
Liver-Specific Overexpression of Prostasin Attenuates High-Fat Diet-Induced Metabolic Dysregulation in Mice

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
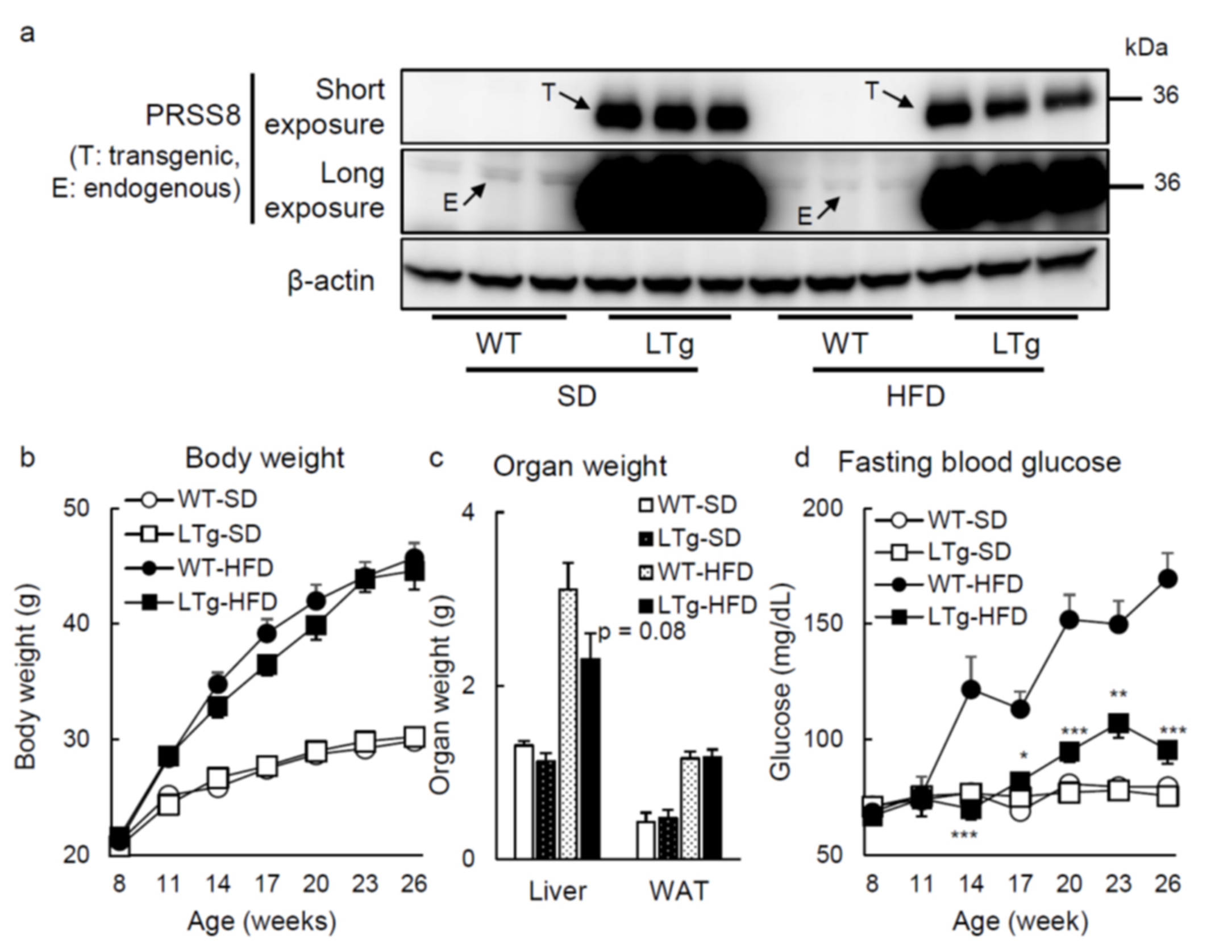
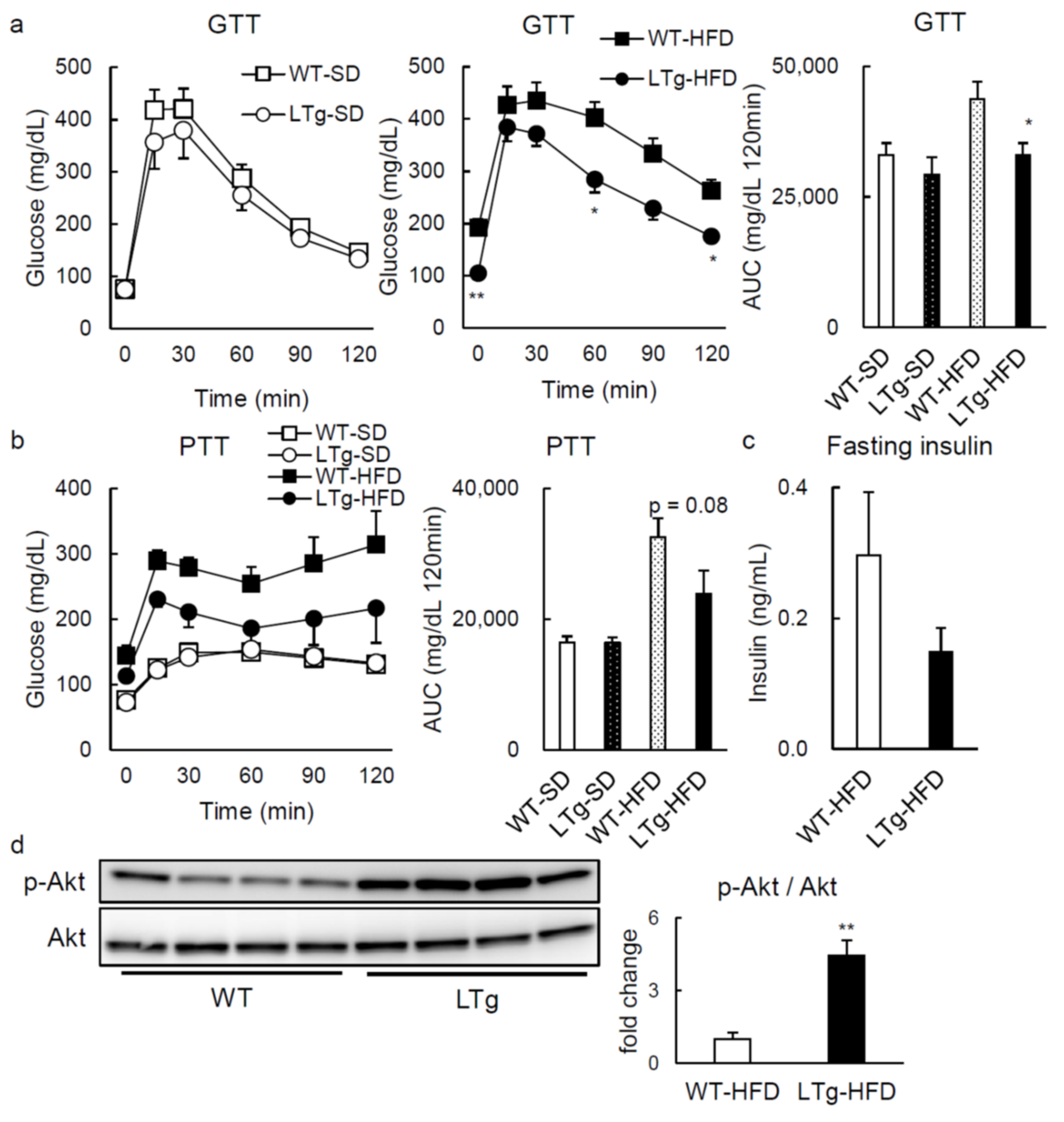
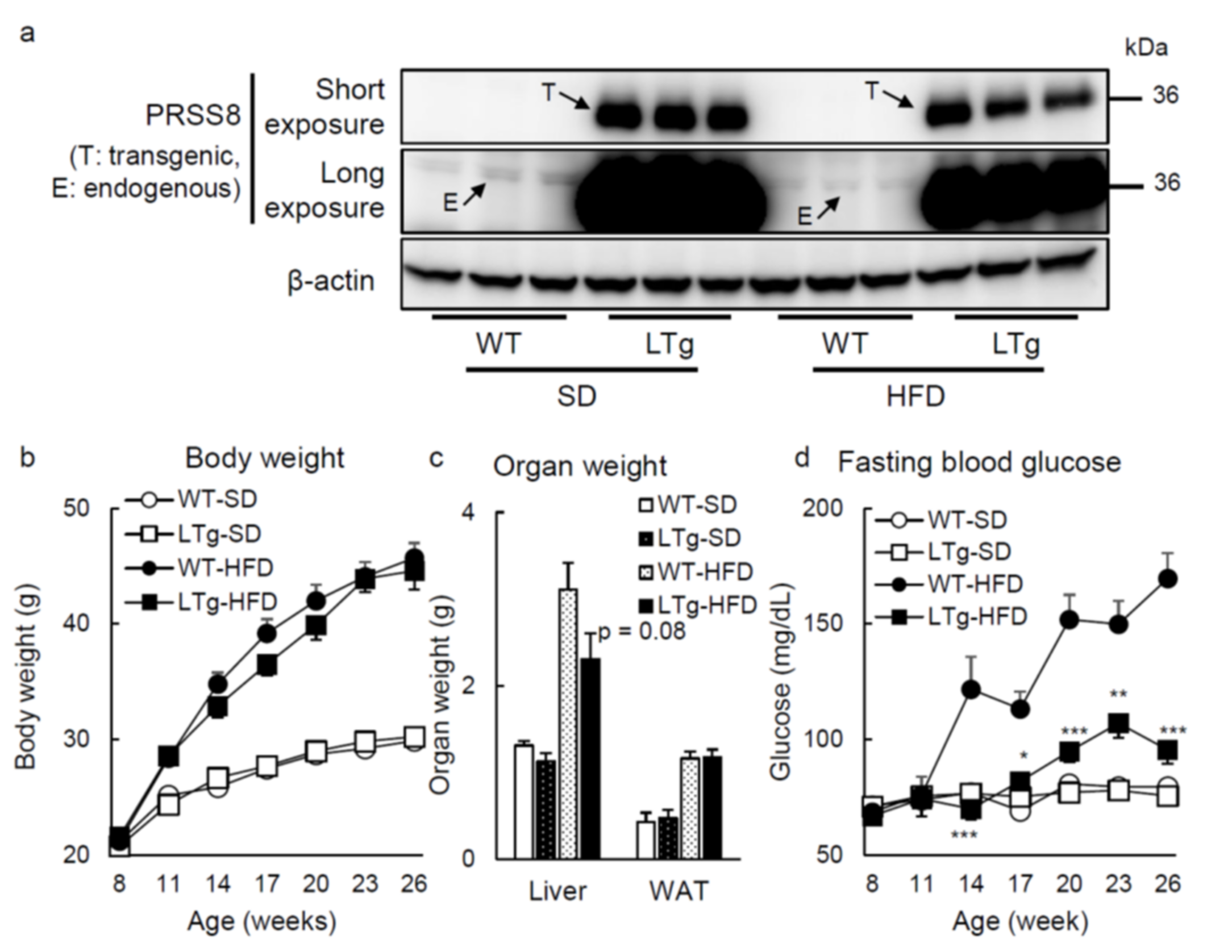
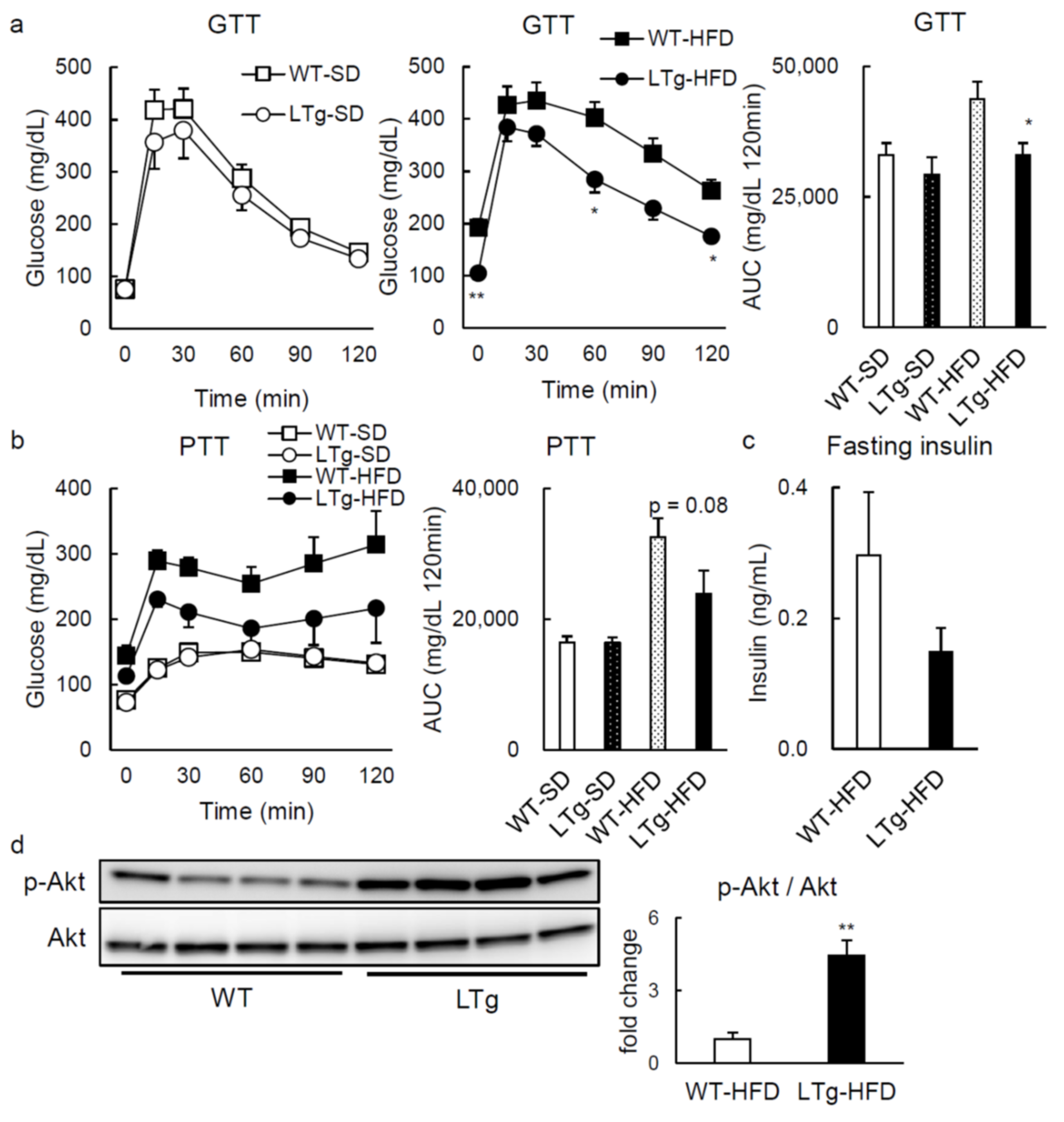
2.1. HFD-Fed LTg Mice Evidenced an Improved Glucose Tolerance Independent of Body Weight
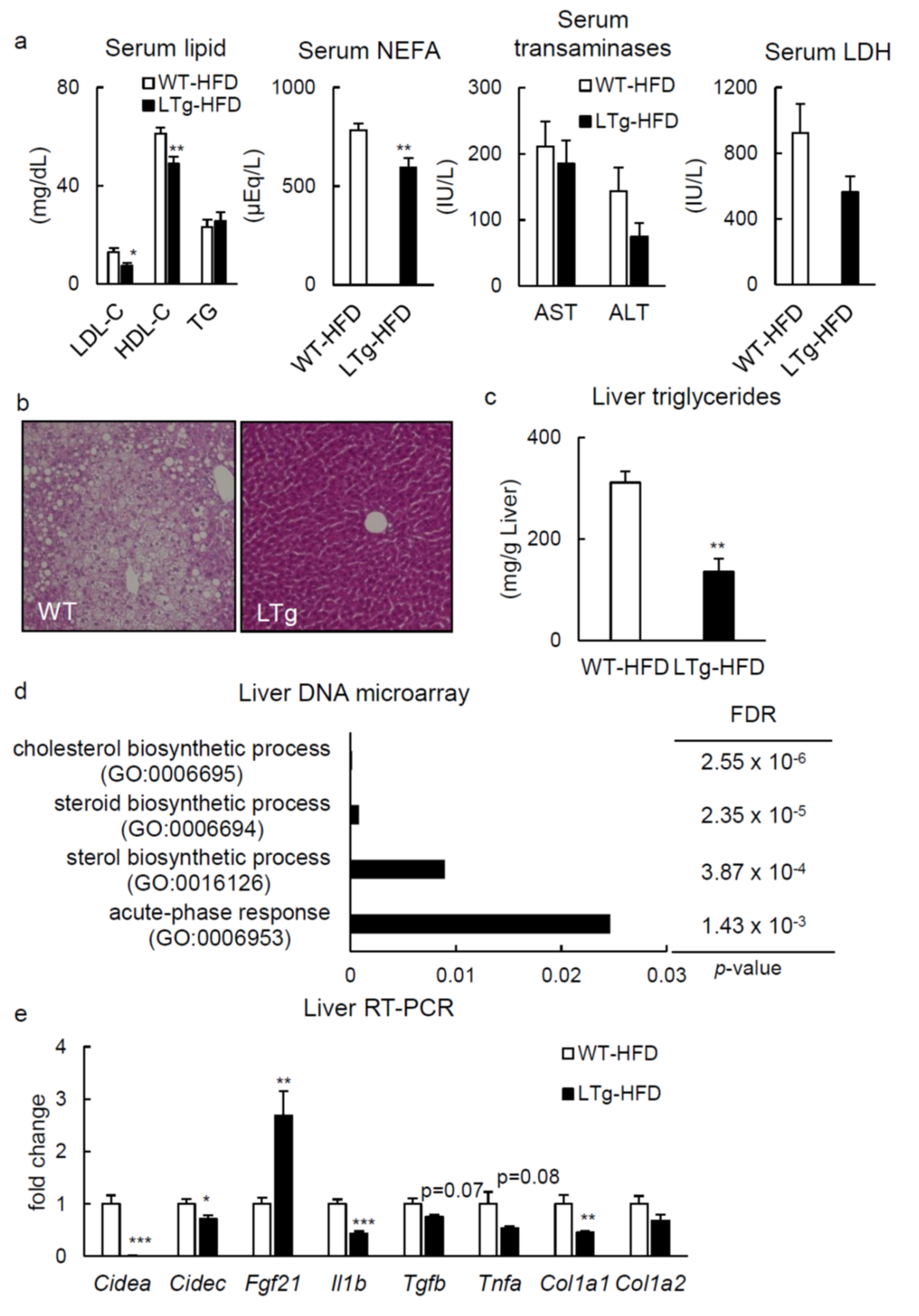
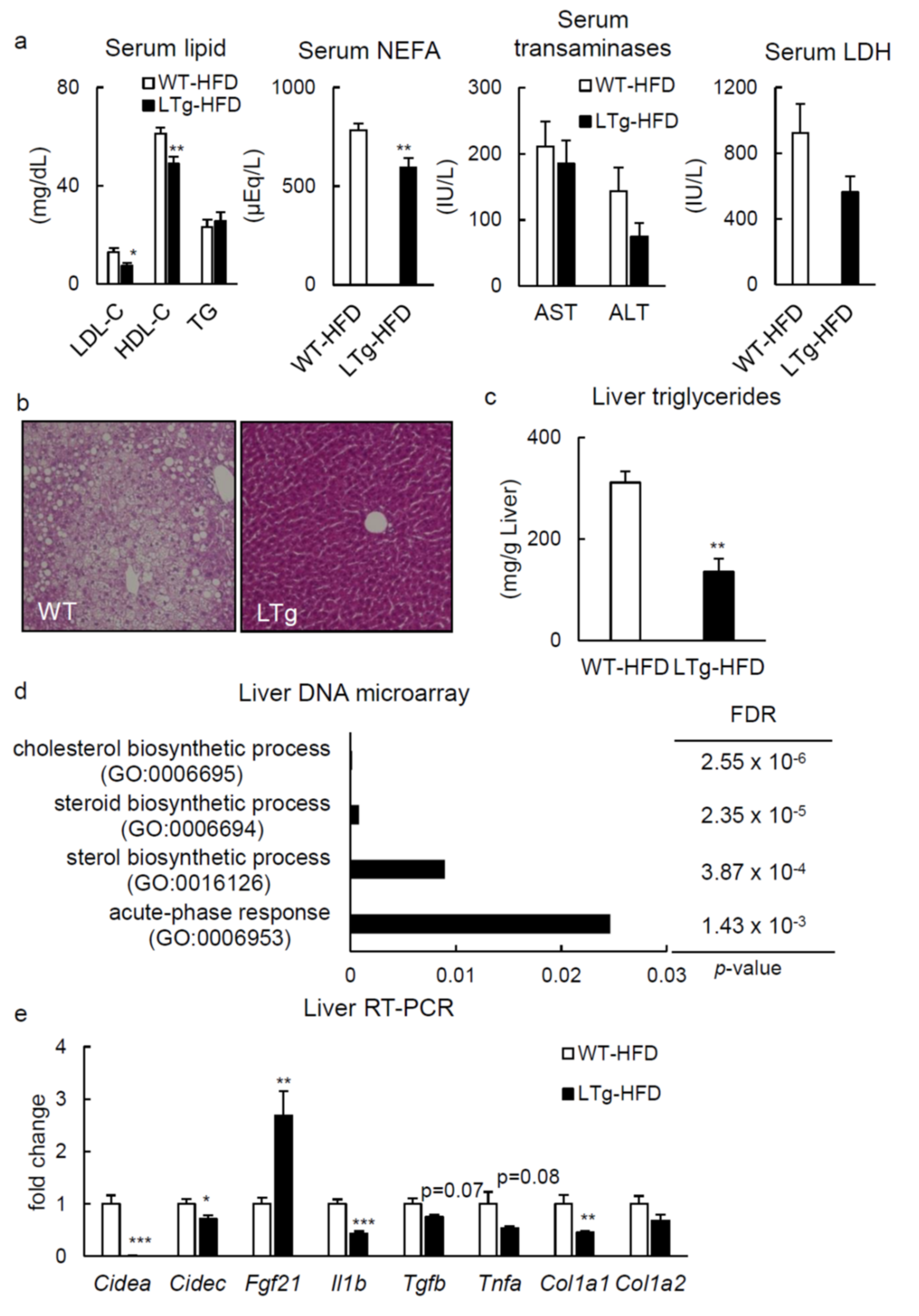
2.2. HFD-Fed LTg Mice Showed Improved Hepatic Steatosis
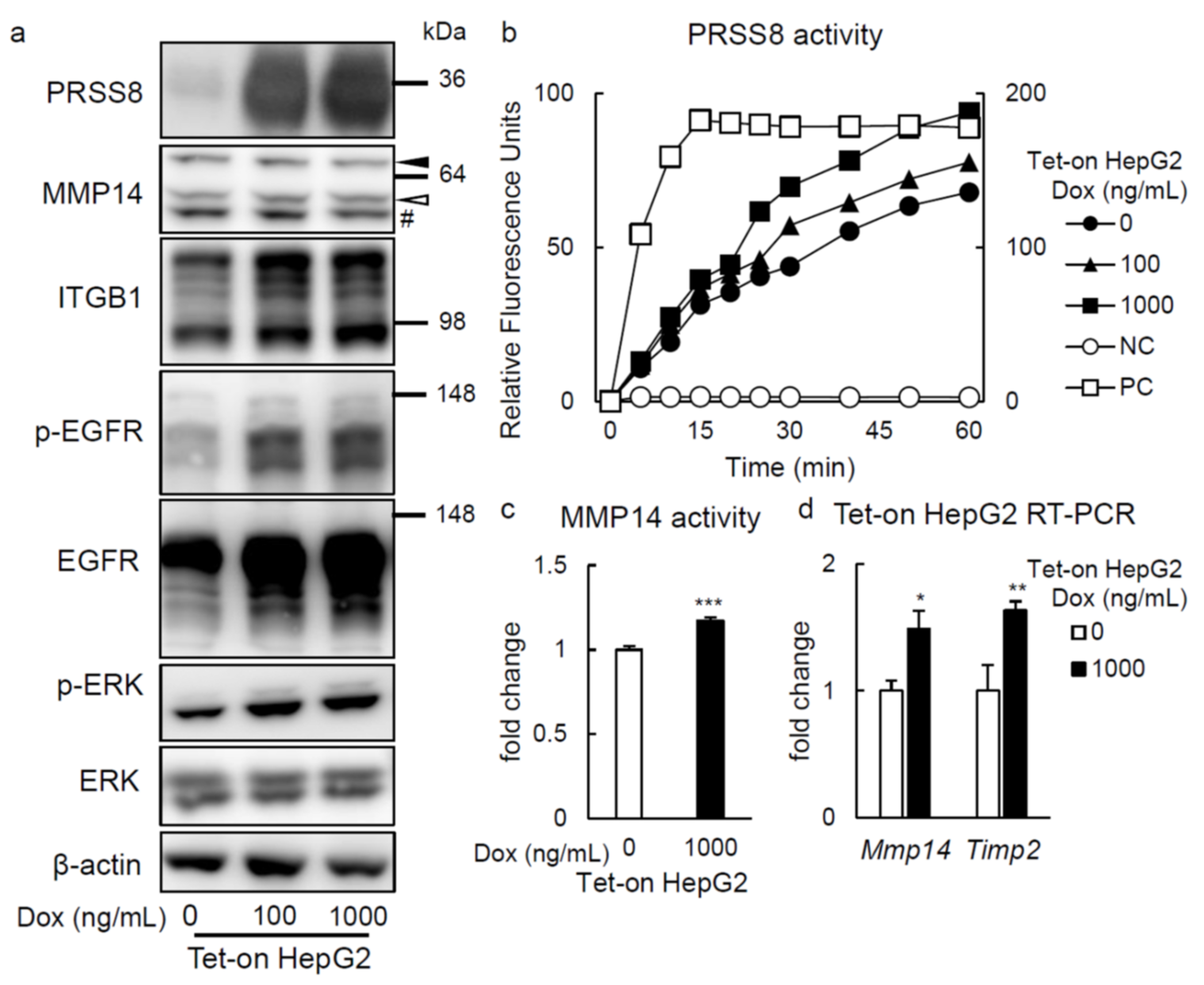
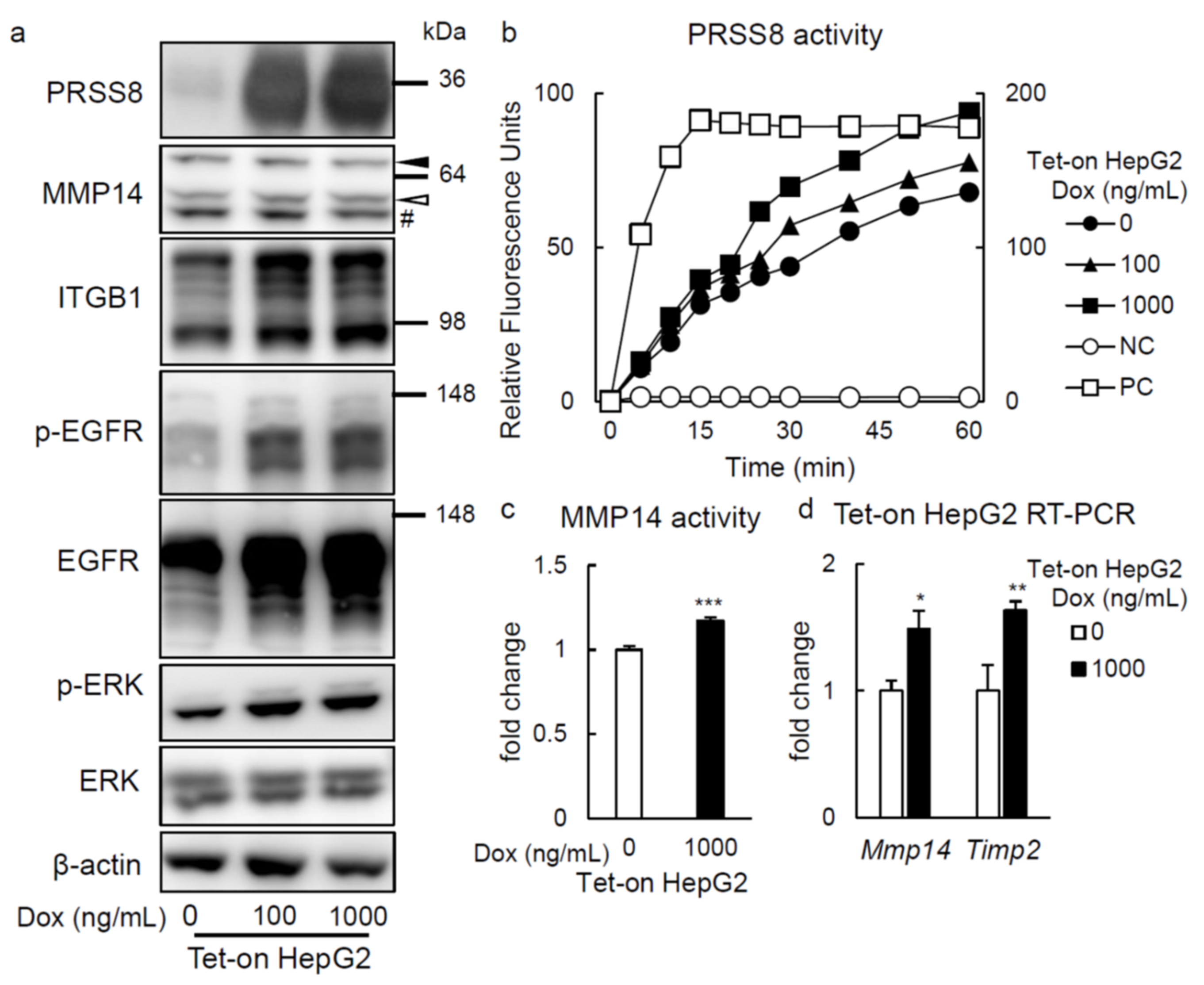
2.3. PRSS8 Amplified ERK Phosphorylation Associated with MMP14 Activation in HepG2 Cells
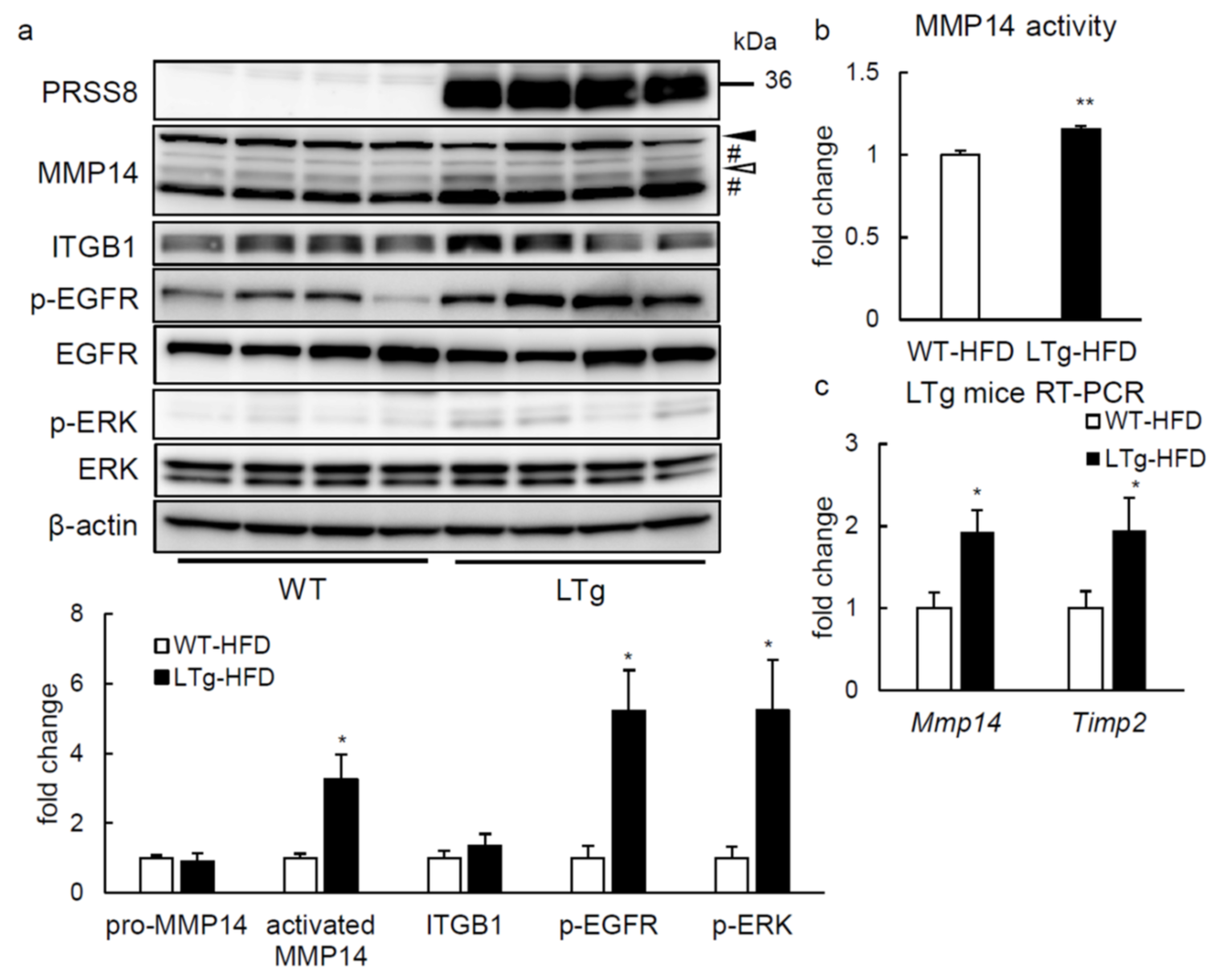
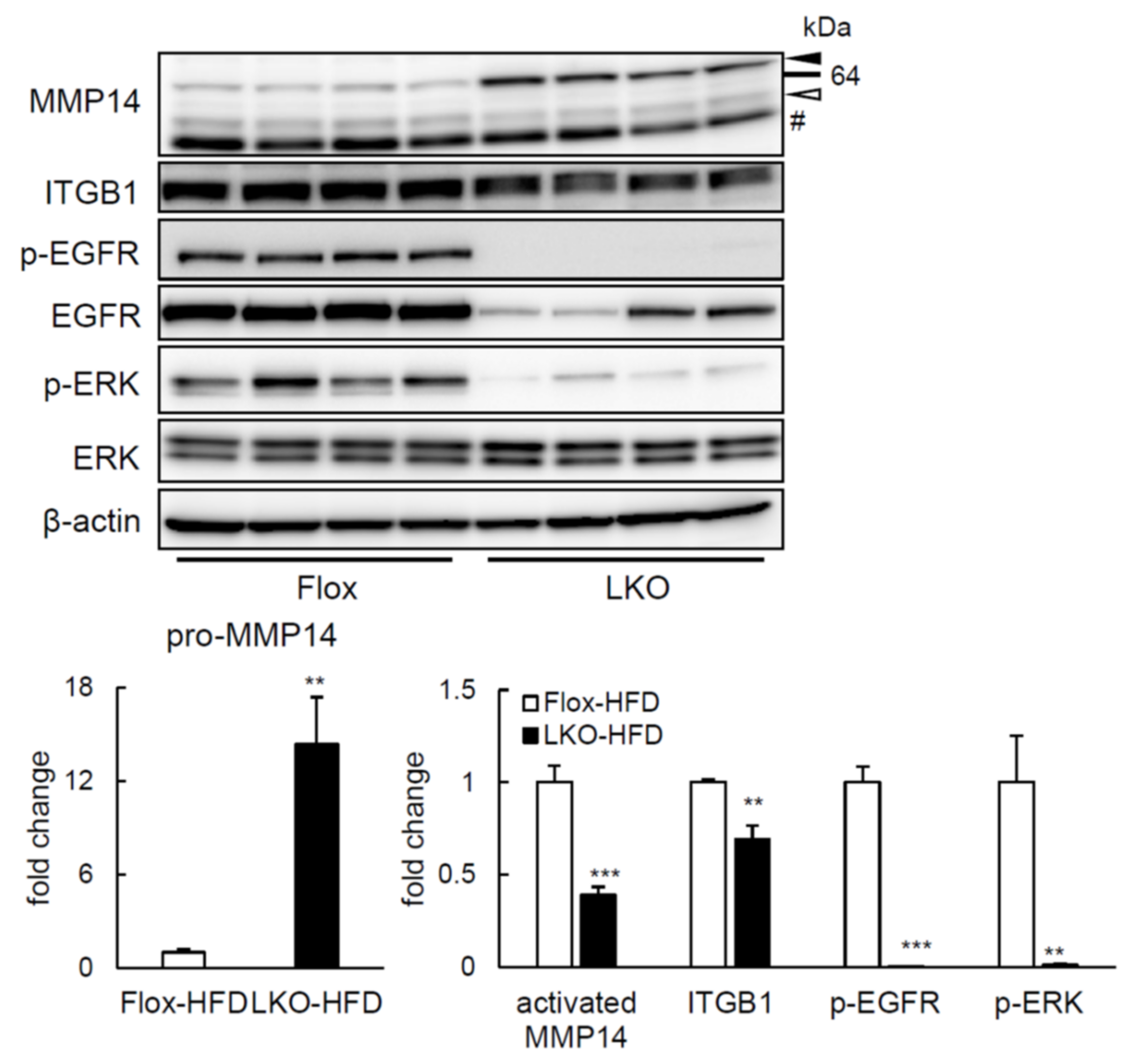
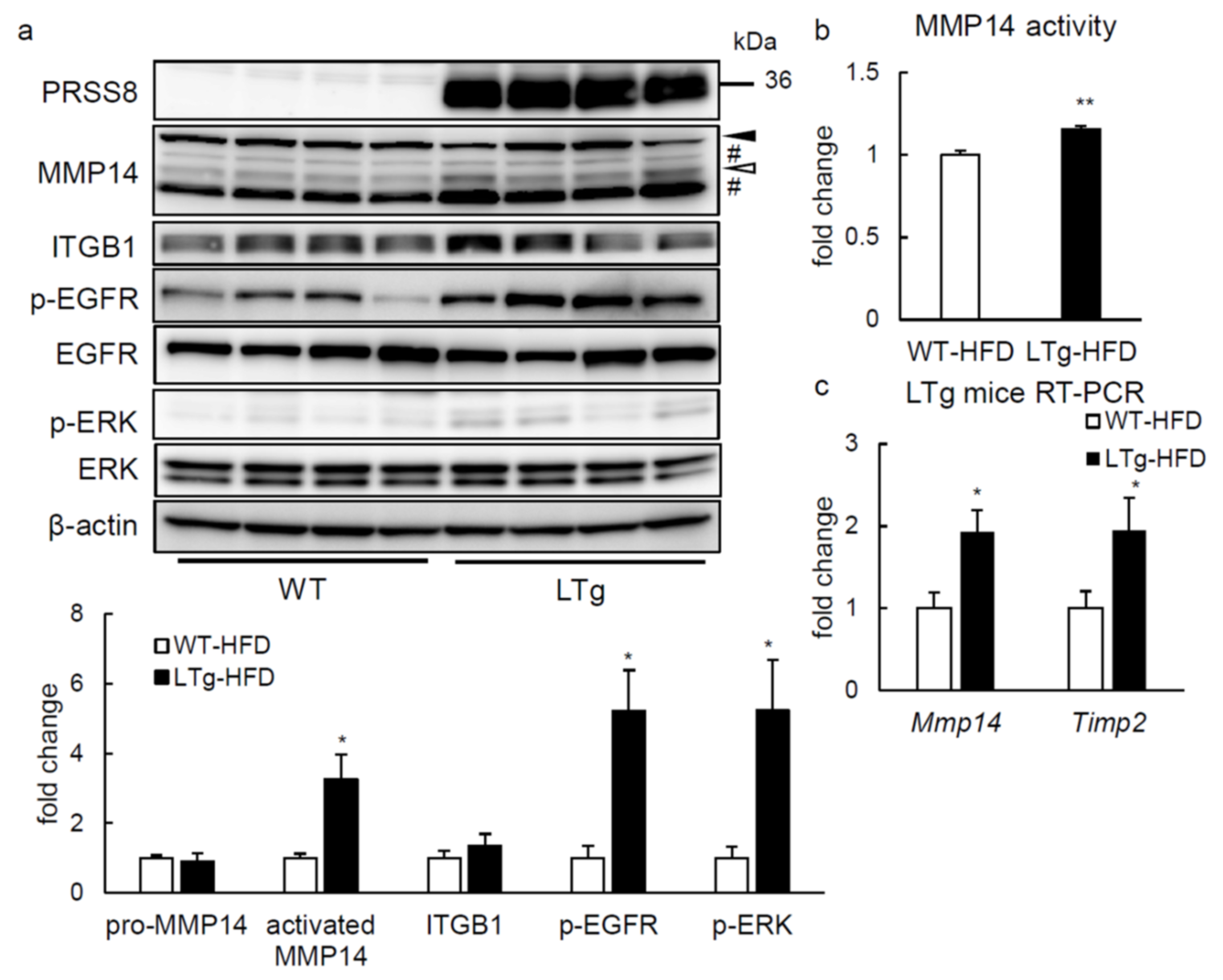
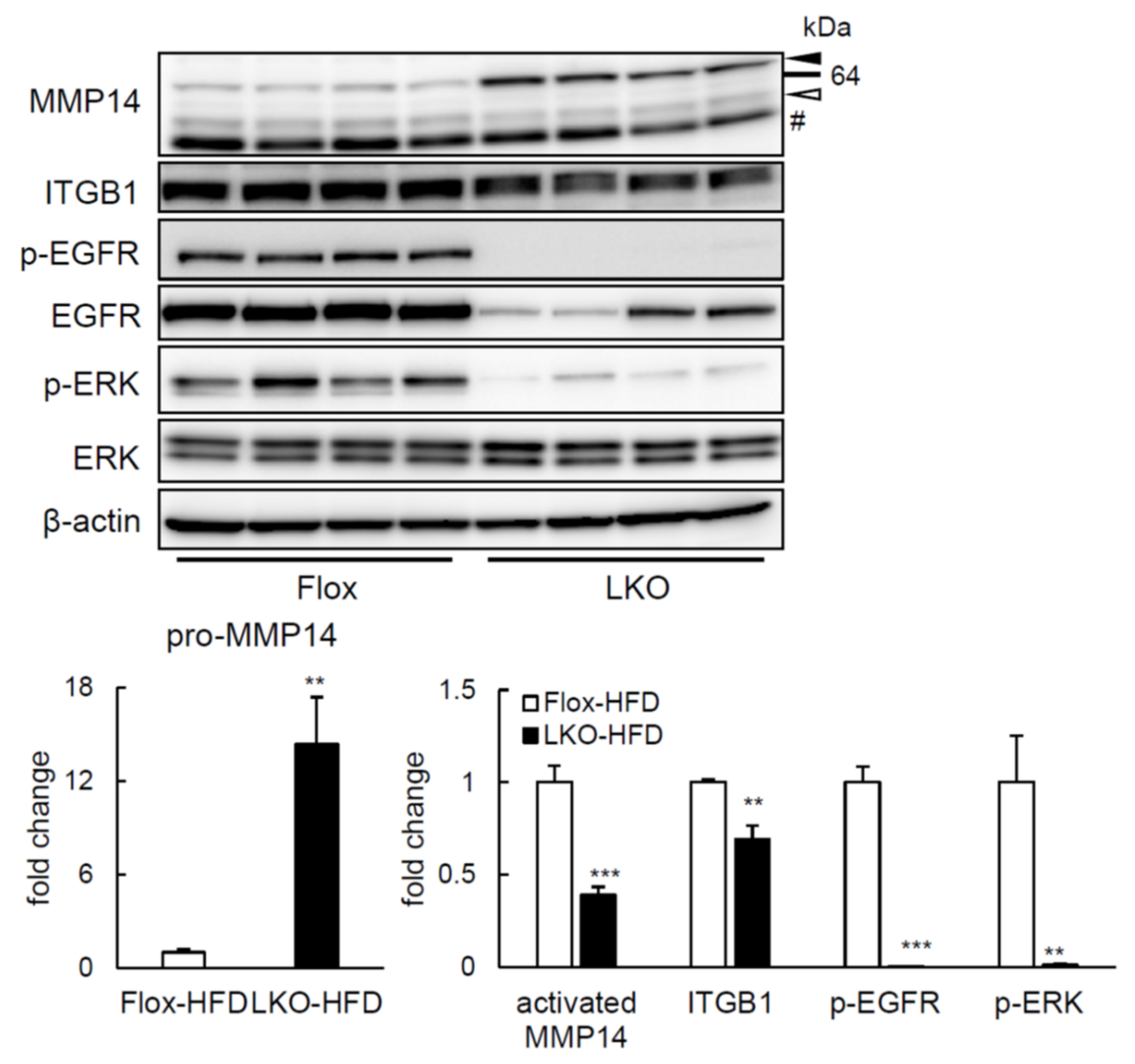
2.4. ERK Phosphorylation Is Elevated in Liver of HFD-fed LTg Mice, Associated with an Increased ITGB1 Expression and MMP14 Activity
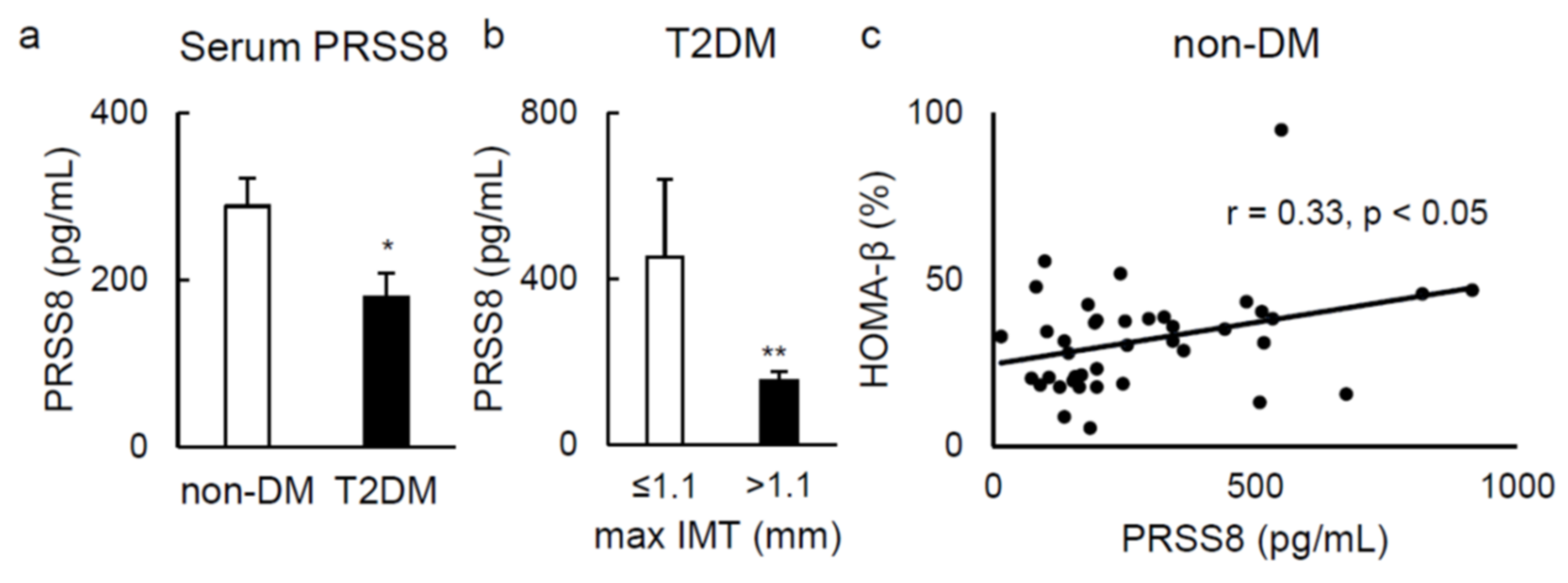
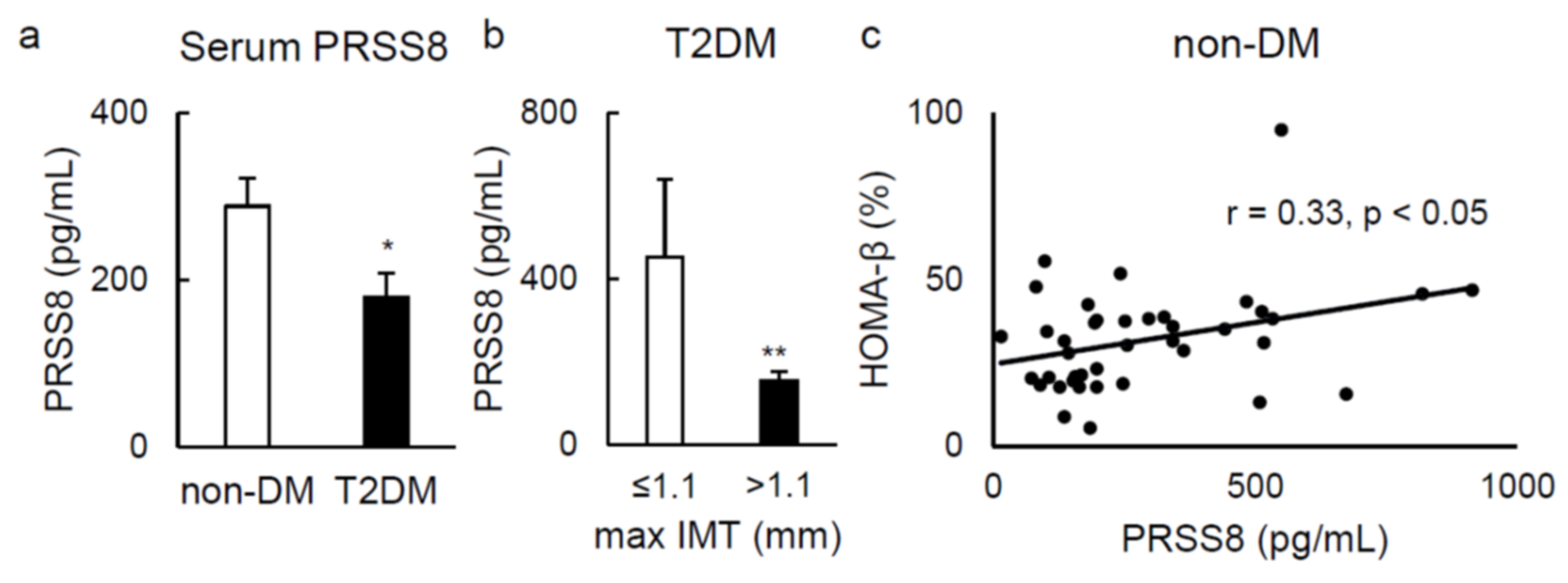
2.5. Serum Soluble PRSS8 Concentration Is Superior in HFD-Fed LTg Mice, Although Being Lessened in Patients with Type 2 Diabetes
3. Discussion
4. Materials and Methods
4.1. Generation of Liver-Specific PRSS8 Overexpressing Transgenic Mice
4.2. Liver-Specific PRSS8 Knockout Mice
4.3. Animal Studies
4.4. Metabolic Tests
4.5. DNA Microarray
4.6. Real-Time Polymerase Chain Reaction
4.7. Western Blotting
4.8. ELISA
4.9. Histologic Examination and Immunohistochemistry
4.10. In Vitro Studies
4.11. PRSS8 Activity Quantification
4.12. Human Studies
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Uchimura, K.; Hayata, M.; Mizumoto, T.; Miyasato, Y.; Kakizoe, Y.; Morinaga, J.; Onoue, T.; Yamazoe, R.; Ueda, M.; Adachi, M.; et al. The serine protease prostasin regulates hepatic insulin sensitivity by modulating TLR4 signalling. Nat. Commun. 2014, 5, 3428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adachi, M.; Kitamura, K.; Miyoshi, T.; Narikiyo, T.; Iwashita, K.; Shiraishi, N.; Nonoguchi, H.; Tomita, K. Activation of epithelial sodium channels by prostasin in Xenopus oocytes. J. Am. Soc. Nephrol. 2001, 12, 1114–1121. [Google Scholar] [CrossRef]
- Saberi, M.; Woods, N.B.; de Luca, C.; Schenk, S.; Lu, J.C.; Bandyopadhyay, G.; Verma, I.M.; Olefsky, J.M. Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metab. 2009, 10, 419–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Invest. 2006, 116, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Suganami, T.; Tanimoto-Koyama, K.; Nishida, J.; Itoh, M.; Yuan, X.; Mizuarai, S.; Kotani, H.; Yamaoka, S.; Miyake, K.; Aoe, S.; et al. Role of the Toll-like receptor 4/NF-kappaB pathway in saturated fatty acid-induced inflammatory changes in the interaction between adipocytes and macrophages. Arter. Thromb. Vasc. Biol. 2007, 27, 84–91. [Google Scholar] [CrossRef] [Green Version]
- Wilson, C.G.; Tran, J.L.; Erion, D.M.; Vera, N.B.; Febbraio, M.; Weiss, E.J. Hepatocyte-Specific Disruption of CD36 Attenuates Fatty Liver and Improves Insulin Sensitivity in HFD-Fed Mice. Endocrinology 2016, 157, 570–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, H.; Kinoshita, T.; Takino, T.; Nakayama, K.; Seiki, M. Activation of a recombinant membrane type 1-matrix metalloproteinase (MT1-MMP) by furin and its interaction with tissue inhibitor of metalloproteinases (TIMP)-2. FEBS Lett. 1996, 393, 101–104. [Google Scholar] [CrossRef] [Green Version]
- Yana, I.; Weiss, S.J. Regulation of membrane type-1 matrix metalloproteinase activation by proprotein convertases. Mol. Biol. Cell 2000, 11, 2387–2401. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.S.; Subramaniam, S.; Dramane, G.; Khelifi, D.; Khan, N.A. ERK1 and ERK2 activation modulates diet-induced obesity in mice. Biochimie 2017, 137, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Kujiraoka, T.; Satoh, Y.; Ayaori, M.; Shiraishi, Y.; Arai-Nakaya, Y.; Hakuno, D.; Yada, H.; Kuwada, N.; Endo, S.; Isoda, K.; et al. Hepatic extracellular signal-regulated kinase 2 suppresses endoplasmic reticulum stress and protects from oxidative stress and endothelial dysfunction. J. Am. Heart Assoc. 2013, 2, e000361. [Google Scholar] [CrossRef] [Green Version]
- Mori, H.; Lo, A.T.; Inman, J.L.; Alcaraz, J.; Ghajar, C.M.; Mott, J.D.; Nelson, C.M.; Chen, C.S.; Zhang, H.; Bascom, J.L.; et al. Transmembrane/cytoplasmic, rather than catalytic, domains of Mmp14 signal to MAPK activation and mammary branching morphogenesis via binding to integrin beta1. Development 2013, 140, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Chen, L.M.; Lin, C.Y.; Chai, K.X. The epidermal growth factor receptor (EGFR) is proteolytically modified by the Matriptase-Prostasin serine protease cascade in cultured epithelial cells. Biochim. Biophys. Acta 2008, 1783, 896–903. [Google Scholar] [CrossRef] [Green Version]
- Speicher, T.; Siegenthaler, B.; Bogorad, R.L.; Ruppert, R.; Petzold, T.; Padrissa-Altes, S.; Bachofner, M.; Anderson, D.G.; Koteliansky, V.; Fassler, R.; et al. Knockdown and knockout of beta1-integrin in hepatocytes impairs liver regeneration through inhibition of growth factor signalling. Nat. Commun. 2014, 5, 3862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, X.; Shi, Z.; Yu, N.; Jiao, Y.; Jin, L.; Zhang, J. The Role of EGFR/ERK/ELK-1 MAP Kinase Pathway in the Underlying Damage to Diabetic Rat Skin. Indian J. Derm. 2013, 58, 101–106. [Google Scholar] [PubMed]
- Nakakuki, T.; Birtwistle, M.R.; Saeki, Y.; Yumoto, N.; Ide, K.; Nagashima, T.; Brusch, L.; Ogunnaike, B.A.; Okada-Hatakeyama, M.; Kholodenko, B.N. Ligand-specific c-Fos expression emerges from the spatiotemporal control of ErbB network dynamics. Cell 2010, 141, 884–896. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Li, Y.; Yang, Z.; Xu, W.; Yang, Y.; Tan, X. ROS mediated EGFR/MEK/ERK/HIF-1alpha Loop Regulates Glucose metabolism in pancreatic cancer. Biochem. Biophys. Res. Commun. 2018, 500, 873–878. [Google Scholar] [CrossRef]
- Cao, W.; Collins, Q.F.; Becker, T.C.; Robidoux, J.; Lupo, E.G., Jr.; Xiong, Y.; Daniel, K.W.; Floering, L.; Collins, S. p38 Mitogen-activated protein kinase plays a stimulatory role in hepatic gluconeogenesis. J. Biol. Chem. 2005, 280, 42731–42737. [Google Scholar] [CrossRef] [Green Version]
- Nakatani, Y.; Kaneto, H.; Kawamori, D.; Hatazaki, M.; Miyatsuka, T.; Matsuoka, T.A.; Kajimoto, Y.; Matsuhisa, M.; Yamasaki, Y.; Hori, M. Modulation of the JNK pathway in liver affects insulin resistance status. J. Biol. Chem. 2004, 279, 45803–45809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.; Liu, H.; Yu, J.; Zhao, Z.; Xiao, F.; Xia, T.; Wang, C.; Li, K.; Deng, J.; Guo, Y.; et al. Activation of ERK1/2 Ameliorates Liver Steatosis in Leptin Receptor-Deficient (db/db) Mice via Stimulating ATG7-Dependent Autophagy. Diabetes 2016, 65, 393–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, Y.; Liu, W.; Cao, H.; Zhang, D.; Yao, X.; Zhang, S.; Xia, H.; Li, D.; Wang, Y.C.; Yan, J.; et al. Hepatic p38alpha regulates gluconeogenesis by suppressing AMPK. J. Hepatol. 2015, 62, 1319–1327. [Google Scholar] [CrossRef]
- Vernia, S.; Cavanagh-Kyros, J.; Garcia-Haro, L.; Sabio, G.; Barrett, T.; Jung, D.Y.; Kim, J.K.; Xu, J.; Shulha, H.P.; Garber, M.; et al. The PPARalpha-FGF21 hormone axis contributes to metabolic regulation by the hepatic JNK signaling pathway. Cell Metab. 2014, 20, 512–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, P.; Xu, X.; Xu, L.; Hui, G.; Wu, S.; Lan, Q. Expression of MMP14 in invasive pituitary adenomas: Relationship to invasion and angiogenesis. Int. J. Clin. Exp. Pathol. 2015, 8, 3556–3567. [Google Scholar]
- Jin, Y.; Liang, Z.Y.; Zhou, W.X.; Zhou, L. High MMP14 expression is predictive of poor prognosis in resectable hepatocellular carcinoma. Pathology 2020, 52, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Lin, Z.; Jiang, J.; Lu, S.; Chen, M.; Que, H.; He, X.; Que, G.; Mao, J.; Xiao, J.; et al. MMP14 regulates cell migration and invasion through epithelial-mesenchymal transition in nasopharyngeal carcinoma. Am. J. Transl. Res. 2015, 7, 950–958. [Google Scholar] [PubMed]
- Zhou, X.; Hovell, C.J.; Pawley, S.; Hutchings, M.I.; Arthur, M.J.; Iredale, J.P.; Benyon, R.C. Expression of matrix metalloproteinase-2 and -14 persists during early resolution of experimental liver fibrosis and might contribute to fibrolysis. Liver Int. 2004, 24, 492–501. [Google Scholar] [CrossRef]
- Shipway, A.; Danahay, H.; Williams, J.A.; Tully, D.C.; Backes, B.J.; Harris, J.L. Biochemical characterization of prostasin, a channel activating protease. Biochem. Biophys. Res. Commun. 2004, 324, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Lehti, K.; Allen, E.; Birkedal-Hansen, H.; Holmbeck, K.; Miyake, Y.; Chun, T.H.; Weiss, S.J. An MT1-MMP-PDGF receptor-beta axis regulates mural cell investment of the microvasculature. Genes Dev. 2005, 19, 979–991. [Google Scholar] [CrossRef] [Green Version]
- Sato, H.; Takino, T.; Okada, Y.; Cao, J.; Shinagawa, A.; Yamamoto, E.; Seiki, M. A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature 1994, 370, 61–65. [Google Scholar] [CrossRef]
- Knauper, V.; Bailey, L.; Worley, J.R.; Soloway, P.; Patterson, M.L.; Murphy, G. Cellular activation of proMMP-13 by MT1-MMP depends on the C-terminal domain of MMP-13. FEBS Lett. 2002, 532, 127–130. [Google Scholar] [CrossRef] [Green Version]
- Mori, H.; Tomari, T.; Koshikawa, N.; Kajita, M.; Itoh, Y.; Sato, H.; Tojo, H.; Yana, I.; Seiki, M. CD44 directs membrane-type 1 matrix metalloproteinase to lamellipodia by associating with its hemopexin-like domain. EMBO J. 2002, 21, 3949–3959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zong, H.; Bastie, C.C.; Xu, J.; Fassler, R.; Campbell, K.P.; Kurland, I.J.; Pessin, J.E. Insulin resistance in striated muscle-specific integrin receptor beta1-deficient mice. J. Biol. Chem. 2009, 284, 4679–4688. [Google Scholar] [CrossRef] [Green Version]
- Bill, H.M.; Knudsen, B.; Moores, S.L.; Muthuswamy, S.K.; Rao, V.R.; Brugge, J.S.; Miranti, C.K. Epidermal growth factor receptor-dependent regulation of integrin-mediated signaling and cell cycle entry in epithelial cells. Mol. Cell Biol. 2004, 24, 8586–8599. [Google Scholar] [CrossRef] [Green Version]
- Frisch, S.M.; Vuori, K.; Ruoslahti, E.; Chan-Hui, P.Y. Control of adhesion-dependent cell survival by focal adhesion kinase. J. Cell Biol. 1996, 134, 793–799. [Google Scholar] [CrossRef] [Green Version]
- Westhoff, M.A.; Serrels, B.; Fincham, V.J.; Frame, M.C.; Carragher, N.O. SRC-mediated phosphorylation of focal adhesion kinase couples actin and adhesion dynamics to survival signaling. Mol. Cell Biol. 2004, 24, 8113–8133. [Google Scholar] [CrossRef] [Green Version]
- Del Sol, A.I.; Moons, K.G.; Hollander, M.; Hofman, A.; Koudstaal, P.J.; Grobbee, D.E.; Breteler, M.M.; Witteman, J.C.; Bots, M.L. Is carotid intima-media thickness useful in cardiovascular disease risk assessment? The Rotterdam Study. Stroke 2001, 32, 1532–1538. [Google Scholar] [CrossRef]
- O’Leary, D.H.; Polak, J.F.; Kronmal, R.A.; Manolio, T.A.; Burke, G.L.; Wolfson, S.K., Jr. Carotid-artery intima and media thickness as a risk factor for myocardial infarction and stroke in older adults. Cardiovascular Health Study Collaborative Research Group. N. Engl. J. Med. 1999, 340, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Matthews, D.R.; Hosker, J.P.; Rudenski, A.S.; Naylor, B.A.; Treacher, D.F.; Turner, R.C. Homeostasis model assessment: Insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985, 28, 412–419. [Google Scholar] [CrossRef] [Green Version]
- Wallace, T.M.; Levy, J.C.; Matthews, D.R. Use and abuse of HOMA modeling. Diabetes Care 2004, 27, 1487–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanda, Y. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transplant. 2013, 48, 452–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sekine, T.; Takizawa, S.; Uchimura, K.; Miyazaki, A.; Tsuchiya, K. Liver-Specific Overexpression of Prostasin Attenuates High-Fat Diet-Induced Metabolic Dysregulation in Mice. Int. J. Mol. Sci. 2021, 22, 8314. https://doi.org/10.3390/ijms22158314
Sekine T, Takizawa S, Uchimura K, Miyazaki A, Tsuchiya K. Liver-Specific Overexpression of Prostasin Attenuates High-Fat Diet-Induced Metabolic Dysregulation in Mice. International Journal of Molecular Sciences. 2021; 22(15):8314. https://doi.org/10.3390/ijms22158314
Chicago/Turabian StyleSekine, Tetsuo, Soichi Takizawa, Kohei Uchimura, Asako Miyazaki, and Kyoichiro Tsuchiya. 2021. "Liver-Specific Overexpression of Prostasin Attenuates High-Fat Diet-Induced Metabolic Dysregulation in Mice" International Journal of Molecular Sciences 22, no. 15: 8314. https://doi.org/10.3390/ijms22158314