
West Nile Virus in Italy: An Update of the Viral Strains Circulating in the Late 2022 Epidemic Season
 , , , , , , , , , , , , ,
, , , , , , , , , , , , ,  and
and
Abstract
:Simple Summary
Abstract
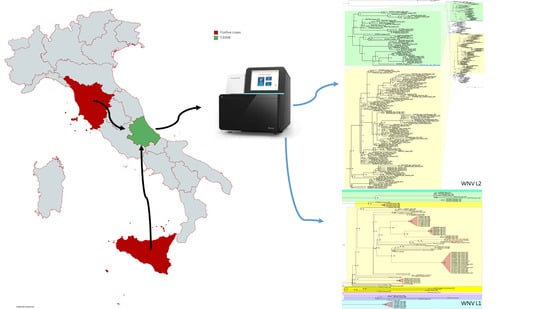
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Real-Time PCR Assay
2.3. Next-Generation Sequencing (NGS)
2.4. Phylogenetic Analyses
3. Results
3.1. WNV L1 Detection, Sequencing and Phylogenetic Analysis
3.2. WNV L2 Detection, Sequencing and Phylogenetic Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rizzoli, A.; Jiménez-Clavero, M.A.; Barzon, L.; Cordioli, P.; Figuerola, J.; Koraka, P.; Martina, B.; Moreno, A.; Nowotny, N.; Pardigon, N.; et al. The challenge of West Nile virus in Europe: Knowledge gaps and research priorities. Eurosurveillance 2015, 20, 21135. [Google Scholar] [CrossRef] [PubMed]
- Savini, G.; Monaco, F.; Calistri, P.; Lelli, R. Phylogenetic analysis of West Nile virus isolated in Italy in 2008. Eurosurveillance 2008, 13, 19048. [Google Scholar] [CrossRef] [PubMed]
- Autorino, G.L.; Battisti, A.; Deubel, V.; Ferrari, G.; Forletta, R.; Giovannini, A.; Lelli, R.; Murri, S.; Scicluna, M.T. West Nile virus Epidemic in Horses, Tuscany Region, Italy. Emerg. Infect. Dis. 2002, 8, 1372–1378. [Google Scholar] [CrossRef] [PubMed]
- National Integrated Surveillance and Response Plan for West Nile Virus. 2016. Available online: https://westnile.izs.it/j6_wnd/docMinisteriale?annoDocumento=2016 (accessed on 6 July 2023).
- Velati, C.; Angelini, P.; Pupella, S. State of the art: Vest Nile Virus circulation surveillance in Italy and transfusion risk early prevention methods. Transfus. Clin. Biol. 2017, 24, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Riccardo, F.; Bella, A.; Monaco, F.; Ferraro, F.; Petrone, D.; Mateo-Urdiales, A.; Andrianou, X.D.; Del Manso, M.; Venturi, G.; Fortuna, C.; et al. Rapid increase in neuroinvasive West Nile virus infections in humans, Italy, July 2022. Eurosurveillance 2022, 27, 2200653. [Google Scholar] [CrossRef] [PubMed]
- West Nile Disease. Available online: https://westnile.izs.it/j6_wnd/home (accessed on 6 July 2023).
- Barzon, L.; Montarsi, F.; Quaranta, E.; Monne, I.; Pacenti, M.; Michelutti, A.; Toniolo, F.; Danesi, P.; Marchetti, G.; Gobbo, F.; et al. Early start of seasonal transmission and co-circulation of West Nile virus lineage 2 and a newly introduced lineage 1 strain, northern Italy, June 2022. Eurosurveillance 2022, 24, 2200548. [Google Scholar] [CrossRef] [PubMed]
- Mencattelli, G.; Iapaolo, F.; Polci, A.; Marcacci, M.; Di Gennaro, A.; Teodori, L.; Curini, V.; Di Lollo, V.; Secondini, B.; Scialabba, S.; et al. West Nile Virus Lineage 2 Overwintering in Italy. Trop. Med. Infect. Dis. 2022, 7, 160. [Google Scholar] [CrossRef]
- Cavrini, F.; Della Pepa, M.E.; Gaibani, P.; Pierro, A.M.; Rossini, G.; Landini, M.P.; Sambri, V. A rapid and specific real-time RT-PCR assay to identify Usutu virus in human plasma, serum, and cerebrospinal fluid. J. Clin. Virol. 2011, 50, 221–223. [Google Scholar] [CrossRef]
- Del Amo, J.; Sotelo, E.; Fernández-Pinero, J.; Gallardo, C.; Llorente, F.; Agüero, M.; Jiménez-Clavero, M.A. A novel quantitative multiplex real-time RT-PCR for the simultaneous detection and differentiation of West Nile virus lineages 1 and 2, and of Usutu virus. J. Virol. Methods 2013, 189, 321–327. [Google Scholar] [CrossRef]
- Vázquez, A.; Herrero, L.; Negredo, A.; Hernández, L.; Sánchez-Seco, M.P.; Tenorio, A. Real time PCR assay for detection of all known lineages of West Nile virus. J. Virol. Methods 2016, 236, 266–270. [Google Scholar] [CrossRef]
- Diagne, M.M.; Ndione, M.H.D.; Mencattelli, G.; Diallo, A.; Ndiaye, E.H.; Di Domenico, M.; Diallo, D.; Kane, M.; Curini, V.; Top, N.M.; et al. Novel Amplicon-Based Sequencing Approach to West Nile Virus. Viruses 2023, 15, 1261. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An amplicon-based sequencing framework for accurately measuring intrahost virus diversity using PrimalSeq and iVar. Genome Biol. 2019, 20, 8. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; The UGENE Team. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Mencattelli, G.; Ndione, M.H.D.; Rosà, R.; Marini, G.; Diagne, C.T.; Diagne, M.M.; Fall, G.; Faye, O.; Diallo, M.; Faye, O.; et al. Epidemiology of West Nile virus in Africa: An underestimated threat. PLoS Negl. Trop. Dis. 2022, 16, e0010075. [Google Scholar] [CrossRef] [PubMed]
- Mencattelli, G.; Iapaolo, F.; Monaco, F.; Fusco, G.; de Martinis, C.; Portanti, O.; Di Gennaro, A.; Curini, V.; Polci, A.; Berjaoui, S.; et al. West Nile Virus Lineage 1 in Italy: Newly Introduced or a Re-Occurrence of a Previously Circulating Strain? Viruses 2021, 14, 64. [Google Scholar] [CrossRef] [PubMed]
- Farooq, Z.; Rocklöv, J.; Wallin, J.; Abiri, N.; Sewe, M.O.; Sjödin, H.; Semenza, J.C. Artificial intelligence to predict West Nile virus outbreaks with eco-climatic drivers. Lancet Reg. Health Eur. 2022, 17, 100370. [Google Scholar] [CrossRef]
- Story Map Integrated Surveillance of West Nile and Usutu Virus. Available online: https://storymaps.arcgis.com/collections/5f04c28b7a264d31b53d9cc676b8a12b (accessed on 6 July 2023).
- Veo, C.; della Ventura, C.; Moreno, A.; Rovida, F.; Percivalle, E.; Canziani, S.; Torri, D.; Calzolari, M.; Baldanti, F.; Galli, M.; et al. Evolutionary Dynamics of the Lineage 2 West Nile Virus That Caused the Largest European Epidemic: Italy 2011–2018. Viruses 2019, 11, 814. [Google Scholar] [CrossRef]
- Bagnarelli, P.; Marinelli, K.; Trotta, D.; Monachetti, A.; Tavio, M.; Del Gobbo, R.; Capobianchi, M.R.; Menzo, S.; Nicoletti, L.; Magurano, F.; et al. Human case of autochthonous West Nile virus lineage 2 infection in Italy, September 2011. Eurosurveillance 2011, 16, 20002. [Google Scholar] [CrossRef]
- Savini, G.; Capelli, G.; Monaco, F.; Polci, A.; Russo, F.; Di Gennaro, A.; Marini, V.; Teodori, L.; Montarsi, F.; Pinoni, C.; et al. Evidence of West Nile virus lineage 2 circulation in Northern Italy. Vet. Microbiol. 2012, 158, 267–273. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valleriani, F.; Polci, A.; Iapaolo, F.; Portanti, O.; Pisciella, M.; Cersini, A.; Guercio, A.; Del Lesto, I.; Curini, V.; Mincarelli, L.F.; et al. West Nile Virus in Italy: An Update of the Viral Strains Circulating in the Late 2022 Epidemic Season. Zoonotic Dis. 2024, 4, 49-56. https://doi.org/10.3390/zoonoticdis4010006
Valleriani F, Polci A, Iapaolo F, Portanti O, Pisciella M, Cersini A, Guercio A, Del Lesto I, Curini V, Mincarelli LF, et al. West Nile Virus in Italy: An Update of the Viral Strains Circulating in the Late 2022 Epidemic Season. Zoonotic Diseases. 2024; 4(1):49-56. https://doi.org/10.3390/zoonoticdis4010006
Chicago/Turabian StyleValleriani, Fabrizia, Andrea Polci, Federica Iapaolo, Ottavio Portanti, Maura Pisciella, Antonella Cersini, Annalisa Guercio, Irene Del Lesto, Valentina Curini, Luana Fiorella Mincarelli, and et al. 2024. "West Nile Virus in Italy: An Update of the Viral Strains Circulating in the Late 2022 Epidemic Season" Zoonotic Diseases 4, no. 1: 49-56. https://doi.org/10.3390/zoonoticdis4010006