
Organocatalysts for the Synthesis of Cyclic Carbonates under the Conditions of Ambient Temperature and Atmospheric CO2 Pressure

Department of Chemistry, Chungbuk National University, Cheongju 28644, Chungbuk, Republic of Korea
*
Authors to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Catalysts 2024, 14(1), 90; https://doi.org/10.3390/catal14010090
Submission received: 31 December 2023
/
Revised: 15 January 2024
/
Accepted: 16 January 2024
/
Published: 22 January 2024
(This article belongs to the Special Issue Catalysis on Stable Molecules (CO2, CO, CH4, N2, NH3) Activation and Their Transformation, 2nd Edition)
Abstract
:2–(1H–1,2,4–Triazol–3–yl)phenol (CAT–1) was used as an organocatalyst for the coupling reaction of CO2 and epoxides at an ambient temperature and atmospheric CO2 pressure (1 bar). This compound has a structure in which a hydrogen bond donor, a hydrogen bond acceptor, and another hydrogen bond donor are adjacent in sequence in a molecule. The binary catalytic system of CAT–1/nBu4NI showed TON = 19.2 and TOF = 1.60 h−1 under 1 bar CO2 at room temperature within 12 h using 2–butyloxirane. Surprisingly, the activity of CAT–1, in which phenol and 1H–1,2,4–triazole are chemically linked, showed a much greater synergistic effect than when simply mixing the same amount of phenol and 1H–1,2,4–triazole under the same reaction conditions. In addition, our system showed a broad terminal and internal epoxide substrate scope.
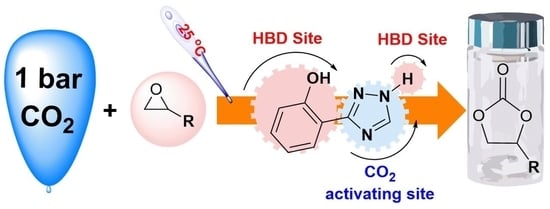
1. Introduction
The cycloaddition of CO2 and epoxides yielding cyclic carbonates, which are used as aprotic polar solvents, electrolytes for lithium–ion batteries, monomers for polymerization, and pharmaceutical intermediates, is one of the most important reactions, alongside the transformation of CO2 as a C1 feedstock due to its atom economy and broad applicability [1,2]. To date, numerous catalytic systems for the synthesis of CO2-based cyclic carbonates, including various types of metal- and organic-based catalysts, have been reported in the literature [3,4,5]. Although organocatalysts have many advantages in terms of cost, toxicity, eco-friendliness, and accessibility, they generally require high reaction temperatures (>100 °C), high CO2 pressures (>10 bar), and high catalyst loadings (>5 mol%) for efficient conversion, and also these reaction conditions are more stringent than those required by metal-based catalysts. To date, several active organocatalysts for this coupling reaction under mild conditions have been reported in the literature [6,7,8]; however, the development of efficient organocatalysts capable of operating at an ambient temperature and atmospheric CO2 pressure is quite difficult, and only a few examples are known [9,10,11,12,13,14,15,16].
Pairs of hydrogen–bond donors (HBDs) as organocatalysts and nucleophiles as cocatalysts are well-known catalytic systems for the synthesis of CO2-based cyclic carbonates under mild conditions [17,18,19]. Phenol and its derivatives are among the most extensively investigated examples of HBDs because they can easily be modified to include substituents that have steric and electronic effects on the other five carbon atoms in the phenyl ring [20,21,22,23,24,25]. In addition, HBDs with more than two vicinal –OH groups, which can interact synergistically with the O atom of the epoxide through H–bonds, exhibit higher activities than HBDs with non-vicinal –OH groups or only one –OH group [21,22,23,24,25]. To the best of our knowledge, HBDs with two different vicinal groups, specifically –OH and –NH groups, have never been used as catalysts for CO2/epoxide coupling reactions, even though organocatalysts with two vicinal NH groups have recently been reported [26,27]. Furthermore, pyridine-like N atoms in the catalyst can activate CO2 to afford carbamate intermediates during the catalytic cycle [15]. Thus, designing simple organocatalysts with cooperative H-bond sites and multiple CO2-activating sites located close to or adjacent to each other within a single molecule is highly desirable. As shown in Figure 1, we demonstrate that 2–(1H–1,2,4–triazol–3–yl)phenol (CAT–1) with cooperative H-bonds from the vicinal –OH group and –NH group and with pyridine-like N atoms that can activate CO2 in a single molecule can be a practical alternative to metal catalysts for the synthesis of CO2-based cyclic carbonates at an ambient temperature and atmospheric CO2 pressure.
2. Results
As mentioned in the Introduction section, we investigated whether the proposed CAT–1 [28] could actually interact with CO2 and epoxides. First, peak assignment in the 1H and 13C NMR spectra was performed through COSY, HSQC, and HMBC experiments (see Supplementary Materials). To determine whether CO2 can effectively bind to CAT–1, CAT–1 (20 μmol) in 0.5 mL of D2O was bubbled with 1 bar CO2 (balloon) at room temperature. As shown in Figure 2a, all 1H NMR peaks were shifted to downfield. In addition, a newly observed peak appeared at 166 ppm in 13C NMR, as shown in Figure 2b, and is known to be a typical carbamate carbon peak [29]. 1H and 13C NMR data show that CAT–1 could readily bind with CO2 to form an adduct even at room temperature.
Next, the binding pattern of CAT–1 and epoxides should be investigated. CAT–1 (20 μmol) was mixed with 1a (40 μmol) in 0.5 mL of CDCl3 at room temperature. The O–H and N–H peaks of CAT–1 in the 1H NMR spectrum shifted to downfield from 11.2 ppm to 11.4 ppm (see Supplementary Materials). The same trend for the methine proton and methylene protons on the three-membered ring carbons in 1a was observed. Like CO2, we found that adducts of CAT–1 and epoxide could be easily made at room temperature.
Prior to the coupling reaction of CO2 with epoxides using CAT–1, we synthesized five organocatalysts such as 2–(1H–pyrazol–3–yl)phenol (CAT–2) [30], 2–(1H–tetrazol–5–yl)pyridine (CAT–3) [30], 2–(2–methyl–2H–tetrazol–5–yl)phenol (CAT–4) [31], 2–(1–phenyl–1H–imidazol–5–yl)phenol (CAT–5) [32], and 2–(2–benzyl–2H–tetrazol–5–yl)phenol (CAT–6), as shown in Figure 3, to compare the catalytic activity with CAT–1.
The logic of catalyst synthesis is as follows. It is necessary to examine the effect of differences in the number of nitrogen atoms, which act as CO2-activating sites, on catalytic acidity. Thus, CAT–2 has one less nitrogen atom in the five-membered ring than CAT–1. In CAT–3, a pyridine group was introduced instead of a phenol group to investigate the role of phenol in CAT–1. Interestingly, the only epoxide-activating site in CAT–3 is the N–H group, which is known to have weaker HBDs than the O–H group. To investigate the importance of the N–H group present in the five-membered ring of CAT–1, we synthesized CAT–4, CAT–5, and CAT–6. They lack N–H groups and have one less HBD than CAT–1 and CAT–2. All compounds were characterized by 1H and 13C NMR spectroscopy (See Supplementary Materials), and single-crystal X-ray diffraction methods were used to confirm the structure of CAT–6.
Initially, the coupling of CO2 with 2–butyloxirane (1a) as a substrate as well as a solvent was performed using the organocatalysts given in Figure 3 in the presence of the nucleophilic cocatalyst nBu4NI. The coupling reaction conditions were fixed at 5.0 mol% organocatalyst and 5.0 mol% nBu4NI loadings, a reaction temperature of 25 °C, 1 bar CO2 (balloon), and a reaction time of 12 h, and the results are summarized in Table 1. As expected, phenol in the presence of nBu4NI showed no catalytic activity for the synthesis of 4–butyl–1,3–dioxolan–2–one (2a) (Table 1, entry 1). Binary system 1H–1,2,4–triazole/nBu4NI (Table 1, entry 2) and ternary system 1H–1,2,4–triazole/phenol/nBu4NI (Table 1, entry 3) showed catalytic activities of 24% and 26%, respectively. This result means that 1H–1,2,4–triazole could have much more contribution to the increase in the catalytic activity than phenol. Surprisingly, even at atmospheric CO2 pressure and ambient temperature, organocatalyst CAT–1/nBu4NI easily converted 1a into 2a, with a high activity of 96% under the same reaction condition (Table 1, entry 4). The dramatic increase in the catalytic activity for CAT–1, a simple form connected between 1H–1,2,4–triazole and phenol by a single bond, may originate from the synergistic effect of the sequential existence of a phenol-based strong HBD site, vicinal hydrogen bond acceptor (HBA) site and N–H-based HBD site for epoxide activation, and their vicinal five-membered N–heterocycle for CO2 activation in a single molecule. As shown in entries 5 and 6 of Table 1, CAT–2, which has one less nitrogen atom in the five-membered ring, showed similar activity to CAT–1; however, the activity of CAT–3, which does not contain the –OH group, was found to decrease rapidly. CAT–4, CAT–5, and CAT–6, without N–H groups in the five-membered ring, showed little activity (Table 1, entries 7–9). These data demonstrate that compounds such as CAT–1, in which HBD, HBA, and other HBDs are adjacent in order within the molecule, can be used as efficient organocatalysts for the synthesis of cyclic carbonates under ambient conditions.
We then screened a series of nucleophilic cocatalysts, such as nBu4NI, bis(triphenylphosphine)iminium chloride (PPNCl), nBu4NBr, nBu4NCl, DMAP, and KI, in the coupling of 1a and CO2 with CAT–1 (Table 2, entries 1–6). The highest activity was achieved when using CAT–1 in combination with nBu4NI (Table 2, entry 1). Sterically hindered phosphonium salts were not as effective as nBu4NI (Table 2, entry 2). The catalytic activity decreased in the order I > Br > Cl for tetrabutylammonium salts at 25 °C and 1 bar CO2 (Table 2, entries 1, 3, and 4). DMAP and KI did not show any catalytic activity (Table 2, entries 5 and 6). Because nBu4NI in conjunction with CAT–1 showed the highest activity, this was selected as the optimal catalytic system for further investigations and epoxide screening.
We next investigated the substrate scope using 5.0 mol% CAT–1 and 5.0 mol% nBu4NI loading at 25 °C and 1 bar CO2 for 24 h, and the results are shown in Figure 3. The tested substrates included eight epoxides, namely, 2–butyloxirane (1a), 2–methyloxirane (1b), 2–ethyloxirane (1c), 2–phenyloxirane (1d), 2–(chloromethyl)oxirane (1e), 2–(methoxymethyl)oxirane (1f), 2–(tert–butoxymethyl)oxirane (1g), and 2–(phenoxymethyl)oxirane (1h). Generally, the reactivity of epoxides with CO2 for the synthesis of cyclic carbonates is highly dependent upon the structure of the epoxide. As shown in Figure 4, very high activity for the synthesis of 2a–c was obtained, regardless of the type of alkyl chain of epoxides. In addition to this fact, the lower activity of 1d and 1e than 1a–c appears to be due to electronic effects rather than the steric hindrance of the pendant groups on the epoxides. Compared with 2a–c, CAT–1/nBu4NI showed noticeably lower catalytic activities for 2f–h because of the presence of heteroatoms in the substituents on the epoxide, and these atoms can compete for the formation of H–bonds with CAT–1. Steric hindrance (OtBu > OMe) and the electronic effect (OtBu > OPh) also influence the activity of epoxides 1f–1h. 2h gave the lowest yield because the phenyl in 1h may cause iodide attack at the benzylic site, resulting in some yield loss.
As shown in Figure 5, we also investigated the synthesis of more challenging cyclic carbonates using 1,2–disubstituted trans–2,3–dimethyloxirane (trans–1i), cis–3,6–dioxabicyclo[3.1.0]hexane (cis–1j), cis–6–oxabicyclo[3.1.0]hexane (cis–1k), and cis–7–oxabicyclo[4.1.0]heptane (cis–1l). Due to the low reactivities of 1i–l, a high temperature of 70 °C and a high CO2 pressure of 10 bar were applied. Due to the strain associated with bicyclic epoxides, 1j–l showed slightly lower reactivity compared with the 1,2–disubstituted epoxide 1i. The yields of bicyclic carbonates 2k and 2l were affected by the ring size of the bicyclic epoxides; 1l, with a six–membered ring, exhibited lower activity than the five–membered (1k) system. All diastereochemically pure epoxides 1i–l produced the corresponding 2i–l that maintained their stereochemistry. In addition, no polymeric side products were observed. Thus, the stereochemical retention of cyclic carbonates obtained from the corresponding epoxides indicated that two consecutive SN2 reactions occurred. This means that there are two inversions of stereochemistry at the carbon atom of the epoxides during the reaction.
As shown in Figure 6, a plausible mechanism for the synthesis of cyclic carbonates from epoxides and CO2 using CAT–1 in the presence of the cocatalyst nBu4NI was proposed. This mechanism is similar to that previously proposed for the synthesis of cyclic carbonates using other HBD-based organocatalysts [14]. The epoxide interacts with CAT–1 via H–bonding to generate intermediate I. The insertion of CO2 into intermediate I and the simultaneous nucleophilic ring opening of the epoxide with the iodide anion of the cocatalyst generate carbamate intermediate II. The displacement of the iodide in intermediate II by the carboxylate anion generates intermediate III, followed by the alkoxide attack of the carbamate carbon with concomitant triazole departure. Finally, cyclic carbonates are produced as final products, and intermediate I is regenerated.
3. Materials and Methods
All chemicals were purchased from commercial sources (purity > 95%) and were used as received unless otherwise indicated. Diethyl ether was purified by a Grubbs solvent purification system under a nitrogen atmosphere and stored over activated molecular sieves (4 Å) [42]. Carbon dioxide (99.999%) was used as received without further purification. All epoxides were purified via treatment with calcium hydride to remove residual water. The 1H NMR and 13C NMR spectra were recorded at ambient temperature with a Bruker DPX–500 MHz NMR spectrometer with standard parameters. All chemical shifts are reported in δ units with regard to the residual CDCl3 (δ 7.24 for 1H NMR; δ 77.00 for 13C NMR), DMSO–d6 (δ 2.50 for 1H NMR; δ 39.52 for 13C NMR) or D2O (δ 4.79 for 1H NMR). High-resolution mass spectrometry (HRMS) data were acquired using a high-resolution Q–TOF mass spectrometer (ionization mode: ESI).
3.1. Synthesis of Known Compounds
3.2. Synthesis of 2–(2–Benzyl–2H–tetrazol–5–yl)phenol (CAT–6)
Benzyl bromide (1.71 g, 10 mmol) was added to a stirred solution of 2–(1H–tetrazol–5–yl)phenol [43] (1.62 g, 10 mmol) in diethyl ether (30 mL). The reaction mixture was stirred at room temperature for 2 h. All volatiles were removed in vacuo, and then the residue was purified via column chromatography using a 1:2 mixture of diethyl ether and hexane as the eluent. CAT–6 was obtained as a colorless powder with 44% yield (1.1 g). 1H NMR (CDCl3): δ 9.84 (s, 1H, –OH), 8.18 (m, 1H), 7.55 (m, 6H), 7.17 (m, 1H), 7.08 (m, 1H), 5.96 (s, 2H, –CH2Ph). 13C NMR (CDCl3): δ 164.3, 156.3, 132.7, 132.2, 129.2, 129.1, 128.4, 127.4, 120.0, 117.5, 111.1, 57.11. HRMS m/z calcd for [C14H12N4O + H] 253.1089. Found: 253.1084.
3.3. Representative Procedure for the Coupling of Terminal Epoxide and CO2 at Ambient Condition
Terminal epoxides 1a–h (10 mmol), CAT–1 (80.6 mg, 0.5 mmol), and nBu4NI (184.7 mg, 0.5 mmol) were charged in a 20 mL round-bottomed flask with a magnetic stirring bar in a glovebox. A rubber balloon containing approximately 2 L of CO2 was connected to the flask, and then the reaction vessel was well sealed. The reaction vessel was stirred at 25 °C for 12 h. After 12 h, an aliquot of the reaction mixture was transferred to an NMR tube, and the conversion was determined by 1H NMR spectroscopy. Synthesized cyclic carbonates 2a–h were purified using column chromatography.
3.4. Representative Procedure for the Coupling of Internal Epoxide and CO2 at Ambient Condition
Internal epoxides trans–1i, cis–1j, cis–1k and cis–1l (10 mmol), CAT–1 (80.6 mg, 0.5 mmol), and nBu4NI (184.7 mg, 0.5 mmol) were charged into a 20 mL stainless steel autoclave with a magnetic stirring bar in a glovebox. The autoclave was pressurized to 10 bar of CO2 and was heated to 70 °C. After 12 h, the reactor was cooled and vented. An aliquot of the reaction mixture was transferred to an NMR tube, and the conversion and stereochemistry of trans–1i into trans–2i was determined via 1H NMR spectroscopy. Synthesized cyclic carbonates 2i–l were purified using column chromatography.
3.5. X-ray Crystallographic Structure Determination
The crystallographic measurement was performed at 293(2) K for CAT–6 using a Bruker Apex II diffractometer with Mo Kα (λ = 0.71073 Å) radiation. Specimens of suitable quality and size were selected, mounted, and centered on the X-ray beam using a video camera. The structures were solved via direct methods and refined by full-matrix least-squares methods using the SHELXTL program package with anisotropic thermal parameters for all non-hydrogen atoms, resulting in the X-ray crystallographic data of CAT–6 being obtained in CIF format. Final refinement based on the reflections (I > 2σ(I)) converged at R1 = 0.0491, wR2 = 0.11193, and GOF = 1.010 for CAT–6. Further details are given in Table S1 (see the Supplementary Materials). CCDC 1900793 (CAT–6) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre.
4. Conclusions
We developed one of the most effective organocatalysts reported to date for the generation of cyclic carbonates via the coupling of epoxides and CO2 under an ambient temperature and CO2 pressure. Among our six rationally designed organocatalysts, the solid-state structure for 2–(2–benzyl–2H–tetrazol–5–yl)phenol was determined by single-crystal X-ray diffraction analysis. Among the organocatalysts, 2–(1H–1,2,4–triazol–3–yl)phenol, which has vicinal –OH and –NH groups as cooperative hydrogen-bond donors and a triazolyl group providing multiple CO2-activating sites, showed the best catalytic activity for the synthesis of cyclic carbonates in the presence of nBu4NI as a cocatalyst. The 2–(1H–1,2,4–Triazol–3–yl)phenol/nBu4NI catalytic system was highly effective in the formation of cyclic carbonates from a wide range of terminal epoxides under ambient conditions. This system could convert the internal epoxides at 70 °C and 10 bar CO2 pressure within 12 h. The cyclic carbonates obtained from the 1,2–disubstituted epoxides showed stereochemical retention due to two consecutive SN2 reactions.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/catal14010090/s1, Table S1: List of known compounds [28,30,31,32,33,34,35,36,37,38,39,40,41]; Table S2: Crystallographic data for CAT–6; Figure S1: X-ray structure for 2–(2–benzyl–2H–tetrazol–5–yl)phenol(CAT–6); Figure S2: 1H NMR spectrum of 2–(1H–1,2,4–triazol–3–yl)phenol (CAT–1) in CDCl3; Figure S3: 1H NMR spectrum of 2–(1H–1,2,4–triazol–3–yl)phenol (CAT–1) in D2O; Figure S4: 13C NMR spectrum of 2–(1H–1,2,4–triazol–3–yl)phenol (CAT–1) in D2O; Figure S5: HR-MS spectrum of 2–(1H–1,2,4–triazol–3–yl)phenol (CAT–1); Figure S6: COSY spectrum of 2–(1H–1,2,4–triazol–3–yl)phenol (CAT–1) in D2O; Figure S7: HSQC spectrum of 2–(1H–1,2,4–triazol–3–yl)phenol (CAT–1) in D2O; Figure S8: HMBC spectrum of 2–(1H–1,2,4–triazol–3–yl)phenol (CAT–1) in D2O; Figure S9: 1H NMR spectrum of CAT–1 before (bottom) and after (top) bubbling CO2 in D2O; Figure S10: 13C NMR spectrum of CAT–1 before (bottom) and after (top) bubbling CO2 in D2O; Figure S11: 1H NMR spectrum of CAT–1 (bottom), 1a (middle), and a mixture of CAT–1 and 1a (top) in CDCl3. (a) O–H and N–H peaks in the 1H NMR spectrum of CAT–1 before (bottom) and after (top) adding 1a in CDCl3; (b) The methine proton and methylene protons on the three-membered ring carbons in the 1H NMR spectrum of 1a before (bottom) and after (top) adding CAT–1 in CDCl3; Figure S12: O–H and N–H peaks in the 1H NMR spectrum of CAT–1 before (bottom) and after (top) adding 1a in CDCl3; Figure S13: The methine proton and methylene protons on the three-membered ring carbons in the 1H NMR spectrum of 1a before (bottom) and after (top) adding CAT–1 in CDCl3; Figure S14: 1H NMR spectrum of 2–(1H–pyrazol–3–yl)phenol (CAT–2) in CDCl3; Figure S15: 13C NMR spectrum of 2–(1H–pyrazol–3–yl)phenol (CAT–2) in CDCl3; Figure S16: 1H NMR spectrum of 2–(1H–tetrazol–5–yl)pyridine (CAT–3) in DMSO–d6; Figure S17: 13C NMR spectrum of 2–(1H–tetrazol–5–yl)pyridine (CAT–3) in DMSO–d6; Figure S18: 1H NMR spectrum of 2–(2–methyl–2H–tetrazol–5–yl)phenol (CAT–4) in CDCl3; Figure S19: 13C NMR spectrum of 2–(2–methyl–2H–tetrazol–5–yl)phenol (CAT–4) in CDCl3; Figure S20: 1H NMR spectrum of 2–(1–phenyl–1H–imidazol–5–yl)phenol (CAT–5) in CDCl3; Figure S21: 13C NMR spectrum of 2–(1–phenyl–1H–imidazol–5–yl)phenol (CAT–5) in CDCl3; Figure S22: 1H NMR spectrum of 2–(2–benzyl–2H–tetrazol–5–yl)phenol (CAT–6) in CDCl3; Figure S23: 13C NMR spectrum of 2–(2–benzyl–2H–tetrazol–5–yl)phenol (CAT–6) in CDCl3; Figure S24: HR-MS spectrum of 2–(2–benzyl–2H–tetrazol–5–yl)phenol (CAT–6); Figure S25: 1H NMR spectrum for an aliquot of the reaction mixture after reaction in Table 1, entry 1; Figure S26: 1H NMR spectrum for an aliquot of the reaction mixture after reaction in Table 1, entry 2; Figure S27: 1H NMR spectrum for an aliquot of the reaction mixture after reaction in Table 1, entry 3; Figure S28: 1H NMR spectrum for an aliquot of the reaction mixture after reaction in Table 1, entry 4; Figure S29: 1H NMR spectrum for an aliquot of the reaction mixture after reaction in Table 1, entry 5; Figure S30: 1H NMR spectrum for an aliquot of the reaction mixture after reaction in Table 1, entry 6; Figure S31: 1H NMR spectrum for an aliquot of the reaction mixture after reaction in Table 1, entry 7; Figure S32: 1H NMR spectrum for an aliquot of the reaction mixture after reaction in Table 1, entry 8; Figure S33: 1H NMR spectrum for an aliquot of the reaction mixture after reaction in Table 1, entry 9; Figure S34: 1H NMR spectrum for an aliquot of the reaction mixture after reaction in Table 2, entry 2; Figure S35: 1H NMR spectrum for an aliquot of the reaction mixture after reaction in Table 2, entry 3; Figure S36: 1H NMR spectrum for an aliquot of the reaction mixture after reaction in Table 2, entry 4; Figure S37: 1H NMR spectrum for an aliquot of the reaction mixture after reaction in Table 2, entry 5; Figure S38: 1H NMR spectrum for an aliquot of the reaction mixture after reaction in Table 2, entry 6; Figure S39: 1H NMR spectrum for an aliquot containing mixtures after reaction for 2b in Figure 3; Figure S40: 1H NMR spectrum for an aliquot containing mixtures after reaction for 2c in Figure 3; Figure S41: 1H NMR spectrum for an aliquot containing mixtures after reaction for 2d in Figure 3; Figure S42: 1H NMR spectrum for an aliquot containing mixtures after reaction for 2e in Figure 3; Figure S43: 1H NMR spectrum for an aliquot containing mixtures after reaction for 2f in Figure 3; Figure S44: 1H NMR spectrum for an aliquot containing mixtures after reaction for 2g in Figure 3; Figure S45: 1H NMR spectrum for an aliquot containing mixtures after reaction for 2h in Figure 3; Figure S46: 1H NMR spectrum for an aliquot containing mixtures after reaction for trans–2i in Figure 4; Figure S47: 1H NMR spectrum for an aliquot containing mixtures after reaction for cis–2j in Figure 4; Figure S48: 1H NMR spectrum for an aliquot containing mixtures after reaction for cis–2k in Figure 4; Figure S49: 1H NMR spectrum for an aliquot containing mixtures after reaction for cis–2l in Figure 4; Figure S50: 1H NMR spectrum of purified 4–butyl–1,3–dioxolan–2–one (2a) in CDCl3; Figure S51: 13C NMR spectrum of purified 4–butyl–1,3–dioxolan–2–one (2a) in CDCl3; Figure S52: 1H NMR spectrum of purified 4–methyl–1,3–dioxolan–2–one (2b) in CDCl3; Figure S53: 13C NMR spectrum of purified 4–methyl–1,3–dioxolan–2–one (2b) in CDCl3; Figure S54: 1H NMR spectrum of purified 4–ethyl–1,3–dioxolan–2–one(2c) in CDCl3; Figure S55: 13C NMR spectrum of purified 4–ethyl–1,3–dioxolan–2–one(2c) in CDCl3; Figure S56: 1H NMR spectrum of purified 4–phenyl–1,3–dioxolan–2–one(2d) in CDCl3; Figure S57: 13C NMR spectrum of purified 4–phenyl–1,3–dioxolan–2–one(2d) in CDCl3; Figure S58: 1H NMR spectrum of purified 4–(chloromethyl)–1,3–dioxolan–2–one(2e) in CDCl3; Figure S59: 13C NMR spectrum of purified 4–(chloromethyl)–1,3–dioxolan–2–one(2e) in CDCl3; Figure S60: 1H NMR spectrum of purified 4–(methoxymethyl)–1,3–dioxolan–2–one(2f) in CDCl3; Figure S61: 13C NMR spectrum of purified 4–(methoxymethyl)–1,3–dioxolan–2–one(2f) in CDCl3; Figure S62: 1H NMR spectrum of purified 4–[(1,1–dimethylethoxy)methyl]–1,3–dioxolan–2–one(2g) in CDCl3; Figure S63: 13C NMR spectrum of purified 4–[(1,1–dimethylethoxy)methyl]–1,3–dioxolan–2–one(2g) in CDCl3; Figure S64: 1H NMR spectrum of purified 4–(phenoxymethyl)–1,3–dioxolan–2–one(2h) in CDCl3; Figure S65: 13C NMR spectrum of purified 4–(phenoxymethyl)–1,3–dioxolan–2–one(2h) in CDCl3; Figure S66: 1H NMR spectrum of purified trans–4,5–dimethyl–1,3–dioxolan–2–one(trans–2i) in CDCl3; Figure S67: 13C NMR spectrum of purified trans–4,5–dimethyl–1,3–dioxolan–2–one(trans–2i) in CDCl3; Figure S68: 1H NMR spectrum of purified cis–tetrahydrofuro[3,4–d][1,3]dioxol–2–one(cis–2j) in CDCl3; Figure S69: 13C NMR spectrum of purified cis–tetrahydrofuro[3,4–d][1,3]dioxol–2–one (cis–2j) in CDCl3; Figure S70: 1H NMR spectrum of purified cis–tetrahydro–4H–cyclopenta[d][1,3]dioxol–2–one (cis–2k) in CDCl3; Figure S71: 13C NMR spectrum of purified cis–tetrahydro–4H–cyclopenta[d][1,3]dioxol–2–one(cis–2k) in CDCl3; Figure S72: 1H NMR spectrum of purified cis–hexahydrobenzo[d][1,3]dioxol–2–one (cis–2l) in CDCl3; Figure S73: 13C NMR spectrum of purified cis–hexahydrobenzo[d][1,3]dioxol–2–one(cis–2l) in CDCl3.
Author Contributions
Conceptualization, Y.K. (Yoseph Kim) and Y.K. (Youngjo Kim); Data curation, S.C.; Investigation, Y.S., S.L., S.C. and Y.K. (Yoseph Kim); Supervision, Y.K. (Youngjo Kim); Validation, Y.K. (Yoseph Kim); Writing—original draft, Y.S., S.L. and Y.K. (Yoseph Kim); Writing—review and editing, Y.K. (Youngjo Kim). All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Research Foundation funded by the Ministry of Science and ICT of Korea (2020R1A2C2006412) and funding for the academic research program of Chungbuk National University in 2022.
Data Availability Statement
The original data are include in the article and Supplementary Materials. Further inquiries can be directly addressed to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Bobbink, F.D.; Van Muyden, A.P.; Dyson, P.J. En route to CO2-containing renewable materials: Catalytic synthesis of polycarbonates and non-isocyanate polyhydroxyurethanes derived from cyclic carbonates. Chem. Commun. 2019, 55, 1360–1373. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.-W.; Zhou, Z.-H.; He, L.-N. Efficient, selective and sustainable catalysis of carbon dioxide. Green Chem. 2017, 19, 3707–3728. [Google Scholar] [CrossRef]
- Weidlich, T.; Kamenická, B. Utilization of CO2-available organocatalysts for reactions with industrially important epoxides. Catalysts 2022, 12, 298. [Google Scholar] [CrossRef]
- Martín, C.; Fiorani, G.; Kleij, A.W. Recent advances in the catalytic preparation of cyclic organic carbonates. ACS Catal. 2015, 5, 1353–1370. [Google Scholar] [CrossRef]
- Alves, M.; Grignard, B.; Mereau, R.; Jerome, C.; Tassaing, T.; Detrembleur, C. Organocatalyzed coupling of carbon dioxide with epoxides for the synthesis of cyclic carbonates: Catalyst design and mechanistic studies. Catal. Sci. Technol. 2017, 7, 2651–2684. [Google Scholar] [CrossRef]
- Cokoja, M.; Wilhelm, M.E.; Anthofer, M.H.; Herrmann, W.A.; Kühn, F.E. Synthesis of cyclic carbonates from epoxides and carbon dioxide by using organocatalysts. ChemSusChem 2015, 8, 2436–2454. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, R.R.; Pornpraprom, S.; D’Elia, V. Catalytic strategies for the cycloaddition of pure, diluted, and waste CO2 to epoxides under ambient conditions. ACS Catal. 2018, 8, 419–450. [Google Scholar] [CrossRef]
- Fiorani, G.; Guo, W.; Kleij, A.W. Sustainable conversion of carbon dioxide: The advent of organocatalysis. Green Chem. 2015, 17, 1375–1389. [Google Scholar] [CrossRef]
- Yingcharoen, P.; Kongtes, C.; Arayachukiat, S.; Suvarnapunya, K.; Vummaleti, S.V.C.; Wannakao, S.; Cavallo, L.; Poater, A.; D’Elia, V. Assessing the pKa–dependent activity of hydroxyl hydrogen bond donors in the organocatalyzed cycloaddition of carbon dioxide to epoxides: Experimental and theoretical study. Adv. Synth. Catal. 2019, 361, 366–373. [Google Scholar] [CrossRef]
- Arayachukiat, S.; Kongtes, C.; Barthel, A.; Vummaleti, S.V.C.; Poater, A.; Wannakao, S.; Cavallo, L.; D’Elia, V. Ascorbic acid as a bifunctional hydrogen bond donor for the Synthesis of cyclic carbonates from CO2 under ambient conditions. ACS Sustain. Chem. Eng. 2017, 5, 6392–6397. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, G.; Kodama, K.; Hirose, T. An efficient metal- and solvent-free organocatalytic system for chemical fixation of CO2 into cyclic carbonates under mild conditions. Green Chem. 2016, 18, 1229–1233. [Google Scholar] [CrossRef]
- Hardman-Baldwin, A.M.; Mattson, A.E. Silanediol-catalyzed carbon dioxide fixation. ChemSusChem 2014, 7, 3275–3278. [Google Scholar] [CrossRef] [PubMed]
- Sperandio, C.; Rodriguez, J.; Quintard, A. Organocatalytic carbon dioxide fixation to epoxides by perfluorinated 1,3,5-triols catalysts. Org. Biomol. Chem. 2020, 18, 2637–2640. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Kim, Y.; Kim, H.; Cho, H.J.; Baik, M.-H.; Kim, Y. Scorpionate catalysts for coupling CO2 and epoxides to cyclic carbonates: A rational design approach for organocatalysts. J. Org. Chem. 2018, 83, 9370–9380. [Google Scholar] [CrossRef]
- Liu, N.; Xie, Y.-F.; Wang, C.; Li, S.-J.; Wei, D.; Li, M.; Dai, B. Cooperative multifunctional organocatalysts for ambient conversion of carbon dioxide into cyclic carbonates. ACS Catal. 2018, 8, 9945–9957. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, G.-X.; Zhang, W.-Z.; Lu, X.-B. CO2 adducts of phosphorus ylides: Highly active organocatalysts for carbon dioxide transformation. ACS Catal. 2015, 5, 6773–6779. [Google Scholar] [CrossRef]
- Wu, X.; Chen, C.; Guo, Z.; North, M.; Whitwood, A.C. Metal- and halide-free catalyst for the synthesis of cyclic carbonates from epoxides and carbon dioxide. ACS Catal. 2019, 9, 1895–1906. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Y. Boronic acids as hydrogen bond donor catalysts for efficient conversion of CO2 into organic carbonate in water. ACS Catal. 2016, 6, 4871–4876. [Google Scholar] [CrossRef]
- Gennen, S.; Alves, M.; Mereau, R.; Tassaing, T.; Gilbert, B.; Detrembleur, C.; Jerome, C.; Grignard, B. Fluorinated alcohols as activators for the solvent–free chemical fixation of carbon dioxide into epoxides. ChemSusChem 2015, 8, 1845–1849. [Google Scholar] [CrossRef]
- Martinez–Rodriguez, L.; Garmilla, J.O.; Kleij, A.W. Cavitand-based polyphenols as highly reactive organocatalysts for the coupling of carbon dioxide and oxiranes. ChemSusChem 2016, 9, 749–755. [Google Scholar] [CrossRef] [PubMed]
- Sopena, S.; Fiorani, G.; Martin, C.; Kleij, A.W. Highly efficient organocatalyzed conversion of oxiranes and CO2 into organic carbonates. ChemSusChem 2015, 8, 3248–3254. [Google Scholar] [CrossRef] [PubMed]
- Alves, M.; Grignard, B.; Gennen, S.; Detrembleur, C.; Jerome, C.; Tassaing, T. Organocatalytic synthesis of bio-based cyclic carbonates from CO2 and vegetable oils. RSC Adv. 2015, 5, 53629–53636. [Google Scholar] [CrossRef]
- Whiteoak, C.J.; Henseler, A.H.; Ayats, C.; Kleij, A.W.; Pericas, M.A. Conversion of oxiranes and CO2 to organic cyclic carbonates using a recyclable, bifunctional polystyrene-supported organocatalysts. Green Chem. 2014, 16, 1552–1559. [Google Scholar] [CrossRef]
- Whiteoak, C.J.; Nova, A.; Maseras, F.; Kleij, A.W. Merging sustainability with organocatalysis in the formation of organic carbonates by using CO2 as a feedstock. ChemSusChem 2012, 5, 2032–2038. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-Q.; Sun, J.; Cheng, W.-G.; Dong, K.; Zhang, X.-P.; Zhang, S.-J. Experimental and theoretical studies on hydrogen bond-promoted fixation of carbon dioxide and epoxides in cyclic carbonates. Phys. Chem. Chem. Phys. 2012, 14, 11021–11026. [Google Scholar] [CrossRef]
- Takaishi, K.; Okuyama, T.; Kadosaki, S.; Uchiyama, M.; Ema, T. Hemisquaramide tweezers as organocatalysts: Synthesis of cyclic carbonates from epoxides and CO2. Org. Lett. 2019, 21, 1397–1401. [Google Scholar] [CrossRef]
- Fan, Y.; Tiffner, M.; Schörgenhumer, J.; Robiette, R.; Waser, M.; Kass, S.R. Synthesis of cyclic organic carbonates using atmospheric pressure CO2 and charge–containing thiourea catalysts. J. Org. Chem. 2018, 83, 9991–10000. [Google Scholar] [CrossRef] [PubMed]
- Pagacz-Kostrzewa, M.; Saldyka, M.; Wierzejewska, M.; Khomenko, D.M.; Doroschuk, R.O. Theoretical DFT and matrix isolation FT IR studies of 2-(1,2,4-triazolyl)phenol isomers. Chem. Phys. Lett. 2016, 657, 156–161. [Google Scholar] [CrossRef]
- Wada, S.; Kushida, T.; Itagaki, H.; Shibue, T.; Kadowaki, H.; Arakawa, J.; Furukawa, Y. 13C NMR study on carbamate hydrolysis reactions in aqueous amine/CO2 solutions. Int. J. Greenh. Gas Control 2021, 104, 103175. [Google Scholar] [CrossRef]
- Kumar, S.; Jaller, D.; Patel, B.; LaLonde, J.M.; DuHadaway, J.B.; Malachowski, W.P.; Prendergast, G.C.; Muller, A.J. Structure based development of phenylimidazole-derived inhibitors of indoleamine 2,3-dioxygenase. J. Med. Chem. 2008, 51, 4968–4977. [Google Scholar] [CrossRef]
- Padwa, A.; Nahm, S.; Sato, E. Intramolecular 1,3-dipolar cycloaddition reactions of alkenyl-substituted nitrile imines. J. Org. Chem. 1978, 43, 1664–1671. [Google Scholar] [CrossRef]
- Castro-Osma, J.A.; Martinez, J.; de la Cruz-Martinez, F.; Caballero, M.P.; Fernandez-Baeza, J.; Rodriguez-Lopez, J.; Otero, A.; Lara-Sanchez, A.; Tejeda, J. Development of hydroxy-containing imidazole organocatalysts for CO2 fixation into cyclic carbonates. Catal. Sci. Technol. 2018, 8, 1981–1987. [Google Scholar] [CrossRef]
- Whiteoak, C.J.; Kielland, N.; Laserna, V.; Escudero-Adán, E.C.; Martin, E.; Kleij, A.W. A powerful aluminum catalyst for the synthesis of highly functional organic carbonates. J. Am. Chem. Soc. 2013, 135, 1228–1231. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.; Shin, M.S.; Hwang, S.; Kim, H.; Kim, M.; Kim, J.G.; Kim, Y. Tertiary amines: A new class of highly efficient organocatalysts for CO2 fixations. J. Ind. Eng. Chem. 2016, 44, 210–215. [Google Scholar] [CrossRef]
- Li, P.-Z.; Wang, X.-J.; Liu, J.; Lim, J.S.; Zou, R.; Zhao, Y. A triazole-containing metal-organic framework as a highly effective and substrate size-dependent catalyst for CO2 conversion. J. Am. Chem. Soc. 2016, 138, 2142–2145. [Google Scholar] [CrossRef]
- Paddock, R.L.; Nguyen, S.T. Chemical CO2 fixation: Cr(III) salen complexes as highly efficient catalysts for the coupling of CO2 and epoxides. J. Am. Chem. Soc. 2001, 123, 11498–11499. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Choi, S.H.; Ahn, D.; Kim, Y.; Ryu, J.Y.; Lee, J.; Kim, Y. Facile synthesis of a dimeric titanium(IV) complex with terminal Ti=O moieties and its application as a catalyst for the cycloaddition reaction of CO2 to epoxides. RSC Adv. 2016, 6, 97800–97807. [Google Scholar] [CrossRef]
- Stewart, J.A.; Drexel, R.; Arstad, B.; Reubsaet, E.; Weckhuysen, B.M.; Bruijnincx, P.C.A. Homogeneous and heterogenized masked N-heterocyclic carbenes for bio-based cyclic carbonate synthesis. Green Chem. 2016, 18, 1605–1618. [Google Scholar] [CrossRef]
- Jose, T.; Canellas, S.; Pericas, M.A.; Kleij, A.W. Polystyrene-supported bifunctional resorcinarenes as cheap, metal-free and recyclable catalysts for epoxides/CO2 coupling reactions. Green Chem. 2017, 19, 5488–5493. [Google Scholar] [CrossRef]
- Saptal, V.B.; Nanda, B.; Parida, K.M.; Bhanage, B.M. Fabrication of amine and zirconia on MCM-41 as acid-base catlysts for the fixation of carbon dioxide. ChemCatChem 2017, 9, 4105–4111. [Google Scholar] [CrossRef]
- Zheng, X.; Luo, S.; Zhang, L.; Cheng, J.-P. Magnetic nanoparticle supported ionic liquid catalysts for CO2 cycloaddition reactions. Green Chem. 2009, 11, 455–458. [Google Scholar] [CrossRef]
- Pangborn, A.B.; Giardello, M.A.; Grubbs, R.H.; Rosen, R.K.; Timmers, F.J. Safe and convenient procedure for solvent purification. Organometallics 1996, 15, 1518–1520. [Google Scholar] [CrossRef]
- Shelkar, R.; Singh, A.; Nagarkar, J. Amberlyst-15 catalyzed synthesis of 5-substituted 1-H-tetrazole via [3+2] cycloaddition of nitriles and sodium azide. Tetrahedron Lett. 2013, 54, 106–109. [Google Scholar] [CrossRef]
Figure 1.
2–(1H–1,2,4–Triazol–3–yl)phenol as a catalyst for the fixation of CO2 under ambient conditions.
Figure 1.
2–(1H–1,2,4–Triazol–3–yl)phenol as a catalyst for the fixation of CO2 under ambient conditions.

Figure 2.
(a) 1H NMR spectrum of CAT–1 before (bottom) and after (top) bubbling CO2 in D2O; (b) 13C NMR spectrum of CAT–1 before (bottom) and after (top) bubbling CO2 in D2O.
Figure 2.
(a) 1H NMR spectrum of CAT–1 before (bottom) and after (top) bubbling CO2 in D2O; (b) 13C NMR spectrum of CAT–1 before (bottom) and after (top) bubbling CO2 in D2O.

Figure 3.
Organocatalysts synthesized and investigated in this study.



Figure 6.
A plausible mechanism for the synthesis of cyclic carbonates from epoxides and CO2 by using CAT–1 in the presence of nBu4NI.
Figure 6.
A plausible mechanism for the synthesis of cyclic carbonates from epoxides and CO2 by using CAT–1 in the presence of nBu4NI.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Screening of various catalysts for the coupling of CO2 to 2–butyloxirane (1a) under ambient conditions in the presence of nBu4NI.
Table 1.
Screening of various catalysts for the coupling of CO2 to 2–butyloxirane (1a) under ambient conditions in the presence of nBu4NI.

| Entry 1 | Catalyst | Conversion (%) 2 | Yield (%) 3 | TON 4 | TOF (h−1) 5 |
|---|---|---|---|---|---|
| 1 | Phenol | 0 | 0 | 0 | 0 |
| 2 | 1H–1,2,4–triazole | 24 | 19 | 4.80 | 0.40 |
| 3 | Phenol+1H–1,2,4–triazole | 26 | 23 | 5.20 | 0.43 |
| 4 | CAT–1 | 96 | 93 | 19.2 | 1.60 |
| 5 | CAT–2 | 88 | 87 | 17.6 | 1.47 |
| 6 | CAT–3 | 38 | 34 | 7.60 | 0.63 |
| 7 | CAT–4 | 3 | 1 | 0.60 | 0.05 |
| 8 | CAT–5 | 4 | 2 | 0.80 | 0.07 |
| 9 | CAT–6 | 4 | 3 | 0.80 | 0.07 |
1 1a (10 mmol), catalyst (0.5 mmol, 5.0 mol%), nBu4NI (0.5 mmol, 5.0 mol%), 25 °C, 1 bar CO2 (balloon), 12 h, no solvent used. 2 Conversion determined by 1H NMR spectroscopy (see the Supplementary Materials). 3 Isolated yield. 4 TON, turnover number. 5 TOF, turnover frequency (TOF = TON/reaction time (h)).
Table 2.
Screening of various cocatalysts for the coupling of CO2 to 1a under ambient conditions using CAT–1.
Table 2.
Screening of various cocatalysts for the coupling of CO2 to 1a under ambient conditions using CAT–1.

| Entry 1 | Cocatalyst | Conversion (%) 2 | Yield (%) 3 | TON 4 | TOF (h−1) 5 |
|---|---|---|---|---|---|
| 1 | nBu4NI | 96 | 93 | 19.2 | 1.60 |
| 2 | PPNCl | 8 | 7 | 1.60 | 0.13 |
| 3 | nBu4NBr | 66 | 61 | 13.2 | 1.10 |
| 4 | nBu4NCl | 17 | 16 | 3.40 | 0.28 |
| 5 | DMAP | 0 | 0 | 0 | 0 |
| 6 | KI | 1 | 1 | 0.20 | 0.02 |
1 1a (10 mmol), CAT–1 (0.5 mmol, 5.0 mol%), cocatalyst (0.5 mmol, 5.0 mol%), 25 °C, 1 bar CO2 (balloon), 12 h, no solvent used. 2 Conversion determined by 1H NMR spectroscopy (see the Supplementary Materials). 3 Isolated yield. 4 TON, turnover number. 5 TOF, turnover frequency (TOF = TON/reaction time (h)).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Seong, Y.; Lee, S.; Cho, S.; Kim, Y.; Kim, Y. Organocatalysts for the Synthesis of Cyclic Carbonates under the Conditions of Ambient Temperature and Atmospheric CO2 Pressure. Catalysts 2024, 14, 90. https://doi.org/10.3390/catal14010090
AMA Style
Seong Y, Lee S, Cho S, Kim Y, Kim Y. Organocatalysts for the Synthesis of Cyclic Carbonates under the Conditions of Ambient Temperature and Atmospheric CO2 Pressure. Catalysts. 2024; 14(1):90. https://doi.org/10.3390/catal14010090
Chicago/Turabian StyleSeong, Yeongju, Sanghun Lee, Seungyeon Cho, Yoseph Kim, and Youngjo Kim. 2024. "Organocatalysts for the Synthesis of Cyclic Carbonates under the Conditions of Ambient Temperature and Atmospheric CO2 Pressure" Catalysts 14, no. 1: 90. https://doi.org/10.3390/catal14010090
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.