
Clinical Characterisation and Comorbidities of Acquired Generalised Lipodystrophy: A 14-Year Follow-Up Study

 ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population and Design
2.2. Clinical Data Collection
2.3. Anthropometry and Body Composition Analysis
2.4. Analytical Measurements
2.5. Molecular Analysis
2.6. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brown, R.-J.; Araujo-Vilar, D.; Cheung, P.-T.; Dunger, D.; Garg, A.; Jack, M.; Mungai, L.; Oral, E.-A.; Patni, N.; Rother, K.-I.; et al. The Diagnosis and Management of Lipodystrophy Syndromes: A Multi-Society Practice Guideline. J. Clin. Endocrinol. Metab. 2016, 101, 4500–4511. [Google Scholar] [CrossRef] [PubMed]
- Chiquette, E.; Oral, E.-A.; Garg, A.; Araújo-Vilar, D.; Dhankhar, P. Estimating the prevalence of generalized and partial lipodystrophy: Findings and challenges. Diabetes Metab. Syndr. Obes. 2017, 10, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Ceccarini, G.; Magno, S.; Gilio, D.; Pelosini, C.; Santini, F. Autoimmunity in lipodystrophy syndromes. Presse Med. 2021, 50, 104073. [Google Scholar] [CrossRef] [PubMed]
- Misra, A.; Garg, A. Clinical features and metabolic derangements in acquired generalized lipodystrophy: Case reports and review of the literature. Medicine 2003, 82, 129–146. [Google Scholar] [CrossRef]
- Araujo-Vilar, D.; Santini, F. Diagnosis and treatment of lipodystrophy: A step-bystep approach. J. Endocrinol. Investig. 2019, 42, 61–73. [Google Scholar] [CrossRef]
- Lawrence, R.-D. Lipodystrophy and hepatomegaly with diabetes, lipaemia, and other metabolic disturbances; a case throwing new light on the action of insulin. Lancet 1946, 1, 773. [Google Scholar] [CrossRef]
- Corvillo, F.; Aparicio, V.; López-Lera, A.; Garrido, S.; Araújo-Vilar, D.; de Miguel, M.-P.; López-Trascasa, M. Autoantibodies Against Perilipin 1 as a Cause of Acquired Generalized Lipodystrophy. Front. Immunol. 2018, 9, 2142. [Google Scholar] [CrossRef]
- Corvillo, F.; Abel, B.-S.; López-Lera, A.; Ceccarini, G.; Magno, S.; Santini, F.; Araújo-Vilar, D.; Brown, R.-J.; Nozal, P.; López-Trascasa, M. Characterization and Clinical Association of Autoantibodies Against Perilipin 1 in Patients with Acquired Generalized Lipodystrophy. Diabetes 2023, 72, 71–84. [Google Scholar] [CrossRef]
- Mandel-Brehm, C.; Vazquez, S.-E.; Liverman, C.; Cheng, M.; Quandt, Z.; Kung, A.-F.; Parent, A.; Miao, B.; Disse, E.; Cugnet-Anceau, C.; et al. Autoantibodies to Perilipin-1 Define a Subset of Acquired Generalized Lipodystrophy. Diabetes 2023, 72, 59–70. [Google Scholar] [CrossRef]
- Jehl, A.; Cugnet-Anceau, C.; Vigouroux, C.; Legeay, A.-L.; Dalle, S.; Harou, O.; Marchand, L.; Lascols, O.; Caussy, C.; Thivolet, C.; et al. Acquired Generalized Lipodystrophy: A New Cause of Anti-PD-1 Immune-Related Diabetes. Diabetes Care 2019, 42, 2008–2010. [Google Scholar] [CrossRef]
- Haddad, N.; Vidal-Trecan, T.; Baroudjian, B.; Zagdanski, A.-M.; Arangalage, D.; Battistella, M.; Gautier, J.-F.; Lebbe, C.; Delyon, J.; PATIO group. Acquired generalized lipodystrophy under immune checkpoint inhibition. Br. J. Dermatol. 2020, 182, 477–480. [Google Scholar] [CrossRef] [PubMed]
- Bedrose, S.; Turin, C.-G.; Lavis, V.-R.; Kim, S.-T.; Thosani, S.-N. A case of acquired generalized lipodystrophy associated with pembrolizumab in a patient with metastatic malignant melanoma. AACE. Clin. Case Rep. 2020, 6, e40–e45. [Google Scholar] [CrossRef] [PubMed]
- Falcao, C.-K.; Cabral, M.-C.-S.; Mota, J.-M.; Arbache, S.-T.; Costa-Riquetto, A.-D.; Muniz, D.-Q.-B.; Cury-Martins, J.; Almeida, M.-Q.; Kaczemorska, P.-C.; Nery, M.; et al. Acquired Lipodystrophy Associated with Nivolumab in a Patient with Advanced Renal Cell Carcinoma. J. Clin. Endocrinol. Metab. 2019, 104, 3245–3248. [Google Scholar] [CrossRef]
- Oral, E.-A.; Simha, V.; Ruiz, E.; Andewelt, A.; Premkumar, A.; Snell, P.; Wagner, A.-J.; DePaoli, A.-M.; Reitman, M.-L.; Taylor, S.-I.; et al. Leptin-replacement therapy for lipodystrophy. N. Engl. J. Med. 2002, 346, 570–578. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association Professional Practice Committee. 2. Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2022. Diabetes Care 2022, 45 (Suppl. S1), S17–S38. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association Professional Practice Committee. 12. Retinopathy, Neuropathy, and Foot Care: Standards of Medical Care in Diabetes-2022. Diabetes Care 2022, 45 (Suppl. S1), S18–S194. [Google Scholar] [CrossRef]
- Azziz, R.; Carmina, E.; Dewailly, D.; Diamanti-Kandarakis, E.; Escobar-Morreale, H.-F.; Futterweit, W.; Janssen, O.-E.; Legro, R.-S.; Norman, R.-J.; Taylor, A.-E.; et al. Androgen Excess Society. Positions statement: Criteria for defining polycystic ovary syndrome as a predominantly hyperandrogenic syndrome: An Androgen Excess Society guideline. J. Clin. Endocrinol. Metab. 2006, 91, 4237–4245. [Google Scholar] [CrossRef]
- Arioglu, E.; Duncan-Morin, J.; Sebring, N.; Rother, K.-I.; Gottlieb, N.; Lieberman, J.; Herion, D.; Kleiner, D.-E.; Reynolds, J.; Premkumar, A.; et al. Efficacy and safety of troglitazone in the treatment of lipodystrophy syndromes. Ann. Intern. Med. 2000, 133, 263–274. [Google Scholar] [CrossRef]
- Bolan, C.; Oral, E.-A.; Gorden, P.; Taylor, S.; Leitman, S.-F. Intensive, long-term plasma exchange therapy for severe hypertriglyceridemia in acquired generalized lipoatrophy. J. Clin. Endocrinol. Metab. 2002, 87, 380–384. [Google Scholar] [CrossRef]
- Garg, A. Lipodystrophies. Am. J. Med. 2000, 108, 143–152. [Google Scholar] [CrossRef]
- Brechtel, K.; Jacob, S.; Machann, J.; Hauer, B.; Nielsen, M.; Meissner, H.-P.; Matthaei, S.; Haering, H.-U.; Claussen, C.-D.; Schick, F. Acquired generalized lipoatrophy (AGL): Highly selective MR lipid imaging and localized (1)H-MRS. J. Magn. Reson. Imaging 2000, 12, 306–310. [Google Scholar] [CrossRef]
- Catalano, P.-M.; Capeless, E.-L.; Simmons, G.-M.; Robbins, D.-C.; Horton, E.-S. Successful pregnancy outcome in association with lipoatrophic diabetes mellitus. Obstet. Gynecol. 1990, 76 Pt 2, 978–979. [Google Scholar] [PubMed]
- Hubler, A.; Abendroth, K.; Keiner, T.; Stocker, W.; Kauf, E.; Hein, G.; Stein, G. Dysregulation of insulin-like growth factors in a case of generalized acquired lipoatrophic diabetes mellitus (Lawrence Syndrome) connected with autoantibodies against adipocyte membranes. Exp. Clin. Endocrinol. Diabetes 1998, 106, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Ono, H.; Nakajima, H.; Sugimoto, J. Lipoatrophic diabetes. J. Dermatol. 1992, 19, 246–249. [Google Scholar] [CrossRef] [PubMed]
- Hussain, I.; Garg, A. Lipodystrophy Syndromes. Endocrinol. Metab. Clin. N. Am. 2016, 45, 783–797. [Google Scholar] [CrossRef] [PubMed]
- Savage, D.-B.; Semple, R.-K.; Clatworthy, M.-R.; Lyons, P.-A.; Morgan, B.-P.; Cochran, E.-K.; Gorden, P.; Raymond-Barker, P.; Murgatroyd, P.-R.; Adams, C.; et al. Complement abnormalities in acquired lipodystrophy revisited. J. Clin. Endocrinol. Metab. 2009, 94, 10–16. [Google Scholar] [CrossRef]
- Eren, E.; Özkan, T.-B.; Çakır, E.-D.; Sağlam, H.; Tarım, Ö. Acquired generalized lipodystrophy associated with autoimmune hepatitis and low serum C4 level. J Clin. Res. Pediatr. Endocrinol. 2010, 2, 39–42. [Google Scholar] [CrossRef]
- Blasco-Patiño, F. Infection as the source and driving force of autoimmune diseases. An. Med. Interna 2002, 19, 44–48. (In Spanish) [Google Scholar]
- Garg, A. Clinical review#: Lipodystrophies: Genetic and acquired body fat disorders. J. Clin. Endocrinol. Metab. 2011, 96, 3313–3325. [Google Scholar] [CrossRef]
- Klein, S.; Jahoor, F.; Wolfe, R.-R.; Stuart, C.-A. Generalized lipodystrophy: In vivo evidence for hypermetabolism and insulin-resistant lipid, glucose, and amino acid kinetics. Metabolism 1992, 41, 893–896. [Google Scholar] [CrossRef]
- Hussain, I.; Patni, N.; Garg, A. Lipodystrophies, dyslipidaemias and atherosclerotic cardiovascular disease. Pathology 2019, 51, 202–212. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Case No. | Age (Years) | Gender | Onset of Phenotype | Consanguinity | Altered Fat Distribution | Clinical Features | Comorbidities |
|---|---|---|---|---|---|---|---|
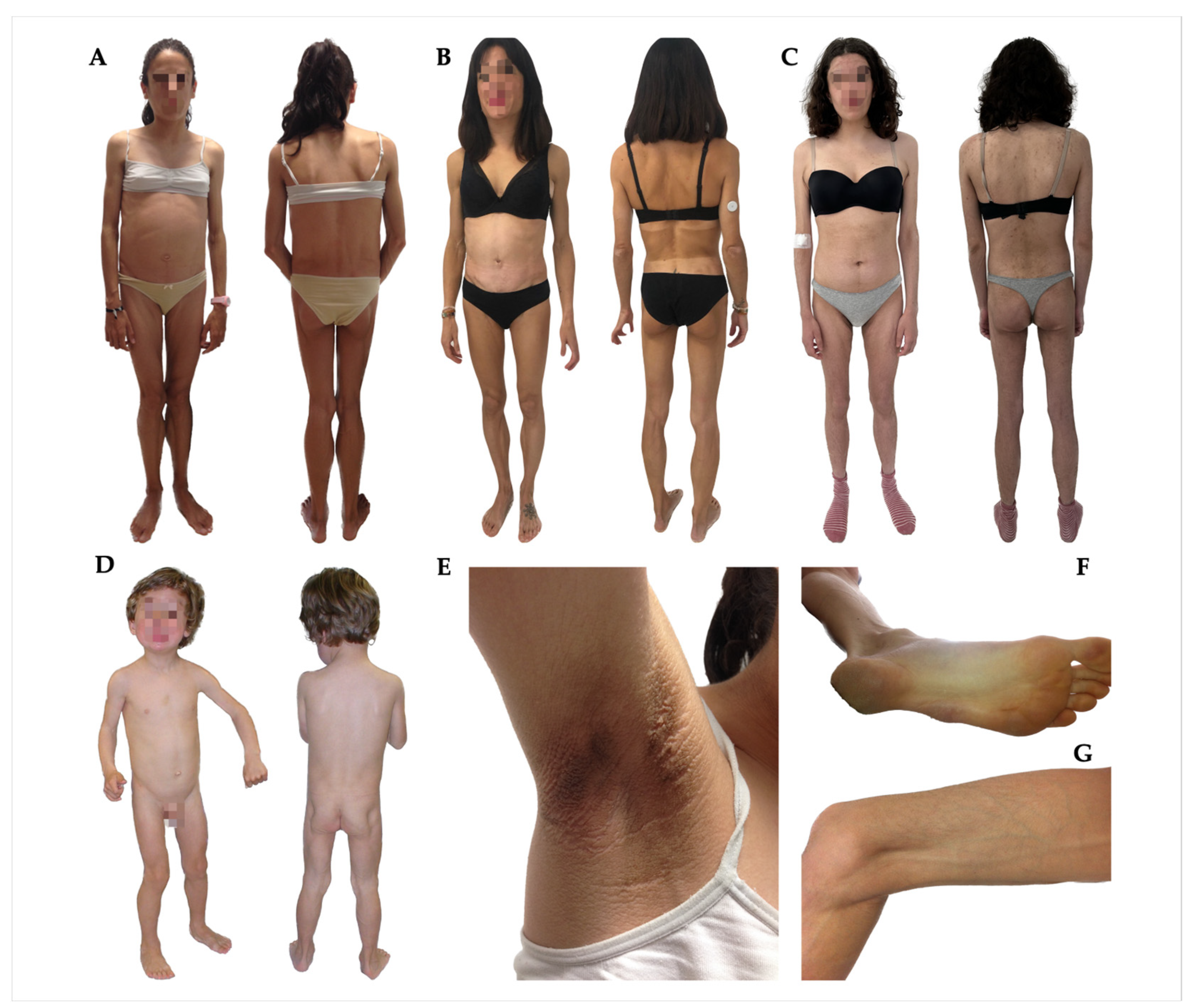
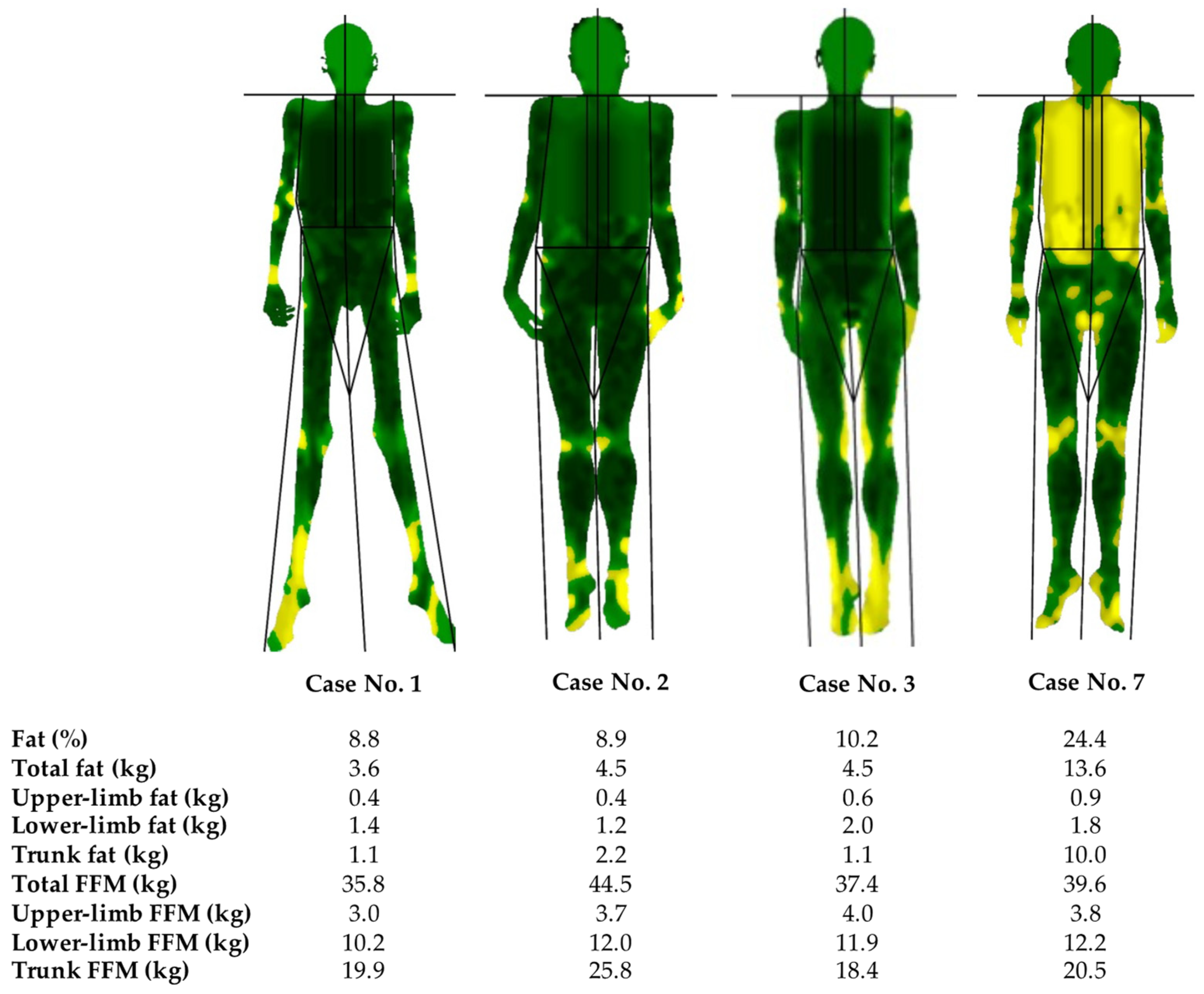
| 1 | 25 | M | Childhood (8 years) | Yes | Generalised loss of fat. Palms and soles fat loss; facial fat loss | Acanthosis nigricans, prognathism, phlebomegaly, umbilical hernia, polyphagia | Type 1 diabetes, albuminuria, diabetic neuropathy, hypertriglyceridaemia, hypertension, hepatomegaly, pancreatitis, hypothyroidism, mesangial proliferative glomerulonephritis, and mucociliary dysfunction |
| 2 | 23 | F | Childhood (6 years) | No | Generalised loss of fat. Palms fat loss, soles, and facial fat preserved | Acanthosis nigricans, phlebomegaly, and umbilical hernia. polyphagia | Type 2 diabetes, albuminuria, hypertriglyceridaemia, hepatomegaly, splenomegaly, and autoimmune hepatitis |
| 3 | 46 | F | Adulthood (35 years) | No | Generalised loss of fat. Palms, soles preserved, and facial fat loss | Phlebomegaly | Amenorrhea, celiac disease |
| 4 | 65 | F | Adulthood (44 years) | No | Generalised loss of fat. Palms, soles, and facial fat preserved | Muscle pain | Splenomegaly, primary hypothyroidism |
| 5 | 44 | F | Childhood (3 years) | No | Generalised loss of fat. Palms and soles fat loss; facial fat loss | Acanthosis nigricans, phlebomegaly, and umbilical hernia | Type 1 diabetes, amenorrhea |
| 6 | 16 | M | Childhood (2 years) | No | Generalised loss of fat. Palms and soles fat loss; facial fat preserved | Phlebomegaly, umbilical hernia | Prediabetes, primary hypothyroidism, and celiac disease |
| 7 | 19 | F | Adolescence (16 years) | Yes | Generalised loss of fat. Palms and soles fat loss; facial fat loss | Acanthosis nigricans, phlebomegaly, and hirsutism | Prediabetes, amenorrhea, and PCOS |
| Case No. 1 | Case No. 2 | Case No. 3 | Case No. 4 | Case No. 5 | Case No. 6 | Case No. 7 | |
|---|---|---|---|---|---|---|---|
| Weight (kg) | 40.1 | 51.5 | 43.8 | 46.5 | 47.0 | 50.0 | 56 |
| BMI (kg/m2) | 12.1 | 19.1 | 15.1 | 21.2 | 17.3 | 17.8 | 19.4 |
| Waist perimeter (cm) | 74 | 81 | 64 | NA | NA | 52 | 81 |
| Hip perimeter (cm) | 78 | 77 | 77 | NA | NA | 52 | 86 |
| Triceps skinfold (mm) | 2.8 | 3.0 | 4.0 | 14.0 | 4.0 | 4.3 | 4.0 |
| Biceps skinfold (mm) | 2.0 | 3.0 | 1.8 | 3.8 | 3.0 | 2.2 | 3.0 |
| Subscapular skinfold (mm) | 4.0 | 6.0 | 4.5 | 6.0 | 7.0 | 5.8 | 11.0 |
| Suprailiac skinfold (mm) | 3.8 | 4.0 | 3.6 | NA | 4.0 | 5.6 | 8.0 |
| Thigh skinfold (mm) | 3.2 | 4.0 | 4.2 | 20.0 | 4.0 | 5.2 | 6.0 |
| Calf skinfold (mm) | 3.2 | 3.5 | 3.0 | 11.0 | 3.0 | 3.9 | 4.5 |
| Case No. | Anti-GAD | Anti-IA2 | Anti-IAA | Anti-TPO | Anti-Tg | ANA | ATA | APCA | ADR | RF |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | + | - | + | - | - | NA | - | NA | + | NA |
| 2 | - | - | - | - | - | - | - | + | NA | NA |
| 3 | - | - | - | - | - | NA | + | - | NA | NA |
| 4 | - | - | - | + | - | + | - | - | - | NA |
| 5 | + | + | - | - | - | + | - | NA | NA | - |
| 6 | + | - | - | + | - | - | + | - | - | NA |
| 7 | - | - | - | - | - | NA | NA | NA | - | + |
| Case No. 1 | Case No. 2 | Case No. 3 | Case No. 4 | Case No. 5 | Case No. 6 | Case No. 7 | |
|---|---|---|---|---|---|---|---|
| Fasting glucose (mg/dL) (70–100) | 186 | 277 | 83 | 87 | 109 | 93 | 95 |
| Insulin (mIU/L) (1.5–18.5) | NA | 101.1 | 3.7 | NA | 13.7 | 9.4 | 144.3 |
| HOMA-IR (<2.0) | NA | 69.1 | 0.8 | NA | 3.7 | 2.2 | 33.8 |
| HbA1c (%) (3.5–5.6) | 8.1 | 10.4 | 5.4 | 5.7 | 7.3 | 5.8 | 5.4 |
| Total cholesterol (mg/dL) (150–255) | 135 | 215 | 154 | 180 | 197 | 141 | 133 |
| LDL-C (mg/dL) (55–125) | 42 | 92 | 71 | 56 | 52 | 35 | 29 |
| HDL-C (mg/dL) (34–91) | 17 | 24 | 71 | 56 | 52 | 35 | 29 |
| Triglycerides (mg/dL) (27–150) | 380 | 1249 | 58 | 48 | 68 | 102 | 256 |
| Total bilirubin (mg/dL) (0.2–1.3) | 0.5 | 0.3 | 0.6 | 0.3 | 0.4 | 0.4 | 0.3 |
| AST (IU/L) (10–40) | 23 | 98 | 21 | 25 | 23 | 24 | 23 |
| ALT (IU/L) (3–41) | 28 | 186 | 29 | 16 | 24 | 23 | 46 |
| GGT (IU/L) (8–73) | 12 | 147 | 17 | 15 | 20 | 18 | 23 |
| Creatinine (mg/dL) (0.4–1.3) | 0.6 | 0.3 | 0.7 | 0.6 | 0.6 | 0.9 | 0.6 |
| Calcium (mg/dL) (8.5–11.0) | 8.6 | 9.4 | 9.2 | 8.8 | 10.4 | 9.6 | 9.1 |
| Phosphate (mg/dL) (2.7–5.0) | NA | 4.9 | NA | 4.6 | 4.5 | 4.7 | 3.6 |
| PTH (pg/mL) (14–72) | 44 | 20 | 29 | NA | 14 | NA | NA |
| 25(OH)D (ng/mL) (deficiency <30) | 5 | 24 | 7 | 12 | 26 | 32 | 8 |
| Leptin (μg/L) (3.60–11.10) | 0.10 | 0.05 | NA | NA | NA | 0.10 | 2.70 |
| Complement C3 (mg/dL) (85–180) | NA | 146.3 | NA | 88.0 | 113.0 | 151.0 | 137.0 |
| Complement C4 (mg/dL) (11–42) | NA | 7.7 | NA | 17.0 | 16.0 | 32.0 | 20.8 |
| Treatment at the time of analysis | Insulin glargine and lispro, levothyroxine | Metformin, insulin glargine and lispro, fenofibrate, enalapril, losartan | No medication | Levothyroxine | Metformin, sitagliptin, vitamin D3 | Metformin, levothyroxine, vitamin D3 | Fenofibrate |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandez-Pombo, A.; Prado-Moraña, T.; Diaz-Lopez, E.J.; Sanchez-Iglesias, S.; Castro, A.I.; Cobelo-Gomez, S.; Araujo-Vilar, D. Clinical Characterisation and Comorbidities of Acquired Generalised Lipodystrophy: A 14-Year Follow-Up Study. J. Clin. Med. 2023, 12, 7344. https://doi.org/10.3390/jcm12237344
Fernandez-Pombo A, Prado-Moraña T, Diaz-Lopez EJ, Sanchez-Iglesias S, Castro AI, Cobelo-Gomez S, Araujo-Vilar D. Clinical Characterisation and Comorbidities of Acquired Generalised Lipodystrophy: A 14-Year Follow-Up Study. Journal of Clinical Medicine. 2023; 12(23):7344. https://doi.org/10.3390/jcm12237344
Chicago/Turabian StyleFernandez-Pombo, Antia, Teresa Prado-Moraña, Everardo Josue Diaz-Lopez, Sofia Sanchez-Iglesias, Ana I. Castro, Silvia Cobelo-Gomez, and David Araujo-Vilar. 2023. "Clinical Characterisation and Comorbidities of Acquired Generalised Lipodystrophy: A 14-Year Follow-Up Study" Journal of Clinical Medicine 12, no. 23: 7344. https://doi.org/10.3390/jcm12237344