

Mechanistic and Compositional Aspects of Industrial Catalysts for Selective CO2 Hydrogenation Processes


1
Department of Civil, Chemical and Environmental Engineering, University of Genoa, Via Opera Pia 15, 16145 Genova, Italy
2
Research Unit of Genoa, National Inter-University Consortium of Science and Technology of Materials (INSTM), Via Dodecaneso 33, 16146 Genova, Italy
3
Department of Chemistry and Industrial Chemistry, University of Genoa, Via Dodecaneso 33, 16146 Genova, Italy
*
Author to whom correspondence should be addressed.
Catalysts 2024, 14(2), 95; https://doi.org/10.3390/catal14020095
Submission received: 16 November 2023
/
Revised: 15 January 2024
/
Accepted: 18 January 2024
/
Published: 24 January 2024
(This article belongs to the Special Issue Application of Catalysts in CO2 Capture, Production and Utilization)
Abstract
:The characteristics of industrial catalysts for conventional water-gas shifts, methanol syntheses, methanation, and Fischer-Tropsch syntheses starting from syngases are reviewed and discussed. The information about catalysts under industrial development for the hydrogenation of captured CO2 is also reported and considered. In particular, the development of catalysts for reverse water-gas shifts, CO2 to methanol, CO2-methanation, and CO2-Fischer-Tropsch is analyzed. The difference between conventional catalysts and those needed for pure CO2 conversion is discussed. The surface chemistry of metals, oxides, and carbides involved in this field, in relation to the adsorption of hydrogen, CO, and CO2, is also briefly reviewed and critically discussed. The mechanistic aspects of the involved reactions and details on catalysts’ composition and structure are critically considered and analyzed.
1. Introduction
E-fuels are, by definition, burnable compounds that are (or, better, will be) produced from captured CO2 and green electrolytic hydrogen [1]. These fuels can be used in conventional internal combustion engines, thus reducing emissions of greenhouse gases of fossil origins, which are the likely causes of global warming. CO2 hydrogenation [2] can also produce, in a renewable way, compounds, e.g., olefins, for application as chemical intermediates in the field of a future green chemical industry.
Most of the compounds that could be produced in the future via CO2 hydrogenation are manufactured today, starting from fossil-derived syngases, i.e., CO-rich COx + H2 mixtures produced mostly either from steam reforming of natural gas [3,4], or from coal gasification [5]. Relevant data on these syngas-based processes and the respective industrial catalysts are reported in Table 1. Such compounds could also be produced, in principle, starting from biomass-derived syngases [6] as biofuels or renewable chemicals. To convert these processes from CO-rich feeds to pure CO2 feedstocks, both catalysts and process modification may be needed.
As it is well known, transition metals are the most catalytically active materials for hydrogenation reactions, at least in sulfur-free environments [7]. In fact, the large majority of catalysts used industrially for hydrogenations are metal-based. However, most of them are supported on metal oxides or, at least, present in composites with metal oxides. This is also true for catalysts for the hydrogenations of both CO and CO2, where, however, metal carbides can be present and act in catalysis. Thus, from the mechanistic point of view, several questions may arise concerning the origins of catalyst activity and selectivity towards one or another product and the role of supports and different catalyst components in a catalyst’s behavior. These points may be relevant also in developing new catalysts for CO2 hydrogenation technologies.
As it will become clear from our review, and is a common fact in industrial catalysis, although an enormous number of different catalyst compositions are objects of investigation in academic research, as well as in applied industrial research, industries converge usually over very few (frequently only one) main catalyst compositions that are found to be most performant and stable in industrial practice. In fact, commercial catalysts offered by different producers for the same reaction frequently differ only for small amounts of promoters or slight morphological differences, being based on the same chemical system. For example, as reported by Gao et al. [8] in 2015 and by Rönsch et al. [9] in 2016, detailed academic studies on COx methanation catalysts concerned at least eleven different metals as active phases (Ru, Ir, Rh, Ni, Co, Os, Pt, Fe, Mo, Pd, Ag), unsupported or supported on at least eight different carrier families (Al2O3, SiO2, TiO2, ZrO2, CeO2, SiC, hexa-aluminates, perovskites), as well as a number of promoters, leading to hundreds of combinations. In contrast, probably as a result of these research efforts, only two systems were applied commercially for this reaction, i.e., Ru/Al2O3- and Ni/Al2O3-based catalysts (see below).
On the other hand, it is also clear that the development of the optimal catalyst is the key to the design of the reactor(s) and product purification sections, i.e., for the development of the entire new process. Indeed, catalyst manufacturing is a key activity in the overall chemical industry: the global catalyst market size was valued at USD 29.7 billion in 2022 and is anticipated to grow at a compound annual growth rate (CAGR) of 4.6% from 2023 to 2030 [10].
In the present paper, an analysis of the state of the art of this technological transition, from syngas conversion to CO2 hydrogenation, is attempted, and related surface chemical and mechanistic aspects are discussed.
2. The Water-Gas Shift and Reverse Water-Gas Shift Equilibrium and the Catalysts
In Figure 1, the standard free energy of relevant reactions (ΔG°) as functions of temperature are reported for both CO (left) and CO2 (right) conversion. The water-gas shift (WGS) reaction and its reverse (rWGS), as well as methanation and methanol synthesis reactions, are considered, with the addition of dimethylether synthesis. The last reaction, which will not be further discussed in the present review, produces an important intermediate in the frame of methanol to chemical processes (methanol to olefins, MTO, as well as methanol to gasoline, MTG), of which its role may grow in the frame of the expected energy transition. In all cases, CO-based reactions are more favored than CO2-based ones. Additionally, COx hydrogenation to methane is always the most favored below ~900 K, while above this temperature, WGS and rWGS, in CO and CO2 cases, respectively, become the least favored in the considered situation.
The water-gas shift (WGS) equilibrium [11] is constituted of two opposite reversible reactions that allow for the modification of the COx/H2 ratio in syngas and to convert CO into CO2 or vice versa.
CO + H2O D CO2 + H2 ΔH°298 = −41.2 kJ/mol ΔG°298 = −28.7 kJ/mol
The reaction is weakly sensitive to pressure. According to the exothermicity of the direct WGS reaction, it can be realized at low temperatures. The ΔG° value increases with temperature and becomes positive at temperatures around 1098 K, above which the reverse reaction rWGS becomes more favored [12]. In the most common case today, the WGS process is applied to syngas produced from the steam reforming of natural gas to convert CO into CO2 (less toxic, more useful, more easily separated from H2, and more easily managed) and to further produce H2 in processes aimed at the production of hydrogen or ammonia synthesis gases. The WGS process is frequently realized in two steps, high-temperature WGS and low-temperature WGS, as usual for ammonia synthesis gas production [3,13], or in a single step at a high [14] or medium temperature [15] if a pressure swing adsorption (PSA) step follows to deeply purify the produced hydrogen.
2.1. High-Temperature Water-Gas Shift (HTWGS) Catalysts
High-temperature water-gas shift is usually carried out at 650–720 K, decreasing the CO concentration in syngas down to 1–3% mol on a dry basis [3,16]. Due to the limited exothermicity of the reaction, a single fixed-bed adiabatic reactor [13,14], or, sometimes, more adiabatic reactors with intermediate cooling, are used. Cheap and robust Fe2O3-based catalysts, frequently stabilized with Cr2O3 and also containing MgO, with a typical composition of 74.2% Fe2O3, 10.0% Cr2O3, and 0.2% MgO, and the remaining volatiles [11], are used. In the most recent formulation, 1–3% copper is added to the catalyst, improving catalytic activity [16]. The Clariant ShiftMax 120 catalyst is reported to have Fe2O3 > 80%, Cr2O3 8.5%, and Cu 2% [17], while the Johnson Matthey KATALCO 71-6 is based on Fe2O3 with ~2.5 wt% CuO and 9 wt% Cr2O3 [18]. Under reaction conditions, Fe2O3 (hematite/maghemite) is reduced to Fe3O4 (magnetite), which is stabilized morphologically and structurally by chromium, producing a spinel structure with a composition of Fe[Fe2−xCrx]O4, with a medium–low specific surface area (10–50 m2/g). It has been concluded that, in this case, copper metal is mainly acting as an activator for iron oxide.
A high steam-to-dry gas ratio is needed in the feed to prevent iron carbide formation on these catalysts, resulting in unwanted by-product formation and reduced catalyst strength. Another limit of this catalytic system is associated with the pyrophoricity of the reduced working catalyst, thus needing a complex procedure for process shutdown. A further drawback is associated with the presence of chromium, which, upon the preparation of the catalyst, may result in the hexavalent state, characterized by high toxicity. For these reasons, Cr-free catalysts are under development [19,20]. The substitution of chromium with Al gives rise to lower activity catalysts. The addition of other elements, such as Zn, Ce, and Mn, is under study. In the case of the two-WGS-step process, the first HTWGS bed also works as a guard bed to protect the more sensitive and expensive catalyst used in the second LTWGS step.
To enlarge the operating steam-to-carbon ratio window (limited to avoid the formation of iron carbides in the case of iron oxide-based catalysts), Topsøe recently developed a new Cr- and Fe-free catalyst based on zinc oxide and zinc aluminum spinel, SK-501 Flex™, which is unique because of its stability under dry conditions [21].
2.2. Low-Temperature Water-Gas Shift (LTWGS) Catalysts
A low-temperature water-gas shift is carried out at 450–573 K using more active, but also more expensive and delicate, copper-based catalysts [16,22]. These catalysts need an absolute absence (<0.1 ppm) of sulfur and chlorine. Due to the small exothermicity of the reaction, a single adiabatic reactor is commonly used [23], although (pseudo)isothermal reactors have also been developed [15]. After the LTWGS reactor, CO concentration in syngas may be as low as 0.1% on a dry basis. The most relevant feature of these catalysts is high selectivity for WGS, reducing the coproduction of methanol, which will be recovered in the process condensate. The most recent LTWGS catalyst from Clariant ShiftMax®® 217 is based on 45% CuO, 44% ZnO, Al2O3 [17], and the addition of a special promoter [24] to limit methanol coproduction. It is used together with the guard catalyst SHIFTGUARD 200, based on 45% CuO, Al2O3, and a proprietary metal oxide, which effectively adsorbs and strongly retains chlorides so that the downstream LTWGS catalyst is protected [24]. Interestingly, the same company (Süd Chemie at that time) previously produced the same application a catalyst which was even richer in copper (ShiftMax®® 240, 57% CuO, 31% ZnO, 11% Al2O3, 1% promoter [25].
The Topsøe LK-823 catalyst, as well as its latest version LK-853 FENCE™, are also based on Cu-ZnO-Al2O3 and are promoted by cesium to limit methanol formation [26,27]. Topsøe et al. also reported data on a guard catalyst constituted by Cu-ZnO-Cr2O3 (14% Cu) to protect copper/zinc/aluminum against chlorine poisoning [28]. Cu-ZnO-Al2O3 and alkali-promoted Cu-ZnO-Al2O3 are also sold by Johnson Matthey. According to these producers [29], alkali doping suppresses methanol formation and also boosts poison pick-up.
The HiFUEL®®W220 LTWGS catalyst (sold by Alfa Aesar, Haverhill, MA, USA) contains CuO 52.5 wt%, ZnO 30.2 wt%, Al2O3 17.0 wt%, and others at 0.3 wt% [30]. A commercial low-temperature shift catalyst, BASF K3-110, was reported to contain nominally 40% CuO, 40% ZnO, 20% Al2O3, BET area 102 m2/g, pore volume 0.35 mL/g, copper area 9.83 m2/g, dispersion 4.8%, and copper crystallite size 219 Ǻ [31]. BASF later patented a new preparation method to produce water-gas shift catalysts that, in the unreduced form, have 50.5–52.3 wt% CuO, 27.1–29.6 wt% ZnO, 18.8–19.7 wt% Al2O3, and 0.08–0.1 wt% Na [32]. These catalysts too have pyrophoric behavior and must be carefully discharged to prevent risks.
It can be noted that LTWGS catalysts are quite similar to methanol synthesis catalysts, which are also based on Cu-ZnO-Al2O3 with quite similar component ratios (see below) [33,34]. Indeed, while reaction temperatures and COx/H2 ratios can be also comparable in the two reactions, LTWGS is realized at a lower pressure and with much more water vapor in the reaction conditions than methanol synthesis. Thus, the real state of the catalyst surface in the two cases could differ significantly. In fact, more pronounced sintering of copper particles is reported to occur upon LTWGS than upon methanol synthesis [35]. In any case, metallic copper particles are the most abundant phase in these catalysts and cannot be considered as “supported” on the ZnO-ZnAl2O4 oxide minor component. Several Transmission electron microscopy studies [36,37,38,39] show that large copper particles are separated from each other by large ZnO-ZnAl2O4 particles, as schematized in Figure 2. However, data show that a strong interaction between substantially unsupported metallic copper and zinc species certainly occurs, leading to the formation of the most active species and modifying the surface of copper particles. This interaction can also involve metallic Zn species or Cu-Zn alloy [40], copper-supported ZnO layers [41], and/or partially oxidized Cu+ sites [42]. The real nature of the most active species in these catalysts is still far from being clearly identified but certainly involves a surface modification of metallic copper by zinc species. Most of the Zn-Al oxide phase, constituted by ZnO where Al ions are present as spinel domains [43], act as structure stabilizer phases against sintering.
2.3. Medium-Temperature Water-Gas Shift (MTWGS) Catalysts
When hydrogen purification is performed later by PSA, medium-temperature WGS (MTWGS) can be realized at about 490 to 550 K down to approx. 0.5% CO on a dry basis at the reactor outlet, realized either in adiabatic reactors or in isothermal water-cooled reactors [15]. Catalysts for MT-WGS are designed to be more thermally stable and manage exothermicity under low steam-to-carbon conditions [44]. Although nickel-based catalysts have also been proposed for MTWGS [45], it seems that industrial catalysts are also based on Cu/ZnO-Al2O3 [46]. In particular, Clariant’s ShiftMax 300 catalyst (Clariant Produkte, Munich, Germany) is a stabilized copper–zinc catalyst reported to be ideally suited for the MTWGS [47]. In order to achieve optimum MTS performance, a composite loading, consisting of different types of Cu/ZnO-Al2O3-based MTS catalysts at the top and the bottom of the reactor can be used, as proposed by Topsøe, with the more active LK-813 catalyst on the top and the less active and more thermostable LK-819 catalyst at the bottom [48].
2.4. Other Industrial Water-Gas Shift Catalysts
Different versions of the WGS process can be applied and need different catalysts. Noble metal-based catalysts, such as Pt/CeO2 [49], have even higher activity and can be preferentially used in miniaturized applications, e.g., for on-board hydrogen production, where more active catalysts and smaller reactors are needed.
2.5. The Reverse Water-Gas Shift Process and Catalysts
The reverse water-gas shift reaction (rWGS) could be used to reduce the amount of hydrogen and CO2 and increase that of CO in syngases with low H2/COx ratios (as those formed by coal gasification or by methane autothermal reforming). However, this was never realized, apparently, up to now: in fact, external CO2 additions or recycling to syngas was a more convenient procedure to obtain the same result.
In contrast, the use of pure CO2 feeds, such as captured CO2, makes rWGS an interesting approach to produce CO or CO-containing syngases as the first step for further conversion [51], e.g., to synthetic fuels from the Fischer-Tropsch process, acetic acid, oxo-alcohols, or methanol, as discussed below. The reverse water-gas shift process is also a candidate technology for water and oxygen production on Mars under the in situ propellant production project [52] due to high (∼95%) atmospheric CO2 concentrations on Mars and the availability of H2 as a byproduct of oxygen generation needed in these conditions.
Evidently, rWGS is an endothermic reaction which is shifted to products only above around 1100 K (Figure 1) when its ΔG° starts to become negative. As expected for the micro-reversibility principle, the same materials catalyzing WGS (such as Cu/ZnO-based LTWGS catalysts) also catalyze rWGS at low temperatures [53]. Noble metal-based catalysts [54] and Cu/CeO2 catalysts [55] are proposed to realize rWGS at medium temperatures (673–873 K). However, to push conversion towards CO and to obtain relatively low H2 to CO ratios (e.g., for the Fischer-Tropsch reaction), rWGS must be realized at definitively higher temperatures (>1200 K), i.e., at temperatures where copper catalysts are unstable due to copper being near its melting point of 1257 K. In these conditions, several metal catalysts show activity for rWGS but with incomplete selectivity due to the coproduction of hydrocarbons and methanol [56], which can occur in small amounts, particularly at high pressures, together with the production of coke.
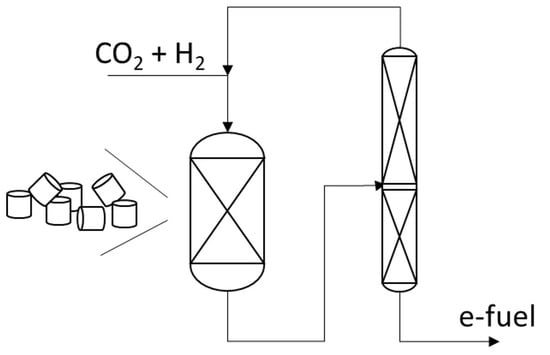
Interest in rWGS is now focusing on Ni-based catalysts, which are stable and commonly used at high temperatures. In particular, several projects were undertaken for the production of sustainable aviation fuel starting from CO2 and electrolytic hydrogen, where rWGS is mostly realized using electrically heated reactors [57], such as the eREACT™ technology [58] used to realize the electrified reverse water-gas shift (eRWGS™), developed by Topsøe [59], in order to avoid CO2 emissions. Using a feed of H2/CO2 in a ratio of 2.25 at 10 barg and high-temperature operation at 1320 K over a Ni/ZrO2 washcoat catalyst, the production of synthesis gas with a H2/CO ratio of 2.0 and no detectable methane, ideal for Fischer-Tropsch synthesis, was obtained. The authors suggest that a sequential CO2 methanation + methane steam reforming reaction scheme may occur. Also, Clariant developed a promoted nickel catalyst, ShiftMax®® 100 RE, for the rWGS shift as the first step in the production of e-fuels in collaboration with Ineratec, Karlsruhe, Germany [60]. According to Clariant, this catalyst shows high resistance against coking, low-methane byproduct formation, long lifetime, and high strength.
Johnson Matthey recently revealed the launch of HyCOgenTM [61], a reverse water-gas shift technology designed to help enable the conversion of captured CO2 and green hydrogen into sustainable aviation fuel (SAF) through the conventional Fischer-Tropsch process (FT CANS technology [62]). Axens, Paul Wurth, and IFP Energies Nouvelles (IFPEN) have signed a co-development agreement for the optimization of the rWGS technology and its integration into e-fuel projects [63]. Indeed, the idea to develop rWGS as a first step to produce methanol from CO2 + H2 feeds was reported already several years ago [64].
3. The Methanol Synthesis and the Catalysts
The relevant reactions in methanol synthesis [65,66] are the following:
CO + 2 H2 D CH3OH ΔH°298 = −90.7 kJ/mol ΔG°298 = −25.3 kJ/mol
CO2 + 3 H2 D CH3OH + H2O ΔH°298 = −49.5 kJ/mol ΔG°298 = +3.3 kJ/mol
Also, the water gas shift equilibrium (Reaction (1)) may be established upon the reaction.
The methanol synthesis from CO (Reaction (1)) is favored (ΔG° < 0) at temperatures below 408 K. Taking into account that the best industrial catalysts have an activity threshold at about 450 K, the reaction is carried out in conditions where ΔG° is positive. To shift to methanol equilibrium, high pressure is needed.
The methanol synthesis from CO2 (Reaction (2)) is favored (ΔG° < 0) only at even lower temperatures than the synthesis from CO, i.e., below 280 K. Thus, thermodynamically, methanol synthesis from CO2 hydrogenation is even less favored (lower C conversion in the same temperature and pressure conditions) than its synthesis from CO (Figure 1). Equilibrium becomes less and less favorable by increasing the CO2/CO molar ratio in the hydrogenation of COx mixtures. Although, at 470 K, theoretical conversion in pure CO hydrogenation to methanol at 75 bar is very high, >95%, in the same conditions, the allowed conversion in pure CO2 hydrogenation to methanol is less than 70% [67]. In Figure 3, the calculated equilibrium compositions starting from stoichiometric mixtures (CO + 2H2 and CO2 + 3H2) at 100 atm show a significant difference in the methanol concentration starting from CO2 hydrogenation with respect to pure CO hydrogenation and the significant amounts of water produced.
3.1. Conventional Methanol Synthesis
Methanol synthesis is classically realized today in Western countries by converting syngas produced from the steam reforming or autothermal reforming of natural gas [68,69,70]. For large-scale methanol synthesis processes, such as the Megamethanol process from AirLiquide-Lurgi [71] and SynCOR MethanolTM from Topsøe [72], autothermal steam reforming with a low steam-to-carbon ratio is considered best suited today. In China, the methanol synthesis gas is mainly produced by coal gasification [70,73]. The obtained syngas is essentially a mixture of CO, CO2, and H2, with a stoichiometry ratio, SR = H2-CO2/CO + CO2 in the range of 1.5–3, and a CO/CO2 ratio in the range of 0.8–1.2. To adjust SR, the water-gas shift reaction (in the case of SR < 2, as typical for gases from autothermal reforming or coal gasification) or the reverse water-gas shift reaction (in the case of SR > 2 from methane steam reforming) could be realized. Instead, a high purging rate with the recovery of reactants from purge is commonly realized.
The reaction is performed at 473–523 K, 50–150 barg, using water-cooled, steam-rising, or gas-cooled reactors, sometimes more than one in series or in series and in parallel [74]. In Figure 4, the reactor system for the Megamethanol process is shown, where a multitubular gas-cooled reactor (GCR) and a multitubular water-cooled reactor (WCR) are used in series, with intermediate cooling and liquid product removal, to push thermodynamics in the GCR [71]. A main practical point concerning methanol synthesis processes is the selectivity of products. The typical byproducts of methanol synthesis from syngases are dimethylether, methylformate, higher alcohols, and hydrocarbons, with selectivities which are usually very low (of the order of hundreds of ppm) but must be minimized to reduce the complexity of the purification section.
The main suppliers all produce catalysts based on Cu/ZnO with the addition of Al2O3 and/or Cr2O3 [74,75], besides additional promoters such as Mg, Ca, Si, and Zr [35]. The catalysts are prepared by the thermal treatment of precursors obtained via coprecipitation, such as Cu-Zn mixed malachite, rosasite, or aurichalcite-type hydroxycarbonates [76], with the typical Cu/Zn atomic ratio being in the 2–3 range and minor alumina amounts [36,77]. According to Waugh [78], the best composition in terms of CuO/ZnO/Al2O3 in the unreduced catalyst is around 60:30:10. The Topsøe MK-121 catalyst was reported to contain, in the unreduced form, >55 wt% CuO, 21–25 wt% ZnO, and 8–10 wt% Al2O3 in the fresh catalyst, graphite, carbonate, and moisture balance [79]. According to Topsøe and researchers, Cu particles act as the active component in this material, while ZnO and Al2O3, more than a support, can be regarded as structural promoters that separate the Cu particles from one another, thereby diminishing particle growth. ZnO also has the additional, even more important role of assisting Cu in producing methanol. The activity boost is related to the migration of Zn atoms onto the surface of the Cu particles: the fraction of surface atoms in the Cu particles substituted by Zn, θZn, is a function of the gas’s reduction potential [80]. The development of the new FENCE™ technology allowed us to produce the more recent MK-151 FENCETM catalyst, where the copper nanoparticles are finely dispersed in the catalyst by creating a separation between them of multiple oxide particles. Topsøe emphasizes the role of impurities in the catalyst, coming from low-purity catalyst raw materials, such as iron, chlorine, and sulfur, for high methanol selectivity. Also, Clariant (previously Süd Chemie) catalysts, denoted as MegaMax®®700 and the newest MegaMax®®800 and MegaMax®®900, are reported to be constituted of CuO/ZnO/Al2O3 [81,82].
A novel catalyst layering technology for enhanced methanol synthesis performance denoted as MegaZone™ Technology from Clariant and Air Liquide has been recently developed. Catalysts with moderate activity are loaded in the hotter zones of the converter to prevent hotspots, while activity-enhanced catalysts are placed further down the reaction pathway to intensify reaction rates in the lower portion of the converter. On the one hand, less thermal stress on catalysts will lead to longer catalyst lifetimes. High activity in the bottom part of the reactor will increase reaction rates and reduce by-product formation by up to 10% [81].
The composition of the Katalko 51 series from Johnson Matthey is 64% CuO, 24% ZnO, 10% Al2O3, and 2% MgO. Magnesium oxide (MgO) is incorporated to maximize the initial dispersion of copper and, therefore, boost the initial activity of the methanol synthesis catalysts. The increase in the copper surface area in a Mg-containing copper-based catalyst can be of the order of 20% versus a Mg-free formulation [83]. The addition of MgO allows for increased selectivity to methanol, reducing the coproduction of the following byproducts: methylformate, ethanol, n-propanol, 2-propanol, and 2-butanol. The addition of Si, in the form of silica, to the formulation improved long-term stability and further reduced ethanol selectivity in the new Katalko 51–102 commercial catalyst [84].
Over commercial catalysts, the maximum reaction rate is obtained with a CO2/CO + CO2 ratio of around 10%, quite typical of gases from coal gasification, while pure CO2 hydrogenation is notably slower than the conversion of typical syngases but significantly faster than the hydrogenation of pure CO to methanol [67,85]. To reduce the emissions of CO2 from methanol synthesis starting from natural gas, BASF patented a process that, in principle, does not emit any CO2, being based on syngas produced by partial oxidation (an exothermic reaction which does not need heated furnaces), and also using CO2 captured from the waste purge gases [86].
3.2. Methanol Synthesis from CO2 through Previous Reverse Water-Gas Shift
As already said, methanol synthesis can be obtained using CO2 feeds with a conventional methanol synthesis process through a previous reverse water-gas shift step [64]. However, most studies suggest that this way is not needed for methanol synthesis from CO2.
3.3. Direct Methanol Synthesis from CO2
Catalysts for conventional methanol synthesis from syngases based on the Cu/ZnO/Al2O3 system are also active for the hydrogenation of pure CO2 to methanol [87]. The methanol synthesis rate on typical Cu-based methanol synthesis catalysts increases significantly by adding CO2 to pure CO by up to 10% (nCO2/nCO + nCO2); then, it slows down again [67]. Thus, the reaction rate is fastest for syngas compositions (in terms of the CO2/CO ratio) similar to that of coal gasification, while it decreases for syngases with higher CO2/CO ratios than those from methane steam reforming and is even slower for pure CO2 feed. In any case, methanol synthesis from CO2 + H2 is faster than methanol synthesis from CO + H2.
Additionally, as obvious, methanol synthesis from COx produces more and more water by increasing the CO2/CO molar feed ratio, which results in faster copper sintering [35]; thus, stronger hydrothermal stability is needed for the catalyst. On the other hand, the water-gas shift equilibrium (Reaction (3)) is also established in typical reaction conditions, leading to an equilibrium between CO and CO2 dependent on the water-to-hydrogen ratio.
As stated, the byproducts of conventional methanol synthesis are dimethylether, methylformate, and higher alcohols and hydrocarbons. In the case of pure CO2 feed, CO can also form as a byproduct through the reverse water-gas shift equilibrium. Higher methanol yields would be obtained with catalysts that catalyze methanol synthesis without catalyzing rWGS under reaction conditions [35]. However, taking into account that CO may be recycled to the reactor together with unreacted CO2 and hydrogen and that CO+CO2 mixtures react faster than CO2, CO coproduction can be seen as a good catalyst feature-accelerating process reaction rate.
In any case, conventional industrial methanol synthesis catalysts, such as the Katalko 51 series from Johnson Matthey, also work as excellent catalysts for CO2 hydrogenation to methanol. The addition of silicon in the composition of Katalko 51 series Johnson Matthey catalysts also improved its isothermal stability for application in the CO2 hydrogenation methanol process [84]. Similarly, Topsøe developed a new catalyst also based on the Cu/ZnO/Al2O3 system, MK-317 SUSTAIN™, which has excellent activity, selectivity, and stability for direct methanol synthesis from CO2 [88].
4. The Methanation Processes and the Catalysts
The relevant reactions in methanation processes are the following:
CO + 3H2 → CH4 + H2O ΔH°298 = −206.63 kJ/mol ΔG°298 = −141.8 kJ/mol
CO2 + 4H2 → CH4 + 2H2O ΔH°298 = −164.9 kJ/mol ΔG°298 = −113.2 kJ/mol
However, they may also involve the WGS equilibrium. According to the strong exothermicity of both reactions, they are strongly favored at low temperatures already at atmospheric pressure and even more at higher pressures [89,90]. They are the reverse of steam methane-reforming reactions that become favored above ~870 K when their ΔG° becomes positive. CO methanation is more favored thermodynamically than CO2 methanation at low temperatures, up to the temperature where their ΔG is negative [91]. Even at atmospheric pressure, the complete conversion of CO is possible until 673 K. At stoichiometric conditions and atmospheric pressure, the allowed CO2 conversion at 673 K is of the order of 80% mol [89] and increases by decreasing temperature. Methane selectivity can be slightly decreased by some coproduction of higher hydrocarbons, of which the amount would increase with pressure and temperature [92]. Starting from CO and H2, CO2, and carbon can also be produced by the Boudouard equilibrium, CO dissociation, and water-gas shift [93], while starting from CO2 and H2, rWGS can result in the production of CO. The methanation reactions are, in principle, also competitive with methanol synthesis but are thermodynamically much more favored at low temperatures. Carbon deposits coming from CO dissociation and Boudouard equilibrium can result in catalyst deactivation.
Two types of methanation processes are currently realized: the low-temperature process to remove residual COx from pure hydrogen and ammonia synthesis gas and the high-temperature process to produce substitute natural gas.
4.1. Low-Temperature Methanation
Low-temperature methanation is usually realized in processes for preparing hydrogen for hydrogenation reactions, or H2/N2 mixtures for ammonia synthesis [3,13,94]. They are realized to reduce the concentration of COx in such gases as much as possible because of the poisoning effect of COx on hydrogenation catalysts. This reaction is realized in highly diluted COx in H2 mixtures (COx concentration, most commonly < 1% mol), with CO being the predominant COx compound in the feed, allowing the COx concentration to be reduced to less than 5ppm in the process of gas leaving the methanator. As a consequence of the small amount of reacting molecules, the evolved heat is low, and adiabatic reactors can be used. The reaction temperature is typically 440–620 K.
Commercial catalysts for methanation in CO-rich syngases are either based on supported ruthenium or based on supported nickel [7,95,96]. A typical Ru-based catalyst is METH 150 Clariant (Süd Chemie) based on 0.3% Ru/Al2O3 [25] for very low-temperature applications (down to 440 K). Ruthenium-based catalysts are most active at low temperatures, do not require pre-reduction, and are not pyrophoric. However, they have found only a limited area of application due to their high costs.
Due to their sufficient activity, stability, and relatively low cost, Ni-based catalysts are predominantly used for conventional low-temperature methanation [3,95,96]. Typical nickel loadings are in the range of 20–45%wt as NiO, over γ-Al2O3 or metal aluminate supports, or Cr2O3 [95]. Clariant produces catalysts with 43% and 25% NiO on support (likely alumina) METH 135 and METH 134 in their oxidized forms [15]. Topsøe produces the pre-reduced PK-7R catalyst, constituted of >23% Ni wt [96] on an alumina carrier, which ensures that CO and CO2 are fully converted to methane at an operating temperature of 460 K [97]. Johnson Matthey sells catalyst precursors (Katalko 11 series) constituted by 35 wt% NiO supported by refractory oxides, such as alumina–silica–lime–magnesia, strengthened with calcium aluminate cement [98]. Nickel catalysts are pyrophoric and cannot be used below 460 K to avoid the formation of Ni(CO)4.
Several studies showed that the methanation of CO2 is faster and more selective to methane than CO methanation, where ethane may form in small amounts over both nickel and ruthenium catalysts [99]. However, in the CO + CO2 mixture, CO strongly inhibits the methanation of CO2 over both Ni catalysts [100,101,102]. In fact, selective (or preferential) CO hydrogenation in the presence of CO2 usually occurs, allowing the possibility of realizing CO abatement in syngases with an alternative process to WGS, e.g., with Ru/Al2O3 catalysts [103].
4.2. High-Temperature Methanation for Substitute Natural Gas Synthesis
High-temperature methanation is the conversion of a CO + CO2-rich syngas, from coal gasification or biomass conversion, to produce a methane-rich gas to be used as a substitute for natural gas (SNG [66,104]). Due to the much higher COx concentration in hydrogen (usually in the 25–35% mol range) and their high conversion degree, heat significantly evolved. A syngas of H2/CO = 3 at 30 bar would result in an adiabatic temperature increase to 1196 K with an inlet temperature of 573 K [105]. To manage the first reactor, the partial recycling of the cooled product gas is realized, thus reducing CO concentration and, in parallel, the temperature increase to what can be handled by a methanation catalyst. Also, very efficient cooling in the reactor system is carried out, and still, the reaction occurs in a high temperature range of up to 973 K. Several different processes have been proposed [104,106]. Most of them use multiple adiabatic fixed-bed reactors with interbed cooling [107]. Real industrial applications of this technology have been very few in the previous century [68,104], but several plants appear to have been recently put in operation or are under construction now, particularly in China [108]. Only Ni-based catalysts are apparently used, taking into account that the extent of the reaction under industrial conditions is more limited in heat and mass transfer rather than kinetics. With these catalysts, selectivity to methane in the full process is very high, although carbon deposition and the formation of higher hydrocarbons occur to a limited extent.
The Topsøe TREMPTM technology [105,109] allows producing SNG from coal gasification syngas using a series of adiabatic fixed-bed reactors with interbed cooling and recycling (Figure 5), with temperatures ranging from 523 to 973 K and pressures of up to 30 bar. T. A previous gas-conditioning step (i.e., water-gas shift) step is also realized to reduce the CO concentration. Up to 85% of the released heat from the methanation reactions is recovered as a high-pressure superheated steam. The product gas contains 94–98% methane, with a low percentage of residual hydrogen and CO2 and CO < 100 ppm. The TREMP process is also part of the RNG technology allowing the production of renewable natural gas starting from biomass via biomass gasification [110].
In Topsøe’s TREMP™ process, the catalyst (MCR2X) is a supported nickel catalyst containing 22 wt% Ni on a stabilized support, possibly based on MgAl2O4 [111], with a surface decreasing from 50 m2/g (fresh) to 30 m2/g (used) [103] and stable activity of up to 970 K [112]. Other nickel-based catalysts, MCR-8 and PK-7R, are used in medium- and low-temperature steps. Catalysts for this process need to be sufficiently active at low temperatures, resistant against sintering at high temperatures as well as to other phenomena producing deactivation, i.e., gum and Ni(CO)4 formation [113].
The reactor system of the competitor process DAVY [114] from Johnson Matthey consists of two adiabatic bulk methanators, recycled to the first reactor, and in a variable number of adiabatic trim methanators in series at the end. The Johnson Matthey CRG catalysts used in the process are apparently based on Ni and refractory supports such as alumina–lanthana–magnesia [115]. They catalyze the methanation reaction and, simultaneously, the WGS reaction, thanks to the water produced by the methanation reaction.
Also, in the case of the VESTA process, several fixed-bed reactors in series are used, with a previous HTWGS step [116,117]. The catalysts, provided by Clariant [118], with a wide operating temperature range (500–973 K), are based on nickel [116]. Also, this process can be fed by syngases produced both by coal and by biomass gasification.
4.3. CO2 Methanation
The methanation of captured CO2 using green hydrogen is considered a possible method to valorize and reuse it, which is also in view of the so-called “power to Gas” technologies [119,120]. Many projects and plants of various sizes have been undertaken in the last fifteen years [121,122]. The new processes under development today for CO2 methanation [105,123,124] are strictly related to the ones described above for SNG production from CO-rich syngases. For example, the VESTA process is also applied as the methanation step of power-to-gas plants [117].
According to its large exothermicity, the theoretical thermodynamic methane yield approaches totality at temperatures of up to 473 K at moderate pressures [89,90,125]. At higher temperatures, CO2 conversions and methane yields from thermodynamics decrease. However, methane synthesis is competitive with the production of CO through rWGS (Reaction –(1)), of which its equilibrium is shifted to CO + H2O more when the temperature is higher. Studies show high selectivities of pure CO2 methanation, with a possible coproduction of small amounts of CO due to the rWGS reaction. In the presence of CO, the conversion of CO2 is inhibited, but the conversion of CO to methane occurs [99,126]. Reactor modeling studies suggest that, despite the high temperature, at moderate pressures, the amount of CO will always be very low in CO2 hydrogenation reactors [111,127].
Although an enormous number of projects are under development [122], information on real industrial applications is scarce. One of the earliest examples of power-to-gas technology was Audi’s e-gas plant in Werlte, Germany, which was constructed by Etogas, to produce renewable synthetic methane from hydrogen and carbon dioxide emissions to achieve low-carbon mobility for Audi’s A3 Sportback g-tron vehicles. Clariant provides the catalyst to be applied in such an application [128]. No detailed information is available on the Clariant’s SNG 100 DCARB methanation catalyst just developed for power-to-gas processes: it seems that it is based on nickel [116]. In fact, taking into account that practical reaction temperatures in the catalyst bed are relatively high, the low-temperature activity of noble metals such as ruthenium is not necessary, at least in main methanation reactors. Thus, Ni-based catalysts are most promising [129]. High-temperature stability is certainly a key feature. A very large number of different catalyst compositions have been proposed in the literature for CO2 methanation. A critical analysis of the scientific literature data concerning catalysts for CO2 methanation allowed us to propose that Ni/Al2O3 catalysts with medium metal loading allowing small metal particles to almost entirely cover the support surface, with additional ceria and/or basic oxide dopants as the stabilizers and activating components, would be very active in methanation and not produce many carbon residues and gums [130].
5. The Production of Higher Hydrocarbons
The most relevant reactions in the production of higher hydrocarbons from COx hydrogenation, usually denoted as the Fischer–Tropsh processes, are the following:
which are strongly exothermic equilibrium reactions favored at low temperatures and high pressures. Reactions (6) and (7) are the reverse of the steam reforming of higher hydrocarbons, while Reaction (8) is the reverse of the dry reforming of higher hydrocarbons. In the low-temperature process with used catalysts, cobalt-based linear paraffins and the corresponding linear olefins are the products. According to thermodynamic analyses [131,132], product distribution is very sensitive to feed composition, and to temperature and pressure to a lesser extent. An increase in the H2/CO feed ratio and temperature results in a shift in selectivity toward low carbon number hydrocarbons, especially towards methane. Conversely, an increase in pressure favors high carbon number hydrocarbons. In fact, the high-temperature process is realized to produce gasoline-range hydrocarbons, olefins, and some oxygenated compounds.
CO + 2H2 → −CH2− + H2O ΔH°298 = −165 kJ/mol
CO2 + 3H2 → −CH2− + 2H2O ΔH°298 = −125 kJ/mol
2CO + H2 → −CH2− + CO2 ΔH°298 = −204 kJ/mol
5.1. Conventional Low-Temperature Fischer-Tropsch (LTFT) Processes
The LTFT process, performed at 473–523 K, 20–30 bar, allows us to convert syngases into middle distillate and hydrocarbon waxes, both applicable to produce a high cetane number, sulfur-free Diesel fuel, and jet fuel [133]. Cobalt-based catalysts are used in most LTFT industrial technologies [134,135,136]. In fact, among the different metals active as FT catalysts (essentially ruthenium, iron, cobalt, and, to a lower extent, nickel), cobalt represents the best choice for this application for its moderate price, good selectivity to high molecular weight paraffins, and low selectivity to methane, olefins, and oxygenates [137]. According to IFPEN researchers, among possible supports, alumina is a good choice because of its availability with texture suitable to slurry conditions in waxy liquids. The cobalt FT catalysts are usually supported on high surface area alumina (150–200 m2/g) and typically contain 15 ÷ 30 wt% of cobalt. This means that cobalt is largely dispersed on the support surface forming, in the reduced state, very small cobalt metal particles [138,139]. To stabilize them and decrease selectivity to methane, these catalysts also contain small amounts of noble metal promoters (typically 0.05–0.1% weight of Ru, Rh, Pt, or Pd) and/or an oxide dopant as well (potash, zirconia, lanthana, cerium oxide, 1–10%) [140]. The role of noble metal doping has been supposed to be to improve the reducibility of cobalt, while the support and the oxide promoters may have a role in controlling the size of the cobalt metal to an optimal value. The slurry phase distillate (SPD) process from Sasol uses a Co-based Sasol proprietary catalyst typically operating at 500 K and 2.5 MPa [133], apparently supported by alumina [141]. Upon working, part of the catalyst is converted into the carbide CoC2, with moderate deactivation [141] but increased olefin production [142,143].
In a recent study, researchers from Johnson Matthey [144] showed that different supports for cobalt may significantly influence the stability of the catalyst and selectivity to products, with the titania- and zirconia-supported catalysts producing predominantly paraffins, whereas CeO2− and ZnO− containing catalysts produce significantly more olefins and alcohols, showing that the product distribution may be tailored depending on the support material. The catalysts used by Shell in their GTL technology were reported to be likely constituted by cobalt on ZrO2-SiO2 support [145]. With cobalt catalysts, the optimum ratio of the reactants CO + CO2/H2 is 1:2, that is, the stoichiometric ratio for FT. CO2 as well is involved in the reaction.
The ability to catalyze hydrocarbon chain growth is a key feature of Fischer-Tropsch catalysts. Although the molecular mechanism for chain growth on cobalt catalysts is still far from ascertained, data indicate that the high pressure of CO favors it and that adsorbed CO is certainly a promoter of chain growth [146].
Water-cooled multi-tubular fixed-bed reactors (FBR) or slurry bubble column reactors (SBCR) are used industrially [135,147]. Recently, Johnson Matthey developed a new reactor technology for its FT CANS process, based on fixed-bed reactors, termed CANSTM’, consisting of modular catalyst containers providing modified reactant flow paths [144].
Iron catalysts can also be applied at low temperatures in an LTFT process, as carried out years ago by Sasol [145]. Due to their high activity in the water-gas shift reaction, iron catalysts are suited for FT syntheses using syngases characterized by a quite low H2/COx ratio (~1.7 mol), as those arising from coal gasification. They produce more olefins and oxygenates than cobalt catalysts. Thus, Co-based catalysts appear to be the most performant, particularly for the synthesis of Diesel-type fuels, and have higher H2/CO ratio syngases (e.g., from natural gas steam reforming). However, Co-based catalysts are less resistant to some poisons, such as ammonia [148], which can be present, e.g., in syngases from biomass gasification.
5.2. Conventional High-Temperature Fischer-Tropsch (HTFT) and Medium-Temperature Fischer-Tropsch (MTFT) Processes
The high-temperature Fischer-Tropsch process (HTFT) [149] consists of the conversion of syngases at 590–630 K and 20–40 bar, aimed at the production of low molecular mass alkenes and liquid products primarily in the gasoline range, with the coproduction of several oxygenate compounds. Quite recently, an MTFT process has been developed in China, working at 530-570 K [150,151]. Both HTFT and MTFT are performed on iron-based catalysts [149,150,152,153]. These processes produce more oxygenates and olefins and a slightly different molecular weight distribution for hydrocarbons compared to Co-based catalysts applied in the LTFTS [149,151].
A typical iron FT catalyst is unsupported but also contains a low percentage of silica, copper, and potassium. Copper is added to aid in the reduction of iron, while silica is a structural promoter added to stabilize the surface area but may also have a chemical effect on the catalyst properties. Potassium is considered to increase the catalytic activity for FT synthesis and water-gas shift reactions, promote CO dissociation, and enhance chain growth, increasing olefin yield and lowering the CH4 fraction [154,155]. Under reaction conditions, the catalyst converts into mixtures of carbides like χ-Fe5C2, ε-Fe2C, θ-Fe3C [156,157], and magnetite Fe3O4, with only small amounts of α-Fe. Studies using isotopically labeled 13CO as a reactant or, the reverse, 13C-labeled catalyst and 12CO as a reactant indicate that the carbon atoms of hydrocarbon products come from the reactant CO, while surface carbon atoms of the carbide may participate in the initiation step of chain growth [158]. In any case, carbidic rather than metallic catalysis should really occur in the case of HTFT synthesis. Also, in the case of iron carbide catalysis, it seems clear that CO is a promoter of chain growth, with a possible role of a CO insertion step in the chain growth mechanism [159].
5.3. Fischer-Tropsch Processes Using Electrolytic Hydrogen and Captured CO2
The Fischer-Tropsch processes can be of interest to convert pure CO2 feed with electrolytic hydrogen to e-fuels, such as e-jet fuel and e-Diesel fuel. This can be accomplished in two ways [160]: (i) a direct way, i.e., feeding directly the CO2 + H2 mixture to the catalyst (the so-called CO2-FTS); (ii) an indirect way, i.e., sequencing the rWGS reaction step producing a CO + CO2 containing syngas with a following conventional FT step, either LTFT or HTFT.
The direct CO2-FTS reaction is slower than CO-FTS and conventional FTS reactions under the same conditions and tends to produce smaller chain hydrocarbons. This is likely related to the main role of carbon monoxide in dissociation-producing active carbide species as well as in the promoting role of CO in the chain growth steps. This, however, influences far more cobalt- than iron-based catalysts. At 490 K and 20 bar, iron catalysts produce long-chain olefins, while cobalt-based catalysts act mostly as methanation catalysts [161]. The different behavior is explained by taking into account that iron catalysts have high WGS/rWGS activity, thus allowing the production of the real reacting and chain growth promoter molecule CO from CO2 in FTS conditions. It has been reported that catalysts based on delafossite, CuFeO2, produce copper-doped iron-carbide-based catalysts for this process, giving rise to high selectivities to hydrocarbons in the Diesel fuel and wax range [162]. When combined with the HZSM5 zeolite, this catalytic material allows the production of gasoline-rich [163] and aromatic-rich [164] hydrocarbon mixtures. Using a Na-containing Co-Fe catalyst, high selectivity to kerosene/jet fuel has been obtained [165]. Apparently, only laboratory tests are available today for this reaction.
On the other hand, the indirect method, through the previous rWGS reaction step, followed by a more or less conventional LTFT step, seems to be the industrial choice today. In fact, several companies, such as Topsøe [56], Johnson Matthey, London, UK [61], Axens, Rueil-Malmaison, France [63], and Clariant with Ineratec [60] and Audi, Zwickau, Germany [166], are developing processes based on this scheme, with a previous high-temperature rWGS step (as mentioned above) and a nearly conventional LTFT step later.
6. Mechanistic Aspects of CO2 Hydrogenations
The mechanisms involved in COx hydrogenation imply the role of metallic phases, as well as of oxide and carbide phases. To have an idea of the role of these phases, a look at the surface chemistry and physics of the different catalyst components is needed. Concerning oxide phases, discussion can be based on their ionicity and acido-basicity [167,168], as well as their reducibility [169]. On the other hand, the catalytic behavior of transition metals in reaction conditions is strongly related to the position of the d-band center, in agreement with the d-band model of Hammer and Nørskov relating adsorption energies to the d-band position, and the adsorption energies to barriers in catalytic reactions [170,171,172]. In Table 2, data concerning the interaction of the most relevant catalytic metals with the reactants of the processes discussed here are summarized.
Another relevant point in developing industrial catalysts is their relative cost. In particular, the use of expensive metals, like platinum group and noble metals, even when they can be used in much lower amounts than cheaper transition metals, due to their great price differences, can be inconvenient when performance advantages are limited. For this reason, today’s price of metals is also added to Table 2.
6.1. The Adsorption and Activation of Hydrogen
A number of oxides that can be used as supports or components of hydrogenation catalysts significantly adsorb hydrogen and show some useful activity as catalysts for hydrogenation and dehydrogenation reactions. This is the case of ZnO, Fe3O4, Cr2O3, Ga2O3, ZrO2, and CeO2 [178]. Besides very weak and reversible molecular adsorption occurring only at very low temperatures, at moderate temperatures, the adsorption of hydrogen on several metal oxides was reported to be heterolytic, occurring on exposed cation–oxide couples. Thus, hydrogen dissociation produces a metal hydride species on cationic centers and a new hydroxyl group on an oxide site. The best-known case is that of ZnO, which has been the subject of a number of studies [179,180,181]. This oxide is an active catalyst in hydrogenations, such as methanol synthesis [182], as well as in alcohol dehydrogenation reactions [178]. Well-evident surface hydride species have been observed to be formed by H2 adsorption at room temperature by vibrational spectroscopies (IR, HREELS, INS), both of the terminal Zn-H type I (1700–1600 cm−1), which is more weakly adsorbed, and of the bridging Zn-H-Zn type II (1450–1200 cm−1), which is more strongly adsorbed, formed together with new OH groups. Spectroscopic studies confirmed that some of the hydride species are indeed active in hydrogenation, e.g., producing CHO formyl species from CO as a possible intermediate in methanol synthesis over ZnO [183]. The heterolytic dissociation of hydrogen-forming active metal hydride species has also been observed on chromia (Cr2O3), a component of hydrodealkylation and paraffin dehydrogenation catalysts [178], where only terminal hydride species were found (νCr-H 1714, 1697 cm−1 [184]) and the combination of ZnO and Cr2O3, i.e., zinc chromite (ZnCr2O4) catalysts [184]. The latter is the active phase of the old BASF industrial catalysts for methanol synthesis with the high temperature and pressure process, operated at above 300 bar and 573–673 K [70], industrially used until the seventies of the last century. On Cr2O3 and on other mixed chromites such as Co-Cr and Mn-Cr oxides, both terminal and bridging species were found [184]. Two terminal hydride species (νGa-H 2003 and 1980 cm−1) have also been found to be formed by hydrogen adsorption on gallium oxide polymorphs [185] and assigned to hydride species bonded to tetrahedral and octahedral Ga ions, respectively. Dissociative adsorption of hydrogen was also observed on zirconia: to terminal hydride species was assigned a sharp νZr-H band at 1562 cm−1, while a band at 1540 cm−1 was assigned to a dihydride H-Zr-H species, and a broad band at 1371 cm−1 was assigned to the bridging species. (ZrHZr) [186,187,188]. The dissociative adsorption of H2 was also observed to occur in small amounts on the defect sites of MgO. Calculations allowed us to assign a band of reversible species at 1325 cm−1 to the terminal Mg-H hydride group on tri-coordinated magnesium. The hydrogen atoms bridging two or three neighboring, low-coordinated Mg atoms of the surface are suggested to be responsible for complex bands in the 1130–880 cm−1 region [189].
A different mechanism has been reported to occur over more easily reducible oxides such as CeO2, or at higher temperatures, i.e., the reductive homolytic adsorption of hydrogen-producing hydroxy groups and near-surface reduced metal centers [190,191]. In this case, an “inverted” version of the so-called Mars–van Krevelen mechanism, or redox mechanism, can occur in hydrogenation catalysis [192], such as in hydrodeoxygenation reactions. The surface, which is reduced by hydrogenation, may be re-oxygenated by the substrate, which is consequently de-oxygenated. DFT calculations have shown that H2 may adsorb through homolytic dissociation on CeO2(111) with a relatively low activation barrier (0.2 eV) and strong exothermicity [193]. More recent studies showed that hydride species may form by hydrogen adsorption on reduced ceria CeO2−x, thus concluding that both heterolytic and homolytic H2 adsorption can occur on ceria [194]. Theoretical studies indicate that a similar conclusion can be obtained for H2/TiO2 systems (anatase and rutile) [195] and for the H2/Fe3O4 system [196], i.e., that both heterolytic and homolytic dissociation can occur and that the heterolytic product and the hydride M-H species can play a role in the homolytic dissociation mechanism. However, experimental data are lacking for these systems. In more recent years, the hydrogenation activity of indium oxide In2O3 has also been established. Also, in this oxide, heterolytic dissociation is supposed to occur, although the resulting species seem to be IR and Raman inactive [197,198]. It is possible that also oxides that adsorb heterolytically hydrogen at low temperatures, undergo reductive adsorption at higher temperatures, heterolytic dissociation being the first step in homolytic dissociation mechanism. On the other hand, several transition metal oxides, can be reduced to lower oxides and to metals by hydrogen at moderately high temperatures [199], thus they are essentially not present in practical hydrogenation catalysis conditions. In practice, iron, cobalt, nickel, and copper oxides do not exist as stable phases in most catalytic hydrogenation conditions, while cerium, zinc, zirconium, and chromium oxides may exist as slightly reduced phases. Typical nonreducible oxides, thus being stable even in significant reducing conditions, are alumina, silica, magnesia, etc.
As for most common insulating supports, the adsorption of hydrogen on silicas is negligible at room or higher temperatures [200] in normal conditions. The adsorption and reactivity of hydrogen on alumina have been the subject of early studies [201,202,203], showing the formation of different species. Adsorbed species stable at high temperatures are observed. In particular, Kramer and Andre [203] showed that atomic hydrogen can be spilled over from metallic centers to alumina and remain stable until 750 K. More recent theoretical and experimental studies suggest that spilled-over hydrogen can be adsorbed over γ-Al2O3-forming hydride species likely bonded on trigonal Al3+ centers [204,205,206]. It must, however, be taken into account that these studies fully neglect the possible role of transition metal impurities unavoidably present on and in γ-Al2O3, such as, in particular, the non-negligible amount of Fe3+ and Cr3+ [207], that can either be reduced by hydrogen or involved in H2 dissociation. On the other hand, hydrogen spillover is far more evident on reducible oxides than on alumina [208].
Quantitatively, the adsorption of hydrogen in non-reducible oxides (γ-Al2O3) and weakly reducible oxides (TiO2 and ZrO2) appears to be essentially negligible at temperatures up to 873 K, while it is considerable on reducible oxides such as on CeO2 above 573 K and on Fe2O3 above 673 K [209]. In the latter case, bulk reduction to Fe3O4, FeO, and metallic Fe also occurs depending on temperature and hydrogen pressure.
On the other hand, hydrogen activation over metal/oxide catalysts is mostly considered to occur on metallic centers [210], and when supports are active in adsorbing it. For example, the amount of hydrogen adsorbed on a ZnO-ZrO2 support is negligible with respect to the hydrogen adsorbed on copper [211]. A similar phenomenon occurs for Pt on several oxides [209]. In fact, hydrogen dissociatively adsorbs very quickly on almost all metal surfaces, with the occupation of on-top, bridge, or hollow sites by atomic hydrogen species [212]. On most metal surfaces, the dissociation of hydrogen is only weakly activated or even barrierless. For example, it has been found that when a H2 molecule chemisorbs on a Pt surface, the antibonding σ* orbital of H2 is completely filled by electrons from platinum, leading to its homolytic dissociation, which is not kinetically hindered [213]. Most hydrogen molecules are expected to stay on the surface because of the relative instability of subsurface hydrogen compared to surface hydrogen on most metals, except Pd [214]. In all cases [215], hydrogen dissociation gives rise to strongly bonded surface atomic hydrogen, mostly occupying hollow sites. Studies mostly devoted to (111) surfaces of face-centered cubic metals, (0001) surfaces of hexagonal close-packed metals, and (110) faces of body-centered cubic metals show the resulting predominant occupancy 3-fold sites, although other adsorption sites, such as bridge sites and top sites, may be competitive with 3-fold sites [216]. Only in the case of Ir(111), top sites appear more favored than hollow sites for surface atomic hydrogen location.
On the other hand, it must be considered that on group 11 metals (Cu, Ag, Au), hydrogen dissociation is significantly activated and endothermic [174]. Adsorbed hydrogen is definitely less stable on copper than on the other metals of interest here (nickel, cobalt, iron) [174]. Hydrogen adsorption strength follows the trend of the position of their d-band (Table 2).
Indeed, the size and shape of metal particles [217] and the nature of the support have effects on the nature of adsorbed hydrogen. DFT calculations [218] show that the adsorption energy of hydrogen should increase for Pt particles on ionic (basic) supports. As for nickel powder, TPD studies [219] evidenced three distinct forms of hydrogen permanently chemisorbed at 100 K: γ-form, located in the subsurface layer and desorbed at about 186 K; β-form, adsorbed in the so-called second layer and evolved at about 327 K; and α-form, fixed directly on nickel surface and desorbed at the 350–670 K range. Alumina and silica supports insignificantly affect hydrogen strongly adsorbed on nickel but significantly affect weakly adsorbed hydrogen [220]. On the other hand, the nature of the supports also affects the metal particle size and stability of nanoparticles, as well as their electronic properties [221].
An additional point concerns the role of the contamination and/or doping of metallic surfaces on hydrogen adsorption. One relevant example is the Cu-ZnOx case, where it seems that the ZnOx species strongly interacting with copper may favor hydrogen dissociation and act as a reservoir of atomic hydrogen [222].
As shown above, metal carbides have a role in the catalysis of Fischer-Tropsch syntheses. Relatively few data are reported on the adsorption of hydrogen on transition metal carbide phases. A DFT study of hydrogen adsorption on the low-index surfaces of four major carbides, TiC, VC, ZrC, and NbC, indicates that it is generally exothermic and occurs predominantly on the surface carbon atoms, producing surface CH bonds [223]. In fact, the stretching vibrations of hydrocarbon species were observed when an iron sample previously carbided by syngas treatment, was treated with H2 flow [224]. A computational study of the interaction between hydrogen and eight primary surfaces of Fe5C2 nanoparticles allowed us to conclude that dissociative adsorption is favorable due to spontaneous dissociation on iron-rich surfaces, as well as with a lower barrier (<0.5 eV) on carbon-rich surfaces. The single hydrogen atom preferred to be located at C sites on C-rich surfaces and Fe sites on iron-rich facets [225].
6.2. The Adsorption and Activation of CO
Carbon monoxide is a very weak base that adsorbs at low temperatures, preferentially on electron-withdrawing centers on oxide surfaces (i.e., both Lewis acidic and Brønsted acidic centers) through the lone pair at its C atom [226,227], although very weak O-bonded species can also sometimes be observed at very low temperatures [228]. Infrared spectroscopic studies of CO adsorption on oxide materials have been reported by many authors because they are very informative for the characterization of active centers on metal oxide surfaces [229,230]. On covalent oxides, such as amorphous silica, very weak reversible adsorption on surface OH groups (silanols) is observed. On ionic oxides, molecular end-on (terminal) adsorption is observed in cationic centers. In the case of d0 cations, the position of the CO band is sensitive to the cation Lewis acid strength that enhances the C-O stretching frequency as a result of electron withdrawal through the σ-bond at the C-end. The CO stretching shifts from the 2140 cm−1 position of unperturbed CO to >2200 cm−1 for CO interacting with the strongest Lewis sites (Al3+ ions of alumina-based materials). Such a terminal CO adsorption is observed at low to very low temperatures and is usually weak and reversible at room or slightly higher temperatures. In the case of cations with partially filled d orbitals, d - π* electron backdonation can also occur, and this tends to shift down the CO stretching frequency but may increase adsorption bond strength significantly. This is the case of, e.g., CO’s interaction with cuprous ions, Cu+, with CO stretching that is found at 2120–2100 cm−1. Polycarbonyl species, e.g., dicarbonyl and tricarbonyl species, can also be observed to sometimes form over metal cations [225]. In any case, the molecular adsorption of CO on metal oxides is quite a weak phenomenon, definitely weaker than on zerovalent metal centers [231].
At moderately high temperatures, CO adsorption on hydroxylated ionic metal oxides may give rise to formate ions through reactions with hydroxy groups with “associative” adsorption mechanisms [232,233]. Only at very high temperatures, carbon deposition from CO is observed on metal oxides together with CO2, attributed to the reverse Boudouard reaction [234].
Only when highly oxidizing cations are present at the surface (like Ru4+, Pd4+, Cu2+, Pt2+, and Co3+ ions), CO oxidative adsorption also gives rise to adsorbed CO2 in its different forms (see below) at very low temperatures. On the other hand, all reducible oxides, such as Fe, Co, Ni, and Cu oxides, are reduced by carbon monoxide to lower oxides and/or to the corresponding metals [235] or converted to metal carbides at relatively high temperatures, producing CO2 as a co-product. In any case, as already stated, reducible oxides do not exist in strongly reducing conditions, such as in most hydrogenation reactions.
Experimental studies show that CO can adsorb on metallic centers in at least three forms: terminal (on-top), bridging, and triply (or multiply) bridging. Also, the terminal end-on adsorption of CO on on-top metallic centers [226,236] is primarily due to the σ-type interaction of the doublet at the C atom with empty orbitals of the metal ion, coupled to the backdonation from filled d orbitals of the metal center to the empty antibonding 2π* orbitals of CO. The latter results in a weakening of the CO bond and a lowering of its stretching frequency falling in the range of 2120–1850 cm−1. The interaction of a CO molecule with multiple metal atoms is associated with an even more efficient M→C π backdonation and results in a further significant weakening of the CO bond down to the range of 1950–1600 cm−1 [237,238]. Backdonation strength increases by decreasing nucleus charge in the transition metal rows (i.e., from right to left in the element table, following the position of the d-band; see Table 2) and also by increasing the principal quantum number of external electrons (i.e., from top to bottom in the elemental table). In fact, the interaction of CO is very weak on metallic copper, where it is dominated by the σ-donation, as the π backdonation is very weak in the case of copper and other group 1b metals, such as Ag and Au [239]. The adsorption of CO on other first-row transition metals, particularly Ni, Co, and Fe, where bridging and triply bridging species may also be observed, is definitely stronger than on Cu [236], where only terminal on-top species form [240]. As for copper, the adsorption of CO is far stronger on Cu+ than on Cu° [241], being a rare case where CO adsorption is stronger on oxide than on metal.
The strong reactivity of CO with some metal surfaces can result in the possible formation and evolution of gaseous carbonyl complexes, like in the case of the production of Ni(CO)4, which may form whenever Ni-based catalysts are reduced and exposed to CO at temperatures below 473 K [242]. A less easy case is the formation of iron and cobalt carbonyls. A possible intermediate state is that of highly dispersed (single) metallic atoms where surface polycarbonyl species may form by CO adsorption, such as for highly dispersed Ni on alumina [243].
Another result of this strong interaction is the occurrence of CO dissociation on the surface, producing carbon and oxygen species [175]. The CO dissociation rate is fundamentally influenced by two main factors [244]: (i) an electronic effect on the dissociation barrier, i.e., the increase in the dissociation barrier from left to right in the periodic table, which has been correlated with the metal d band center [170,171,172] (see Table 2); (ii) an effect of particle and surface morphology, being lower the dissociation barrier on steps, kinks, and defects than on terraces. As a common feature in catalysis, the activation energy for CO dissociation is correlated inversely with the adsorption strength, which is definitely larger for CO on platinum group metals than on group 1 elements (Cu, Ag, and Au [236]), and, for the metals of interest here, follows the trend Fe > Ni > Co >> Cu.
The formation of carbon species can result in the bulk conversion of metal into carbide. According to Chai et al. [245], Raney iron CO dissociation occurs already starting from 120 K, while partial carburization in pure CO occurs from 490 K, first producing χ-Fe5C2. To obtain full carburization, H2 is needed to remove oxygen coming from CO dissociation as H2O. Such reactions can be favored by opportune doping: in particular, potassium doping favors CO dissociation on iron and its carburization [246]. Also, cobalt can be carburized to CoC2 using pure CO at 490–500 K, while only partial carburization occurs using syngas [142,247]. Only the partial carburization of nickel to Ni3C was found with pure CO at 538 K [248].
6.3. The Adsorption and Activation of CO2
CO2 can adsorb on solid oxides in a molecular form or in a reactive form [249,250]. Molecular adsorption occurs through one of the electron doublets at an oxygen end in a linear way. This adsorption may occur on hydroxy groups through hydrogen bonding or at Lewis acid sites. This interaction is definitely weak and reversible at room or slightly higher temperatures. On ionic metal oxides, CO2 may also adsorb in a reactive way, producing weakly adsorbed but strongly perturbed CO2 molecules, and carbonate and bicarbonate (monohydrogencarbonate) species with different geometries and adsorption strength. Bicarbonate species are well evident on strongly hydroxylated and weakly basic oxides, such as alumina, while different types of carbonate species, i.e., monodentate, bidentate, or bridging and polydentate form on more basic surfaces. On very basic materials such as alkali earth oxides, the interaction of CO2 can result in bulk carbonation even at very low temperatures. The desorption of carbonate from surfaces (including subsurface and bulk) can be taken as a measure of the basicity of the oxide [168]. Considering most common oxide carriers, CO2 adsorption is practically non-occurring on silica, as on most covalent oxides (e.g., V2O5, MoO3, WO3), quite weak on alumina (mainly in the form of bicarbonates) and titania, and definitely stronger on zinc oxide, zirconia, ceria, magnesia, in all cases forming strongly bonded carbonates and less strongly bonded bicarbonates, while bulk carbonation easily occurs on strongly basic oxides such as CaO.
CO2 adsorption on metal surfaces has been the subject of early spectroscopic studies on monocrystal surfaces [251,252,253]. These studies report strong spectroscopic evidence for the formation, besides physisorbed CO2 only at a very low temperature, of negatively charged bent CO2δ−, which, depending on the nature of the metal, may dissociate into CO and O or transform into CO32− + CO. The presence of surface adatoms may dramatically influence the adsorption and reactivity of CO2. In particular, alkali adatoms increase the binding energy of adsorbed CO2 and promote the dissociation and/or the transformation of CO2 into CO + O. Theoretical studies of CO2 adsorption on transition metals [176,254,255] indicate that CO2 exothermic adsorption strength forming bent CO2δ−, the charge of this species, and the exothermic dissociation energy to produce CO-O follow the trend Cu < Ni < Co < Fe, i.e., again, the trend of d-band position (Table 2). In fact, CO2 adsorption was reported to be only molecular and very weak, also in the presence of hydrogen, on flat copper surfaces [256], but dissociation is reported to occur on stepped surfaces [257]. A recent ambient-pressure X-ray photoelectron spectroscopy study [258] indicated that carbonate species, adsorbed CO, and graphitic carbon form on both Ni(111) and Ni(100) surfaces in the presence of 0.2 Torr CO2. However, more than 90% of adsorption species on the Ni(111) surface are carbonate, whereas the Ni(100) surface is mainly covered by adsorbed CO and graphitic carbon.
7. Mechanistic Aspects and the Role of the Different Catalyst’s Components
The mechanism of the WGS reactions is still controversial [12]. Regarding the HTWGS reaction over Fe3O4-based catalysts, it seems that there is a general agreement concerning the so-called redox mechanism, where carbon monoxide can first adsorb end-on on iron ions and then reduce Fe3+ ions, while water would reoxidize them in the second step.
where the second step is the reverse of the reductive homolytic dissociative adsorption of hydrogen on iron oxides (see above). As seen, new catalysts based on ZnAl2O4 have also been developed for HTWGS to be used at low steam-to-carbon ratios. In this case, the so-called associative mechanism, through formate ions, seems to be most likely:
where, again, the second step is the reverse of the heterolytic dissociation of hydrogen on ZnO (see above) [259]. In both cases, oxide catalysis certainly occurs.
2Fe3+ + O2− + CO -> 2Fe2+ + CO2
2Fe2+ + H2O -> 2Fe3+ + H2 + O2−
Zn2+ + −OH + CO -> Zn2+ + HCOO− -> Zn2+ −H + CO2
Zn2+ −H + H2O -> Zn2+ + −OH + H2
More controversy exists concerning the mechanism of LTWGS on Cu-ZnO-Al2O3 catalysts, which are very similar to methanol synthesis catalysts, with mechanisms that are supposed to be strongly related [260]. As said, the real catalysts, with copper contents that are usually > 50 wt%, are essentially constituted by unsupported copper particles modified by zinc species, likely in the form of ZnO or partially reduced ZnOx (x < 1) species strongly interacting with it [41,218]. Most of the ZnO-Al2O3 phase acts as a stabilizing agent for copper metal particles against sintering. Zinc-modified copper particles appear to represent the real active phase for both WGS and methanol synthesis from syngas, with a possible common role of formate species as surface intermediates [33]. It is worth noting that pure copper surfaces very weakly adsorb both CO and hydrogen, as well as CO2 (Table 2), while the addition of the Zn species strongly increases its activity [261]. The mechanism of the WGS reaction over Cu/ZnO can be similar to that described above for ZnAl2O4 (Reactions (11) and (12)), with a likely activating effect of Cu+ species which is known to adsorb CO strongly, thus favoring the formation of formate species on ZnOx, and suggest a role of metallic copper favoring hydrogen recombination and desorption. On the other hand, the hydrogenations of both CO and CO2 are certainly favored by the activation of hydrogen on the Cu-ZnO system where H2 dissociation would occur on metallic copper, with the formation of Zn hydride species by some kind of spillover [218]. Such species may react with CO2-producing formate ions that can either decompose to CO + −OH at a lower hydrogen pressure (rWGS) or be further hydrogenated to methoxy groups and finally methanol at a higher hydrogen pressure (methanol synthesis from CO2). CO hydrogenation by hydride species can occur through the formation of formyl species (formally −CHO) [262] that can be hydrogenated to methoxy groups directly or through formate species too. The faster reaction of CO2 with respect to CO in methanol synthesis can be associated with the strong nucleophilicity of the carbon atom of CO2 (much stronger than that of CO), possibly further increased by its previous interaction with cations through oxygen atoms, coupled with the strong electrophilicity of the hydride species of Zn-H. On the other hand, the promotional effect of CO in the hydrogenation of CO2 over these catalysts can be associated with the occurrence of the WGS reaction with water produced by methanol synthesis from CO2 [260].
It is clear that dissociative adsorption neither of CO nor of CO2 (which, in fact, do not occur on copper and the other components of these catalysts) is needed for methanol synthesis. Additionally, the activation of hydrogen as nucleophilic hydride ions can be the key for Cu/ZnO-based systems that, in fact, are used for the hydrogenation of electrophilic carbon-containing molecules, such as CO2 and ketones [7]. Finally, it is worth noticing that, although commercial LTWGS catalysts and methanol synthesis catalysts are mainly based on metallic copper, it seems likely that catalysis for these reactions is non-metallic, mainly based on metal-supported surface Cu-Zn oxide species.
The production of hydrocarbons from COx hydrogenation occurs on metals that more strongly adsorb, but not too much, carbon monoxide and that also easily adsorb hydrogen [148]. These metals are reported to be active in CO dissociation, producing carbon species or metal carbides, as well as in CO2 dissociation. They can catalyze hydrocarbon synthesis both as bulk metals and when supported. However, while the chain growth mechanism is very efficient on cobalt and iron catalysts, it is less for nickel catalysts [263], which, in fact, are used for methanation but are less efficient for Fischer-Tropsch due to limited chain growth.
In the case of nickel catalysts for methanation and cobalt catalysts for LTFTS, ionic oxides act as the best carriers, alumina seeming to be the choice support. Other ionic oxide components are usually present too, acting as activators. The amount of metal loaded is similar to that allowing the full coverage of the support surface, finally producing very small metal particles strongly interacting with the support. This is a key factor in reducing the probability of producing volatile metal carbonyl molecules (in particular, Ni(CO)4 in the case of nickel catalysts) and also in limiting the formation of carbon and the carburization of the metal. In fact, data suggest that carbon species preferentially form on large metal particles.
Studies provide evidence of the possible role of the support in hosting and activating surface-oxidized intermediates such as formate and methoxy species over both cobalt/alumina and nickel/alumina catalysts [264], with a possible role of hydrogen spillover from the metal to the supporting oxide and a oxygenate mechanism occurring on the support surface for both methanation and FTS. However, mechanisms via oxygenate intermediates (carboxylate, carbonate, formate, formyl, and methoxy) could also occur at nickel surfaces [265,266]. On the other hand, the availability of the support surface in the case of this system is very small, being the surface covered by dispersed metal particles. On the other hand, most of the data strongly converge for a predominant role of CO dissociation over the metal surface and carbide mechanisms for both reactions when carried out with syngas as a reactant [100]. Thus, the main role of the ionic support for Ni and Co catalysts for syngas conversion is to produce and stabilize very small metal particles resistant to sintering and carbon deactivation.
Nevertheless, the relatively strong adsorption of CO2 over ionic oxides used as catalyst carriers, e.g., on aluminas doped with components with a basic character, such as K-doped alumina [267], MgO-Al2O3 [268] and La2O3/Al2O3 [269] may have a role in CO2 methanation. In fact, a positive effect of support basicity on CO2 methanation on supported nickel has been reported [130].
8. Conclusions
The data summarized in this review paper provide examples of how complex the science of catalysis and catalyst engineering are, even for reactions realized with the same or related reactants and in quite similar conditions. Oxide-supported metal catalysis, metal-supported oxide catalysis, as well as pure metal and pure oxide catalysis, and carbide catalysis as well, are involved in the conversion of syngases and the hydrogenations of carbon oxides. The data concerning catalysis in practical conditions can be successfully interpreted by taking into account surface chemistry and physical chemistry studies and DFT calculations, showing the usefulness of surface physics and chemistry studies. It is also clear that the cost of the catalyst may also have some relevance and that the use of the very expensive platinum group and noble metals is avoided when increases in performance are not very relevant.
The metals which play a key role in the reactions considered here are those that adsorb hydrogen and carbon monoxide with intermediate strength, in agreement with the Sabatier principle. On the other hand, the industrial catalysts for the reactions considered here are first-row transition metals (Cu, Ni, Co, and Fe), which are far cheaper than platinum group metals because, for these reactions, these metals are sufficiently competitive. Among these metals, those that more strongly adsorb CO are more performant in the Fischer-Tropsch reaction, where CO dissociation certainly plays a central role. On the other hand, oxide supports also have a main role from different points of view. The main role of support is to optimize the size and shape of the supported metallic particles, particularly in the case of alumina as the support of nickel catalysts for methanation and cobalt catalysts for LTFT, probably with the result of limiting the tendency to carburization of the corresponding metals. As for the reactions that do not imply the full breaking of the C-O bond of carbon oxides, the need for strong CO adsorption vanishes. On the other hand, we have emphasized that, although the metallic nature of copper is likely useful to provide hydrogen easy adsorption, the activity of methanol synthesis and LTWGS catalysts, both based on Cu-ZnO is (strictly speaking) not metallic. In fact, these catalysts are essentially constituted by unsupported copper modified by surface ZnOx species and likely work as copper-supported Cu-Zn oxides. In particular, the weaker interaction of CO with cationic centers and the more nucleophilic character of surface hydride species may be the key features for catalyzing these reactions. On the other hand, the HTWGS reaction catalysis implies fully oxidic catalysts such as those based on Fe3O4 and those based on ZnAl2O4.
It is clear that the catalytic conversion of captured CO2 with green hydrogen to hydrocarbons and alcohols (methanol) is a possible method of producing sustainable e-fuels, such as e-methanol, e-methane, e-gasoline, e-jet fuel, and e-Diesel fuel. The development of optimized catalysts for these reactions is already at a nearly industrial level, and this allows the development of reactors and the corresponding manufacturing processes. According to the data discussed above, a slight modification of catalysts used for syngas conversion is apparently a successful approach for developing new CO2-based processes. A different situation is that of reverse WGS processes. In fact, this reaction, although strictly related to direct WGS processes, needs much higher temperatures to be realized in significant amounts. Thus, very different catalysts practically act for rWGS with particular respect to LTWGS.
Although, as said, the state of the art seems to indicate already clear ways to optimize the new processes for the production of e-fuels and renewable chemicals from CO2 hydrogenation, extensive research is still needed to clarify the details of the mechanisms of these reactions and the roles of the different catalyst’ components, which are really still incompletely known. This work is likely needed to further improve the catalytic systems and might also allow us to discover more efficient materials.
In any case, it is clear that heterogeneous catalysis will be a key tool for the production of sustainable fuels, particularly e-fuels, just as it has been in the ending era of fossil hydrocarbon industrial chemistry.
Author Contributions
G.B.: conceptualization, validation, writing—review and editing; E.S.: thermodynamic calculations, conceptualization, validation; P.R.: conceptualization, validation, writing—review and editing; G.G.: funding acquisition, conceptualization, validation, writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
G.B., P.R. and G.G. acknowledge the PROMETH2eus project, PNRR project of MASE italian ministry (missione 2 “Rivoluzione verde e transizione ecologica”, componente 2 “Energia rinnovabile, idrogeno, rete e mobilità sostenibile”, investimento 3.5 “Ricerca e sviluppo sull’idrogeno”). E.S. acknowledges National Recovery and Resilience Plan (NRRP), Mission 4 Component 2 Investment 1.3—Call for tender No. 1561 of 11.10.2022 of Ministero dell’Uni-versità e della Ricerca (MUR); funded by the European Union—NextGenerationEU Project title “Network 4 Energy Sustainable Transition—NEST” (project code: PE0000021).
Data Availability Statement
The data in this review paper are from the cited references.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Nemmour, A.; Inayat, A.; Janajreh, I.; Ghenai, C. Green hydrogen-based E-fuels (E-methane, E-methanol, E-ammonia) to support clean energy transition: A literature review. Int. J. Hydrogen Energy 2023, 48, 29011–29033. [Google Scholar] [CrossRef]
- Cui, L.; Liu, C.; Yao, B.; Edwards, P.P.; Xiao, T.; Cao, F. A review of catalytic hydrogenation of carbon dioxide: From waste to hydrocarbons. Front. Chem. 2022, 10, 1037997. [Google Scholar] [CrossRef] [PubMed]
- Aasberg-Petersen, K.; Dybkjær, I.; Ovesen, C.V.; Schjødt, N.C.; Sehested, J.; Thomsen, S.G. Natural gas to synthesis gas—Catalysts and catalytic processes. J. Nat. Gas Sci. Eng. 2011, 3, 423–459. [Google Scholar] [CrossRef]
- Boretti, A.; Banik, B.K. Advances in Hydrogen Production from Natural Gas Reforming. Adv. Energy Sustain. Res. 2021, 2, 2100097. [Google Scholar] [CrossRef]
- Dai, F.; Zhang, S.; Luo, Y.; Wang, K.; Liu, Y.; Ji, X. Recent Progress on Hydrogen-Rich Syngas Production from Coal Gasification. Processes 2023, 11, 1765. [Google Scholar] [CrossRef]
- José Juan Bolívar Caballero, J.J.; Zaini, I.N.; Yan, W. Reforming processes for syngas production: A mini-review on the current status, challenges, and prospects for biomass conversion to fuels. Appl. Energy Comb. Sci. 2022, 10, 100064. [Google Scholar] [CrossRef]
- Busca, G. Metal catalysts in hydrogenation and dehydrogenation reactions. In Heterogeneous Catalytic Materials; Busca, G., Ed.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 297–343. [Google Scholar]
- Gao, J.; Liu, Q.; Gu, F.; Liu, B.; Zhong, Z.; Su, F. Recent advances in methanation catalysts for the production of synthetic natural gas. RSC Adv. 2015, 5, 22759–22776. [Google Scholar] [CrossRef]
- Rönsch, S.; Schneider, J.; Matthischke, S.; Schlüter, M.; Götz, M.; Lefebvre, J.; Prabhakaran, P.; Bajohr, S. Review on methanation—From fundamentals to current projects. Fuel 2016, 166, 276–296. [Google Scholar] [CrossRef]
- Available online: https://www.grandviewresearch.com/industry-analysis/catalyst-market (accessed on 30 October 2023).
- Newsome, D.S. The Water-Gas Shift Reaction. Catal. Rev. 1980, 21, 275–318. [Google Scholar] [CrossRef]
- Baraj, E.; Ciahotný, K.; Hlinčík, T. The water gas shift reaction: Catalysts and reaction mechanism. Fuel 2021, 288, 119817. [Google Scholar] [CrossRef]
- Pattabathula, V.; Richardson, J. Introduction to Ammonia Production. Chem. Eng. Prog. 2016, 112, 69–75. [Google Scholar]
- ThyssenKrupp: Hydrogen Key for Any Refinery. Available online: https://d2zo35mdb530wx.cloudfront.net/_binary/UCPthyssenkruppUhde/6e7007a8-2bd4-47c1-b215-8a4d268c80ed/brochure-hydrogen_scr.pdf (accessed on 30 October 2023).
- Hydrogen, Linde. Available online: https://www.linde-engineering.com/en/images/H2_1_1_e_12_150dpi_NB_tcm19-4258.pdf (accessed on 30 October 2023).
- Ratnasamy, C.; Wagner, J.P. Water Gas Shift Catalysis. Catal. Rev. 2009, 51, 325–440. [Google Scholar] [CrossRef]
- Clariant, Catalysts and Adsorbents for Syngas. Available online: https://www.clariant.com/en/Business-Units/Catalysts/Syngas-Catalysts (accessed on 30 October 2023).
- Lukashuk, L.; van de Water, L.G.A.; van Dijk, H.A.J.; Cobden, P.D.; Dodds, D.L.; Hyde, T.I.; Watson, M.J. A new application of the commercial high temperature water gas shift catalyst for reduction of CO2 emissions in the iron and steel industry: Lab-scale catalyst evaluation. Int. J. Hydrogen Energy 2021, 46, 39023–39035. [Google Scholar] [CrossRef]
- Lee, D.W.; Lee, M.S.; Lee, J.Y.; Kim, S.; Eom, H.-J.; Moon, D.Y.; Lee, K.Y. The review of Cr-free Fe-based catalysts for high-temperature water-gas shift reactions. Catal. Today 2013, 210, 2–9. [Google Scholar] [CrossRef]
- Sourav, S.; Wachs, I.E. Cr-Free, Cu Promoted Fe Oxide-Based Catalysts for High-Temperature Water-Gas Shift (HT-WGS) Reaction. Catalysts 2020, 10, 305. [Google Scholar] [CrossRef]
- Available online: https://www.topsoe.com/our-resources/knowledge/our-products/catalysts/sk-501-flextm (accessed on 30 October 2023).
- Pal, D.B.; Chand, R.; Upadhyay, S.N.; Mishra, P.K. Performance of water gas shift reaction catalysts: A review. Renew. Sustain. Energy Rev. 2018, 93, 549–556. [Google Scholar] [CrossRef]
- Amadeo, N.E.; Laborde, M.A. Hydrogen production from the low-temperature water-gas shift reaction: Kinetics and simulation of the industrial reactor. Int. J. Hydrogen Energy 1995, 20, 949–956. [Google Scholar] [CrossRef]
- Available online: https://www.clariant.com/en/Solutions/Products/2019/02/15/09/47/ShiftMax-217 (accessed on 30 October 2023).
- Süd Chemie, General Catalyst Catalogue. Available online: www.sudchemie.com (accessed on 12 December 2011).
- Available online: https://www.topsoe.com/our-resources/knowledge/our-products/catalysts/lk-823 (accessed on 30 October 2023).
- Available online: https://www.topsoe.com/our-resources/knowledge/our-products/catalysts/lk-853-fencetm (accessed on 30 October 2023).
- Haldor Topsøe, Brochure “Leading Edge Performance LTS Catalysts”. Available online: http://www.topsoe.com (accessed on 12 December 2011).
- KATALKOJM Low Temperature Shift Catalysts. Available online: https://matthey.com/products-and-markets/chemicals/low-temperature-shift-catalysts (accessed on 30 October 2023).
- Ozcan, O.; Akın, A.N. Methanol steam reforming kinetics using a commercial CuO/ZnO/Al2O3 catalyst: Simulation of a reformer integrated with HT-PEMFC system. Int. J. Hydrogen Energy 2023, 48, 22777–22790. [Google Scholar] [CrossRef]
- Peppley, B.A.; Amphlett, J.C.; Kearns, L.M.; Mann, R.F. Methanol–steam reforming on Cu/ZnO/Al2O3. Part 1: The reaction network. Appl. Catal. A Gen. 1999, 179, 21–29. [Google Scholar] [CrossRef]
- Madon, R.J.; Nagel, P. Low Temperature Water Gas Shift Catalyst. US Patent 0102278, 11 June 2009. [Google Scholar]
- Wilkinson, S.K.; van de Water, L.G.A.; Miller, B.; Simmons, M.J.H.; Stitt, E.H.; Watson, M.J. Understanding the generation of methanol synthesis and water gas shift activity over copper-based catalysts—A spatially resolved experimental kinetic study using steady and non-steady state operation under CO/CO2/H2 feeds. J. Catal. 2016, 337, 208–220. [Google Scholar] [CrossRef]
- Lacerda de Oliveira Campos, B.; Herrera Delgado, K.; Wild, S.; Studt, F.; Pitter, S.; Sauer, J. Surface reaction kinetics of the methanol synthesis and the water gas shift reaction on Cu/ZnO/Al2O3. React. Chem. Eng. 2021, 6, 868–887. [Google Scholar] [CrossRef]
- Sehested, J. Industrial and scientific directions of methanol catalyst development. J. Catal. 2019, 371, 368–375. [Google Scholar] [CrossRef]
- Behrens, M. Meso- and nano-structuring of industrial Cu/ZnO/(Al2O3) catalysts. J. Catal. 2009, 267, 24–29. [Google Scholar] [CrossRef]
- Etim, U.J.; Song, Y.; Zhong, Z. Improving the Cu/ZnO-Based Catalysts for Carbon Dioxide Hydrogenation to Methanol, and the Use of Methanol as a Renewable Energy Storage Media. Front. Energy Res. 2020, 8, 545431. [Google Scholar] [CrossRef]
- Cui, X.; Liu, Y.; Yan, W.; Xue, Y.; Mei, Y.; Li, J.; Gao, X.; Zhang, H.; Zhu, S.; Niu, Y.; et al. Enhancing methanol selectivity of commercial Cu/ZnO/Al2O3 catalyst in CO2 hydrogenation by surface silylation. Appl. Catal. B Environ. 2023, 339, 123099. [Google Scholar] [CrossRef]
- Huang, X.; Beck, A.; Fedorov, A.; Frey, H.; Zhang, B.; Klötzer, B.; vanBokhoven, J.A.; Copéret, C.; Willinger, M.G. Visualizing Structural and Chemical Transformations of an IndustrialCu/ZnO/Al2O3 Pre-catalyst during Activation and CO2 Reduction. ChemCatChem 2022, 14, e202201280. [Google Scholar] [CrossRef]
- Divins, N.J.; Kordus, D.; Timoshenko, J.; Sinev, I.; Zegkinoglou, I.; Bergmann, A.; Chee, S.W.; Widrinna, S.; Karslıoğlu, O.; Mistry, H.; et al. Operando high-pressure investigation of size-controlled CuZn catalysts for the methanol synthesis reaction. Nat. Commun. 2021, 12, 1435. [Google Scholar] [CrossRef] [PubMed]
- Lunkenbein, T.; Schumann, J.; Behrens, M.; Schlögl, R.; Willinger, M.G. Self-assembled catalyst promotion by overgrowth of layered ZnO in industrial Cu/ZnO/Al2O3 catalysts. Angew. Chem. Int. Ed. 2015, 54, 4544–4548. [Google Scholar] [CrossRef]
- Ahn, S.Y.; Kim, K.J.; Kim, B.J.; Shim, J.O.; Jang, W.J.; Roh, H.S. Unravelling the active sites and structure-activity relationship on Cu–ZnO–Al2O3 based catalysts for water-gas shift reaction. Appl. Catal. B Environ. 2023, 325, 122320. [Google Scholar] [CrossRef]
- Montanari, T.; Sisani, M.; Nocchetti, M.; Vivani, R.; Herrera Delgado, M.C.; Ramis, G.; Busca, G.; Costantino, U. Zinc–aluminum hydrotalcites as precursors of basic catalysts: Preparation, characterization and study of the activation of methanol. Catal. Today 2010, 152, 104–109. [Google Scholar] [CrossRef]
- Available online: https://matthey.com/products-and-markets/chemicals/water-gas-shift-catalysts (accessed on 30 October 2023).
- Xu, K.; Ma, C.; Yan, H.; Gu, H.; Wang, W.W.; Li, S.Q.; Meng, Q.L.; Shao, W.P.; Ding, G.H.; Wang, F.R.; et al. Catalytically efficient Ni-NiOx-Y2O3 interface for medium temperature water-gas shift reaction. Nat. Commun. 2022, 13, 2443. [Google Scholar] [CrossRef] [PubMed]
- Antoniak-Jurak, K.; Kowalik, P.; Bicki, R.; Michalska, K.; Prochniak, W.; Wiercioch, P. Cu substituted ZnAl2O4 ex-LDH catalysts for medium-temperature WGS: Effect of Cu/Zn ratio and thermal treatment on catalyst efficiency. Int. J. Hydrogen Energy 2019, 44, 27390–27400. [Google Scholar] [CrossRef]
- Clariant’s ShiftMax® 300 MTS Catalyst Delivers Excellent Performance at BHCC Hydrogen unit in Zhejiang, China, Press Release, 2 June 2020. Available online: https://www.clariant.com/en/Corporate/News/2020/06/Clariantrsquos-ShiftMaxreg-300-MTS-catalyst-delivers-excellent-performance-at-BHCC-hydrogen-unit-in (accessed on 30 October 2023).
- Available online: https://www.topsoe.com/our-resources/knowledge/our-products/catalysts/lk-813 (accessed on 20 October 2023).
- Li, Y.; Kottwitz, M.; Vincent, J.L.; Enright, M.J.; Liu, Z.; Zhang, L.; Huang, J.; Senanayake, S.D.; Yang, W.C.D.; Crozier, P.A.; et al. Dynamic structure of active sites in ceria-supported Pt catalysts for the water gas shift reaction. Nat. Commun. 2021, 12, 914. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.topsoe.com/our-resources/knowledge/our-products/catalysts/ssk-10?hsLang=en (accessed on 20 October 2023).
- Bown, R.M.; Joyce, M.; Zhang, Q.; Rmirez Reina, T.; Duya, M.S. Identifying Commercial Opportunities for the Reverse Water Gas Shift Reaction. Energy Technol. 2021, 9, 2100554. [Google Scholar] [CrossRef]
- Available online: https://ntrs.nasa.gov/citations/20020050609 (accessed on 20 October 2023).
- Stone, F.S.; Waller, D. Cu-ZnO and Cu-ZnO/Al2O3 catalysts for the reverse water-gas shift reaction. The effect of the Cu/Zn ratio on precursor characteristics and on the activity of the derived catalysts. Top. Catal. 2003, 22, 305–318. [Google Scholar] [CrossRef]
- Zhu, M.; Ge, Q.; Zhu, X. Catalytic Reduction of CO2 to CO via Reverse Water Gas Shift Reaction: Recent Advances in the Design of Active and Selective Supported Metal Catalysts. Trans. Tianjin Univ. 2020, 26, 172–187. [Google Scholar] [CrossRef]
- Liu, H.X.; Li, S.Q.; Wang, W.W.; Yu, W.Z.; Zhang, W.J.; Ma, C.; Jia, C.J. Partially sintered copper–ceria as excellent catalyst for the high-temperature reverse water gas shift reaction. Nat. Commun. 2022, 13, 2022. [Google Scholar] [CrossRef] [PubMed]
- Topsoe, eFuels Technology for Converting CO2 and Renewable Electricity to Renewable Synthetic Fuels. Available online: https://info.topsoe.com/en/dp-wp-efuels-technology (accessed on 30 October 2023).
- Adelung, S.; Dietrich, R.U. Impact of the reverse water-gas shift operating conditions on the Power-to-Liquid fuel production cost. Fuel 2022, 317, 123440. [Google Scholar] [CrossRef]
- Available online: https://www.topsoe.com/our-resources/knowledge/our-products/equipment/e-react-fuels (accessed on 30 October 2023).
- Wismann, S.T.; Larsen, K.-E.; Mortensen, P.M. Electrical Reverse Shift: Sustainable CO2 Valorization for Industrial Scale. Angew. Chem. Int. Ed. 2022, 61, e202109696. [Google Scholar] [CrossRef]
- Available online: https://www.hydrocarbonengineering.com/clean-fuels/26052022/ineratec-and-clariant-join-forces-for-a-cleaner-future/ (accessed on 30 October 2023).
- Available online: https://matthey.com/en/news/2022/hycogen (accessed on 30 October 2023).
- Available online: https://matthey.com/products-and-markets/chemicals/fischer-tropsch-technology (accessed on 30 October 2023).
- Available online: https://www.hydrocarbonprocessing.com/news/2023/08/e-fuels-axens-paul-wurth-ifpen-agree-to-co-develop-reverse-water-gas-shift-tech/ (accessed on 30 October 2023).
- Joo, O.-S.; Jung, K.-D.; Moon, I.; Rozovskii, A.Y.; Lin, G.I.; Han, S.-H.; Uhm, S.-J. Carbon dioxide hydrogenation to form methanol via a reverse-water-gas-shift reaction (the CAMERE process). Ind. Eng. Chem. Res. 1999, 38, 1808–1812. [Google Scholar] [CrossRef]
- Skrzypek, J.; Lachowska, M.; Serafin, D. Methanol synthesis from CO2 and H2: Dependence of equilibrium conversions and exit equilibrium concentrations of components on the main process variables. Chem. Eng. Sci. 1990, 45, 89–96. [Google Scholar] [CrossRef]
- Iyer, S.S.; Renganathan, T.; Pushpavanam, S.; Kumar, M.V.; Kaisare, N. Generalized thermodynamic analysis of methanol synthesis: Effect of feed composition. J. CO2 Util. 2015, 10, 95–104. [Google Scholar] [CrossRef]
- Topsoe, The Role of the Methanol-Synthesis Catalyst in the Transition towards Green Methanol. Available online: https://video.topsoe.com/webinar-the-role-of-the (accessed on 30 October 2023).
- Wender, I. Reactions of synthesis gas. Fuel Proc. Technol. 1996, 48, 189–297. [Google Scholar] [CrossRef]
- Lange, J.P. Methanol synthesis: A short review of technology improvement. Catal. Today 2001, 64, 3–8. [Google Scholar] [CrossRef]
- Sheldon, D. Methanol Production—A Technical History. Johns. Matthey Technol. Rev. 2017, 61, 172–182. [Google Scholar] [CrossRef]
- Available online: https://engineering.airliquide.com/technologies/methanol (accessed on 30 October 2023).
- Available online: https://www.topsoe.com/processes/methanol (accessed on 30 October 2023).
- Xu, X.; Liu, Y.; Zhang, F.; Di, W.; Zhang, Y. Clean coal technologies in China based on methanol platform. Catal. Today 2017, 298, 61–66. [Google Scholar] [CrossRef]
- Dieterich, V.; Buttler, A.; Hanel, A.; Spliethoff, H.; Fendt, S. Power-to-liquid via synthesis of methanol, DME or Fischer-Tropsch-fuels: A review. Energy Environ. Sci. 2020, 13, 3207–3252. [Google Scholar] [CrossRef]
- Xiao, K.; Wang, Q.; Qi, X.; Zhong, L. For Better Industrial Cu/ZnO/Al2O3 Methanol Synthesis Catalyst: A Compositional Study. Catal. Lett. 2017, 147, 1581–1591. [Google Scholar] [CrossRef]
- Behrens, M.; Girgsdies, F.; Trunschke, A.; Schlögl, R. Minerals as Model Compounds for Cu/ZnO Catalyst Precursors: Structural and Thermal Properties and IR Spectra of Mineral and Synthetic (Zincian) Malachite, Rosasite and Aurichalcite and a Catalyst Precursor Mixture. Eur. J. Inorg. Chem. 2009, 2009, 1347–1357. [Google Scholar] [CrossRef]
- Liu, X.M.; Lu, G.Q.; Yan, Z.F.; Beltramini, J. Recent Advances in Catalysts for Methanol Synthesis via Hydrogenation of CO and CO2. Ind. Eng. Chem. Res. 2003, 42, 6518–6530. [Google Scholar] [CrossRef]
- Waugh, K.C. Methanol synthesis. Catal. Lett. 2012, 142, 1153–1166. [Google Scholar] [CrossRef]
- Topsoe, High Activity Methanol Synthesis Catalyst. 2014. Available online: https://www.topsoe.com/our-resources/knowledge/our-products/process-licensing/syncor-methanoltm?hsLang=en (accessed on 30 October 2023).
- Kuld, S.; Thorhauge, M.; Falsig, H.; Elkjær, C.F.; Helveg, S.; Chorkendorff, I.; Sehested, J. Quantifying the promotion of Cu catalysts by ZnO for methanol synthesis. Science 2016, 352, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Osborne, S.; Schwartz, H.; Ringe, N.; Reitmeier, S. A flexible tool for methanol symthesis. Nitrogen Syngas 2021, 373, 44–50. [Google Scholar]
- Available online: https://www.clariant.com/en/Business-Units/Catalysts/Syngas-Catalysts/Methanol (accessed on 30 October 2023).
- Johnson Matthey, Latest Catalyst Provides More Methanol for Longer, Nitrogen & Syngas September/October 2021. Available online: https://matthey.com/documents/161599/440829/Reprint+-+N%2BS+-+Latest+catalyst+provides+more+methanol+for+longer.pdf/b8751743-9309-5b50-1259-8428455aba9d?t=1668002379563 (accessed on 30 October 2023).
- Available online: https://matthey.com/documents/161599/162780/An-update-on-KATALCO-51-102-synthesis-catalyst.pdf/cccb1b34-d35e-e62f-d481-9fe2d8f1568f (accessed on 30 October 2023).
- Nielsen, N.D.; Jensen, A.D.; Christensen, J.M. The roles of CO and CO2 in high pressure methanol synthesis over Cu-based catalysts. J. Catal. 2021, 393, 324–334. [Google Scholar] [CrossRef]
- Available online: https://www.basf.com/global/en/media/news-releases/2019/05/p-19-218.html (accessed on 30 October 2023).
- Ruland, H.; Song, H.; Laudenschleger, D.; Stürmer, S.; Schmidt, S.; He, J.; Kähler, K.; Muhler, M.; Schlög, R. CO2 Hydrogenation with Cu/ZnO/Al2O3: A Benchmark Study. ChemCatChem 2020, 12, 3216–3222. [Google Scholar] [CrossRef]
- Available online: https://info.topsoe.com/hyoctane-leaflet-dlp (accessed on 30 October 2023).
- Gao, J.; Wang, Y.; Ping, Y.; Hu, D.; Xu, G.; Gu, F.; Su, F. A thermodynamic analysis of methanation reactions of carbon oxides for the production of synthetic natural gas. RSC Adv. 2012, 2, 2358–2368. [Google Scholar] [CrossRef]
- Stangeland, K.; Kalai, D.; Li, H.; Yu, Z. CO2 methanation: The effect of catalysts and reaction conditions. Energy Procedia 2017, 105, 2022–2027. [Google Scholar] [CrossRef]
- Katla, D.; Węcel, D.; Jurczyk, M.; Skorek-Osikowska, A. Preliminary experimental study of a methanation reactor for conversion of H2 and CO2 into synthetic natural gas (SNG). Energy 2023, 263, 125881. [Google Scholar] [CrossRef]
- Torrente-Murciano, L.; Mattia, D.; Jones, M.D.; Plucinski, P.K. Formation of hydrocarbons via CO2 hydrogenation—A thermodynamic study. J. CO2 Util. 2014, 6, 34–39. [Google Scholar] [CrossRef]
- Lim, J.Y.; McGregor, J.; Sederman, A.J.; Dennis, J.S. The role of the Boudouard and water-gas shift reactions in the methanation of CO or CO2 over Ni/γ-Al2O3 catalyst. Chem. Eng. Sci. 2016, 152, 754–766. [Google Scholar] [CrossRef]
- Pearce, B.B.; Twigg, M.V.; Woodward, C. Methanation. In Catalyst Handbook, 2nd ed.; Twigg, M.V., Ed.; Wolfe Pub.: London, UK, 1989; pp. 340–383. [Google Scholar]
- Golosman, E.Z.; Efremov, V.N. Industrial Catalysts for the Hydrogenation of Carbon Oxides. Catal. Ind. 2012, 4, 267–283. [Google Scholar] [CrossRef]
- Miguel, C.V.; Mendes, A.; Madeira, L.M. Intrinsic kinetics of CO2 methanation over an industrial nickel-based catalyst. J. CO2 Util. 2018, 25, 128–136. [Google Scholar] [CrossRef]
- Available online: http://www.topsoe.com/business_areas/ammonia/processes/methanation.aspx (accessed on 30 October 2023).
- Johnson Matthey Methanation Catalysts. Available online: https://matthey.com/documents/161599/440146/JM+Methanation+product+brochure+%28c2020%29.pdf/56eaef18-4782-776f-ed2a-c332f5f16659?t=1653488109779 (accessed on 30 October 2023).
- Fujita, S.I.; Takezawa, N. Difference in the selectivity of CO and CO2 methanation reactions. Chem. Eng. J. 1997, 68, 63–68. [Google Scholar] [CrossRef]
- Schmider, D.; Maier, L.; Deutschmann, O. Reaction Kinetics of CO and CO2 Methanation over Nickel. Ind. Eng. Chem. Res. 2021, 60, 5792–5805. [Google Scholar] [CrossRef]
- Burger, T.; Donaubauera, P.; Hinrichsen, O. On the kinetics of the co-methanation of CO and CO2 on a co-precipitated Ni-Al catalyst. Appl. Catal. B Environ. 2021, 282, 119408. [Google Scholar] [CrossRef]
- Schumacher, J.; Meyer, D.; Friedland, J.; Güttel, R. Methanation of CO/CO2 mixtures: Evaluation of pellet size effect on methane formation rate and reactant selectivity. Chem. Eng. J. 2023, 463, 142451. [Google Scholar] [CrossRef]
- Dagle, R.A.; Wang, Y.; Xia, G.G.; Strohm, J.J.; Holladay, J.; Palo, D.R. Selective CO methanation catalysts for fuel processing applications. Appl. Catal. A Gen. 2007, 326, 213–218. [Google Scholar] [CrossRef]
- Kopyscinski, J.; Schildhauer, T.J.; Biollaz, S.M.A. Production of synthetic natural gas (SNG) from coal and dry biomass e a technology review from 1950 to 2009. Fuel 2010, 89, 1763–1783. [Google Scholar] [CrossRef]
- Røstrup-Nielsen, J.R.; Pedersen, K.; Sehested, J. High temperature methanation. Sintering and structure sensitivity. Appl Catal. A Gen 2007, 330, 134–138. [Google Scholar] [CrossRef]
- Bolt, A.; Dincer, I.; Agelin-Chaab, M. A critical review of synthetic natural gas production techniques and technologies. J. Nat. Gas Sci. Eng. 2020, 84, 103670. [Google Scholar] [CrossRef]
- Schaaf, T.; Grünig, J.; Schuster, M.R.; Rothenfluh, T.; Orth, A. Methanation of CO2—Storage of renewable energy in a gas distribution system. Energy Sustain. Soc. 2014, 4, 2. [Google Scholar] [CrossRef]
- Li, J.; Tian, Y.; Yan, X.; Yang, J.; Wang, Y.; Xu, W.; Xie, K. Approach and potential of replacing oil and natural gas with coal in China. Front. Energy 2020, 14, 419–431. [Google Scholar] [CrossRef]
- Available online: https://www.netl.doe.gov/sites/default/files/netl-file/tremp-2009.pdf (accessed on 30 October 2023).
- Available online: https://www.topsoe.com/processes/rng (accessed on 30 October 2023).
- Torcida, M.F.; Curto, D.; Martín, M. Design and optimization of CO2 hydrogenation multibed reactors. Chem. Eng. Res. Des. 2022, 181, 89–100. [Google Scholar] [CrossRef]
- Available online: http://www.topsoe.com/products/CatalystPortfolio.aspx (accessed on 30 October 2023).
- Nguyen, T.T.M.; Wissing, L.; Skjøth-Rasmussen, M.S. High temperature methanation: Catalyst considerations. Catal. Today 2013, 215, 233–238. [Google Scholar] [CrossRef]
- Available online: http://www.oilngasprocess.com/petrochemical/substitute-natural-gas-sng-process-by-davy-process-technology-uk.html (accessed on 30 October 2023).
- Ross, J.R.H. Nickel Catalysts for C1 Reactions: Recollections from a Career in Heterogeneous Catalysis. Top. Catal. 2021, 64, 896–906. [Google Scholar] [CrossRef]
- Romano, L.; Ruggeri, F. Methane from syngas—Status of Amec Foster Wheeler VESTA technology development. Energy Procedia 2015, 81, 249–254. [Google Scholar] [CrossRef]
- Depetri, V.; Collodi, G.; Mancuso, L.; Ruggeri, F. VESTA Methanation Applications for Small Scale, Multipurpose, Green SNG Production. Chem. Eng. Trans. 2018, 65, 415–420. [Google Scholar]
- Available online: https://www.clariant.com/en/Corporate/News/2016/07/VESTA-Oncethrough-Methanation-New-Technology-with-Wide-H2CO-Flexibility-Successfully-Passes-Pilot-Te (accessed on 30 October 2023).
- Götz, M.; Lefebvre, J.; Mörs, F.; Koch, A.M.; Graf, F.; Bajohr, S.; Reimert, R.; Kolb, T. Renewable Power-to-Gas: A Technological and Economic Review. Renew. Energy 2016, 85, 1371–1390. [Google Scholar] [CrossRef]
- Vega Puga, E.; Moumin, G.; Neumann, N.C.; Roeb, M.; Ardone, A.; Sattler, C. Holistic View on Synthetic Natural Gas Production: A Technical, Economic and Environmental Analysis. Energies 2022, 15, 1608. [Google Scholar] [CrossRef]
- Thema, M.; Bauer, F.; Sterner, M. Sterner Power-to-Gas: Electrolysis and methanation status review. Renew. Sustain. Energy Rev. 2019, 112, 775–778. [Google Scholar] [CrossRef]
- Pintér, G. The development of global power-to-methane potentials between 2000 and 2020: A comparative overview of international projects. Appl. Energy 2024, 353, 122094. [Google Scholar] [CrossRef]
- Tripodi, A.; Conte, F.; Rossetti, I. Carbon Dioxide Methanation: Design of a Fully Integrated Plant. Energy Fuels 2020, 34, 7242–7256. [Google Scholar] [CrossRef]
- Uddin, Z.; Bor-Yih Yu, B.Y.; Lee, H.Y. Evaluation of alternative processes of CO2 methanation: Design, optimization, control, techno-economic and environmental analysis. J. CO2 Util. 2022, 60, 101974. [Google Scholar] [CrossRef]
- Lee, W.J.; Li, C.; Prajitno, H.; Yoo, J.; Patel, J.; Yang, Y.; Lim, S. Recent trend in thermal catalytic low temperature CO2 methanation: A critical review. Catal. Today 2021, 368, 2–19. [Google Scholar] [CrossRef]
- Mebrahtu, C.; Nohl, M.; Dittrich, L.; Foit, S.R.; de Haart, L.G.L.; Eichel, R.A.; Palkovits, R. Integrated Co-Electrolysis and Syngas Methanation for the Direct Production of Synthetic Natural Gas from CO2 and H2O. ChemSusChem 2021, 14, 2295–2302. [Google Scholar] [CrossRef] [PubMed]
- Schlereth, D.; Hinrichsen, O. A fixed-bed reactor modeling study on the methanation of CO2. Chem. Eng. Res. Des. 2014, 92, 702–712. [Google Scholar] [CrossRef]
- Available online: https://www.clariant.com/en/Business-Units/Catalysts/Energy-Transition/Carbon-Capture-and-Utilization (accessed on 30 October 2023).
- Mohd Ridzuan, N.D.; Shaharun, M.S.; Anawar, M.A.; Ud-Din, I. Ni-Based Catalyst for Carbon Dioxide Methanation: A Review on Performance and Progress. Catalysts 2022, 12, 469. [Google Scholar] [CrossRef]
- Busca, G.; Spennati, E.; Riani, P.; Garbarino, G. Looking for an Optimal Composition of Nickel-Based Catalysts for CO2 Methanation. Energies 2023, 16, 5304. [Google Scholar] [CrossRef]
- Stenger, H.G., Jr.; Askonas, C.F. Thermodynamic Product Distributions for the Fischer-Tropsch Synthesis. Ind. Eng. Chem. Fundamen. 1986, 25, 410–413. [Google Scholar] [CrossRef]
- Chen, J.; Yang, C. Thermodynamic Equilibrium Analysis of Product Distribution in the Fischer−Tropsch Process Under Different Operating Conditions. ACS Omega 2019, 4, 22237–22244. [Google Scholar] [CrossRef]
- Leckel, D. Diesel Production from Fischer−Tropsch: The Past, the Present, and New Concepts. Energy Fuels 2009, 23, 2342–2358. [Google Scholar] [CrossRef]
- Zennaro, R.; Hugues, F.; Caprani, E. The Eni—IFP/Axens GTL Technology: From R&D to a Successful Scale-Up. In Proceedings of the DGMK/SCI-Conference “Synthesis Gas Chemistry”, Dresden, Germany, 4–6 October 2006; Available online: https://www.osti.gov/etdeweb/servlets/purl/20840759 (accessed on 30 October 2023).
- Martinelli, M.; Gnanamani, M.K.; LeViness, S.; Jacobs, G.; Shafer, W.D. An overview of Fischer-Tropsch Synthesis: XtL processes, catalysts and reactors. Appl. Catal. A Gen. 2020, 608, 11774. [Google Scholar] [CrossRef]
- Niu, C.; Xia, M.; Chen, C.; Ma, Z.; Jia, L.; Hou, B.; Li, D. Effect of process conditions on the product distribution of Fischer-Tropsch synthesis over an industrial cobalt-based catalyst using a fixed-bed reactor. Appl. Catal. A Gen. 2020, 601, 117630. [Google Scholar] [CrossRef]
- Khodakov, A.Y.; Chu, W.; Fongarland, P. Development of Novel Cobalt Fischer−Tropsch Catalysts. Chem. Rev. 2007, 107, 1692–1744. [Google Scholar] [CrossRef] [PubMed]
- Cornaro, U.; Rossini, S.; Montanari, T.; Finocchio, E.; Busca, G. K-doping of Co/Al2O3 low temperature Fischer-Tropsch catalysts. Catal. Today 2012, 197, 101–108. [Google Scholar] [CrossRef]
- Spennati, E.; Garbarino, G.; Riani, P.; Busca, G. Alumina-supported cobalt catalysts in the hydrogenation of CO2 at atmospheric pressure. Int. J. Hydrogen Energy 2023, 48, 25006–25015. [Google Scholar] [CrossRef]
- Diehl, F.; Khodakov, A.Y. Promotion of Cobalt Fischer-Tropsch Catalysts with Noble Metals: A Review. Oil Gas Sci. Technol. Rev. IFP 2009, 64, 11–24. [Google Scholar] [CrossRef]
- Claeys, M.; Dry, M.E.; van Steen, E.; du Plessis, E.; van Berge, P.J.; Saib, A.M.; Moodley, D.J. In situ magnetometer study on the formation and stability of cobalt carbide in Fischer-Tropsch synthesis. J. Catal. 2014, 318, 193–202. [Google Scholar] [CrossRef]
- Hazemann, P.; Decottignies, D.; Maury, S.; Humbert, S.; Meunier, F.C.; Schuurman, Y. Selectivity loss in Fischer-Tropsch synthesis: The effect of cobalt carbide formation. J. Catal. 2021, 397, 1–12. [Google Scholar] [CrossRef]
- Shiba, N.C.; Liu, X.; Yao, Y. Advances in lower olefin production over cobalt-based catalysts via the Fischer-Tropsch process. Fuel Proc. Technol. 2022, 238, 107489. [Google Scholar] [CrossRef]
- Peacock, M.; Paterson, J.; Reed, L.; Davies, S.; Carter, S.; Coe, A.; Clarkson, J. Innovation in Fischer-Tropsch: Developing Fundamental Understanding to Support Commercial Opportunities. Top. Catal. 2020, 63, 328–339. [Google Scholar] [CrossRef]
- Espinoza, R.L.; Steynberg, A.P.; Jager, B.; Vosloo, A.C. Low temperature Fischer-Tropsch synthesis from a Sasol perspective. Appl. Catal. A Gen. 1999, 186, 13–26. [Google Scholar] [CrossRef]
- Weststrate, C.J.; Sharma, D.; Garcia Rodriguez, D.; Gleeson, M.A.; Fredriksson, H.O.A.; Niemantsverdriet, J.M. Mechanistic insight into carbon-carbon bond formation on cobalt under simulated FischerTropsch synthesis conditions. Nat. Commun. 2020, 11, 750. [Google Scholar] [CrossRef] [PubMed]
- Perego, C.; Bortolo, R.; Zennaro, R. Gas to liquids technologies for natural gas reserves valorization: The Eni experience. Catal. Today 2009, 142, 9–16. [Google Scholar] [CrossRef]
- Ma, W.; Jacobs, G.; Sparks, D.E.; Todic, B.; Bukur, D.B.; Davis, B.H. Quantitative comparison of iron and cobalt based catalysts for the Fischer-Tropsch synthesis under clean and poisoning conditions. Catal. Today 2020, 343, 125–136. [Google Scholar] [CrossRef]
- Steynberg, A.P.; Espinoza, R.L.; Jager, B.; Vosloo, A.C. High temperature Fischer-Tropsch synthesis in commercial practice. Appl. Catal. A Gen. 1999, 186, 41–54. [Google Scholar] [CrossRef]
- Xu, J.; Yang, Y.; Li, Y. Fischer-Tropsch synthesis process development: Steps from fundamentals to industrial processes. Curr. Opin. Chem. Eng. 2013, 2, 354–363. [Google Scholar] [CrossRef]
- Xu, J.; Yang, Y.; Li, Y.W. Recent development in converting coal to clean fuels in China. Fuel 2015, 152, 122–130. [Google Scholar] [CrossRef]
- Zhang, J.; Abbas, M.; Chen, J. The Evolution of Fe Phases of a Fused Iron Catalyst during Reduction and Fischer-Tropsch Synthesis. Catal. Sci. Technol. 2017, 7, 3626–3636. [Google Scholar] [CrossRef]
- Lyu, S.; Wang, L.; Li, Z.; Yin, S.; Chen, J.; Zhang, Y.; Li, J.; Wang, Y. Stabilization of ε-iron carbide as high-temperature catalyst under realistic Fischer-Tropsch synthesis conditions. Nat. Commun. 2020, 11, 6219. [Google Scholar] [CrossRef]
- Luo, M.; Hamdeh, H.; Davis, B.H. Fischer-Tropsch Synthesis: Catalyst activation of low alpha iron catalyst. Catal. Today 2009, 140, 127–134. [Google Scholar] [CrossRef]
- Gaube, J.; Klein, H.F. The promoter effect of alkali in Fischer-Tropsch iron and cobalt catalysts. Appl. Catal. A Gen. 2008, 350, 126–132. [Google Scholar] [CrossRef]
- De Smit, E.; Cinquini, F.; Beale, A.M.; Safonova, O.V.; van Beek, W.; Sautet, P.; Weckhuysen, B.M. Stability and Reactivity of ϵ-χ-θ Iron Carbide Catalyst Phases in Fischer-Tropsch Synthesis: Controlling μC. J. Am. Chem. Soc. 2010, 132, 14928–14941. [Google Scholar] [CrossRef]
- Lu, F.; Chen, X.; Lei, Z.; Wen, L.; Zhang, Y. Revealing the activity of different iron carbides for Fischer-Tropsch synthesis. Appl. Catal. B Environ. 2021, 281, 119521. [Google Scholar] [CrossRef]
- Ordomsky, V.V.; Legras, B.; Cheng, K.; Paul, S.; Khodakov, A.Y. The role of carbon atoms of supported iron carbides in Fischer-Tropsch synthesis. Catal. Sci. Technol. 2015, 5, 1433–1437. [Google Scholar] [CrossRef]
- Pham, T.H.; Qi, Y.; Yang, J.; Duan, X.; Qian, G.; Zhou, X.; Chen, D.; Yuan, W. Insights into Hägg Iron-Carbide-Catalyzed, D. Fischer−Tropsch Synthesis: Suppression of CH4 Formation and Enhancement of C−C Coupling on χ-Fe5C2(510). ACS Catal. 2015, 5, 2203–2208. [Google Scholar] [CrossRef]
- Atsonios, K.; Li, J.; Inglezakis, V.J. Process analysis and comparative assessment of advanced thermochemical pathways for e-kerosene production. Energy 2023, 278, 127868. [Google Scholar] [CrossRef]
- Visconti, C.G.; Martinelli, M.; Falbo, L.; Fratalocchi, L.; Lietti, L. CO2 hydrogenation to hydrocarbons over Co and Fe-based Fischer-Tropsch catalysts. Catal. Today 2016, 277, 161–170. [Google Scholar] [CrossRef]
- Choi, Y.H.; Jang, Y.J.; Park, H.; Kim, W.Y.; Lee, Y.H.; Choi, S.H.; Lee, J.S. Carbon dioxide Fischer-Tropsch synthesis: A new path to carbon-neutral fuels. Appl. Catal. B Environ. 2017, 202, 605–610. [Google Scholar] [CrossRef]
- Wei, J.; Ge, Q.; Yao, R.; Wen, Z.; Fang, C.; Guo, L.; Xu, H.; Sun, J. Directly converting CO2 into a gasoline fuel. Nat. Commun. 2017, 8, 15174. [Google Scholar] [CrossRef]
- Cheng, Y.; Chen, Y.; Zhang, S.; Wu, X.; Chen, C.; Shi, X.; Qing, M.; Li, J.; Liu, C.L.; Dong, W.S. High-yield production of aromatics over CuFeO2/hierarchical HZSM-5 via CO2 Fischer-Tropsch synthesis. Green Chem. 2023, 25, 3570–3584. [Google Scholar] [CrossRef]
- Zhang, L.; Dang, Y.; Zhou, X.; Gao, P.; van Bavel, A.P.; Wang, H.; Li, S.; Shi, L.; Yang, Y.; Vovk, E.I.; et al. Direct conversion of CO2 to a jet fuel over CoFe alloy catalysts. Innovation 2021, 2, 100170. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, F. Science Alert, 27 April 2015. Available online: https://www.sciencealert.com/audi-have-successfully-made-diesel-fuel-from-air-and-water (accessed on 30 October 2023).
- Busca, G. The surface acidity of solid oxides and its characterization by IR spectroscopic methods. An attempt at systematization. Phys. Chem. Chem. Phys. 1999, 1, 723–736. [Google Scholar] [CrossRef]
- Busca, G. Bases and basic materials in industrial and environmental chemistry. Liquid versus solid basicity. Chem. Rev. 2010, 110, 2217–2249. [Google Scholar] [CrossRef] [PubMed]
- Helali, Z.; Jedidi, A.; Syzgantseva, O.A.; Calatayud, M.; Minot, C. Scaling reducibility of metal oxides. Theor. Chem. Acc. 2017, 136, 100. [Google Scholar] [CrossRef]
- Hammer, B.; Nørskov, J.K. Why gold is the noblest of all the metals. Nature 1995, 376, 238. [Google Scholar] [CrossRef]
- Hammer, B.; Nørskov, J.K. Theoretical surface science and catalysis—Calculations and concepts. Adv. Catal. 2000, 45, 71–129. [Google Scholar]
- Nørskov, J.K.; Abild-Pedersen, F.; Studt, F.; Bligaard, T. Density functional theory in surface chemistry and catalysis. Proc. Natl. Acad. Sci. USA 2011, 108, 937–943. [Google Scholar] [CrossRef]
- Takigawa, I.; Shimizu, K.; Tsuda, K.; Takakusa, S. Machine-learning prediction of the d-band center for metals and bimetals. RSC Adv. 2016, 6, 52587. [Google Scholar] [CrossRef]
- Kristinsdóttir, L.; Skúlason, E. A systematic DFT study of hydrogen diffusion on transition metal surfaces. Surf. Sci. 2012, 606, 1400–1404. [Google Scholar] [CrossRef]
- Vázquez-Parga, D.; Jurado, A.; Roldan, A.; Viñes, F. A computational map of the probe CO molecule adsorption and dissociation on transition 672 metal low Miller indices surfaces. Appl. Surf. Sci. 2023, 618, 156581. [Google Scholar] [CrossRef]
- Jin, W.; Wang, Y.; Liu, T.; Ding, C.; Guo, H. CO2 chemisorption and dissociation on flat and stepped transition metal surfaces. Appl. Surf. Sci. 2022, 599, 154024. [Google Scholar] [CrossRef]
- Available online: www.dailymetalprice.com (accessed on 14 November 2023).
- Busca, G. Catalysts for hydrogenations, dehydrogenations and methathesis: Sulphides and oxides. In Heterogeneous Catalytic Materials; Busca, G., Ed.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 345–374. [Google Scholar]
- Ghiotti, G.; Chiorino, A.; Boccuzzi, F. Surface chemistry and electronic effects of H2 (D2) on two different microcrystalline ZnO powders. Surf. Sci. 1993, 287–288, 228–234. [Google Scholar] [CrossRef]
- French, S.A.; Sokol, A.A.; Bromley, S.T.; Catlow, C.R.A.; Rogers, S.C.; Sherwood, P. Assignment of the Complex Vibrational Spectra of the Hydrogenated ZnO Polar Surfaces Using QM/MM Embedding. J. Chem. Phys. 2003, 118, 317–320. [Google Scholar] [CrossRef]
- Kiss, J.; Witt, A.; Meyer, B.; Marx, D. Methanol synthesis on ZnO(0001): I. Hydrogen coverage, charge state of oxygen vacancies, and chemical reactivity. J. Chem. Phys. 2009, 130, 184706. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, M.; Strunk, J.; Hinrichsen, O.; Muhler, M.; Fink, K.; Meyer, B.; Wöll, C. Active sites on oxide surfaces: ZnO-catalyzed synthesis of methanol from CO and H2. Angew. Chem. 2005, 44, 2790–2794. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, C.; Saussey, J.; Lavalley, J.C.; Djega-Mariadassou, G. Definition of polycrystalline ZnO catalytic sites and their role in CO hydrogenation. Appl. Catal. 1986, 25, 59–68. [Google Scholar] [CrossRef]
- Busca, G. Fourier transform-infrared spectroscopic study of the adsorption of hydrogen on chromia and on some metal chromites. J. Catal. 1989, 120, 303–313. [Google Scholar] [CrossRef]
- Collins, S.E.; Baltanás, M.A.; Bonivardi, A.L. Hydrogen chemisorption on gallium oxide polymorphs. Langmuir 2005, 21, 962–970. [Google Scholar] [CrossRef]
- Kondo, J.N.; Sakata, Y.; Domen, K.; Maruya, K.; Onishi, T. Infrared study of hydrogen adsorbed on ZrO2. J. Chem. Soc. Farad. Trans. 1990, 86, 397–401. [Google Scholar] [CrossRef]
- Hofmann, A.; Clark, S.J.; Oppel, M.; Hahndorf, I. Hydrogen adsorption on the tetragonal ZrO2(101) surface: A theoretical study of an important catalytic reactant. Phys. Chem. Chem. Phys. 2002, 4, 3500–3508. [Google Scholar] [CrossRef]
- Syzgantseva, O.; Calatayud, M.; Minot, C. Revealing the Surface Reactivity of Zirconia by Periodic DFT Calculations. J. Phys. Chem. C 2010, 114, 11918–11923. [Google Scholar] [CrossRef]
- Cavalleri, M.; Pelmenschikov, A.; Morosi, G.; Gamba, A.; Coluccia, S.; Martra, G. Dissociative adsorption of H2 on defect sites of MgO: A combined IR spectroscopic and quantum chemical study. Stud. Surf. Sci. Catal. 2001, 140, 131–139. [Google Scholar]
- Binet, C.; Daturi, M.; Lavalley, J.C. IR study of polycrystalline ceria properties in oxidised and reduced states. Catal. Today 1999, 50, 207–225. [Google Scholar] [CrossRef]
- Vilé, G.; Bridier, B.; Wichert, J.; Pérez-Ramírez, J. Ceria in hydrogenation catalysis: High selectivity in the conversion of alkynes to olefins. Angew. Chem. 2012, 51, 8620–8623. [Google Scholar] [CrossRef]
- Doornkamp, C.; Ponec, V. The universal character of the Mars and Van Krevelen mechanism. J. Mol. Catal. A Chem. 2000, 162, 19–32. [Google Scholar] [CrossRef]
- Chen, H.T.; Choi, Y.M.; Liu, M.; Lin, M.C. A Theoretical Study of Surface Reduction Mechanisms of CeO2(111) and (110) by H2. ChemPhysChem 2007, 8, 849–855. [Google Scholar] [CrossRef]
- Li, Z.; Werner, K.; Chen, L.; Jia, A.; Qian, K.; Zhong, J.Q.; You, R.; Wu, L.; Zhang, L.; Pan, H.; et al. Interaction of Hydrogen with Ceria: Hydroxylation, Reduction, and Hydride Formation on the Surface and in the Bulk. Chem. Eur. J. 2021, 27, 5268–5276. [Google Scholar] [CrossRef]
- Wei, B.; Calatayud, M. Hydrogen activation on Anatase TiO2: Effect of surface termination. Catal. Today 2022, 397–399, 113–120. [Google Scholar] [CrossRef]
- Yua, X.; Zhang, X.; Wang, S. High coverage hydrogen adsorption on the Fe3O4(1 1 0) surface. Appl. Surf. Sci. 2015, 353, 973–978. [Google Scholar] [CrossRef]
- Bielz, T.; Lorenz, H.; Jochum, W.; Kaindl, R.; Klauser, F.; Klötzer, B.; Penner, S. Hydrogen on In2O3: Reducibility, Bonding, Defect Formation, and Reactivity. J. Phys. Chem. C 2010, 114, 9022–9029. [Google Scholar] [CrossRef]
- Posada-Borbón, A.; Grönbeck, H. Hydrogen adsorption on In2O3(111) and In2O3(110). Phys. Chem. Chem. Phys. 2020, 22, 16193–16202. [Google Scholar] [CrossRef] [PubMed]
- Rukini, A.; Rhamdhani, M.A.; Brooks, G.A.; Van den Bulck, A. Metals Production and Metal Oxides Reduction Using Hydrogen: A Review. J. Sustain. Metall. 2022, 8, 1–24. [Google Scholar] [CrossRef]
- Sheppard, D.A.; Buckley, C.E. Hydrogen adsorption on porous silica. Int. J. Hydrogen Energy 2008, 33, 1688–1692. [Google Scholar] [CrossRef]
- Amenomiya, Y. Adsorption of Hydrogen and H2-D2 Exchange Reaction on Alumina. J. Catal. 1971, 22, 109–121. [Google Scholar] [CrossRef]
- Weller, S.W.; Montagna, A.A. Studies of Alumina I. Reaction with Hydrogen at Elevated Temperatures. J. Catal. 1971, 21, 303–311. [Google Scholar] [CrossRef]
- Kramer, R.; Andre, M. Adsorption of Atomic Hydrogen on Alumina by Hydrogen Spillover. J. Catal. 1979, 58, 287–295. [Google Scholar] [CrossRef]
- Digne, M.; Sautet, P.; Raybaud, P.; Euzen, P.; Toulhoat, H. Use of DFT to achieve a rational understanding of acid–basic properties of γ-alumina surfaces. J. Catal. 2004, 226, 54–68. [Google Scholar] [CrossRef]
- Wischert, R.; Laurent, P.; Copéret, C.; Delbecq, F.; Sautet, P. γ-Alumina: The essential and unexpected role of water for the structure, stability, and reactivity of “defect” sites. J. Am. Chem. Soc. 2012, 134, 14430–14449. [Google Scholar] [CrossRef]
- Karim, W.; Spreafico, C.; Kleibert, A.; Gobrecht, J.; VandeVondele, J.; Ekinci, Y.; van Bokhoven, J.A. Catalyst support effects on hydrogen spillover. Nature 2017, 541, 68–71. [Google Scholar] [CrossRef]
- Garbarino, G.; Travi, I.; Pani, M.; Carnasciali, M.M.; Busca, G. Pure vs ultra-pure γ-alumina: A spectroscopic study and catalysis of ethanol conversion. Catal. Commun. 2015, 70, 77–81. [Google Scholar] [CrossRef]
- Shen, H.; Li, H.; Yang, Z.; Li, C. Magic of hydrogen spillover: Understanding and application. Green Energy Environ. 2022, 7, 1161–1198. [Google Scholar] [CrossRef]
- Beck, A.; Rzepka, P.; Marshall, K.P.; Stoian, D.; Willinger, M.G.; van Bokhoven, J.A. Hydrogen Interaction with Oxide Supports in the Presence and Absence of Platinum. J. Phys. Chem. C 2022, 126, 17589–17597. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhou, M.; Wang, A.; Zhang, T. Selective Hydrogenation over Supported Metal Catalysts: From Nanoparticles to Single Atoms. Chem. Rev. 2020, 120, 683–733. [Google Scholar] [CrossRef] [PubMed]
- Arena, F.; Italiano, G.; Barbera, K.; Bordiga, S.; Bonura, G.; Spadaro, L.; Frusteri, F. Solid-state interactions, adsorption sites and functionality of Cu-ZnO/ZrO2 catalysts in the CO2 hydrogenation to CH3OH. Appl. Catal. A Gen. 2008, 350, 16–23. [Google Scholar] [CrossRef]
- Paul, J.F.; Sautet, P. Comparison of the nature of the hydrogen-metal bond on Pd(111) and Ni(111) by a periodic density functional method. Surf. Sci. 1996, 356, L403–L409. [Google Scholar] [CrossRef]
- Huda, M.N.; Kleinman, L. Hydrogen adsorption and dissociation on small platinum clusters: An electronic structure density functional study. Phys. Rev. B 2006, 74, 195407. [Google Scholar] [CrossRef]
- Ferrin, P.; Kandoi, S.; Nilekar, A.U.; Mavrikakis, M. Hydrogen adsorption, absorption and diffusion on and in transition metal surfaces: A DFT study. Surf. Sci. 2012, 606, 679–689. [Google Scholar] [CrossRef]
- Greeley, J.; Marikakis, M. Surface and subsurface hydrogen: Adsorption properties on transition metals and near-surface alloys. J. Phys. Chem. B 2005, 109, 3460–3471. [Google Scholar] [CrossRef]
- Chizallet, C.; Bonnard, C.; Krebs, E.; Bisson, L.; Thomazeau, C.; Raybaud, P. Thermodynamic Stability of Buta-1,3-diene and But-1-ene on Pd(111) and (100) Surfaces under H2 Pressure: A DFT Study. J. Phys. Chem. C 2011, 115, 12135–12149. [Google Scholar] [CrossRef]
- Maki-Arvela, P.; Murzin, D.Y. Effect of metal particle shape on hydrogen assisted reactions. Appl. Catal. A Gen. 2021, 618, 118140. [Google Scholar] [CrossRef]
- Oudenhuijzen, M.K.; van Bokhoven, J.A.; Miller, J.T.; Ramaker, D.E.; Koningsberger, D.C. Three-site model for hydrogen adsorption on supported platinum particles: Influence of support ionicity and particle size on the hydrogen coverage. J. Am. Chem. Soc. 2005, 127, 1530–1540. [Google Scholar] [CrossRef] [PubMed]
- Znak, L.; Zieliński, J. Interaction of hydrogen with unsupported and supported nickel. Langmuir 2006, 22, 8758. [Google Scholar] [CrossRef] [PubMed]
- Znak, L.; Zieliński, J. Effects of support on hydrogen adsorption/desorption on nickel. Appl. Catal. A Gen. 2008, 334, 268–276. [Google Scholar] [CrossRef]
- Wang, Y.; Winter, L.R.; Chen, J.G.; Yan, B.Y. CO2 hydrogenation over heterogeneous catalysts at atmospheric pressure: From electronic properties to product selectivity. Green Chem. 2021, 23, 249–267. [Google Scholar] [CrossRef]
- Zabilskiy, M.; Sushkevich, V.L.; Palagin, D.; Newton, M.A.; Krumeich, F.; van Bokhoven, J.A. The unique interplay between copper and zinc during catalytic carbon dioxide hydrogenation to methanol. Nat. Commun. 2020, 11, 2409. [Google Scholar] [CrossRef] [PubMed]
- Silveri, F.; Quesne, M.G.; Roldan, A.; de Leeuw, N.H.; Catlow, C.R.A. Hydrogen adsorption on transition metal carbides: A DFT study. Phys. Chem. Chem. Phys. 2019, 21, 5335–5343. [Google Scholar] [CrossRef]
- Bian, G.; Oonuki, A.; Kobayashi, Y.; Koizumi, N.; Yamada, M. Syngas adsorption on precipitated iron catalysts reduced by H2, syngas or CO and on those used for high pressure FT synthesis by in situ diffuse reflectance FTIR spectroscopy. Appl. Catal. A 2001, 219, 13–24. [Google Scholar] [CrossRef]
- Bai, Y.; Liu, J.; Wang, T.; Song, J.F.; Yang, Y.; Li, Y.F.; Wen, X. Theoretical insight into the interaction between hydrogen and Hägg carbide (χ-Fe5C2) surfaces. Appl. Surf. Sci. 2022, 583, 152538. [Google Scholar] [CrossRef]
- Blyholder, G. Molecular orbital view of chemisorbed carbon monoxide. J. Phys. Chem. 1964, 68, 2772–2777. [Google Scholar] [CrossRef]
- Lupinetti, A.J.; Fau, S.; Frenking, G.; Strauss, S.H. Theoretical Analysis of the Bonding between CO and Positively Charged Atoms. J. Phys. Chem. A 1997, 101, 9551–9559. [Google Scholar] [CrossRef]
- Sthorozhev, P.Y.; Yanko, V.S.; Tsyganenko, A.A.; Rodriguez-Delgado, G.; Otero Areán, C. Isomeric states of polar molecules on ionic surfaces:electrostatic model and FTIR studies. Appl. Surf. Sci. 2004, 238, 390–394. [Google Scholar] [CrossRef]
- Hadjiivanov, K.; Vayssilov, G. Characterization of Oxide Surfaces and Zeolites by Carbon Monoxide as an IR Probe Molecule. Adv. Catal. 2002, 47, 307–511. [Google Scholar]
- Busca, G. Infrared (IR) Spectroscopy. In Springer Handbook of Advanced Catalyst Characterization; Bañares, M.A., Wachs., I., Eds.; Springer Nature: Cham, Switzerland, 2023; pp. 1–30. [Google Scholar]
- Blomqvist, J.; Lehman, L.; Salo, P. CO adsorption on metal-oxide surfaces doped with transition-metal adatoms. Phys. Status Solid. 2012, 249, 1046–1057. [Google Scholar] [CrossRef]
- Gopal, P.G.; Schneider, R.L.; Watters, K.L. Evidence for Production of Surface Formate upon Direct Reaction of CO with Alumina and Magnesia. J. Catal. 1987, 105, 366–372. [Google Scholar] [CrossRef]
- Shido, T.; Asakura, K.; Iwasawa, Y. Reactant-promoted reaction mechanism for catalytic water-gas shift reaction on MgO. J. Catal. 1990, 122, 55–67. [Google Scholar] [CrossRef]
- Kogler, M.; Köck, E.M.; Klötzer, B.; Schachinger, T.; Wallisch, W.; Henn, R.; Huck, C.W.; Hejny, C.; Penner, S. High-Temperature Carbon Deposition on Oxide Surfaces by CO Disproportionation. J. Phys. Chem. C 2016, 120, 1795–1807. [Google Scholar] [CrossRef]
- Dang, J.; Chou, K. A Model for the Reduction of Metal Oxides by Carbon Monoxide. ISIJ Int. 2018, 58, 585–593. [Google Scholar] [CrossRef]
- Abild-Pedersen, F.; Andersson, M.P. CO adsorption energies on metals with correction for high coordination adsorption sites—A density functional study. Surf. Sci. 2007, 601, 1747–1753. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Sheppard, N. Advances in Infrared and Raman Spectroscopy; Hester, R.E., Clark, R.H.J., Eds.; Heyden: London, UK, 1982; Volume 5, pp. 86–95. [Google Scholar]
- Fielicke, A.; Gruene, P.; Meijer, G.; Rayner, D.M. The adsorption of CO on transition metal clusters: A case study of cluster surface chemistry. Surf. Sci. 2009, 603, 1427–1433. [Google Scholar] [CrossRef]
- Lemire, C.; Meyer, R.; Shaikhutdinov, S.K.; Freund, H.-J. CO adsorption on oxide supported gold: From small clusters to monolayer islands and three-dimensional nanoparticles. Surf. Sci. 2004, 552, 27–34. [Google Scholar] [CrossRef]
- Wadayama, T.; Sasaki, Y.; Shiomitsu, K.; Hatta, A. Infrared reflection absorption study of carbon monoxide adsorption on Cu(100)-(2 · 2)p4g-Pd ordered alloy surface. Surf. Sci. 2005, 592, 72–82. [Google Scholar] [CrossRef]
- Sanchez-Escribano, V.; Arrighi, L.; Riani, P.; Marazza, R.; Busca, G. Characterization of Pd-Cu Alloy Nanoparticles on γ-Al2O3-Supported Catalysts. Langmuir 2006, 22, 9214–9219. [Google Scholar] [CrossRef] [PubMed]
- Aghamiria, S.; Ghobeity, A. Nickel carbonyl formation in a fluidized bedreactor: Experimental investigation and modeling. J. Chem. Technol. Biotechnol. 2020, 95, 2921–2929. [Google Scholar] [CrossRef]
- Garbarino, G.; Campodonico, S.; Romero Perez, A.; Carnasciali, M.M.; Riani, P.; Finocchio, E.; Busca, G. Spectroscopic characterization of Ni/Al2O3 catalytic materials for the steam reforming of renewables. Appl. Catal. A Gen. 2013, 452, 163–173. [Google Scholar] [CrossRef]
- Liu, Z.P.; Hu, P. General trends in CO dissociation on transition metal surfaces. J. Chem. Phys. 2001, 114, 8244–8247. [Google Scholar] [CrossRef]
- Chai, J.; Pestman, R.; Chen, W.; Dugulan, A.I.; Feng, B.; Men, Z.; Wang, P.; Hensen, E.J.M. The role of H2 in Fe carburization by CO in Fischer-Tropsch catalysts. J. Catal. 2021, 400, 93–102. [Google Scholar] [CrossRef]
- Gong, J.; Cao, C.; Sun, R.; Cui, L.; Gao, r.; Hao, H. A DFT Insight into the Tuning Effect of Potassium Promoter on the Formation of Carbon Atoms via Carburization Gases Dissociation on Iron-Based Catalysts. Catalysts 2020, 10, 527. [Google Scholar] [CrossRef]
- Hofer, L.J.E.; Peebles, W.C. Preparation and X-ray Diffraction Studies of a New Cobalt Carbide. J. Am. Chem. Soc. 1947, 69, 893–899. [Google Scholar] [CrossRef]
- Struis, R.P.W.J.; Bachelin, D.; Ludwig, C.; Wokaun, A. Studying the Formation of Ni3C from CO and Metallic Ni at T = 265 °C in Situ Using Ni K-Edge X-ray Absorption Spectroscopy. J. Phys. Chem. C 2009, 113, 2443–2451. [Google Scholar] [CrossRef]
- Busca, G.; Lorenzelli, V. Infrared spectroscopic identification of species arising from reactive adsorption of carbon oxides on metal oxide surfaces. Mat. Chem. 1982, 7, 89–126. [Google Scholar] [CrossRef]
- Ramis, G.; Busca, G.; Lorenzelli, V. Low-temperature CO2 adsorption on metal oxides: Spectroscopic characterization of some weakly adsorbed species. Mat. Chem. Phys. 1991, 29, 425–435. [Google Scholar] [CrossRef]
- Solymosi, F. The bonding, structure and reactions of CO2 adsorbed on clean and promoted metal surfaces. J. Mol. Catal. 1991, 65, 337–358. [Google Scholar] [CrossRef]
- Freund, H.-J.; Roberts, M.W. Surface chemistry of carbon dioxide. Surf. Sci. Rep. 1996, 25, 225–273. [Google Scholar] [CrossRef]
- Taifan, W.; Boily, J.F.; Baltrusaitis, J. Surface chemistry of carbon dioxide revisited. Surf. Sci. Rep. 2016, 71, 595–671. [Google Scholar] [CrossRef]
- Liu, C.; Cundari, T.R.; Wilson, A.K. CO2 Reduction on Transition Metal (Fe, Co, Ni, and Cu) Surfaces: In Comparison with Homogeneous Catalysis. J. Phys. Chem. C 2012, 116, 5681–5688. [Google Scholar] [CrossRef]
- Liu, X.; Sun, L.; Deng, W.Q. Theoretical Investigation of CO2 Adsorption and Dissociation on Low Index Surfaces of Transition Metals. J. Phys. Chem. C 2018, 122, 8306–8314. [Google Scholar] [CrossRef]
- Ernst, K.-H.; Schlatterbeck, D.; Christmann, K. Adsorption of carbon dioxide on Cu(110) and on hydrogen and oxygen covered Cu(110) surfaces. Phys. Chem. Chem. Phys. 1999, 1, 4105–4112. [Google Scholar] [CrossRef]
- Bonicke, I.A.; Kirstein, W.; Thieme, F. A study on CO2 dissociation on a stepped (332) copper surface. Surf. Sci. 1994, 307–309, 177. [Google Scholar] [CrossRef]
- Cai, J.; Han, Y.; Chen, S.; Crumlin, E.J.; Yang, B.; Li, Y.; Liu, Z. CO2 Activation on Ni(111) and Ni(100) Surfaces in the Presence of H2O: An Ambient-Pressure X-ray Photoelectron Spectroscopy Study. J. Phys. Chem. C 2019, 123, 12176–12182. [Google Scholar] [CrossRef]
- Cong, V.Y.; Son, N.V.; Diem, D.Q.; Pham, S.Q.T. A comparison of water-gas shift reaction on ZnO (1010) surface and 6Cu cluster deposited over ZnO (1010) surface using density functional theory studies. J. Mol. Model. 2022, 28, 84. [Google Scholar] [CrossRef] [PubMed]
- Grabow, L.C.; Mavrikakis, M. Mechanism of Methanol Synthesis on Cu through CO2 and CO Hydrogenation. ACS Catal. 2011, 1, 365–384. [Google Scholar] [CrossRef]
- Palomino, R.M.; Ramírez, P.J.; Liu, Z.; Hamlyn, R.; Waluy, I.; Mahapatra, M.; Orozco, I.; Hunt, A.; Simonovis, J.P.; Senanayake, S.D.; et al. Hydrogenation of CO2 on ZnO/Cu(100) and ZnO/Cu(111) Catalysts: Role of Copper Structure and Metal–Oxide Interface in Methanol Synthesis. J. Phys. Chem. B 2018, 122, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Saussey, J.; Lavalley, J.C.; Lamotte, J.; Rais, T. I.r. spectroscopic evidence of formyl species formed by CO and H2 Co-adsorption on ZnO and Cu–ZnO. J. Chem. Soc. Chem. Commun. 1982, 278–279. [Google Scholar] [CrossRef]
- Enger, B.C.; Holmen, A. Nickel and Fischer-Tropsch Synthesis. Catal. Rev. 2012, 54, 437–488. [Google Scholar] [CrossRef]
- Sanchez-Escribano, V.; Larrubia Vargas, M.A.; Finocchio, E.; Busca, G. On the mechanisms and the selectivity determining steps in syngas conversion over supported metal catalysts: An IR study. Appl. Catal. A Gen. 2007, 316, 68–74. [Google Scholar] [CrossRef]
- Heine, C.; Lechner, B.A.J.; Bluhm, H.; Salmeron, M. Recycling of CO2: Probing the Chemical State of The Ni(111) Surface During the Methanation Reaction with Ambient-pressure X-ray Photoelectron Spectroscopy. J. Am. Chem. Soc. 2016, 138, 13246–13252. [Google Scholar] [CrossRef]
- Huang, M.-X.; Liu, F.; He, C.-C.; Yang, S.-Q.; Chen, W.Y.; Ouyang, L.; Zhao, L.; Yu, J. Interface promoted CO2 methanation: A theoretical study of Ni/La2O3. Chem. Phys. Lett. 2021, 768, 138396. [Google Scholar] [CrossRef]
- Montanari, T.; Castoldi, L.; Lietti, L.; Busca, G. Basic catalysis and catalysis assisted by basicity: FT-IR and TPD characterization of potassium-doped alumina. Appl. Catal. A Gen. 2011, 400, 61–69. [Google Scholar] [CrossRef]
- Fasolini, A.; Spennati, E.; Atakoohi, S.E.; Percivale, M.; Busca, G.; Basile, F.; Garbarino, G. A study of CO2 hydrogenation over Ni-MgAlOx catalysts derived from hydrotalcite precursors. Catal. Today 2003, 423, 114271. [Google Scholar] [CrossRef]
- Garbarino, G.; Wang, C.; Valsamakis, I.; Chitsazan, S.; Riani, P.; Finocchio, E.; Flytzani-Stephanopoulos, M.; Busca, G. Acido-basicity of lanthana/alumina catalysts and their activity in ethanol conversion. Appl. Catal. B Environ. 2017, 200, 458–468. [Google Scholar] [CrossRef]
Figure 1.
Standard Gibbs free energy vs. T for selected CO (left) and CO2 (right) reactions.

Figure 2.
Schematics of TEM images of Cu/ZnO(Al2O3) catalysts. Al ions are in the ZnO phase, while the surface of copper particles is modified by additional Zn species.
Figure 2.
Schematics of TEM images of Cu/ZnO(Al2O3) catalysts. Al ions are in the ZnO phase, while the surface of copper particles is modified by additional Zn species.

Figure 3.
Equilibrium composition of methanol synthesis from CO (left) and CO2 (right) with stoichiometric feeds, at 100 atm. Calculated using a Gibbs reactor and Soave–Redlich–Kwong equation of state and allowing a calculated phase for the reaction.
Figure 3.
Equilibrium composition of methanol synthesis from CO (left) and CO2 (right) with stoichiometric feeds, at 100 atm. Calculated using a Gibbs reactor and Soave–Redlich–Kwong equation of state and allowing a calculated phase for the reaction.

Figure 4.
Reactor system in the Megamethanol process, see ref. [71]. BFW = boiler feed water.
Figure 4.
Reactor system in the Megamethanol process, see ref. [71]. BFW = boiler feed water.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Catalysts and conditions of current commercial processes for the conversion of syngases (for references, see the text).
Table 1.
Catalysts and conditions of current commercial processes for the conversion of syngases (for references, see the text).
| Process | Main Component | Other Components | P Range | T Range | Feed Composition | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CO | H2 | CO2 | H2O | Other | |||||||||
| Phase | wt% | Comp. | Role | wt% | Bar | K | mol% | mol% | mol% | mol% | mol% | ||
| WGS | HT, high SR a | Fe3O4 | 70–90 | Cr2O3, | stabilizer | <10 | 30–50 | 650–720 | 5–10 | 30–40 | 3–8 | 30–40 | N2 < 15 |
| Cu | promoter | <3 | CH4 < 0.5 | ||||||||||
| MgO | promoter | <1 | Ar < 0.5 | ||||||||||
| HT, low SR a | ZnO-ZnAl2O4 | ~100 | 6–12 | 45–50 | 4–10 | 15–20 | N2 < 18 | ||||||
| CH4 < 0.6 | |||||||||||||
| Ar < 0.6 | |||||||||||||
| LT | Cu | >40 | ZnO | Activity promoter, stabilizer | 50–30 | 30–50 | 450–573 | 2–2.5 | 35–50 | 9–15 | 20–40 | N2 12–16 | |
| Al2O3 (in ZnO) | stabilizer | <20 | CH4 < 0.5 | ||||||||||
| Cs2O, Na2O | Selectivity promoter | <1 | Ar < 0.5 | ||||||||||
| Methanol synthesis | Cu | >50 | ZnO | Activity promoter, stabilizer | ~40 | 50–150 | 473–523 | 10–35 | 40–75 | 1–13 | <2 | CH4 < 15 | |
| Al2O3 (in ZnO) | stabilizer | <10 | |||||||||||
| MgO | promoter | ~2 | |||||||||||
| SiO2 | stabilizer | ||||||||||||
| Methanation | LT a | Ru | 0.3 | γ-Al2O3 | support | 99 | 30–50 | 440–550 | 0.5 | 75 | 0.2 | --- | N2 24 |
| Ni | 20–50 | γ-Al2O3 | support | <80 | 30–50 | 470–620 | CH4 0.2 | ||||||
| MgO, CaO, La2O3 | stabilizer | <20 | Ar 0.3 | ||||||||||
| HT | Ni | >20 | MgAl2O4 La2O3-Al2O3 CaO-Al2O3 | support, stabilizer | <80 | >30 | 500–970 | 25–35 | 35–75 | 1–30 | --- | CH4 < 10 | |
| FTS | LT | Co | 15–30 | γ-Al2O3 | support | <80 | 20–30 | 473–523 | 25–45 | 45–70 | 0–5 | <1 | N2 < 10 CH4 < 10 |
| Ru, Rh, Pt or Pd | activator | <0.1 | |||||||||||
| ZrO2, CeO2, La2O3 | promoter, stabilizer | <10 | |||||||||||
| CoC2 | Inert b | <6 | |||||||||||
| HT | Fe | 90 | SiO2 | promoter | <5 | 20–40 | 590–630 | 35–60 | 35–60 | 0–5 | <1 | N2 < 10 CH4 < 10 | |
| Cu | promoter | <5 | |||||||||||
| K2O | promoter | <5 | |||||||||||
| Fe carbides | active phases b | ||||||||||||
| Fe3O4 | |||||||||||||
a Data for the processes realized for the preparation of ammonia synthesis gas. b Phases formed under reaction.
Table 2.
Surface physical chemistry data concerning the interaction of catalytic transition metals with carbon oxides and hydrogen, and the d-band center value.
Table 2.
Surface physical chemistry data concerning the interaction of catalytic transition metals with carbon oxides and hydrogen, and the d-band center value.
| d-Band Center | Hydrogen Adsorption | Strongest CO Adsorption | Fastest CO Dissociation | Strongest CO2 Adsorption | Price | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ΔEFCC | ΔEontop | Eads | Face | Geometry | Eads | Eatt | Face | Eads | Face | Site | ||||
| eV | eV | eV | eV | USD/lb | ||||||||||
| Cu | Fcc | −2.67 | 0.07 | 0.62 | −0.68 | 011 | short bridge | 1.81 | 2.68 | 011 | 0.25 | 332 | terrace | 3.67 |
| Ni | Fcc | −1.29 | −0.37 | 0.19 | −1.84 | 111 | threefold | −0.07 | 1.64 | 001 | −0.38 | 332 | terrace | 7.79 |
| Co | Hcp | −1.17 | −0.31 | 0.28 | −1.58 | 11–20 | short bridge | 0.40 | 1.33 | 11–20 | −0.59 | 015 | terrace | 15.16 |
| Fe | Bcc | −0.92 | −0.54 | 0.23 | −2.20 | 011 | threefold | −1.38 | 1.08 | 011 | −1.23 | 321 | step | <<1 |
| Pd | Fcc | −1.83 | −0.42 | 0.11 | −1.77 | 111 | threefold | 1.40 | 2.83 | 001 | −0.10 | 332 | terrace | 16,289.28 |
| Ru | Hcp | −1.41 | −0.41 | −0.10 | −1.95 | 10–10 | terminal | 0.18 | 1.38 | 11–20 | −0.90 | 015 | step | 7440.00 |
| Pt | Fcc | −2.25 | −0.37 | −0.38 | −1.81 | 001 | bridge | 2.27 | 4.09 | 111 | −0.07 | 332 | terrace | 14,206.72 |
| Ref. | [173] | [174] | [174] | [175] | [175] | [175] | [175] | [175] | [175] | [176] | [176] | [176] | [177] | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Busca, G.; Spennati, E.; Riani, P.; Garbarino, G. Mechanistic and Compositional Aspects of Industrial Catalysts for Selective CO2 Hydrogenation Processes. Catalysts 2024, 14, 95. https://doi.org/10.3390/catal14020095
AMA Style
Busca G, Spennati E, Riani P, Garbarino G. Mechanistic and Compositional Aspects of Industrial Catalysts for Selective CO2 Hydrogenation Processes. Catalysts. 2024; 14(2):95. https://doi.org/10.3390/catal14020095
Chicago/Turabian StyleBusca, Guido, Elena Spennati, Paola Riani, and Gabriella Garbarino. 2024. "Mechanistic and Compositional Aspects of Industrial Catalysts for Selective CO2 Hydrogenation Processes" Catalysts 14, no. 2: 95. https://doi.org/10.3390/catal14020095
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.