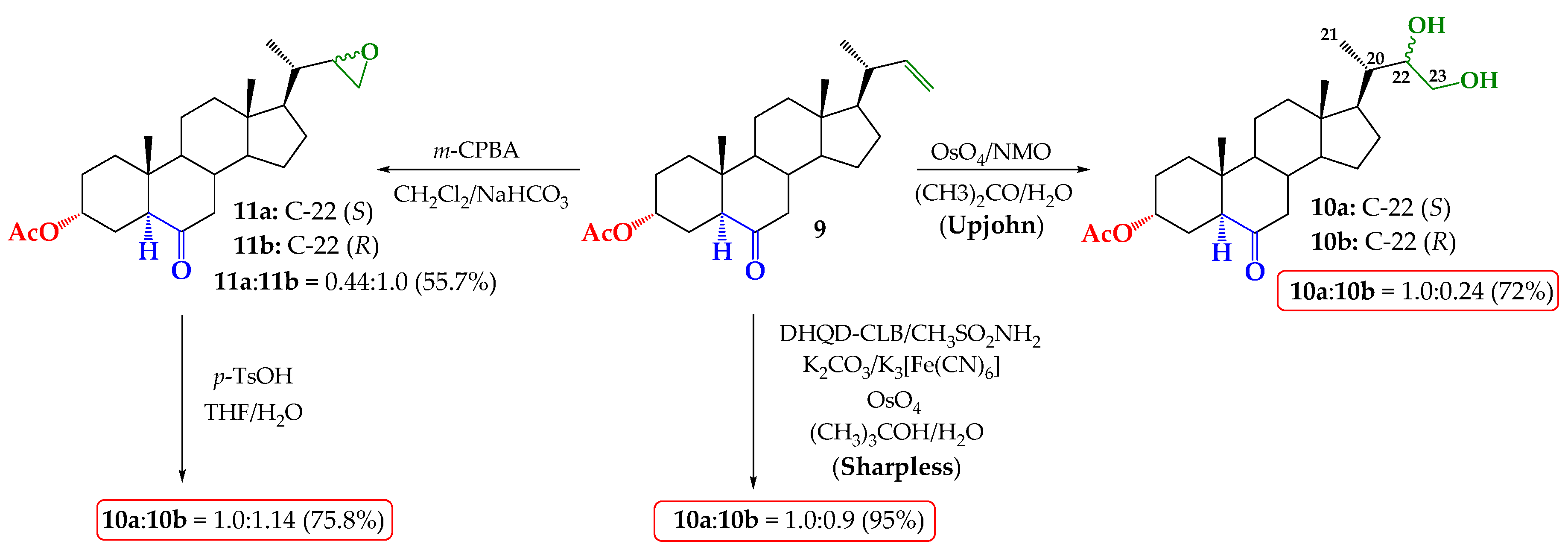
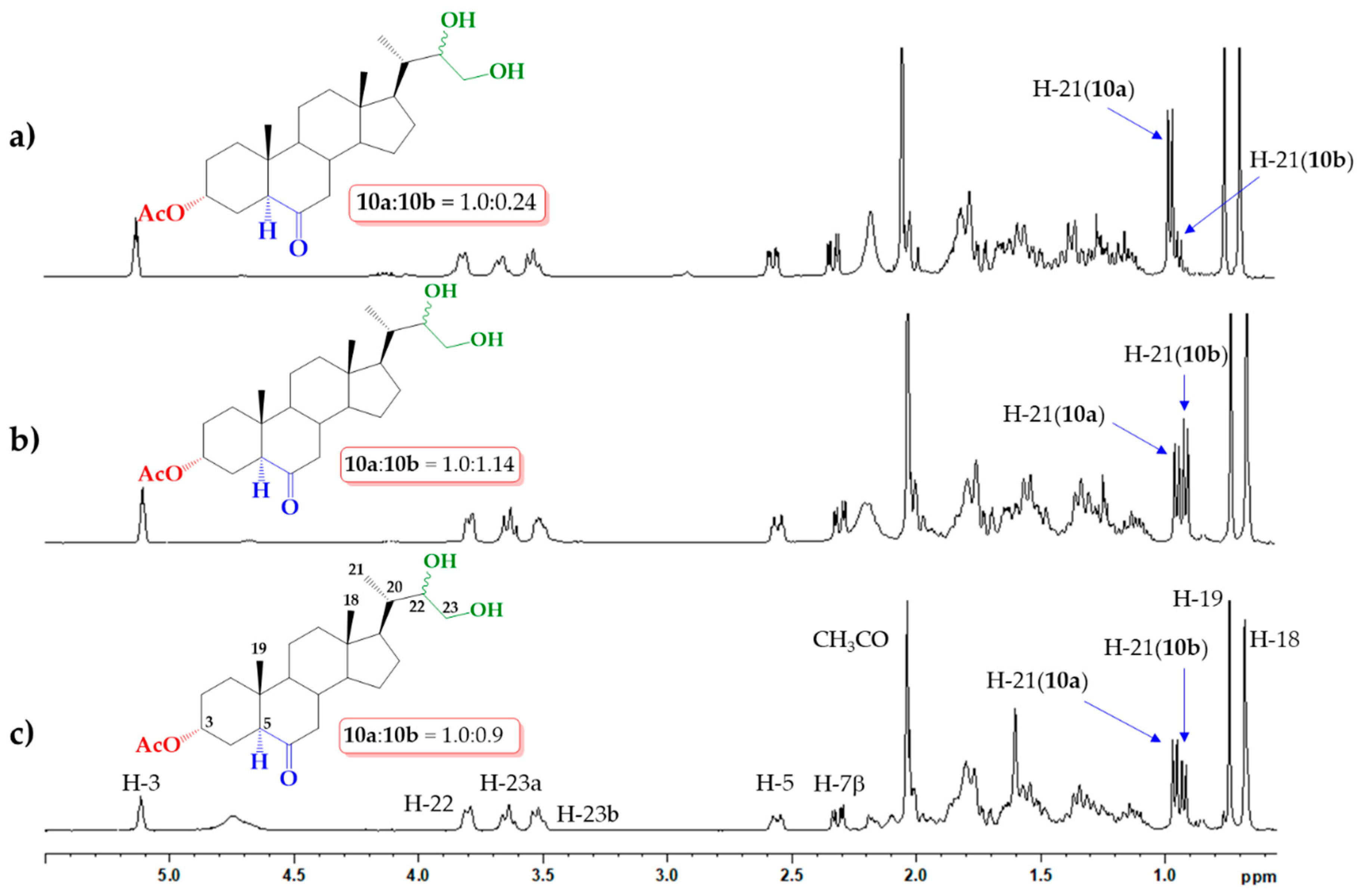
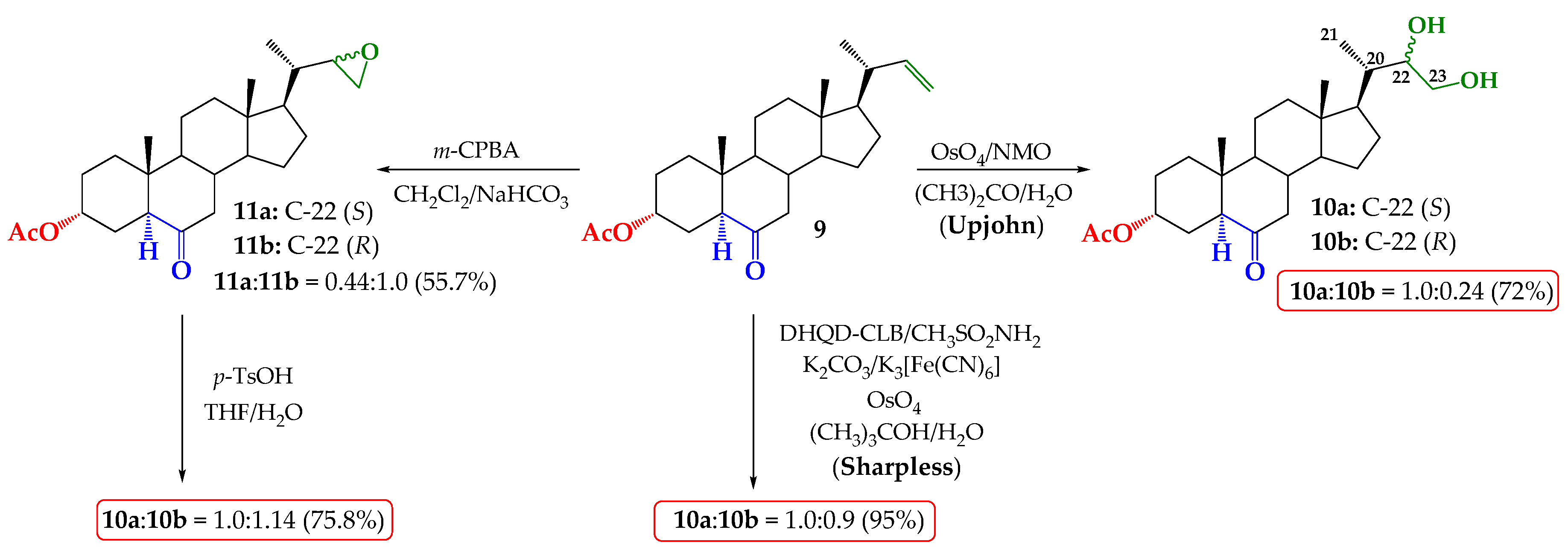
2.1. Direct Upjohn Dihydroxylation
2.2. Epoxidation and Subsequent Opening of Epoxide Ring in Acid Medium
2.3. Sharpless Asymmetric Dihydroxylation
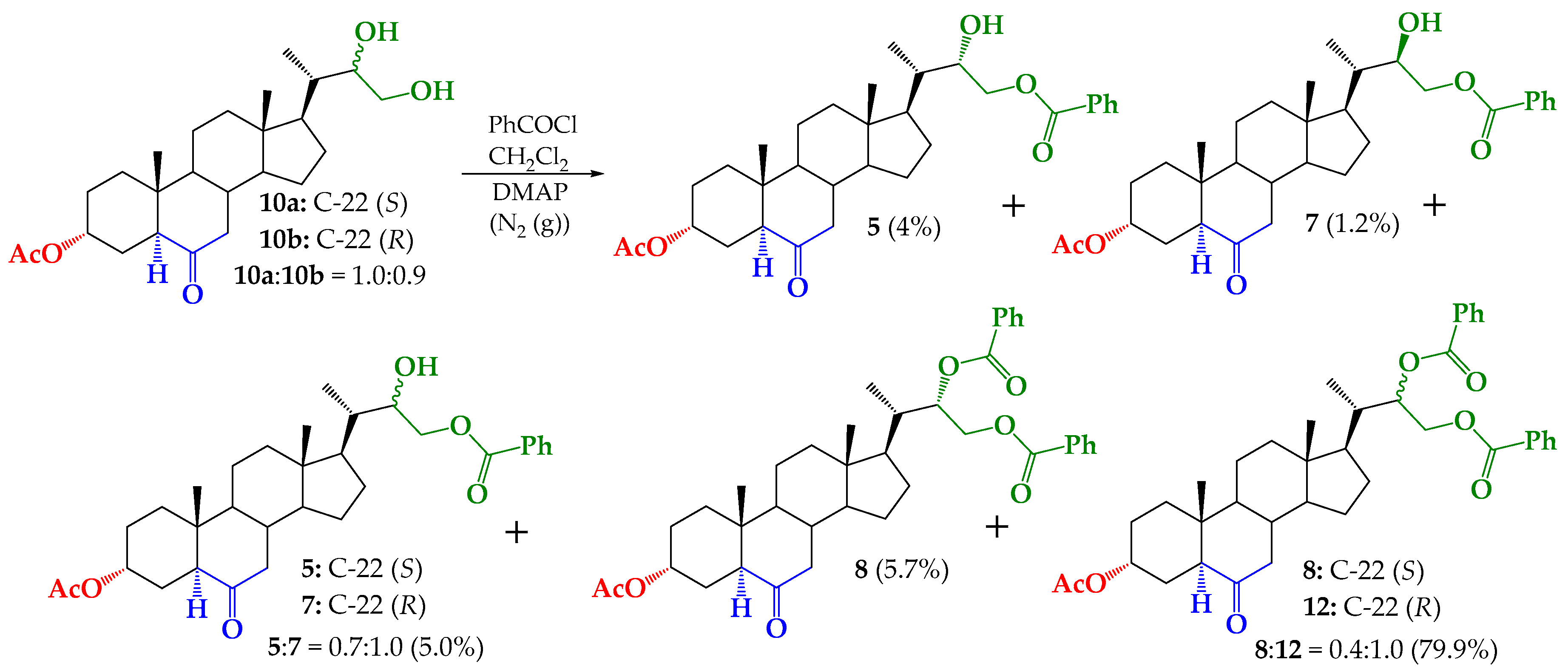
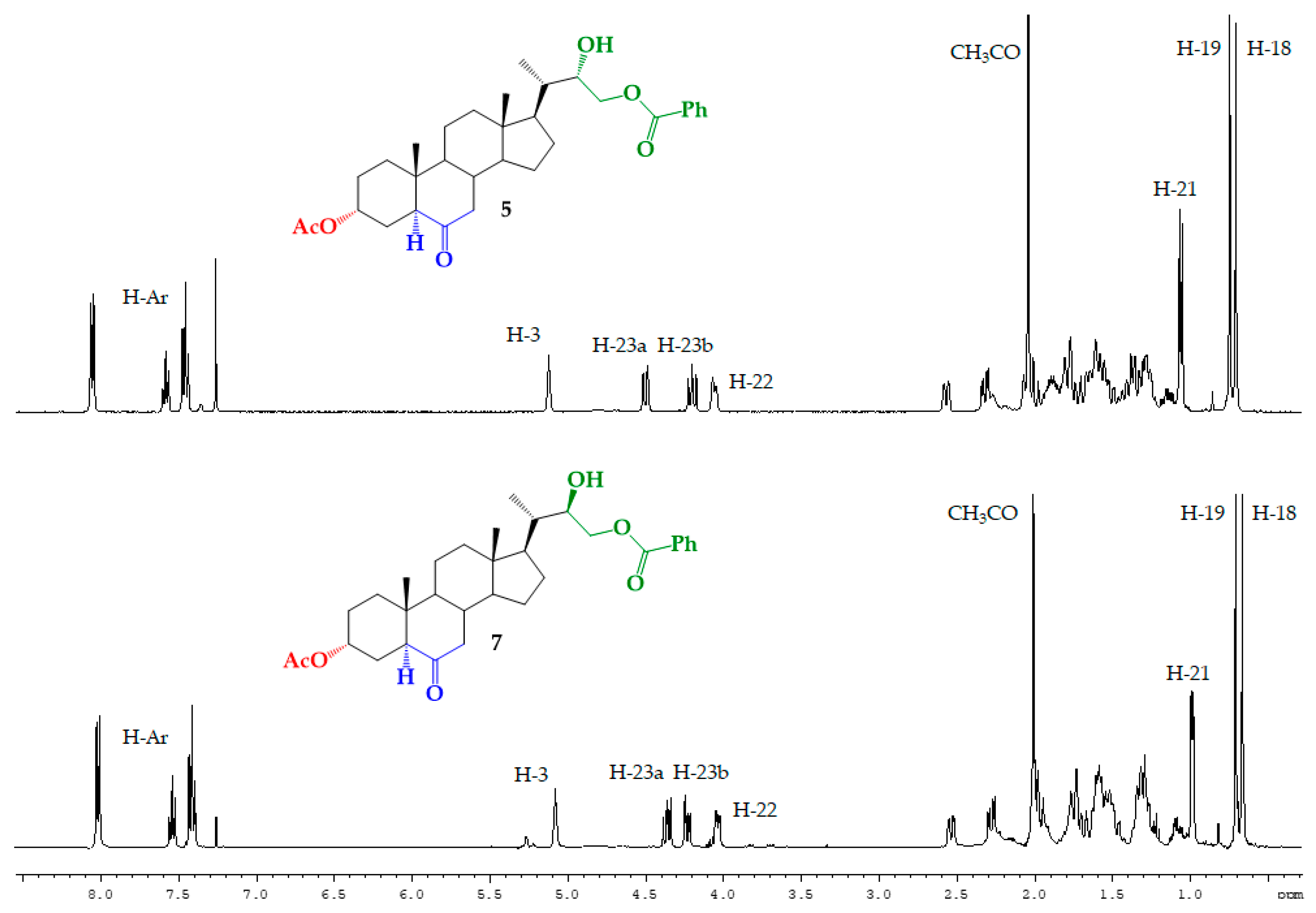
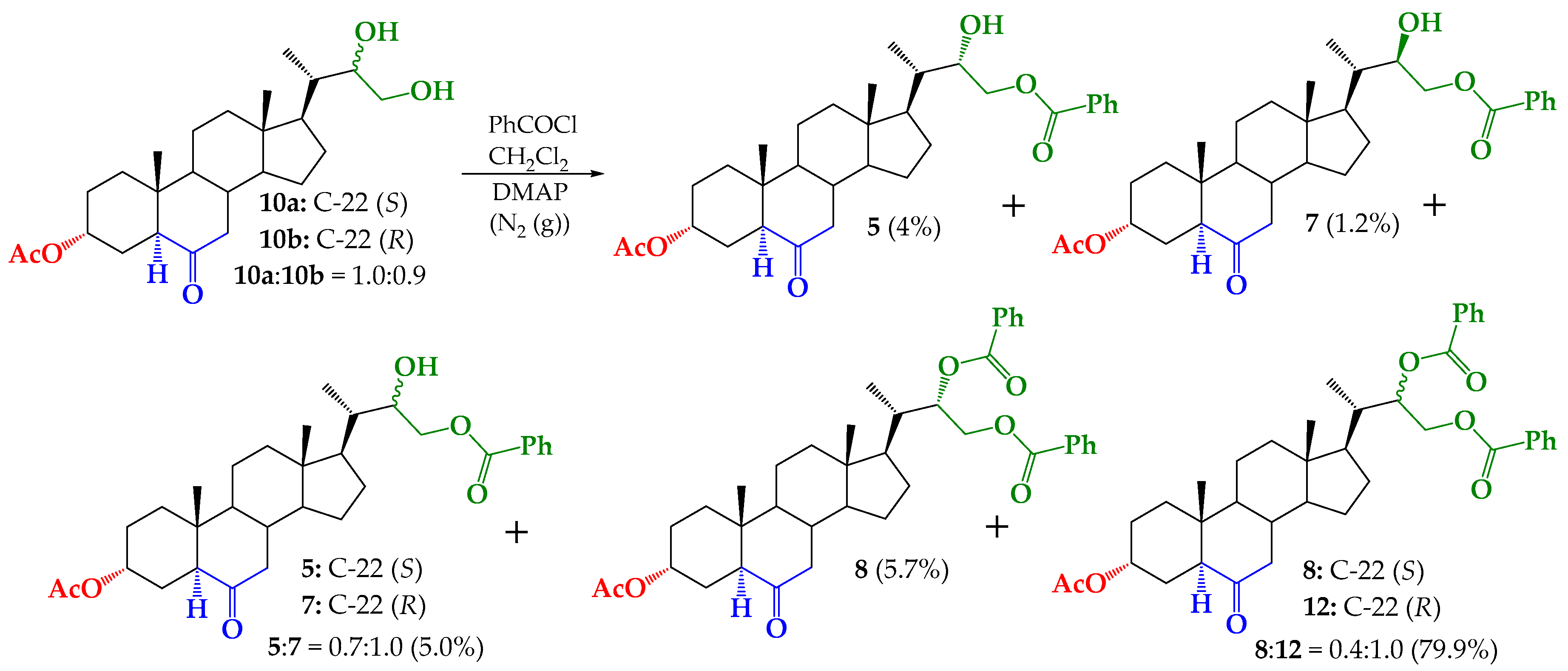
2.4. Synthesis of Benzoylated Derivatives
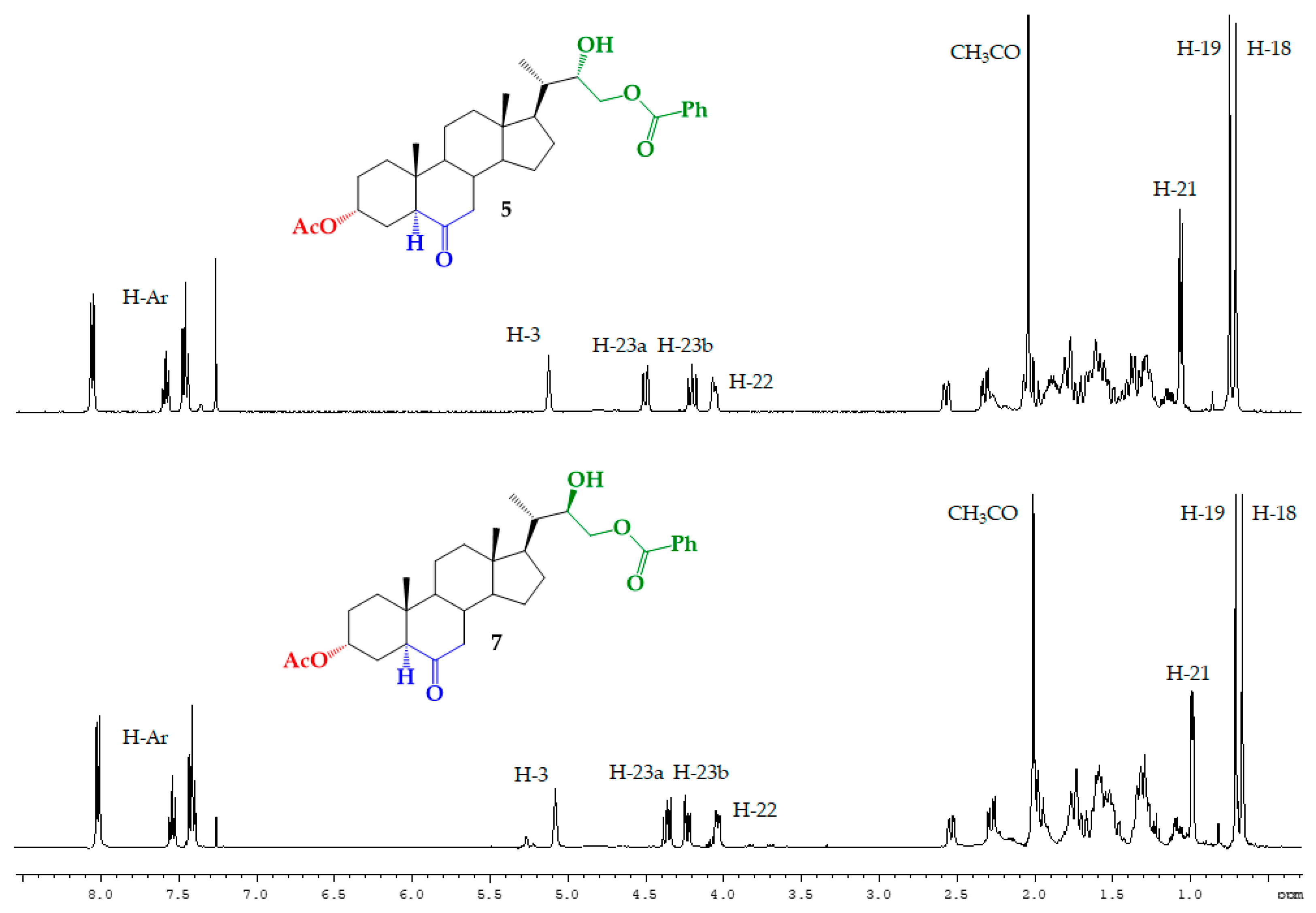
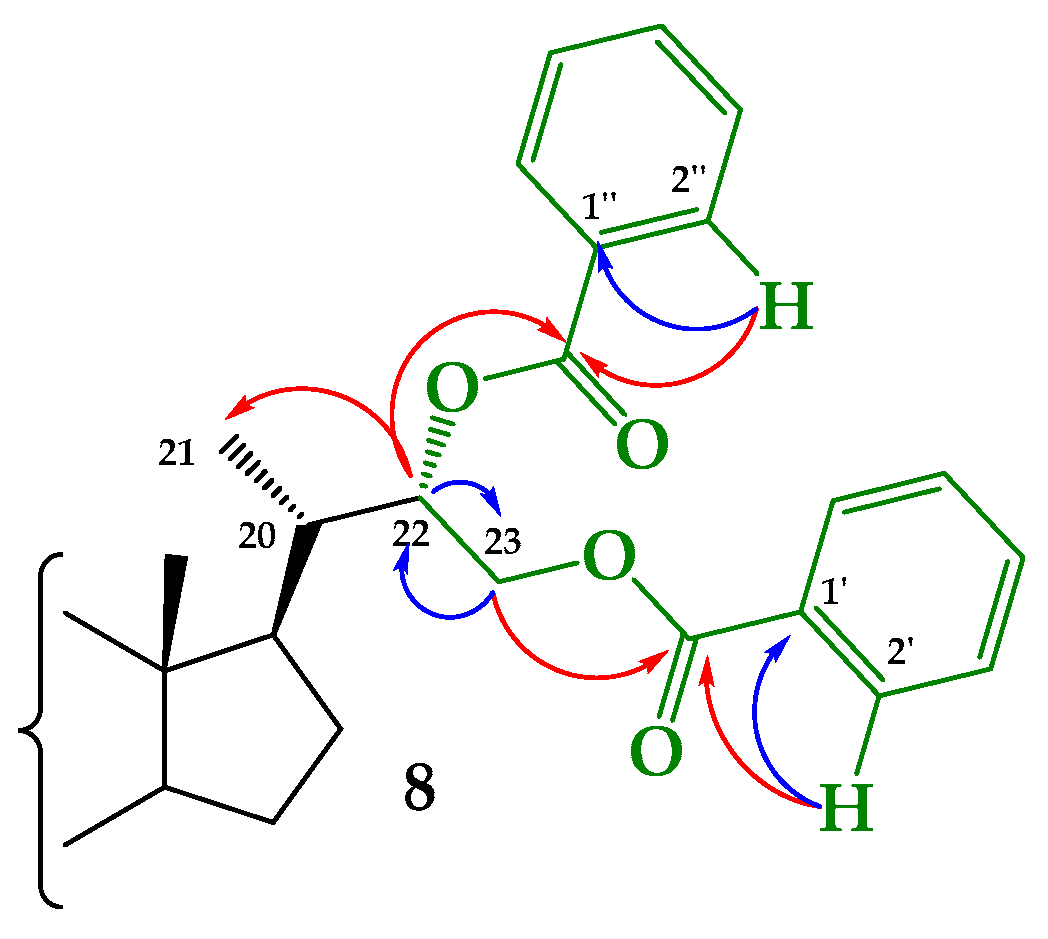
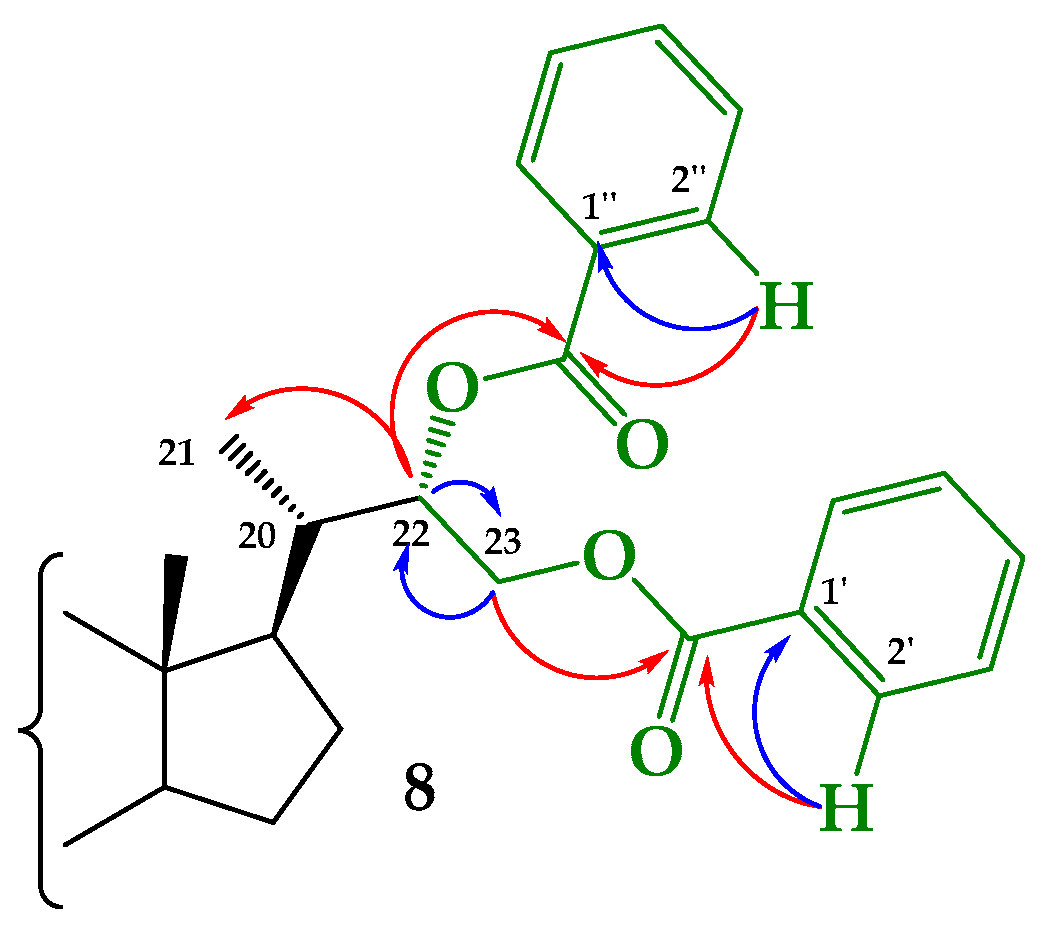
Structural Determination of Derivatives 5, 7, and 8
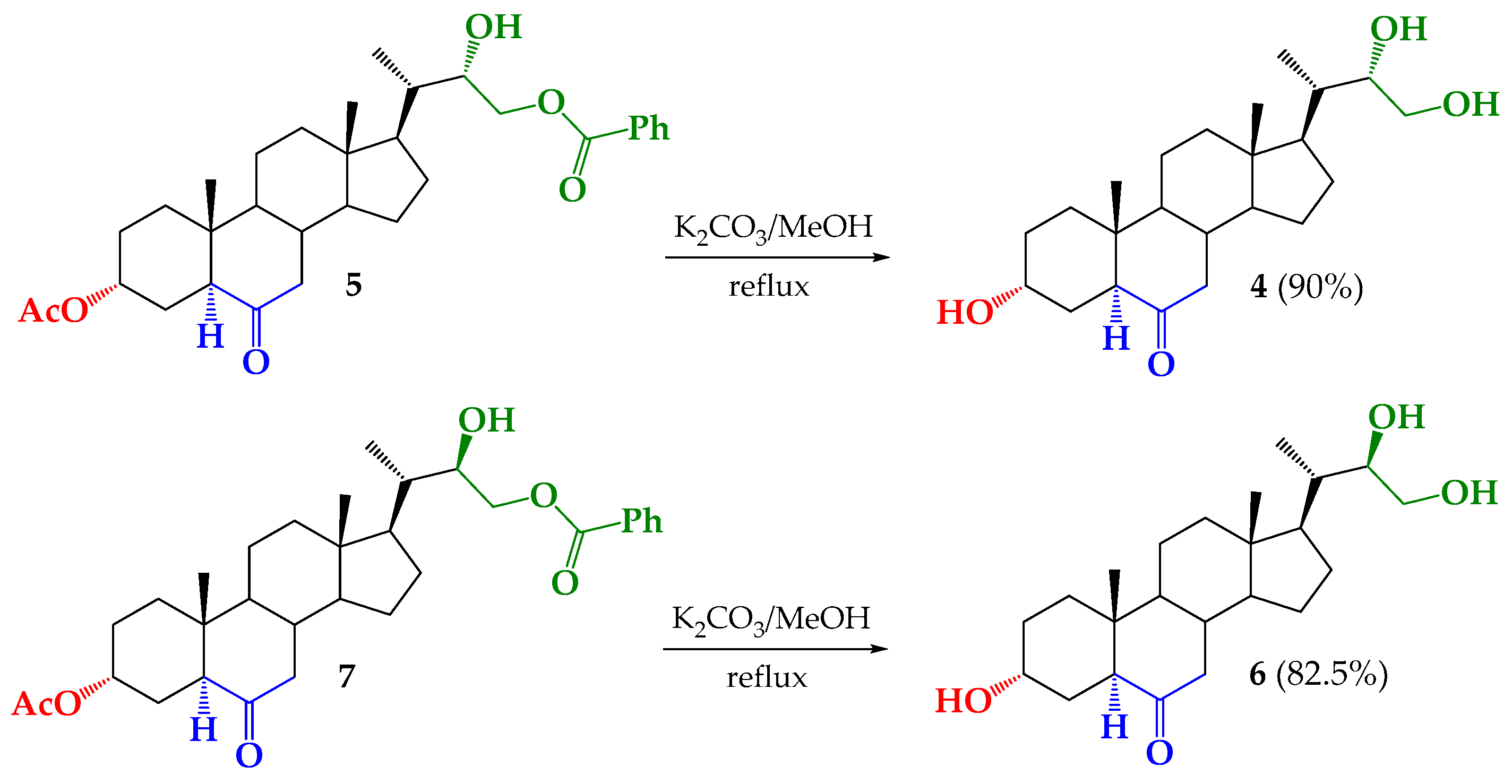
2.5. Synthesis of BRs Analogs
3. Materials and Methods
3.1. General Experimental Methods
3.2. Synthesis
3.2.1. Upjohn Dihydroxylation of Alkene 9. Obtaining (22S)-22,23-dihydroxy-6-oxo-24-nor-5α-cholan-3α-yl acetate (10a) and (22R)-22,23-dihydroxy-6-oxo-24-nor-5α-cholan-3α-yl acetate (10b)
3.2.2. Epoxidation of Alkene 9. Obtaining (22S)-6-oxo-22,23-epoxy-24-nor-5α-cholan-3α-yl acetate and (11a) and (22R)-6-oxo-22,23-epoxy-24-nor-5α-cholan-3α-yl acetate (11b)
3.2.3. Opening of Epoxide Ring of Mixture 11a/11b in Acid Medium. Obtaining (22S)-22,23-dihydroxy-6-oxo-24-nor-5α-cholan-3α-yl acetate (10a) and (22R)-22,23-dihydroxy-6-oxo-24-nor-5 α-cholan-3 α-yl acetate (10b)
3.2.4. Direct Asymmetric Sharpless Dihydroxylation of Alkene 9. Obtaining (22S)-22,23-dihydroxy-6-oxo-24-nor-5α-cholan-3α-yl acetate (10a) and (22R)-22,23-dihydroxy-6-oxo-24-nor-5α-cholan-3α-yl acetate (10b)
3.2.5. Synthesis of (22S)-22-hydroxy-6-oxo-24-nor-5α-cholan-3α,23-diyl 3-acetate 23-benzoate (5), (22R)-22-hydroxy-6-oxo-24-nor-5α-cholan-3α,23-diyl 3-acetate 23-benzoate (7) and (22S)-6-oxo-24-nor-5α-cholan-3α,22,23-triyl 3-acetate 22,23-dibenzoate (8)
3.2.6. Synthesis of (22S)-24-nor-5α-cholan-6-oxo-3α,22,23-triyl 3-acetate 22,23-dibenzoate (8) from 5
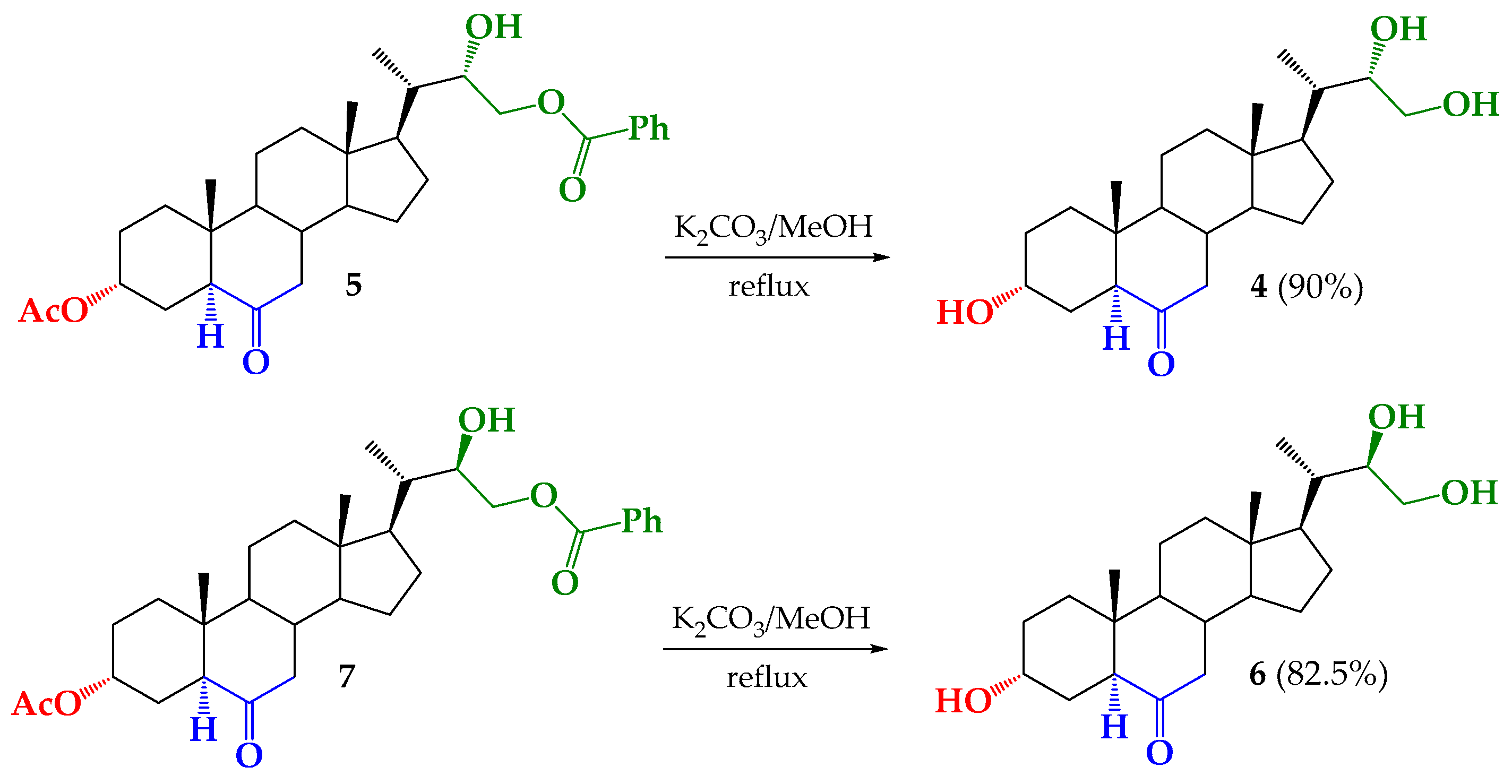
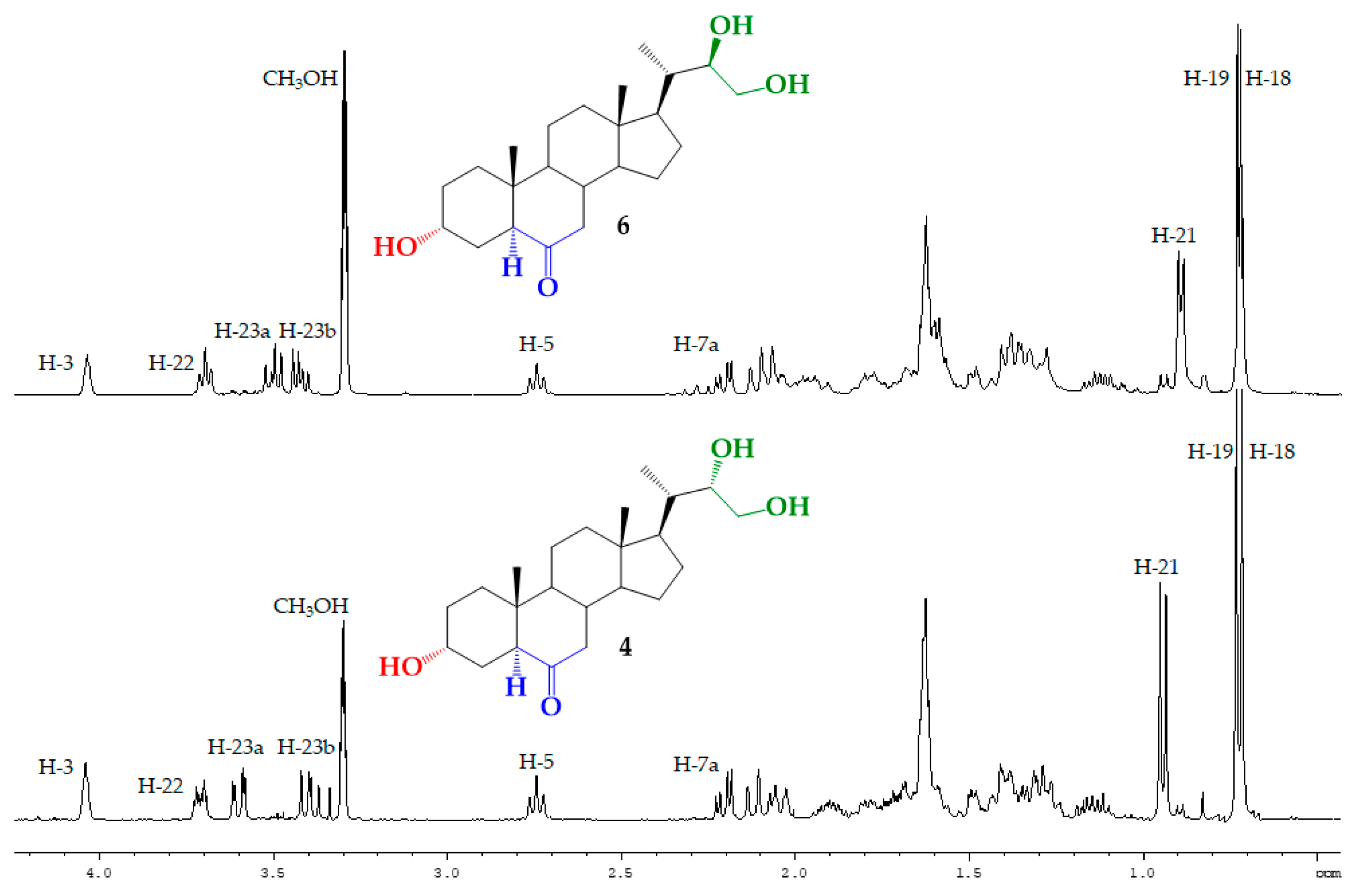
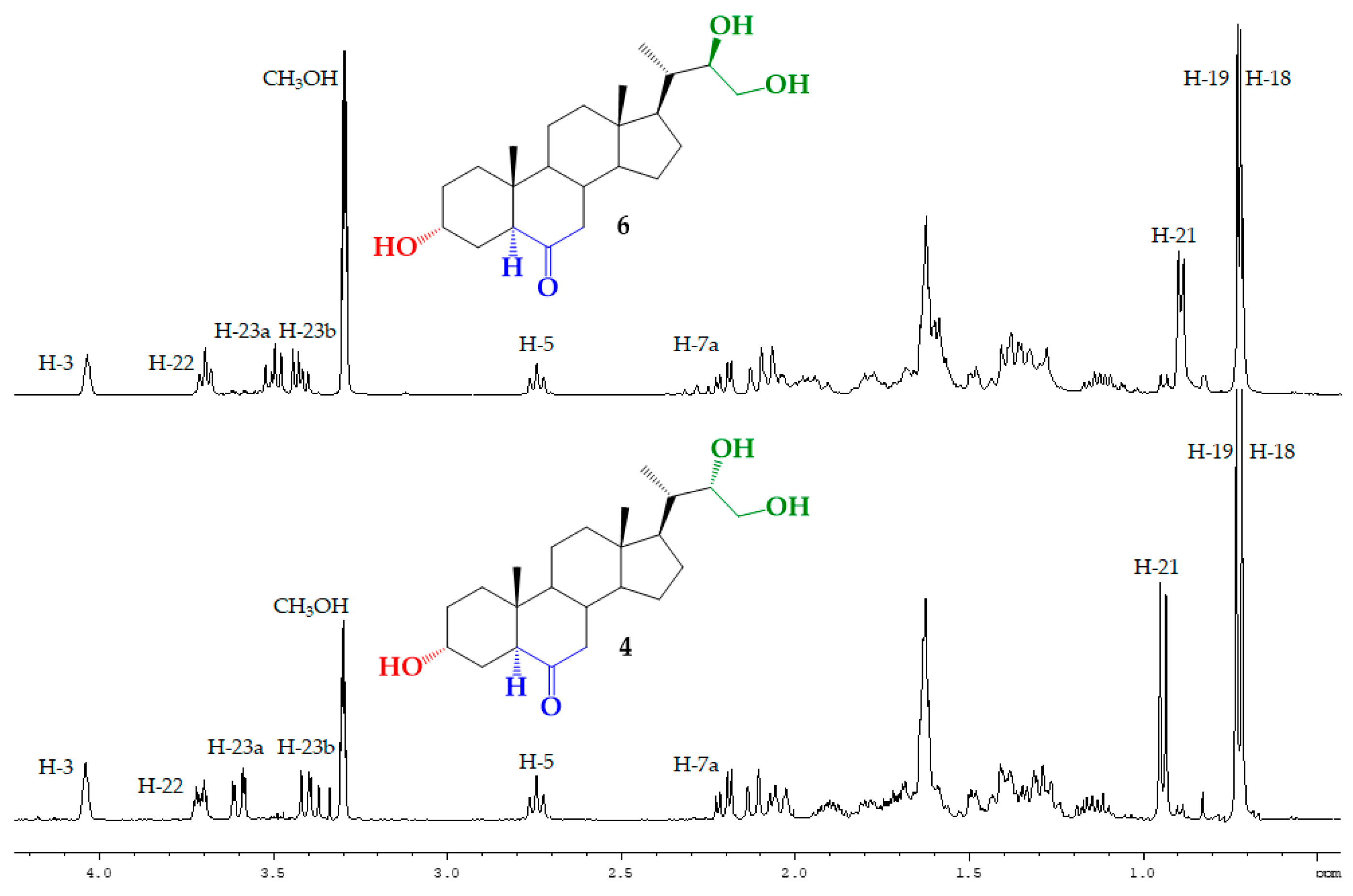
3.2.7. Synthesis of (22S)-3α,22,23-trihydroxy-24-nor-5α-cholan-6-one (4) from 5
3.2.8. Synthesis of (22R)-3α,22,23-trihydroxy-24-nor-5α-cholan-6-one (6) from 7
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mitchell, J.W.; Mandava, N.; Worley, J.F.; Plimmer, J.R.; Smith, M.V. Brassins - A New Family of Plant Hormones from Rape Pollen. Nature 1970, 225, 1065–1066. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, S. Natural Occurrence of Brassinosteroids in the Plant Kingdom. In Brassinosteroids: Steroidal Plant Hormones; Sakurai, A., Yokota, T., Clouse, S.D., Eds.; Springer-Verlag: Tokyo, Japan, 1999; pp. 21–45. [Google Scholar]
- Bajguz, A. Brassinosteroids – Occurence and Chemical Structures in Plants. In Brassinosteroids: A Class of Plant Hormone; Hayat, S., Ahmad, A., Eds.; Springer: Dordrecht, The Netherlands, 2016; pp. 1–27. [Google Scholar]
- Liu, J.; Zhang, D.; Sun, X.; Ding, T.; Lei, B.; Zhang, C. Structure-Activity Relationship of Brassinosteroids and their Agricultural Practical Usages. Steroids 2017, 124, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Peres, A.L.G.L.; Soares, J.S.; Tavares, R.G.; Righetto, G.; Zullo, M.A.T.; Mandava, N.B.; Menossi, M. Brassinosteroids, the Sixth Class of Phytohormones: A Molecular View from the Discovery to Hormonal Interactions in Plant Development and Stress Adaptation. Int. J. Mol. Sci. 2019, 20, 331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oklestkova, J.; Rarova, L.; Kvasnica, M.; Strnad, M. Brassinosteroids: Synthesis and Biological Activities. Phytochem. Rev. 2015, 14, 1053–1072. [Google Scholar] [CrossRef]
- Clouse, S.D. A History of Brassinosteroid Research from 1970 through 2005: Thirty-Five Years of Phytochemistry, Physiology, Genes, and Mutants. J. Plant. Growth. Regul. 2015, 34, 828–844. [Google Scholar] [CrossRef]
- Bajguz, A.; Hayat, S. Effects of Brassinosteroids on the Plant Responses to Environmental Stresses. Plant. Physiol. Biochem. 2009, 47, 1–8. [Google Scholar] [CrossRef]
- Hayat, S.; Alyemeni, M.N.; Hasan, S.A. Foliar Spray of Brassinosteroid Enhances Yield and Quality of Solanum Lycopersicum under Cadmium Stress. Saudi. J. Biol. Sci. 2012, 19, 325–335. [Google Scholar] [CrossRef] [Green Version]
- Černý, V.; Strnad, M.; Kamínek, M. Preparation of 2α,3α-dihydroxy-7-oxa-6-oxo-23,24-dinor-B-homo-5α-Cholanic Acid, its Esters and Amides as Brassinolide Analogues. Collect. Czech. Chem. Commun. 1986, 51, 687–697. [Google Scholar] [CrossRef]
- Kvasnica, M.; Oklestkova, J.; Bazgier, V.; Rarova, L.; Berka, K.; Strnad, M. Biological Activities of New Monohydroxylated Brassinosteroid Analogues with a Carboxylic Group in the Side Chain. Steroids 2014, 85, 58–64. [Google Scholar] [CrossRef]
- Uesusuki, S.; Watanabe, B.; Yamamoto, S.; Otsuki, J.; Nakagawa, Y.; Miyagawa, H. Synthesis of Brassinosteroids of Varying Acyl Side Chains and Evaluation of Their Brassinolide-like Activity. Biosci. Biotechnol. Biochem. 2004, 68, 1097–1105. [Google Scholar] [CrossRef] [Green Version]
- Šíša, M.; Hniličková, J.; Swaczynová, J.; Kohout, L. Syntheses of New Androstane Brassinosteroids with 17β-Ester Groups—Butyrates, Heptafluorobutyrates, and Laurates. Steroids 2005, 70, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Gil, R.P.; Iglesias-Arteaga, M.A.; Martinez, C.P.; Manchado, F.C.; Garcia, D.C.; Rosado, A. Synthesis of Analogues of Brassinosteroids From Chenodeoxycholic Acid. Eur. J. Org. Chem. 1998, 2405–2407. [Google Scholar]
- Iglesias-Arteaga, M.; Gil, R.P.; Leliebre-Lara, V.; Martinez, C.S.P.; Manchado, F. Synthesis and Biological Activity of (22R,25R)-5 alpha-furostan-2 alpha,3 alpha,26-triol. J. Chem. Res. 1996, 504–505. [Google Scholar]
- Iglesias-Arteaga, M.; Gil, R.; Leliebre-Lara, V.; Manchado, F.; Pérez, C.S.; Rosado, A. Synthesis of (25R)-5α-Spirostan-2α,3α,6β-triol Triacetate. Synth. Commun. 1998, 28, 75–81. [Google Scholar] [CrossRef]
- Iglesias-Arteaga, M.; Gil, R.P.; Leliebre-Lara, V.; Martinez, C.S.P.; Manchado, F.; Perez, A.R.; Rios, L.P. Synthesis of (22R,25R)-3 beta,26-dihydroxy-5 alpha-furostan-6-one. Synth. Commun. 1998, 28, 1381–1386. [Google Scholar] [CrossRef]
- Iglesias-Arteaga, M.; Gil, R.P.; Leliebre-Lara, V.; Martinez, C.S.P.; Manchado, F. Synthesis of (22R,25R)-2 alpha,3 alpha,26-trihydroxy-5 alpha-furostanaone-6-one. Synth. Commun. 1998, 28, 1779–1784. [Google Scholar] [CrossRef]
- Iglesias-Arteaga, M.A.; PérezGil, R.; LeliebreLara, V.; CollManchado, F.; PérezMartínez, C.S. Synthesis of (25R)-2α,3α-Epoxy-5α-Spirostan-6,23-Dione. Synth. Commun. 1998, 28, 4387–4392. [Google Scholar] [CrossRef]
- Iglesias-Arteaga, M.A.; Martinez, C.S.P.; Manchado, F.C. Synthesis and Characterization of (25R)-2 alpha,3 alpha-epoxy-5 alpha-spirostan-12,23-dione. Synth. Commun. 1999, 29, 1811–1818. [Google Scholar] [CrossRef]
- Iglesias Arteaga, M.A.; Gil, R.P.; Pérez Martínez, C.S.; Manchado, F.C. Synthetic Steroidal Sapogenins. Part III1 23-Ketohecogenin and 23-Ketoisochiapagenin. Synth. Commun. 2000, 30, 163–170. [Google Scholar] [CrossRef]
- Huang, L.F.; Zhou, W.S. Studies on Steroidal Plant-Growth Regulators. Part 33. Novel Method for Construction of the Side-Chain of 23-Arylbrassinosteroids Via Heck Arylation and Asymmetric Dihydroxylation As Key Steps. J. Chem. Soc. Perkin. Trans. 1 1994, 3579–3585. [Google Scholar] [CrossRef]
- Kvasnica, M.; Oklestkova, J.; Bazgier, V.; Rárová, L.; Korinkova, P.; Mikulík, J.; Budesinsky, M.; Béres, T.; Berka, K.; Lu, Q.; et al. Design, Synthesis and Biological Activities of New Brassinosteroid Analogues with a Phenyl Group in the Side Chain. Org. Biomol. Chem. 2016, 14, 8691–8701. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.S.; Tian, W.S. The Synthesis of Steroids Containing Structural Unit of A, B Ring of Brassinolide and Ecdysone from Hyodesoxycholic Acid. Acta. Chim. Sinica. 1984, 42, 1173–1177. [Google Scholar]
- Back, T.; Pharis, R. Structure-Activity Studies of Brassinosteroids and the Search for Novel Analogues and Mimetics with Improved Bioactivity. J. Plant. Growth. Regul. 2004, 22, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Kovganko, N.V.; Ananich, S.K. Advances in the Chemical Synthesis of Brassinosteroids. Chem. Nat. Compd. 1997, 33, 389–416. [Google Scholar] [CrossRef]
- Korinkova, P.; Bazgier, V.; Oklestkova, J.; Rarova, L.; Strnad, M.; Kvasnica, M. Synthesis of Novel Aryl Brassinosteroids through Alkene Cross-Metathesis and Preliminary Biological Study. Steroids 2017, 127, 46–55. [Google Scholar] [CrossRef]
- Duran, M.I.; Gonzalez, C.; Acosta, A.; Olea, A.F.; Diaz, K.; Espinoza, L. Synthesis of Five Known Brassinosteroid Analogs from Hyodeoxycholic Acid and Their Activities as Plant-Growth Regulators. Int. J. Mol. Sci. 2017, 18, 516. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Jiang, B.; Shen, J. Synthesis of Cholesteric Lactones and Analogs as Plant Growth Regulators. Patent CN 1184113 A, 10 June 1998. [Google Scholar]
- Tian, W.S.; Zhou, W.S. Studies on Steroidal Plant Growth Hormone VI. Conversion of Methyl 3,6-Diketoallocholanate into Methyl Δ^2-6-ketoallocholenate. Acta. Chim. Sinica. 1988, 46, 824–826. [Google Scholar]
- Brosa, C.; Capdevila, J.M.; Zamora, I. Brassinosteroids: A New Way to Define the Structural Requirements. Tetrahedron 1996, 52, 2435–2448. [Google Scholar] [CrossRef]
- Brosa, C.; Soca, L.; Terricabras, E.; Ferrer, J.C.; Alsina, A. New Synthetic Brassinosteroids: A 5 alpha-hydroxy-6-ketone Analog with Strong Plant Growth Promoting Activity. Tetrahedron 1998, 54, 12337–12348. [Google Scholar] [CrossRef]
- Thompson, M.J.; Meudt, W.J.; Mandava, N.B.; Dutky, S.R.; Lusby, W.R.; Spaulding, D.W. Synthesis of Brassinosteroids and Relationship of Structure to Plant Growth-Promoting Effects. Steroids 1982, 39, 89–105. [Google Scholar] [CrossRef]
- Takatsuto, S.; Yazawa, N.; Ikekawa, N.; Takematsu, T.; Takeuchi, Y.; Koguchi, M. Structure Activity Relationship of Brassinosteroids. Phytochem 1983, 22, 2437–2441. [Google Scholar] [CrossRef]
- Carvajal, R.; Gonzalez, C.; Olea, A.F.; Fuentealba, M.; Espinoza, L. Synthesis of 2-Deoxybrassinosteroids Analogs with 24-nor, 22(S)-23-Dihydroxy-Type Side Chains from Hyodeoxycholic Acid. Molecules 2018, 23, 1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishiguro, M.; Kajikawa, A.; Haruyama, T.; Ogura, Y.; Okubayashi, M.; Morisaki, M.; Ikekawa, N. Synthetic studies of Withanolides. Part 1. Synthesis of 5,6beta-epoxy-4beta-hydroxy-5beta-cholest-2-en-1-one and Related Compounds. J. Chem. Soc. Perkin. Trans. 1 1975, 2295–2302. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, L.; Cortes, M. Synthesis and biological Activities of Two New Brassinosteroids Functionalized in Ring C. Bol. Soc. Chil. Quim. 2002, 47, 335–347. [Google Scholar]
- Espinoza, L.; Cortes, M. Synthesis and Biological Activity of Brassinosteroids Analogues. Bol. Soc. Chil. Quim. 2002, 47, 511–516. [Google Scholar]
- Espinoza, L. Synthesis of Four New Brassinosteroids Analogues 11-Oxo-Functionalized on C Ring, with 24-Nor Side Chain and Containing 5â-Cholanic Acid Skeleton. Organic. Chem.Curr.Res. 2015, 4, 1–6. [Google Scholar]
- Yang, Y.X.; Zheng, L.T.; Shi, J.J.; Gao, B.; Chen, Y.K.; Yang, H.C.; Chen, H.L.; Li, Y.C.; Zhen, X.C. Synthesis of 5 alpha-cholestan-6-one Derivatives and their Inhibitory Activities of NO Production in Activated Microglia: Discovery of A Novel Neuroinflammation Inhibitor. Bioorg. Med. Chem. Lett. 2014, 24, 1222–1227. [Google Scholar] [CrossRef]
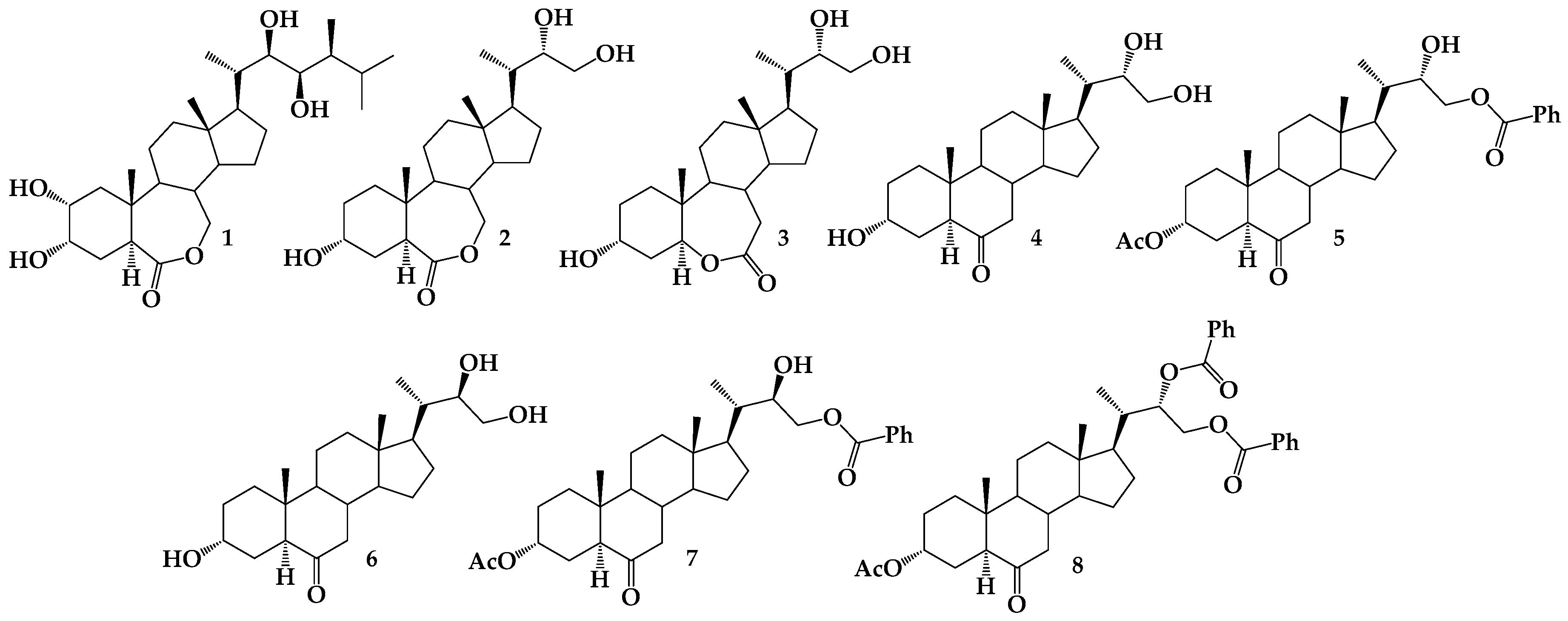
Sample Availability: Samples of the compounds 4–8 are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H/C Signal | Compound 5 | Compound 7 |
|---|---|---|
| H-21 | 1.06 ppm (3H, d, J = 6.9 Hz) | 0.990 ppm (3H, d, J = 6.1 Hz) |
| H-22 | 4.06 ppm (1H, m) | 4.04 ppm (1H, dd, J = 8.4 and 3.6 Hz) |
| H-23a | 4.50 ppm (1H, dd, J = 11.4 and 1.7 Hz) | 4.36 ppm (1H, dd, J = 11.2 and 8.4 Hz) |
| H-23b | 4.20 ppm (1H, dd, J = 11.4 and 4.2 Hz) | 4.23 ppm (1H, dd, J = 11.2 and 3.6 Hz) |
| C21 | 12.90 ppm | 12.30 ppm |
| C22 | 71.77 ppm | 71.40 ppm |
| C23 | 66.39 ppm | 68.84 ppm |
| H/C Signal | Triol 4 | Triol 6 |
|---|---|---|
| H-21 | 0.943 ppm (3H, d, J = 6.9 Hz) | 0.925 ppm (3H, d, J = 6.3 Hz) |
| H-22 | 3.71 ppm (1H, dt, J = 8.9 and 3.2 Hz) | 3.73 ppm (1H, ta, J = 6.5 Hz) |
| H-23a | 3.60 ppm (1H, dd, J = 11.3 and 2.7 Hz) | 3.53 ppm (1H, dd, J = 10.9 and 7.1 Hz) |
| H-23b | 3.40 ppm (1H, dd, J = 11.3 and 8.9 Hz) | 3.46 ppm (1H, dd, J = 10.9 and 6.5 Hz) |
| C21 | 13.42 ppm | 12.37 ppm |
| C22 | 75.19 ppm | 74.45 ppm |
| C23 | 63.19 ppm | 65.53 ppm |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oyarce, J.; Aitken, V.; González, C.; Ferrer, K.; Olea, A.F.; Parella, T.; Espinoza Catalán, L. Synthesis and Structural Determination of New Brassinosteroid 24-Nor-5α-Cholane Type Analogs. Molecules 2019, 24, 4612. https://doi.org/10.3390/molecules24244612
Oyarce J, Aitken V, González C, Ferrer K, Olea AF, Parella T, Espinoza Catalán L. Synthesis and Structural Determination of New Brassinosteroid 24-Nor-5α-Cholane Type Analogs. Molecules. 2019; 24(24):4612. https://doi.org/10.3390/molecules24244612
Chicago/Turabian StyleOyarce, Jocelyn, Vanessa Aitken, César González, Karoll Ferrer, Andrés F. Olea, Teodor Parella, and Luis Espinoza Catalán. 2019. "Synthesis and Structural Determination of New Brassinosteroid 24-Nor-5α-Cholane Type Analogs" Molecules 24, no. 24: 4612. https://doi.org/10.3390/molecules24244612