Novel Structural Insight into Inhibitors of Heme Oxygenase-1 (HO-1) by New Imidazole-Based Compounds: Biochemical and In Vitro Anticancer Activity Evaluation

 ,
,  ,
,  ,
,  ,
,  , , ,
, , ,  ,
,  , and
, and 
Abstract
:
1. Introduction
2. Results and Discussion
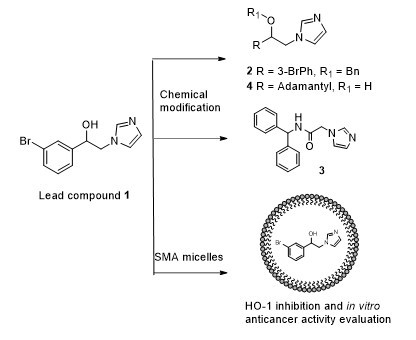
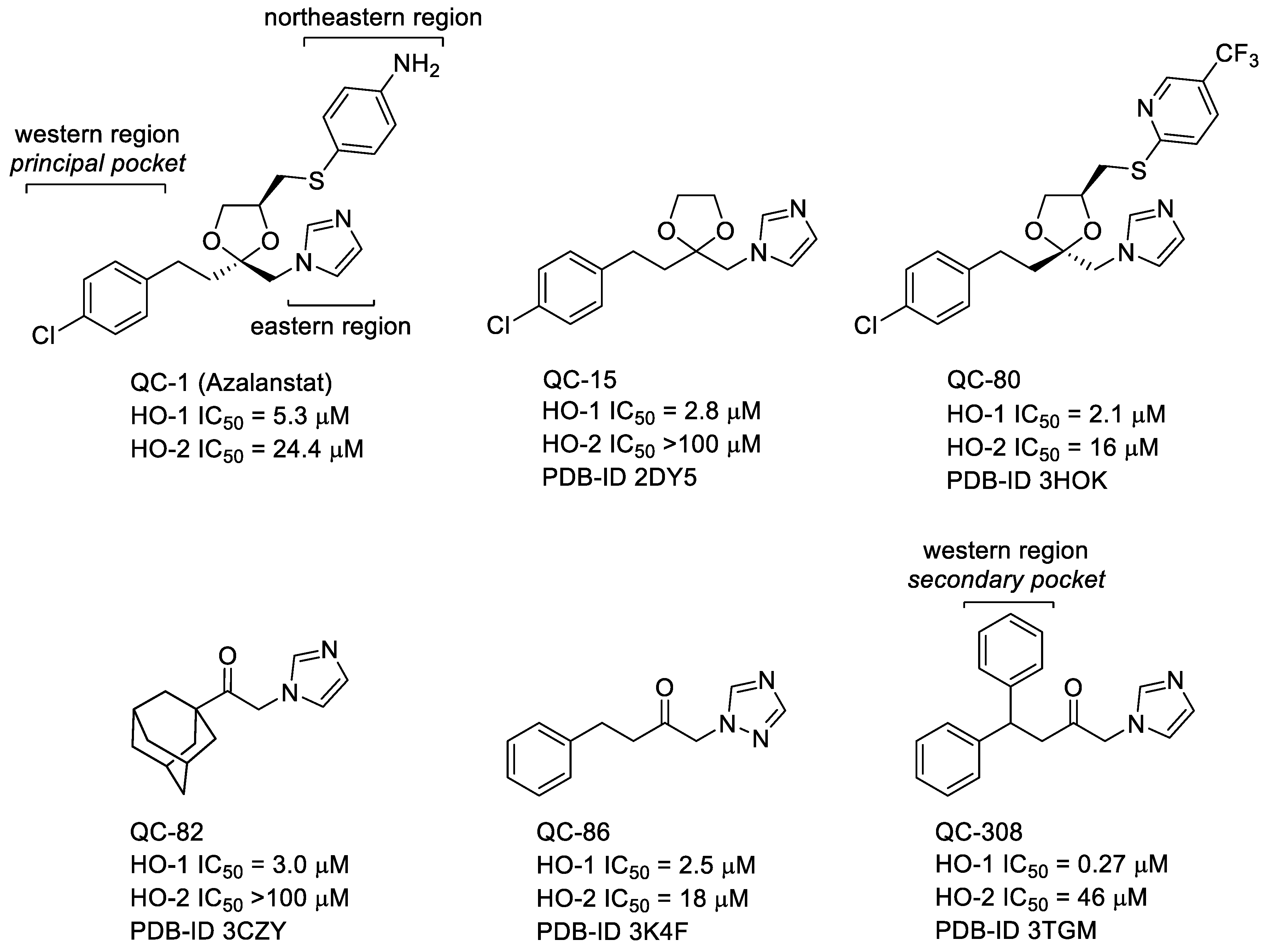
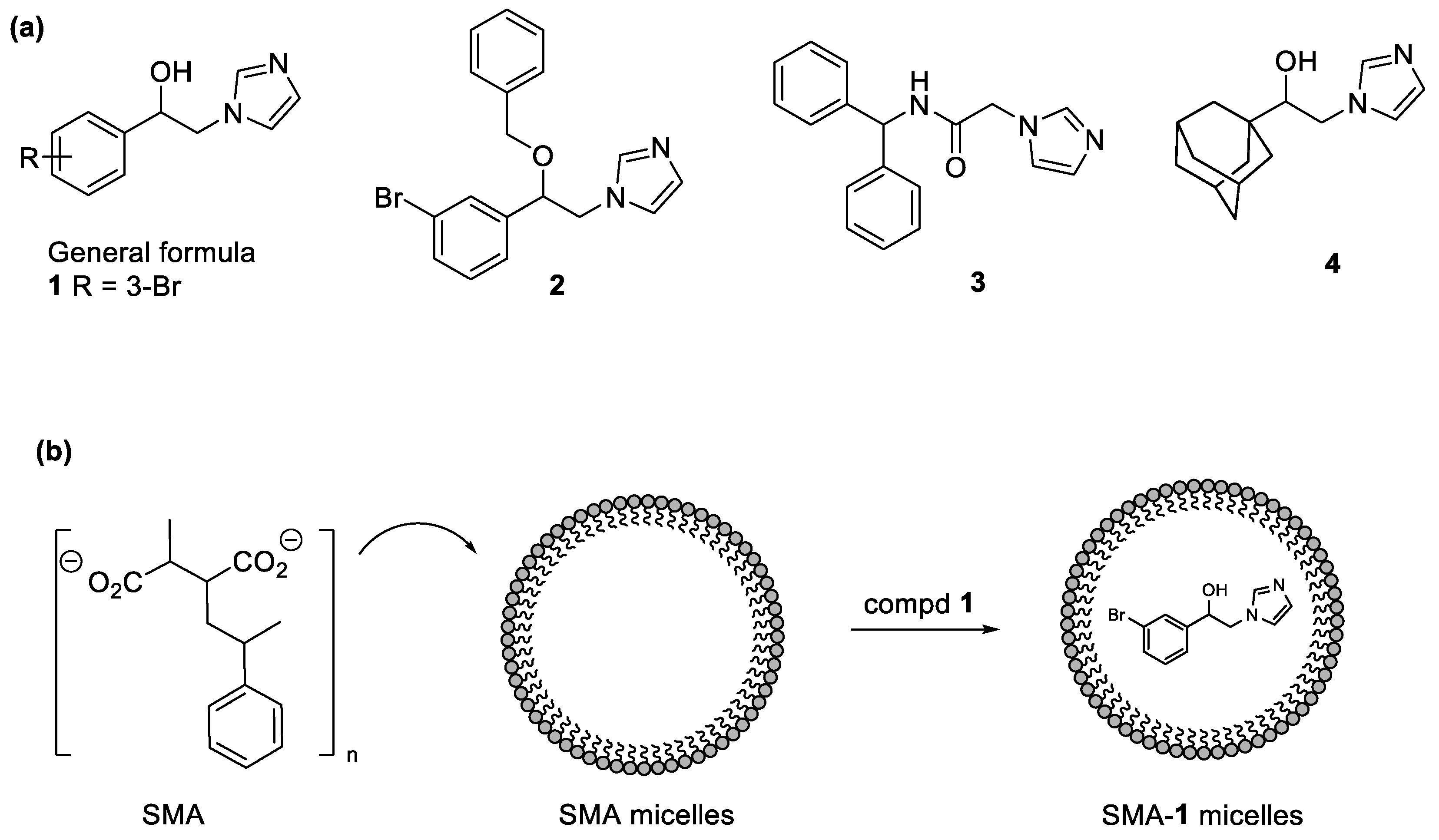
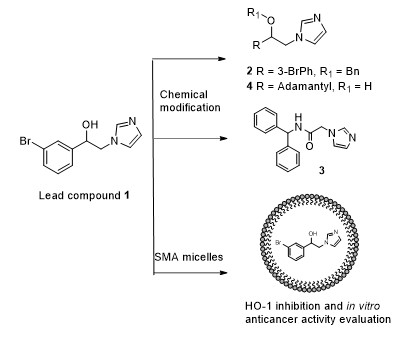
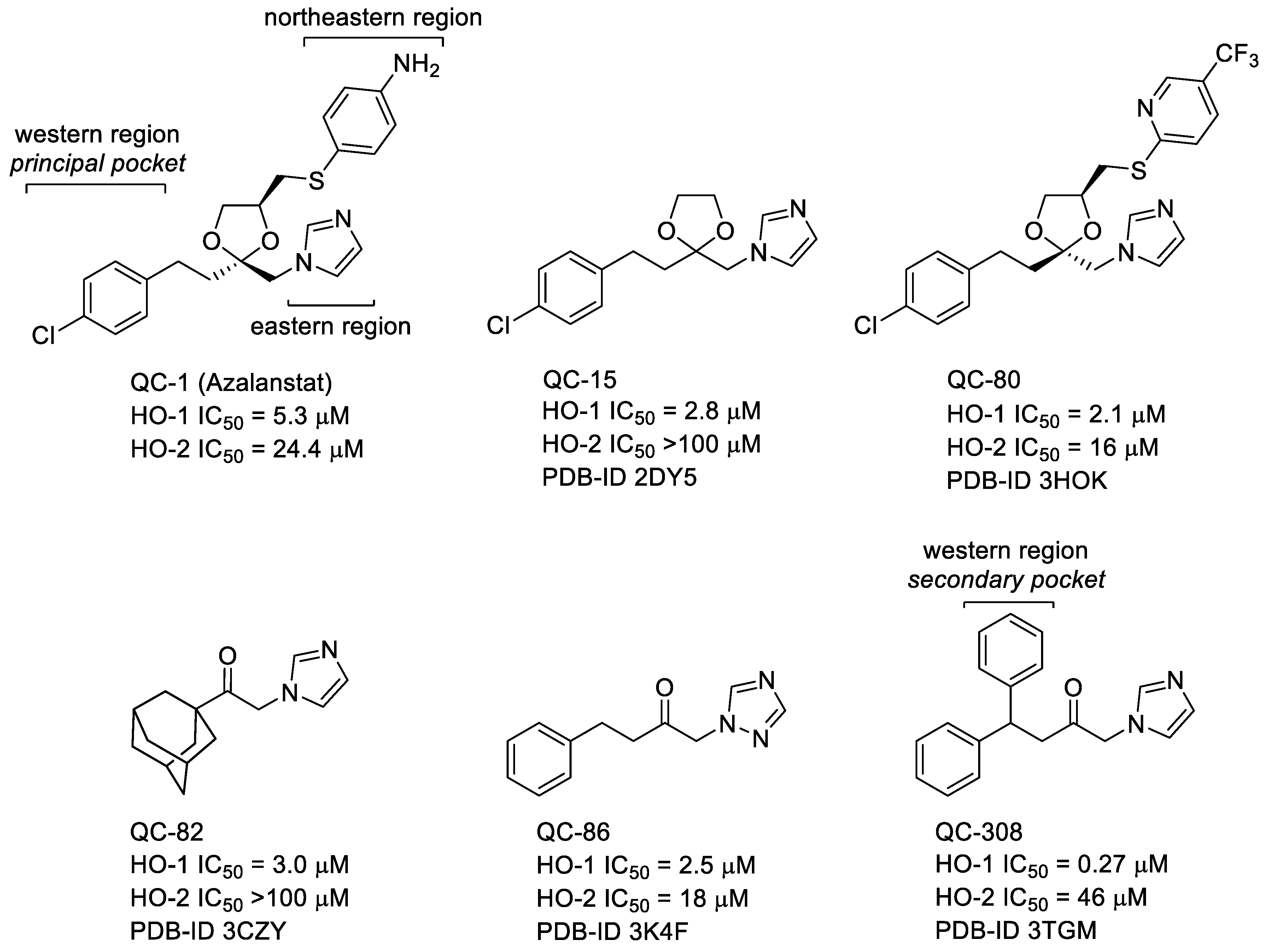
2.1. Design
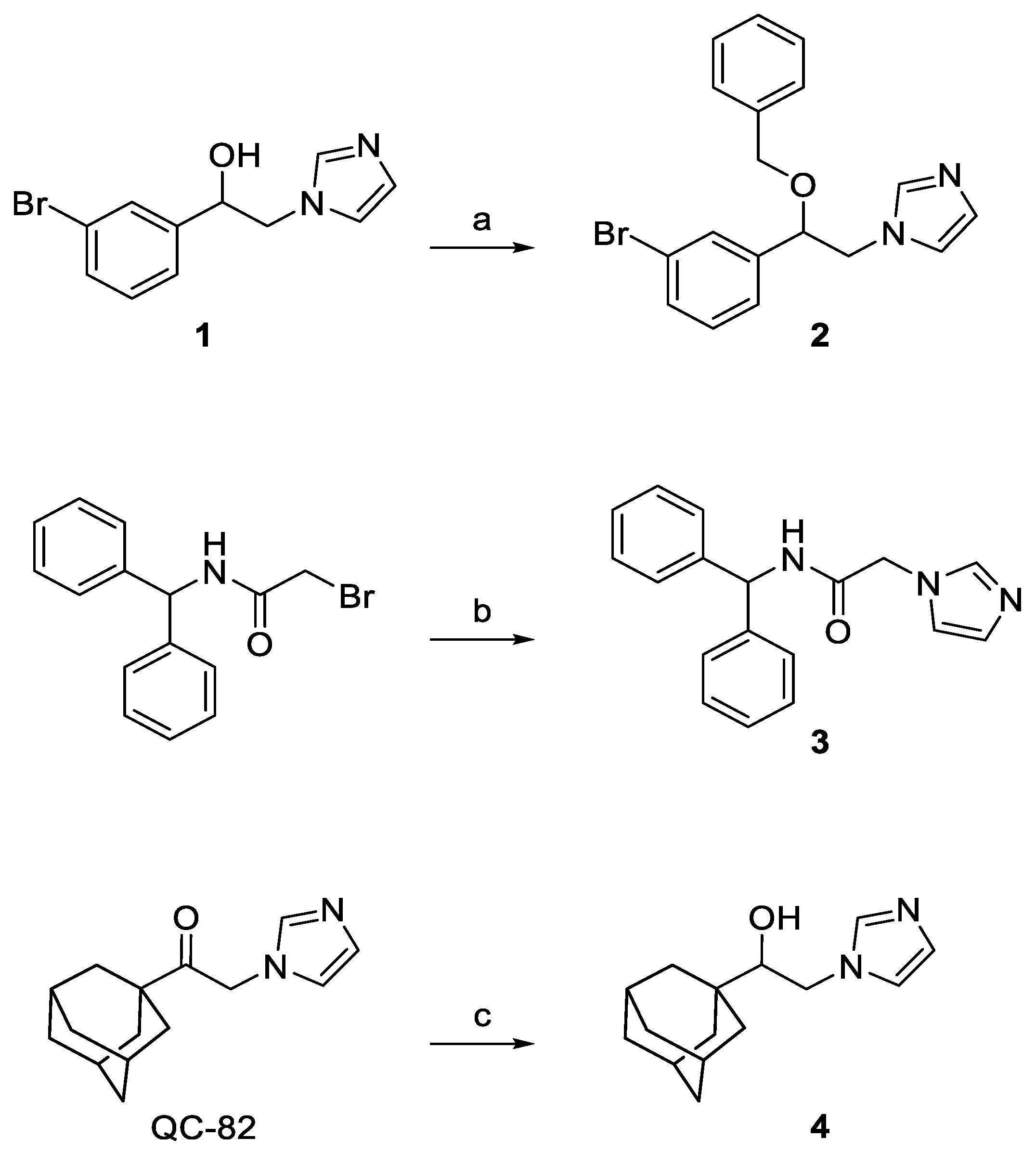
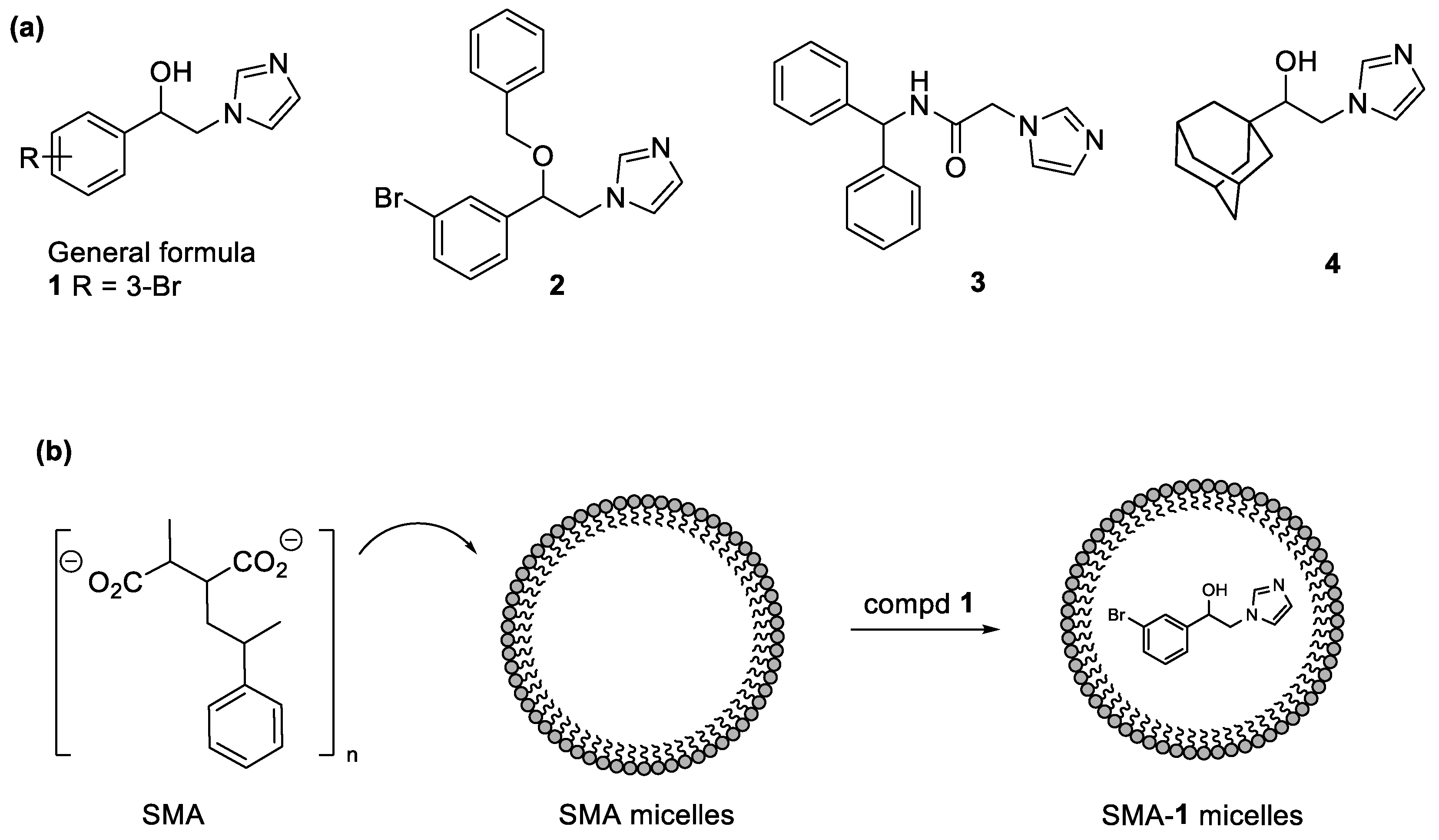
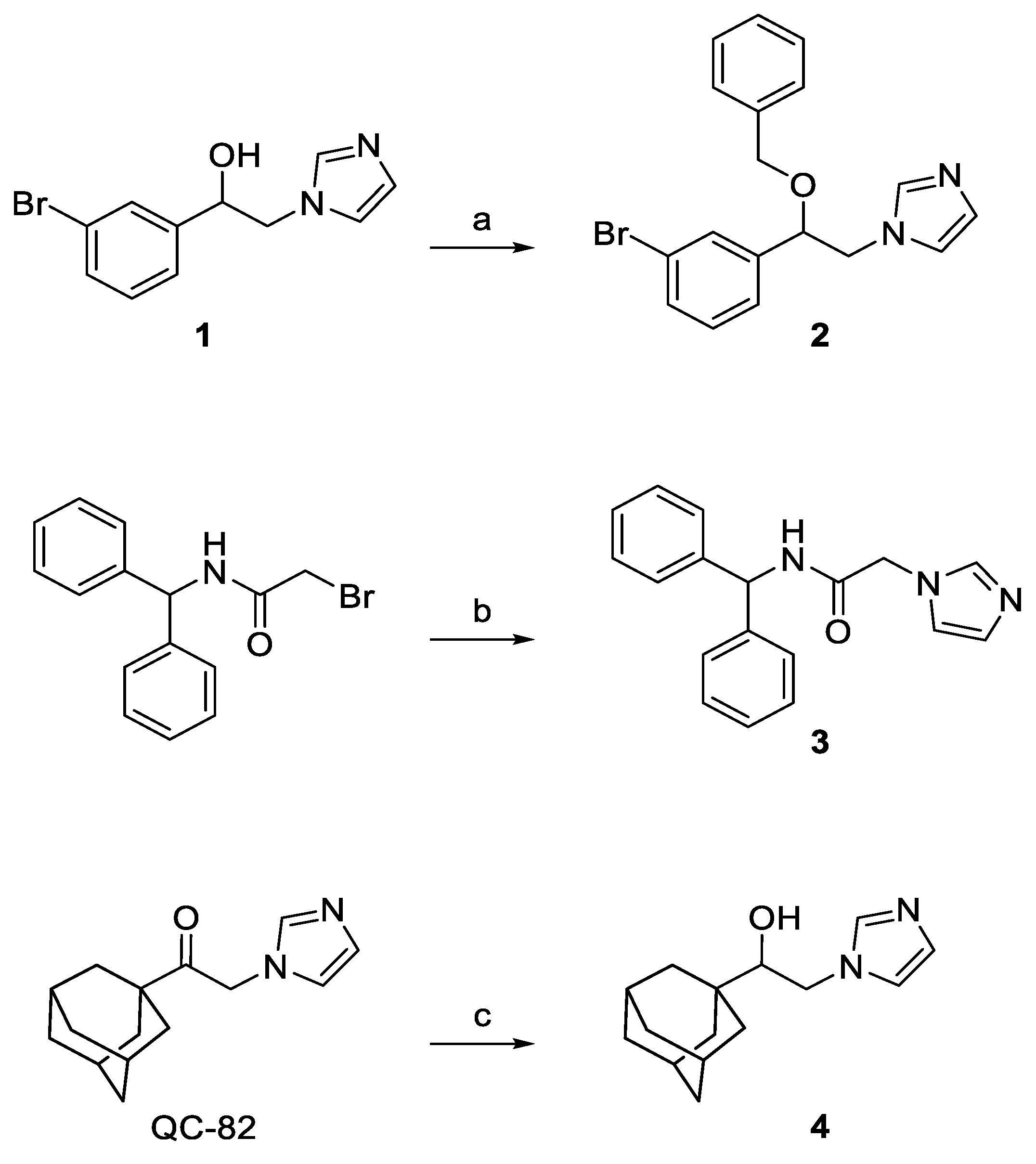
2.2. Chemistry
2.3. Characterization of SMA-1 Micelles
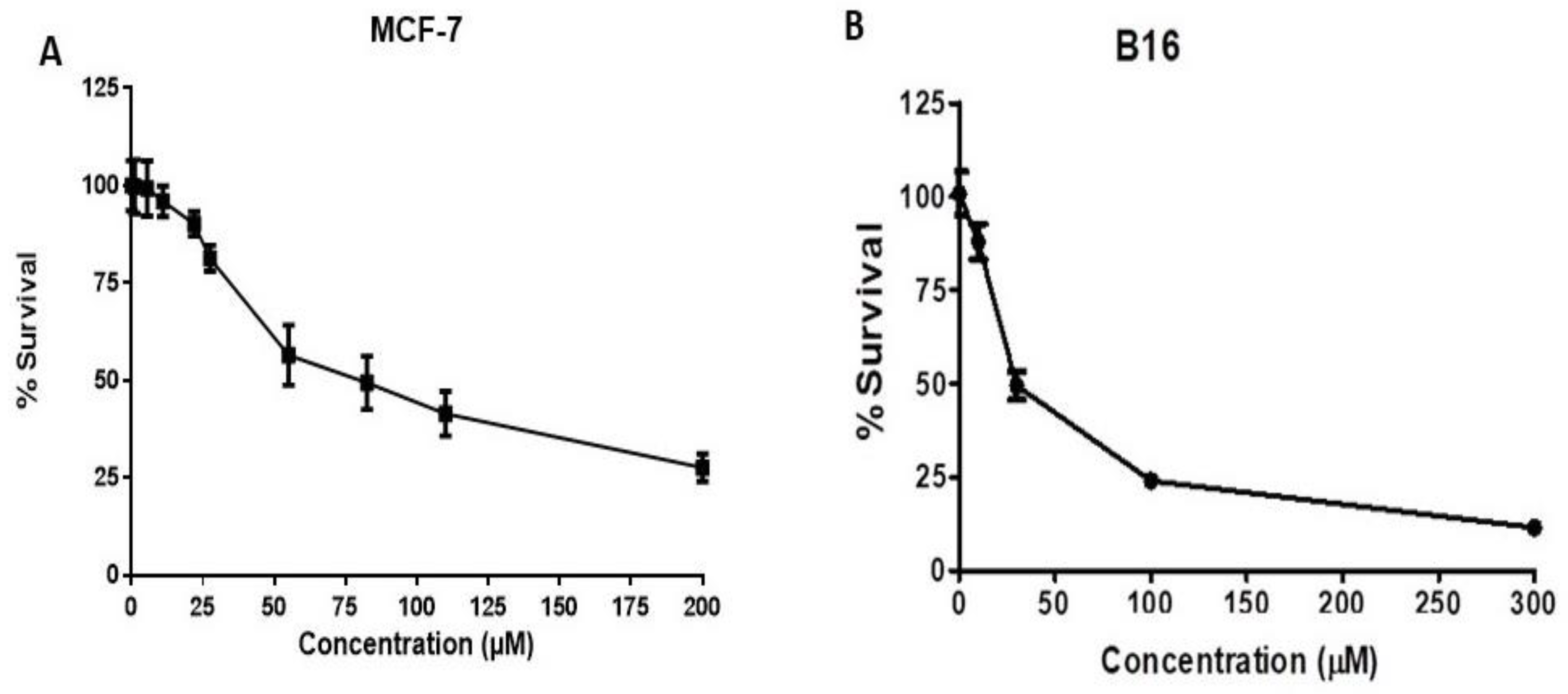
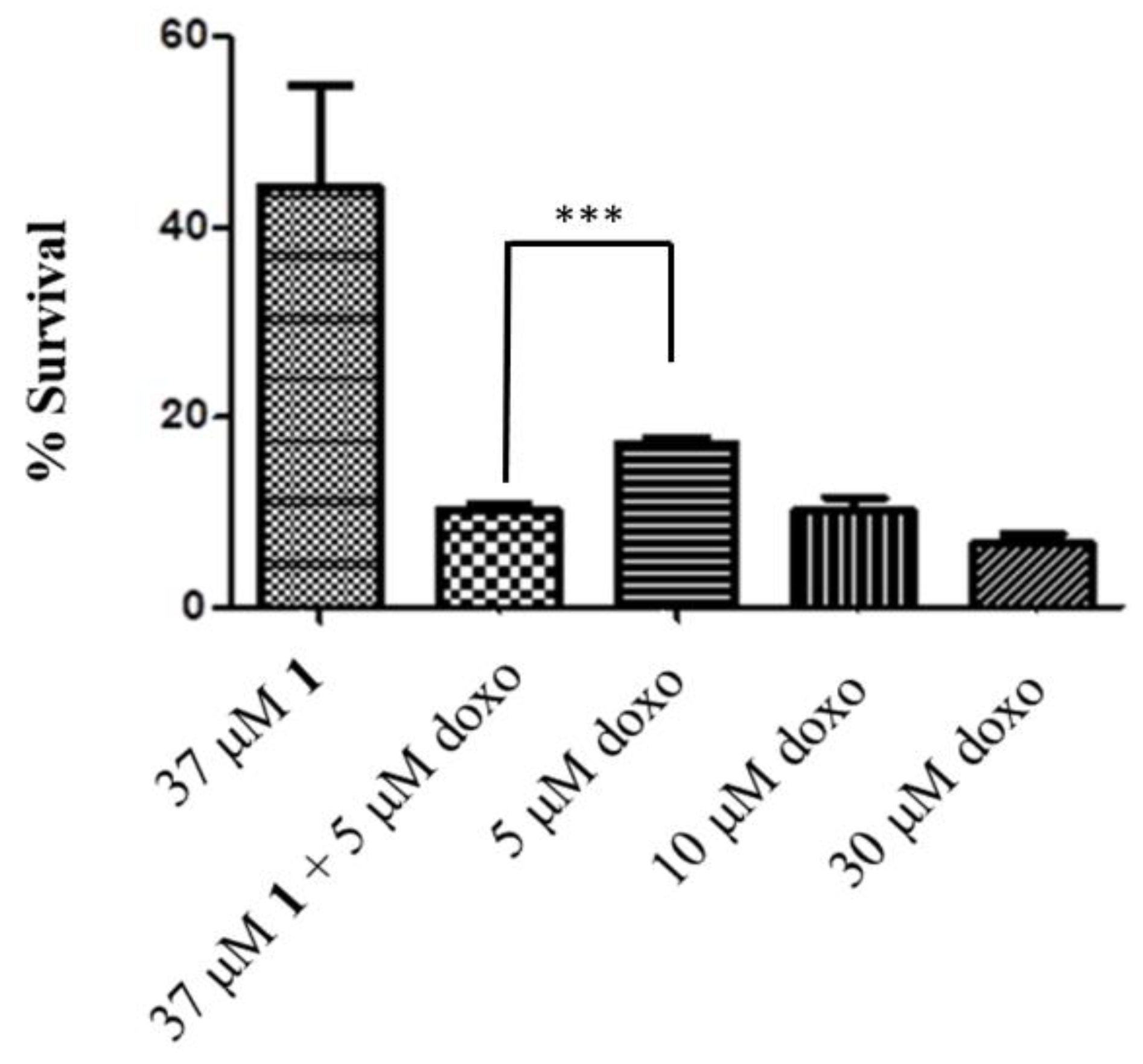
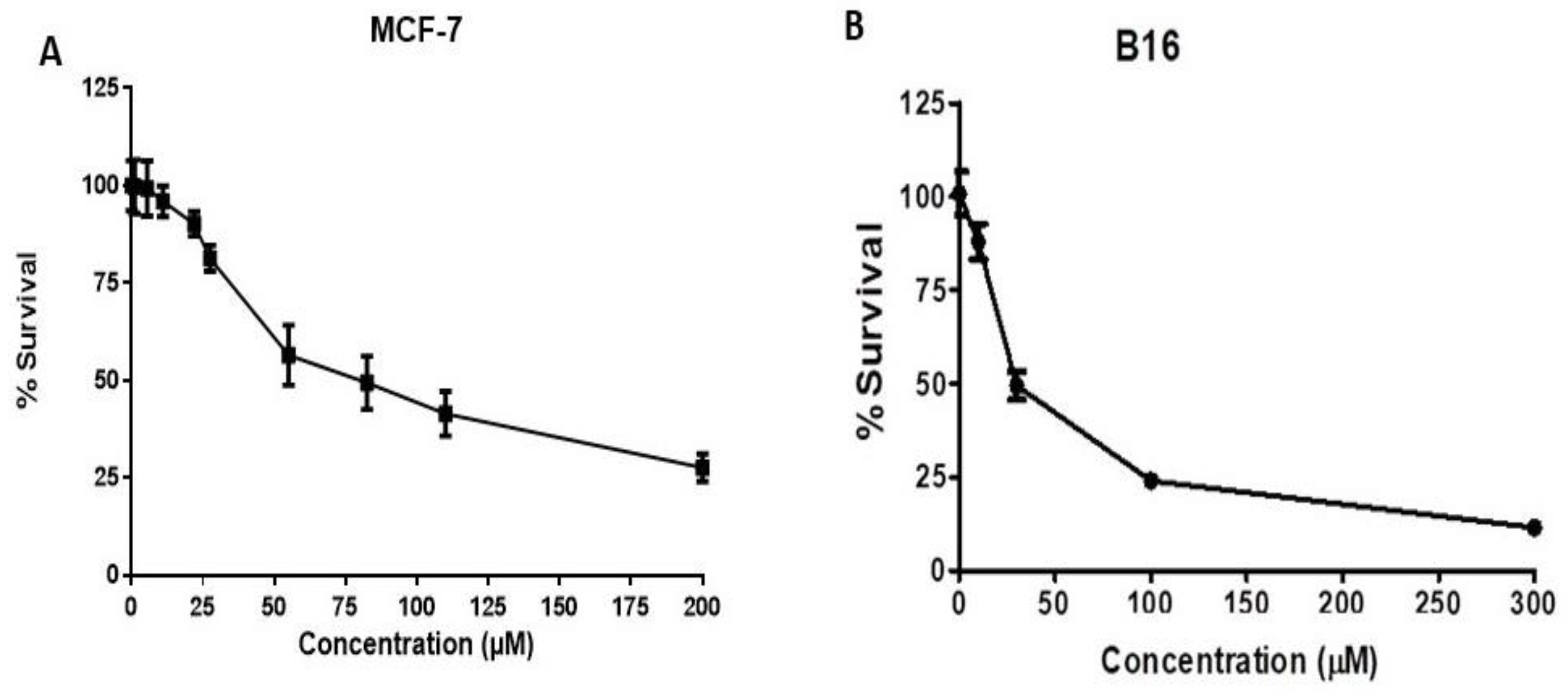
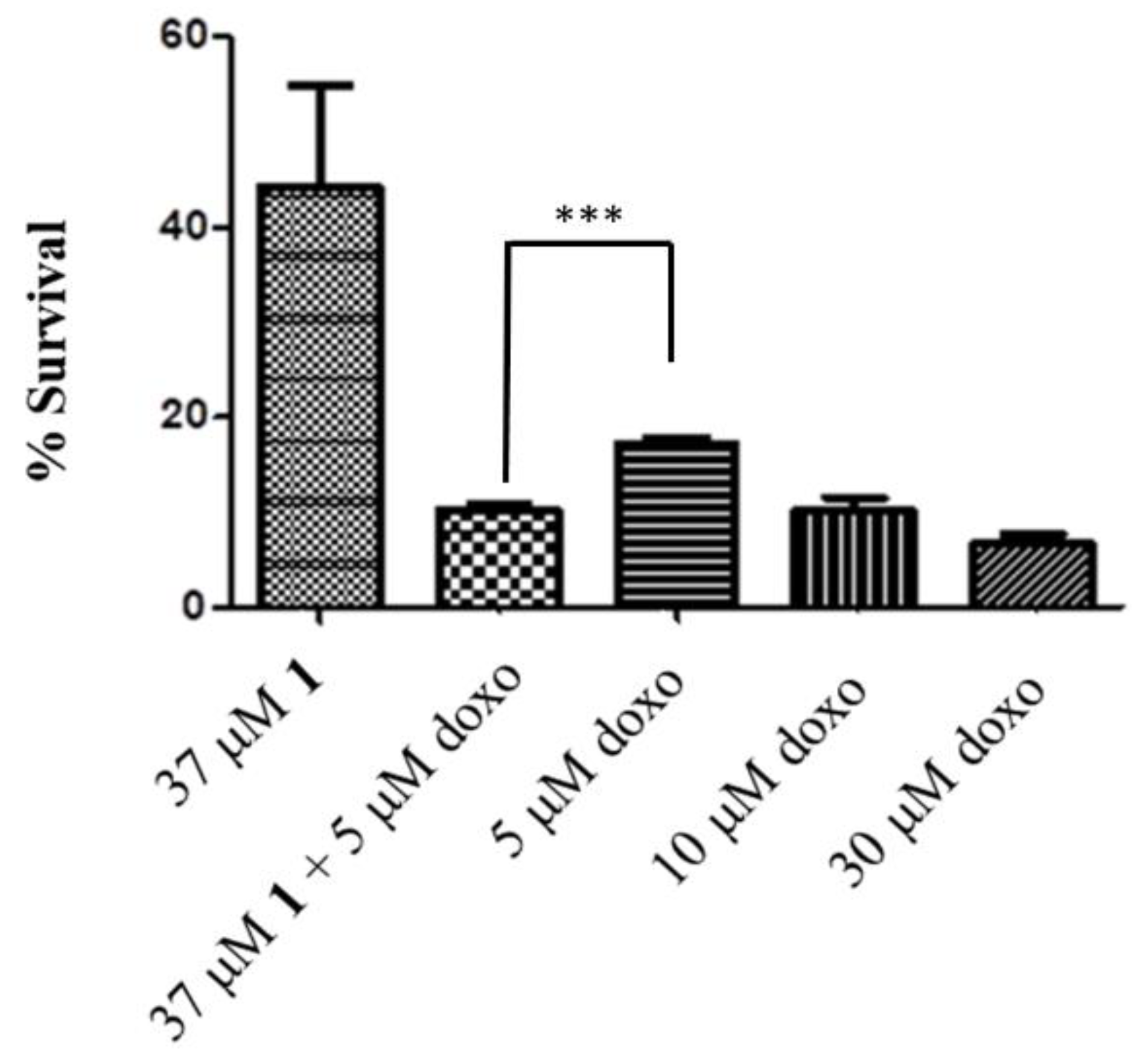
2.4. HO-1 Inhibition and Cytotoxicity Activity
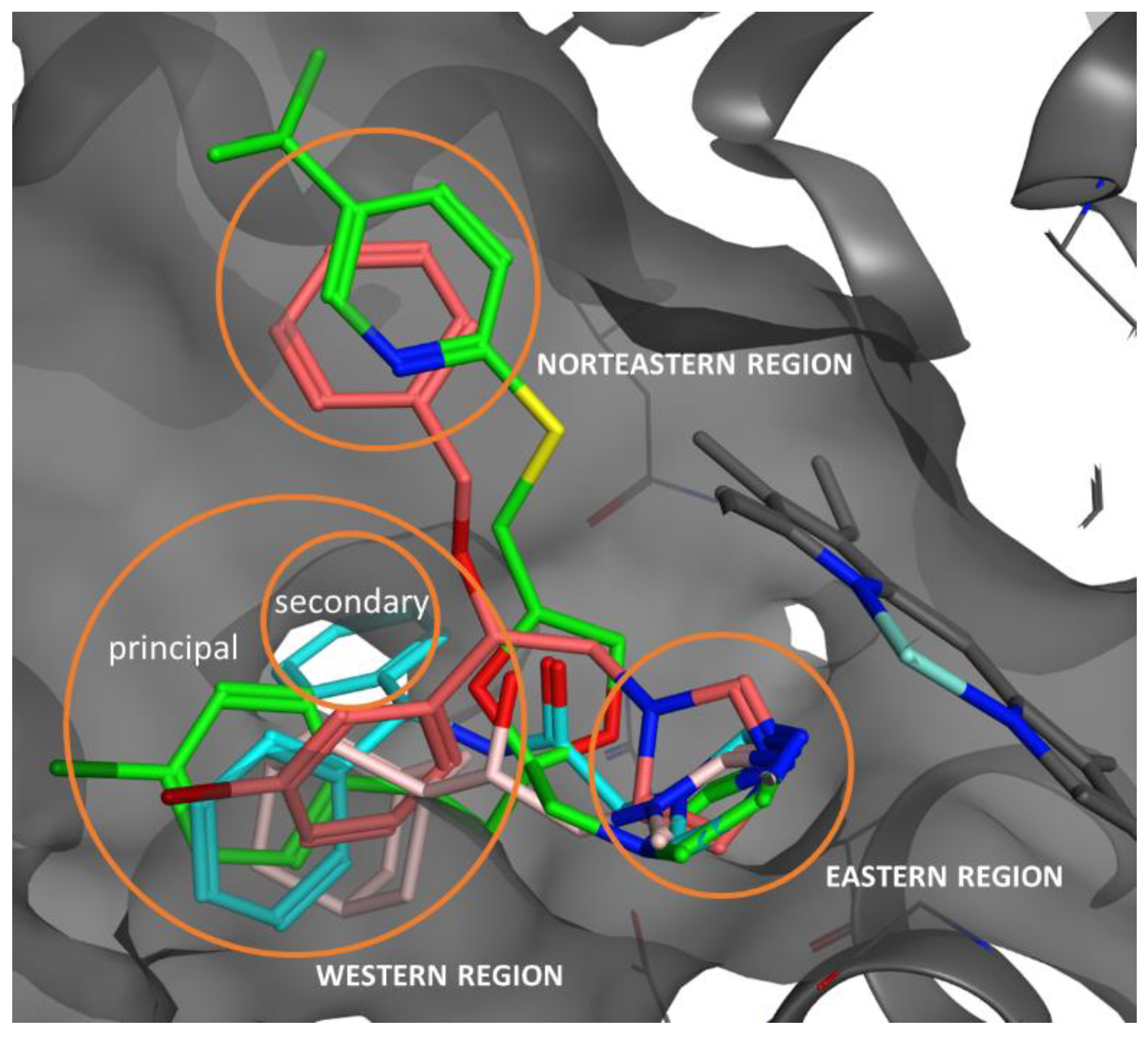
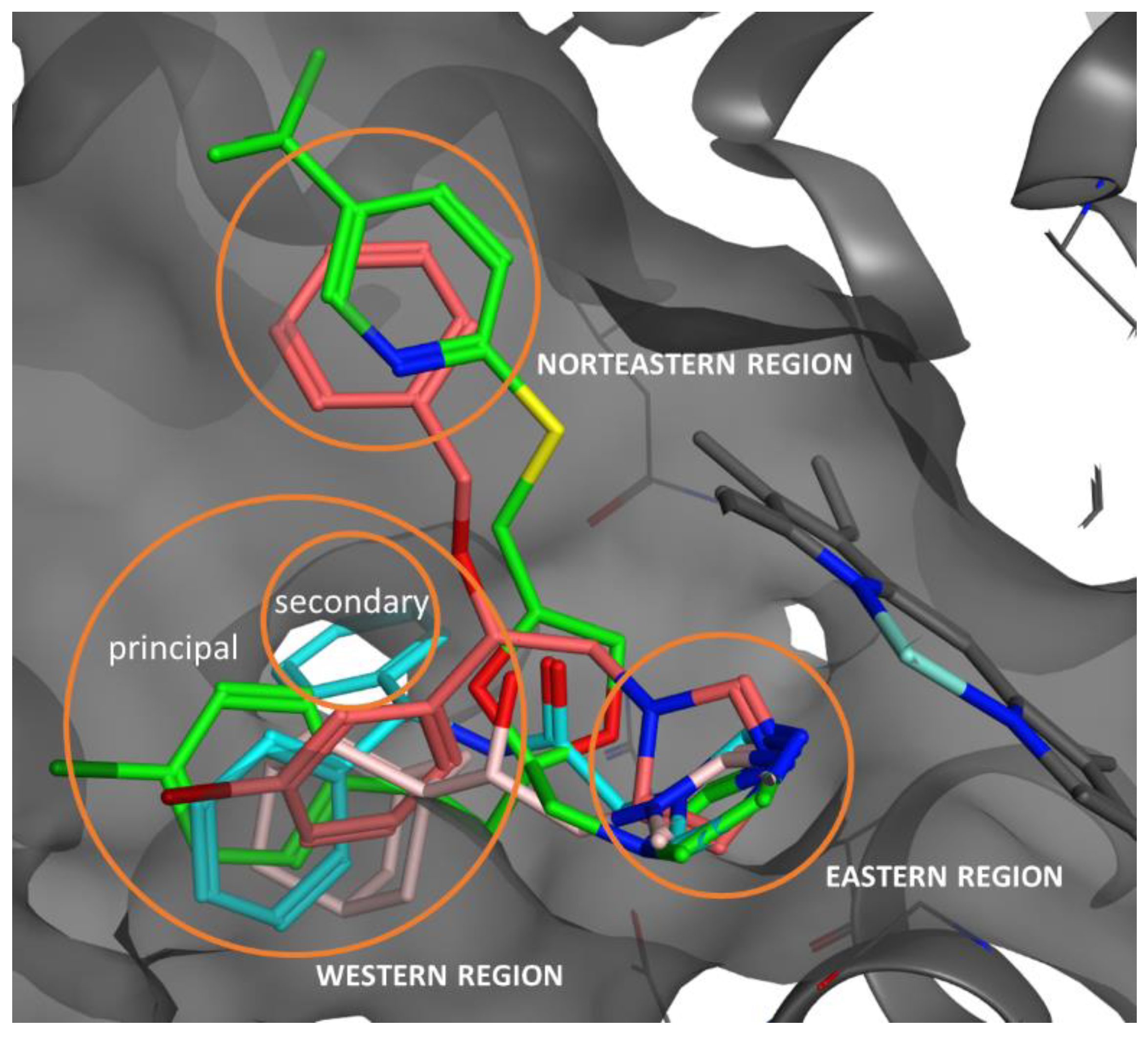
2.5. Docking Studies, ADME, and Toxicity Risk Assessment
3. Materials and Methods
3.1. Chemistry
3.1.1. 1-[2-(3-Bromophenyl)-2-(phenylmethoxy)ethyl]-1H-imidazole (2)
3.1.2. N-Benzhydryl-2-(1H-imidazol-1-yl)acetamide (3)
3.1.3. 2-(1H-Imidazol-1-yl)-1-tricyclo[3.3.1.13,7]dec-1-yl-ethanol (4)
3.2. Synthesis of SMA-1 Micelles
3.2.1. Loading of the SMA-1 Micelles
3.2.2. Size, PDI, and Zeta Potential Determination of SMA Micelles
3.3. Biology
3.3.1. Preparation of Spleen and Brain Microsomal Fractions
3.3.2. Preparation of Biliverdin Reductase
3.3.3. Measurement of HO-1 and HO-2 Enzymatic Activities in Microsomal Fraction of Rat Spleen and Brain
3.3.4. Cell Cultures
3.3.5. In Vitro Cytotoxicity of HO Inhibitors against Breast and Prostate Cancer Cell Lines, Murine Melanoma, and HEK Cells
3.4. Docking
3.4.1. Preparation of Ligands
3.4.2. Docking Protocol
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Abraham, N.G.; Kappas, A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol. Rev. 2008, 60, 79–127. [Google Scholar] [CrossRef] [PubMed]
- Tenhunen, R.; Marver, H.S.; Schmid, R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. USA 1968, 61, 748–755. [Google Scholar] [CrossRef] [PubMed]
- McCoubrey, W.K., Jr.; Maines, M.D. The structure, organization and differential expression of the gene encoding rat heme oxygenase-2. Gene 1994, 139, 155–161. [Google Scholar] [CrossRef]
- Amata, E.; Pittalà, V.; Marrazzo, A.; Parenti, C.; Prezzavento, O.; Arena, E.; Nabavi, S.M.; Salerno, L. Role of the Nrf2/HO-1 axis in bronchopulmonary dysplasia and hyperoxic lung injuries. Clin. Sci. 2017, 131, 1701–1712. [Google Scholar] [CrossRef] [PubMed]
- Pittalà, V.; Vanella, L.; Salerno, L.; Di Giacomo, C.; Acquaviva, R.; Raffaele, M.; Romeo, G.; Modica, M.N.; Prezzavento, O.; Sorrenti, V. Novel caffeic acid phenethyl ester (Cape) analogues as inducers of heme oxygenase-1. Curr. Pharm. Des. 2017, 23, 2657–2664. [Google Scholar] [CrossRef] [PubMed]
- Pittalà, V.; Vanella, L.; Salerno, L.; Romeo, G.; Marrazzo, A.; Di Giacomo, C.; Sorrenti, V. Effects of polyphenolic derivatives on heme oxygenase-system in metabolic dysfunctions. Curr. Med. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Battino, M.; Giampieri, F.; Pistollato, F.; Sureda, A.; de Oliveira, M.R.; Pittalà, V.; Fallarino, F.; Nabavi, S.F.; Atanasov, A.G.; Nabavi, S.M. Nrf2 as regulator of innate immunity: A molecular Swiss army knife! Biotechnol. Adv. 2017, 36, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Dichiara, M.; Prezzavento, O.; Marrazzo, A.; Pittalà, V.; Salerno, L.; Rescifina, A.; Amata, E. Recent advances in drug discovery of phototherapeutic non-porphyrinic anticancer agents. Eur. J. Med. Chem. 2017, 142, 459–485. [Google Scholar] [CrossRef] [PubMed]
- Na, H.K.; Surh, Y.J. Oncogenic potential of Nrf2 and its principal target protein heme oxygenase-1. Free Radic. Biol. Med. 2014, 67, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Ozen, M.; Wong, R.J.; Stevenson, D.K. Heme oxygenase-1 in pregnancy and cancer: Similarities in cellular invasion, cytoprotection, angiogenesis, and immunomodulation. Front. Pharmacol. 2014, 5, 295. [Google Scholar] [CrossRef] [PubMed]
- Chau, L.Y. Heme oxygenase-1: Emerging target of cancer therapy. J. Biomed. Sci. 2015, 22, 22. [Google Scholar] [CrossRef] [PubMed]
- Ignarro, L.J.; Ballot, B.; Wood, K.S. Regulation of soluble guanylate cyclase activity by porphyrins and metalloporphyrins. J. Biol. Chem. 1984, 259, 6201–6207. [Google Scholar] [PubMed]
- Luo, D.; Vincent, S.R. Metalloporphyrins inhibit nitric oxide-dependent cGMP formation in vivo. Eur. J. Pharmacol. 1994, 267, 263–267. [Google Scholar] [CrossRef]
- Pistarà, V.; Rescifina, A.; Punzo, F.; Greco, G.; Barbera, V.; Corsaro, A. Design, synthesis, molecular docking and crystal structure prediction of new azasugar analogues of a-glucosidase inhibitors. Eur. J. Org. Chem. 2011, 36, 7278–7287. [Google Scholar] [CrossRef]
- Vreman, H.J.; Wong, R.J.; Stevenson, D.K. Carbon Monoxide and Cardiovascular Function; CRC: Boca Raton, FL, USA, 2002; Chapter 15; p. 273. [Google Scholar]
- Rahman, M.N.; Vukomanovic, D.; Vlahakis, J.Z.; Szarek, W.A.; Nakatsu, K.; Jia, Z. Structural insights into human heme oxygenase-1 inhibition by potent and selective azole-based compounds. J. R. Soc. Interface 2013, 10, 20120697. [Google Scholar] [CrossRef] [PubMed]
- Pittalà, V.; Salerno, L.; Romeo, G.; Modica, M.N.; Siracusa, M.A. A focus on heme oxygenase-1 (HO-1) inhibitors. Curr. Med. Chem. 2013, 20, 3711–3732. [Google Scholar] [CrossRef] [PubMed]
- Salerno, L.; Pittalà, V.; Romeo, G.; Modica, M.N.; Siracusa, M.A.; Di Giacomo, C.; Acquaviva, R.; Barbagallo, I.; Tibullo, D.; Sorrenti, V. Evaluation of novel aryloxyalkyl derivatives of imidazole and 1,2,4-triazole as heme oxygenase-1 (HO-1) inhibitors and their antitumor properties. Bioorg. Med. Chem. 2013, 21, 5145–5153. [Google Scholar] [CrossRef] [PubMed]
- Sorrenti, V.; Guccione, S.; Di Giacomo, C.; Modica, M.N.; Pittalà, V.; Acquaviva, R.; Basile, L.; Pappalardo, M.; Salerno, L. Evaluation of imidazole-based compounds as heme oxygenase-1 inhibitors. Chem. Biol. Drug Des. 2012, 80, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Salerno, L.; Pittalà, V.; Romeo, G.; Modica, M.N.; Marrazzo, A.; Siracusa, M.A.; Sorrenti, V.; Di Giacomo, C.; Vanella, L.; Parayath, N.N.; et al. Novel imidazole derivatives as heme oxygenase-1 (HO-1) and heme oxygenase-2 (HO-2) inhibitors and their cytotoxic activity in human-derived cancer cell lines. Eur. J. Med. Chem. 2015, 96, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Salerno, L.; Amata, E.; Romeo, G.; Marrazzo, A.; Prezzavento, O.; Floresta, G.; Sorrenti, V.; Barbagallo, I.; Rescifina, A.; Pittalà, V. Potholing of the hydrophobic heme oxygenase-1 western region for the search of potent and selective imidazole-based inhibitors. Eur. J. Med. Chem. 2018, 148, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Pevarello, P.; Fancelli, D.; Vulpetti, A.; Amici, R.; Villa, M.; Pittalà, V.; Vianello, P.; Cameron, A.; Ciomei, M.; Mercurio, C.; et al. 3-Amino-1,4,5,6-tetrahydropyrrolo[3,4-c]pyrazoles: A new class of CDK2 inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 1084–1090. [Google Scholar] [CrossRef] [PubMed]
- Bindi, S.; Fancelli, D.; Alli, C.; Berta, D.; Bertrand, J.A.; Cameron, A.D.; Cappella, P.; Carpinelli, P.; Cervi, G.; Croci, V.; et al. Thieno[3,2-c]pyrazoles: A novel class of Aurora inhibitors with favorable antitumor activity. Bioorg. Med. Chem. 2010, 18, 7113–7120. [Google Scholar] [CrossRef] [PubMed]
- Forte, G.; Fortuna, C.G.; Salerno, L.; Modica, M.N.; Siracusa, M.A.; Cardile, V.; Romeo, G.; Bulbarelli, A.; Lonati, E.; Pittalà, V. Antitumor properties of substituted (αE)-α-(1H-indol-3-ylmethylene)benzeneacetic acids or amides. Bioorg. Med. Chem. 2013, 21, 5233–5245. [Google Scholar] [CrossRef] [PubMed]
- Nastasi, G.; Miceli, C.; Pittalà, V.; Modica, M.N.; Prezzavento, O.; Romeo, G.; Rescifina, A.; Marrazzo, A.; Amata, E. S2RSLDB: A comprehensive manually curated, internet-accessible database of the sigma-2 receptor selective ligands. J. Cheminform. 2017, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Floresta, G.; Rescifina, A.; Marrazzo, A.; Dichiara, M.; Pistarà, V.; Pittalà, V.; Prezzavento, O.; Amata, E. Hyphenated 3D-QSAR statistical model-scaffold hopping analysis for the identification of potentially potent and selective sigma-2 receptor ligands. Eur. J. Med. Chem. 2017, 139, 884–891. [Google Scholar] [CrossRef] [PubMed]
- Amata, E.; Marrazzo, A.; Dichiara, M.; Modica, M.N.; Salerno, L.; Prezzavento, O.; Nastasi, G.; Rescifina, A.; Romeo, G.; Pittalà, V. Heme oxygenase database (HemeOxDB) and QSAR analysis of isoform 1 inhibitors. ChemMedChem 2017, 12, 1873–1881. [Google Scholar] [CrossRef] [PubMed]
- Amata, E.; Marrazzo, A.; Dichiara, M.; Modica, M.N.; Salerno, L.; Prezzavento, O.; Nastasi, G.; Rescifina, A.; Romeo, G.; Pittalà, V. Comprehensive data on a 2D-QSAR model for heme oxygenase isoform 1 inhibitors. Data Brief 2017, 15, 281–299. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.K.; Greish, K.; Fang, J.; Murakami, R.; Maeda, H. High-loading nanosized micelles of copoly(styrene-maleic acid)-zinc protoporphyrin for targeted delivery of a potent heme oxygenase inhibitor. Biomaterials 2007, 28, 1871–1881. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Nakamura, H.; Maeda, H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv. Drug Deliv. Rev. 2011, 63, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Roman, G.; Rahman, M.N.; Vukomanovic, D.; Jia, Z.; Nakatsu, K.; Szarek, W.A. Heme oxygenase inhibition by 2-oxy-substituted 1-azolyl-4-phenylbutanes: Effect of variation of the azole moiety. X-ray crystal structure of human heme oxygenase-1 in complex with 4-phenyl-1-(1H-1,2,4-triazol-1-yl)-2-butanone. Chem. Biol. Drug Des. 2010, 75, 68–90. [Google Scholar] [CrossRef] [PubMed]
- Roman, G.; Riley, J.G.; Vlahakis, J.Z.; Kinobe, R.T.; Brien, J.F.; Nakatsu, K.; Szarek, W.A. Heme oxygenase inhibition by 2-oxy-substituted 1-(1H-imidazol-1-yl)-4-phenylbutanes: Effect of halogen substitution in the phenyl ring. Bioorg. Med. Chem. 2007, 15, 3225–3234. [Google Scholar] [CrossRef] [PubMed]
- Roman, G.; Vlahakis, J.Z.; Vukomanovic, D.; Nakatsu, K.; Szarek, W.A. Heme oxygenase inhibition by 1-aryl-2-(1H-imidazol-1-yl/1H-1,2,4-triazol-1-yl)ethanones and their derivatives. ChemMedChem 2010, 5, 1541–1555. [Google Scholar] [CrossRef] [PubMed]
- Fortuna, C.G.; Forte, G.; Pittalà, V.; Giuffrida, A.; Consiglio, G. Could 2,6-bis((E)-2-(furan-2-yl)vinyl)-1-methylpyridinium iodide and analog compounds intercalate DNA? A first principle prediction based on structural and electronic properties. Comput. Theor. Chem. 2012, 985, 8–13. [Google Scholar] [CrossRef]
- Salerno, L.; Siracusa, M.; Guerrera, F.; Romeo, G.; Pittalà, V.; Modica, M.; Mennini, T.; Russo, F. Synthesis of new 5-phenyl[1,2,4]triazole derivatives as ligands for the 5-HT1A serotonin receptor. Arkivoc 2004, 5, 312–324. [Google Scholar]
- Pittalà, V.; Siracusa, M.A.; Modica, M.N.; Salerno, L.; Pedretti, A.; Vistoli, G.; Cagnotto, A.; Mennini, T.; Romeo, G. Synthesis and molecular modeling of 1H-pyrrolopyrimidine-2,4-dione derivatives as ligands for the α1-adrenoceptors. Bioorg. Med. Chem. 2011, 19, 5260–5276. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, M. Polymeric micelles as drug carriers: Their lights and shadows. J. Drug Target 2014, 22, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, S.; Parayath, N.N.; Taurin, S.; Greish, K. Polymeric nano-micelles: Versatile platform for targeted delivery in cancer. Ther. Deliv. 2014, 5, 1101–1121. [Google Scholar] [CrossRef] [PubMed]
- Alaoui-Jamali, M.A.; Bismar, T.A.; Gupta, A.; Szarek, W.A.; Su, J.; Song, W.; Xu, Y.; Xu, B.; Liu, G.; Vlahakis, J.Z.; et al. A novel experimental heme oxygenase-1-targeted therapy for hormone-refractory prostate cancer. Cancer Res. 2009, 69, 8017–8024. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.Y.; Chen, Y.C.; Shih, C.M.; Lin, C.M.; Cheng, C.H.; Chen, K.C.; Lin, C.W. The induction of heme oxygenase-1 suppresses heat shock protein 90 and the proliferation of human breast cancer cells through its byproduct carbon monoxide. Toxicol. Appl. Pharmacol. 2014, 274, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Bahmani, P.; Hassanshahi, G.; Halabian, R.; Roushandeh, A.M.; Jahanian-Najafabadi, A.; Roudkenar, M.H. The expression of heme oxygenase-1 in human-derived cancer cell lines. Iran J. Med. Sci. 2011, 36, 260–265. [Google Scholar] [PubMed]
- Noh, S.J.; Bae, J.S.; Jamiyandorj, U.; Park, H.S.; Kwon, K.S.; Jung, S.H.; Youn, H.J.; Lee, H.; Park, B.H.; Chung, M.J.; et al. Expression of nerve growth factor and heme oxygenase-1 predict poor survival of breast carcinoma patients. BMC Cancer 2013, 13, 516. [Google Scholar] [CrossRef] [PubMed]
- Gueron, G.; De Siervi, A.; Ferrando, M.; Salierno, M.; De Luca, P.; Elguero, B.; Meiss, R.; Navone, N.; Vazquez, E.S. Critical role of endogenous heme oxygenase 1 as a tuner of the invasive potential of prostate cancer cells. Mol. Cancer Res. 2009, 7, 1745–1755. [Google Scholar] [CrossRef] [PubMed]
- Was, H.; Cichon, T.; Smolarczyk, R.; Rudnicka, D.; Stopa, M.; Chevalier, C.; Leger, J.J.; Lackowska, B.; Grochot, A.; Bojkowska, K.; et al. Overexpression of heme oxygenase-1 in murine melanoma: Increased proliferation and viability of tumor cells, decreased survival of mice. Am. J. Pathol. 2006, 169, 2181–2198. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kim, S.J.; Kim, B.J.; Rah, S.Y.; Chung, S.M.; Im, M.J.; Kim, U.H. Doxorubicin-induced reactive oxygen species generation and intracellular Ca2+ increase are reciprocally modulated in rat cardiomyocytes. Exp. Mol. Med. 2006, 38, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Greish, K.; Mathur, A.; Bakhiet, M.; Taurin, S. Nanomedicine: Is it lost in translation? Ther. Deliv. 2018, 9, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Taurin, S.; Nehoff, H.; Greish, K. Anticancer nanomedicine and tumor vascular permeability; Where is the missing link? J. Control. Release 2012, 164, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Pistarà, V.; Lombardo, G.M.; Rescifina, A.; Bacchi, A.; D’Andrea, F.; Punzo, F. Experimental and in silico characterization of a biologically active inosose. Struct. Chem. 2013, 24, 955–965. [Google Scholar] [CrossRef]
- Greish, K.; Sawa, T.; Fang, J.; Akaike, T.; Maeda, H. SMA-doxorubicin, a new polymeric micellar drug for effective targeting to solid tumours. J. Control. Release 2004, 97, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Maines, M.D. Heme oxygenase: Function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 1988, 2, 2557–2568. [Google Scholar] [CrossRef] [PubMed]
- Trakshel, G.M.; Kutty, R.K.; Maines, M.D. Resolution of the rat brain heme oxygenase activity: Absence of a detectable amount of the inducible form (HO-1). Arch. Biochem. Biophys. 1988, 260, 732–739. [Google Scholar] [CrossRef]
- Vlahakis, J.Z.; Lazar, C.; Roman, G.; Vukomanovic, D.; Nakatsu, K.; Szarek, W.A. Heme oxygenase inhibition by α-(1H-imidazol-1-yl)-ω-phenylalkanes: Effect of introduction of heteroatoms in the alkyl linker. ChemMedChem 2012, 7, 897–902. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.W.; Cui, W.J.; Zhang, X.H.; Shen, Q.X.; Wang, J.; Li, Y.Z.; Chen, S.N.; Yu, S.C. Analysis of heme oxygenase isomers in rat. World J. Gastroenterol. 2002, 8, 1123–1128. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Alam, J.; Choi, A.M. Heme oxygenase-1/carbon monoxide: From basic science to therapeutic applications. Physiol. Rev. 2006, 86, 583–650. [Google Scholar] [CrossRef] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [PubMed]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Allouche, A.R. Gabedit-A graphical user interface for computational chemistry softwares. J. Comput. Chem. 2011, 32, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.J.P. MOPAC2016. Available online: http://OpenMOPAC.net (accessed on 17 May 2018).
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Micelle | Recovery | Loading (wt/wt) | Size (nm) | PDI | Zeta Potential (mV) |
|---|---|---|---|---|---|
| SMA-1 | 80% | 18% | 180.6 ± 12.3 | 0.211 | −0.11 |
| Compound | IC50 (μM) a | |
|---|---|---|
| HO-1 | HO-2 | |
| (R/S)-1 | 0.40 ± 0.01 b | 32.0 ± 2.2 b |
| (R/S)-2 | 80.0 ± 3.3 | ND c |
| 3 | 28.8 ± 1.4 | 14.4 ± 0.9 |
| (R/S)-4 | 67.6 ± 2.1 | ND c |
| Azalanstat b | 5.3 ± 0.4 | 24.4 ± 0.8 |
| Compound | IC50 (μM) a | ||||||
|---|---|---|---|---|---|---|---|
| MDA-MB-231 | MCF-7 | DU145 | PC3 | LnCap | B16 | HEK | |
| 1 | 82.60 ± 0.57 | 52.55 ± 3.76 | 137.60 ± 2.7 | 164.73 ± 2.8 | 158.03 ± 2.3 | 37.00 ± 7.25 | 363.5 ± 21.3 |
| SMA-1 | >100 | >100 | >200 | >200 | >200 | >100 | >300 |
| Compound | ΔGB Calcd. (kcal/mol) | Ki Calcd. (μM) | Exp. IC50 (μM) HO-1 |
|---|---|---|---|
| 2 | −6.24 | 26.5 | 80.0 |
| 3 | −7.04 | 6.87 | 28.8 |
| 4 | −6.75 | 11.2 | 67.6 |
| Compound | Absorption | Distribution | ||
|---|---|---|---|---|
| HIA (%) | In Vitro Caco-2 Cell Permeability (nm s−1) | In Vitro PPB (%) | In Vivo BBB Penetration (Cbrain/Cblood) | |
| 2 | 100.0 | 57.31 | 94.39 | 2.01 |
| 3 | 95.86 | 36.43 | 100 | 0.57 |
| 4 | 95.30 | 26.33 | 67.75 | 0.77 |
| Compound | Mutagenic | Tumorigenic | Reproductive Effects | Irritant | Drug-Likeness | Drug-Score |
|---|---|---|---|---|---|---|
| 2 | none | none | None | none | 2.90 | 0.78 |
| 3 | none | none | None | none | 5.03 | 0.91 |
| 4 | none | none | None | none | 5.10 | 0.93 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Greish, K.F.; Salerno, L.; Al Zahrani, R.; Amata, E.; Modica, M.N.; Romeo, G.; Marrazzo, A.; Prezzavento, O.; Sorrenti, V.; Rescifina, A.; et al. Novel Structural Insight into Inhibitors of Heme Oxygenase-1 (HO-1) by New Imidazole-Based Compounds: Biochemical and In Vitro Anticancer Activity Evaluation. Molecules 2018, 23, 1209. https://doi.org/10.3390/molecules23051209
Greish KF, Salerno L, Al Zahrani R, Amata E, Modica MN, Romeo G, Marrazzo A, Prezzavento O, Sorrenti V, Rescifina A, et al. Novel Structural Insight into Inhibitors of Heme Oxygenase-1 (HO-1) by New Imidazole-Based Compounds: Biochemical and In Vitro Anticancer Activity Evaluation. Molecules. 2018; 23(5):1209. https://doi.org/10.3390/molecules23051209
Chicago/Turabian StyleGreish, Khaled F., Loredana Salerno, Reem Al Zahrani, Emanuele Amata, Maria N. Modica, Giuseppe Romeo, Agostino Marrazzo, Orazio Prezzavento, Valeria Sorrenti, Antonio Rescifina, and et al. 2018. "Novel Structural Insight into Inhibitors of Heme Oxygenase-1 (HO-1) by New Imidazole-Based Compounds: Biochemical and In Vitro Anticancer Activity Evaluation" Molecules 23, no. 5: 1209. https://doi.org/10.3390/molecules23051209
APA StyleGreish, K. F., Salerno, L., Al Zahrani, R., Amata, E., Modica, M. N., Romeo, G., Marrazzo, A., Prezzavento, O., Sorrenti, V., Rescifina, A., Floresta, G., Intagliata, S., & Pittalà, V. (2018). Novel Structural Insight into Inhibitors of Heme Oxygenase-1 (HO-1) by New Imidazole-Based Compounds: Biochemical and In Vitro Anticancer Activity Evaluation. Molecules, 23(5), 1209. https://doi.org/10.3390/molecules23051209