
Redox Chemistry of Pt(II) Complex with Non-Innocent NHC Bis(Phenolate) Pincer Ligand: Electrochemical, Spectroscopic, and Computational Aspects

 , , , , , , ,
, , , , , , ,
Abstract
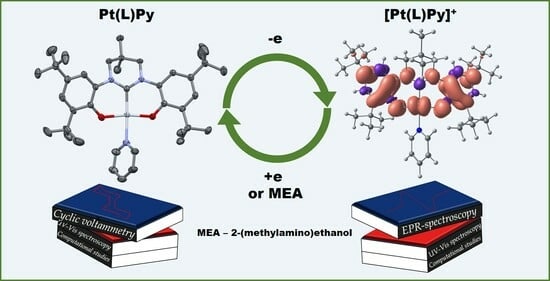
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General Considerations
3.2. X-ray Structure Determination
3.3. Cyclic Voltammetry (CV) and Differential Pulse Voltammetry (DPV)
3.4. Experimental Procedures and Product Characterization
3.4.1. Synthesis of Pt(L)Py
3.4.2. Synthesis of [Pt(L)Py][BF4]
3.5. Quantum-Chemical Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Broere, D.L.J.; Plessius, R.; Van Der Vlugt, J.I. New Avenues for Ligand-Mediated Processes-Expanding Metal Reactivity by the Use of Redox-Active Catechol, o-Aminophenol and o-Phenylenediamine Ligands. Chem. Soc. Rev. 2015, 44, 6886–6915. [Google Scholar] [CrossRef]
- Kaim, W.; Paretzki, A. Interacting Metal and Ligand Based Open Shell Systems: Challenges for Experiment and Theory. Coord. Chem. Rev. 2017, 344, 345–354. [Google Scholar] [CrossRef]
- Broere, D.L.J.; Mercado, B.Q.; Bill, E.; Lancaster, K.M.; Sproules, S.; Holland, P.L. Alkali Cation Effects on Redox-Active Formazanate Ligands in Iron Chemistry. Inorg. Chem. 2018, 57, 9580–9591. [Google Scholar] [CrossRef] [PubMed]
- Queyriaux, N. Redox-Active Ligands in Electroassisted Catalytic H+ and CO2 Reductions: Benefits and Risks. ACS Catal. 2021, 11, 4024–4035. [Google Scholar] [CrossRef]
- Singh, K.; Kundu, A.; Adhikari, D. Ligand-Based Redox: Catalytic Applications and Mechanistic Aspects. ACS Catal. 2022, 12, 13075–13107. [Google Scholar] [CrossRef]
- Kinzel, N.W.; Demirbas, D.; Bill, E.; Weyhermüller, T.; Werlé, C.; Kaeffer, N.; Leitner, W. Systematic Variation of 3d Metal Centers in a Redox-Innocent Ligand Environment: Structures, Electrochemical Properties, and Carbon Dioxide Activation. Inorg. Chem. 2021, 60, 19062–19078. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, L.A.; Vargo, N.P.; May, C.V.; Crockett, M.P.; Hyre, A.S.; McNeely, J.; Elinburg, J.K.; Brown, A.M.; Robinson, J.R.; Rheingold, A.L.; et al. Thiolate-Thione Redox-Active Ligand with a Six-Membered Chelate Ring via Template Condensation and Its Pt(II) Complexes. Inorg. Chem. 2021, 60, 13376–13387. [Google Scholar] [CrossRef]
- Mondal, R.; Guin, A.K.; Chakraborty, G.; Paul, N.D. Metal-Ligand Cooperative Approaches in Homogeneous Catalysis Using Transition Metal Complex Catalysts of Redox Noninnocent Ligands. Org. Biomol. Chem. 2022, 20, 296–328. [Google Scholar] [CrossRef]
- Luca, O.R.; Crabtree, R.H. Redox-Active Ligands in Catalysis. Chem. Soc. Rev. 2013, 42, 1440–1459. [Google Scholar] [CrossRef]
- Paul, N.D.; Rana, U.; Goswami, S.; Goswami, S.; Mondal, T.K. Azo Anion Radical Complex of Rhodium as a Molecular Memory Switching Device: Isolation, Characterization, and Evaluation of Current-Voltage Characteristics. J. Am. Chem. Soc. 2012, 134, 6520–6523. [Google Scholar] [CrossRef]
- Chirik, P.J.; Wieghardt, K. Radical Ligands Confer Nobility on Base-Metal Catalysts. Science 2010, 327, 794–795. [Google Scholar] [CrossRef]
- Suarez, A.I.O.; Lyaskovskyy, V.; Reek, J.N.H.; Van Der Vlugt, J.I.; De Bruin, B. Complexes with Nitrogen-Centered Radical Ligands: Classification, Spectroscopic Features, Reactivity, and Catalytic Applications. Angew. Chem.—Int. Ed. 2013, 52, 12510–12529. [Google Scholar] [CrossRef]
- Hoyt, J.M.; Schmidt, V.A.; Tondreau, A.M.; Chirik, P.J. Iron-Catalyzed Intermolecular [2+2] Cycloadditions of Unactivated Alkenes. Science 2015, 349, 960–963. [Google Scholar] [CrossRef]
- Fujita, D.; Sugimoto, H.; Morimoto, Y.; Itoh, S. Noninnocent Ligand in Rhodium(III)-Complex-Catalyzed C–H Bond Amination with Tosyl Azide. Inorg. Chem. 2018, 57, 9738–9747. [Google Scholar] [CrossRef]
- Jacquet, J.; Cheaib, K.; Ren, Y.; Vezin, H.; Orio, M.; Blanchard, S.; Fensterbank, L.; Desage-El Murr, M. Circumventing Intrinsic Metal Reactivity: Radical Generation with Redox-Active Ligands. Chem.—A Eur. J. 2017, 23, 15030–15034. [Google Scholar] [CrossRef]
- Demir, S.; Jeon, I.-R.; Long, J.R.; Harris, T.D. Radical Ligand-Containing Single-Molecule Magnets. Coord. Chem. Rev. 2015, 289–290, 149–176. [Google Scholar] [CrossRef]
- Jeon, I.R.; Sun, L.; Negru, B.; Van Duyne, R.P.; Dinca, M.; Harris, T.D. Solid-State Redox Switching of Magnetic Exchange and Electronic Conductivity in a Benzoquinoid-Bridged MnII Chain Compound. J. Am. Chem. Soc. 2016, 138, 6583–6590. [Google Scholar] [CrossRef] [PubMed]
- Degayner, J.A.; Wang, K.; Harris, T.D. A Ferric Semiquinoid Single-Chain Magnet via Thermally-Switchable Metal-Ligand Electron Transfer. J. Am. Chem. Soc. 2018, 140, 6550–6553. [Google Scholar] [CrossRef] [PubMed]
- Shimazaki, Y.; Yajima, T.; Tani, F.; Karasawa, S.; Fukui, K.; Naruta, Y.; Yamauchi, O. Syntheses and Electronic Structures of One-Electron-Oxidized Group 10 Metal(II)-(Disalicylidene)Diamine Complexes (Metal = Ni, Pd, Pt). J. Am. Chem. Soc. 2007, 129, 2559–2568. [Google Scholar] [CrossRef] [PubMed]
- Asami, K.; Tsukidate, K.; Iwatsuki, S.; Tani, F.; Karasawa, S.; Chiang, L.; Storr, T.; Thomas, F.; Shimazaki, Y. New Insights into the Electronic Structure and Reactivity of One-Electron Oxidized Copper(II)-(Disalicylidene)Diamine Complexes. Inorg. Chem. 2012, 51, 12450–12461. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, J.W. Free Radical Catalysis by Galactose Oxidase. Chem. Rev. 2003, 103, 2347–2363. [Google Scholar] [CrossRef] [PubMed]
- Lyons, C.T.; Stack, T.D.P. Recent Advances in Phenoxyl Radical Complexes of Salen-Type Ligands as Mixed-Valent Galactose Oxidase Models. Coord. Chem. Rev. 2013, 257, 528–540. [Google Scholar] [CrossRef]
- Shimazaki, Y.; Arai, N.; Dunn, T.J.; Yajima, T.; Tani, F.; Ramogida, C.F.; Storr, T. Influence of the Chelate Effect on the Electronic Structure of One-Electron Oxidized Group 10 Metal(Ii)-(Disalicylidene)Diamine Complexes. Dalt. Trans. 2011, 40, 2469–2479. [Google Scholar] [CrossRef] [PubMed]
- Thomas, F. Ligand-Centred Oxidative Chemistry in Sterically Hindered Salen Complexes: An Interesting Case with Nickel. Dalt. Trans. 2016, 45, 10866–10877. [Google Scholar] [CrossRef] [PubMed]
- Mustieles Marín, I.; Cheisson, T.; Singh-Chauhan, R.; Herrero, C.; Cordier, M.; Clavaguéra, C.; Nocton, G.; Auffrant, A. Electronic Structures of Mono-Oxidized Copper and Nickel Phosphasalen Complexes. Chem.—A Eur. J. 2017, 23, 17940–17953. [Google Scholar] [CrossRef] [PubMed]
- Oshita, H.; Yoshimura, T.; Mori, S.; Tani, F.; Shimazaki, Y.; Yamauchi, O. Characterization of the One-Electron Oxidized Cu(II)-Salen Complexes with a Side Chain Aromatic Ring: The Effect of the Indole Ring on the Cu(II)-Phenoxyl Radical Species. J. Biol. Inorg. Chem. 2018, 23, 51–59. [Google Scholar] [CrossRef]
- Colomban, C.; Philouze, C.; Molton, F.; Leconte, N.; Thomas, F. Copper(II) Complexes of N3O Ligands as Models for Galactose Oxidase: Effect of Variation of Steric Bulk of Coordinated Phenoxyl Moiety on the Radical Stability and Spectroscopy. Inorg. Chim. Acta 2018, 481, 129–142. [Google Scholar] [CrossRef]
- Smith, A.L.; Hardcastle, K.I.; Soper, J.D. Redox-Active Ligand-Mediated Oxidative Addition and Reductive Elimination at Square Planar Cobalt(III): Multielectron Reactions for Cross-Coupling. J. Am. Chem. Soc. 2010, 132, 14358–14360. [Google Scholar] [CrossRef]
- Dzik, W.I.; Van Der Vlugt, J.I.; Reek, J.N.H.; De Bruin, B. Ligands That Store and Release Electrons during Catalysis. Angew. Chem.—Int. Ed. 2011, 50, 3356–3358. [Google Scholar] [CrossRef]
- Borré, E.; Dahm, G.; Aliprandi, A.; Mauro, M.; Dagorne, S.; Bellemin-Laponnaz, S. Tridentate Complexes of Group 10 Bearing Bis-Aryloxide N-Heterocyclic Carbene Ligands: Synthesis, Structural, Spectroscopic, and Computational Characterization. Organometallics 2014, 33, 4374–4384. [Google Scholar] [CrossRef]
- Romain, C.; Miqueu, K.; Sotiropoulos, J.M.; Bellemin-Laponnaz, S.; Dagorne, S. Non-Innocent Behavior of a Tridentate NHC Chelating Ligand Coordinated onto a Zirconium(IV) Center. Angew. Chem.—Int. Ed. 2010, 49, 2198–2201. [Google Scholar] [CrossRef] [PubMed]
- Gafurov, Z.N.; Kantyukov, A.O.; Kagilev, A.A.; Kagileva, A.A.; Sakhapov, I.F.; Mikhailov, I.K.; Yakhvarov, D.G. Recent Advances in Chemistry of Unsymmetrical Phosphorus-Based Pincer Nickel Complexes: From Design to Catalytic Applications. Molecules 2021, 26, 4063. [Google Scholar] [CrossRef]
- Lee, M.T.; Hu, C.H. Density Functional Study of N-Heterocyclic and Diamino Carbene Complexes: Comparison with Phosphines. Organometallics 2004, 23, 976–983. [Google Scholar] [CrossRef]
- Lummiss, J.A.M.; Higman, C.S.; Fyson, D.L.; McDonald, R.; Fogg, D.E. The Divergent Effects of Strong NHC Donation in Catalysis. Chem. Sci. 2015, 6, 6739–6746. [Google Scholar] [CrossRef] [PubMed]
- Gandara, C.; Philouze, C.; Jarjayes, O.; Thomas, F. Coordination Chemistry of a Redox Non-Innocent NHC Bis(Phenolate) Pincer Ligand with Nickel(II). Inorg. Chim. Acta 2018, 482, 561–566. [Google Scholar] [CrossRef]
- Kunert, R.; Philouze, C.; Jarjayes, O.; Thomas, F. Stable M(II)-Radicals and Nickel(III) Complexes of a Bis(Phenol) N-Heterocyclic Carbene Chelated to Group 10 Metal Ions. Inorg. Chem. 2019, 58, 8030–8044. [Google Scholar] [CrossRef]
- Taakili, R.; Canac, Y. NHC Core Pincer Ligands Exhibiting Two Anionic Coordinating Extremities. Molecules 2020, 25, 2231. [Google Scholar] [CrossRef]
- Harris, C.F.; Bayless, M.B.; Van Leest, N.P.; Bruch, Q.J.; Livesay, B.N.; Bacsa, J.; Hardcastle, K.I.; Shores, M.P.; De Bruin, B.; Soper, J.D. Redox-Active Bis(Phenolate) N-Heterocyclic Carbene [OCO] Pincer Ligands Support Cobalt Electron Transfer Series Spanning Four Oxidation States. Inorg. Chem. 2017, 56, 12421–12435. [Google Scholar] [CrossRef]
- Goswami, M.; Lyaskovskyy, V.; Domingos, S.R.; Buma, W.J.; Woutersen, S.; Troeppner, O.; Ivanović-Burmazović, I.; Lu, H.; Cui, X.; Zhang, X.P.; et al. Characterization of Porphyrin-Co(III)-’nitrene Radical’ Species Relevant in Catalytic Nitrene Transfer Reactions. J. Am. Chem. Soc. 2015, 137, 5468–5479. [Google Scholar] [CrossRef]
- Rosenthal, A.J.; Vogt, M.; De Bruin, B.; Grützmacher, H. A Diolefin Diamide Rhodium(I) Complex and Its One-Electron Oxidation Resulting in a Two-Center, Three-Electron Rh-N Bond. Eur. J. Inorg. Chem. 2013, 2013, 5831–5835. [Google Scholar] [CrossRef]
- Luconi, L.; Gafurov, Z.; Rossin, A.; Tuci, G.; Sinyashin, O.; Yakhvarov, D.; Giambastiani, G. Palladium(II) Pyrazolyl–Pyridyl Complexes Containing a Sterically Hindered N-Heterocyclic Carbene Moiety for the Suzuki-Miyaura Cross-Coupling Reaction. Inorg. Chim. Acta 2018, 470, 100–105. [Google Scholar] [CrossRef]
- Luconi, L.; Garino, C.; Cerreia Vioglio, P.; Gobetto, R.; Chierotti, M.R.; Yakhvarov, D.; Gafurov, Z.N.; Morozov, V.; Sakhapov, I.; Rossin, A.; et al. Halogen-Bonding Interactions and Electrochemical Properties of Unsymmetrical Pyrazole Pincer NiII Halides: A Peculiar Behavior of the Fluoride Complex (PCN)NiF. ACS Omega 2019, 4, 1118–1129. [Google Scholar] [CrossRef]
- Luconi, L.; Tuci, G.; Gafurov, Z.N.; Mercuri, G.; Kagilev, A.A.; Pettinari, C.; Morozov, V.I.; Yakhvarov, D.G.; Rossin, A.; Giambastiani, G. Unsymmetrical Nickel (PCN) Pincer Complexes with a Benzothiazole Side-Arm: Synthesis, Characterization and Electrochemical Properties. Inorg. Chim. Acta 2020, 517, 120182. [Google Scholar] [CrossRef]
- Gafurov, Z.N.; Kantyukov, A.O.; Kagilev, A.A.; Sakhapov, I.F.; Luconi, L.; Rossin, A.; Giambastiani, G.; Babaev, V.M.; Islamov, D.R.; Usachev, K.S.; et al. Electrochemical Generation of Pyrazolyl-Pyridyl N-Heterocyclic Carbene Complexes of Nickel. Russ. J. Electrochem. 2021, 57, 134–140. [Google Scholar] [CrossRef]
- Gafurov, Z.N.; Kagilev, A.A.; Kantyukov, A.O.; Balabaev, A.A.; Sinyashin, O.G.; Yakhvarov, D.G. Classification and Synthesis of Nickel Pincer Complexes. Russ. Chem. Bull. 2018, 67, 385–394. [Google Scholar] [CrossRef]
- Gafurov, Z.N.; Bekmukhamedov, G.E.; Kagilev, A.A.; Kantyukov, A.O.; Sakhapov, I.F.; Mikhailov, I.K.; Khayarov, K.R.; Zaripov, R.B.; Islamov, D.R.; Usachev, K.S.; et al. Unsymmetrical Pyrazole-Based PCN Pincer NiII Halides: Reactivity and Catalytic Activity in Ethylene Oligomerization. J. Organomet. Chem. 2020, 912, 121163. [Google Scholar] [CrossRef]
- Gafurov, Z.N.; Zueva, E.M.; Bekmukhamedov, G.E.; Kagilev, A.A.; Kantyukov, A.O.; Mikhailov, I.K.; Khayarov, K.R.; Petrova, M.M.; Dovzhenko, A.P.; Rossin, A.; et al. Benzothiazole- vs. Pyrazole-Based Unsymmetrical (PCN) Pincer Complexes of Nickel(II) as Homogeneous Catalysts in Ethylene Oligomerization. J. Organomet. Chem. 2021, 949, 121951. [Google Scholar] [CrossRef]
- Long, J.; Lyubov, D.M.; Gurina, G.A.; Nelyubina, Y.V.; Salles, F.; Guari, Y.; Larionova, J.; Trifonov, A.A. Using N-Heterocyclic Carbenes as Weak Equatorial Ligands to Design Single-Molecule Magnets: Zero-Field Slow Relaxation in Two Octahedral Dysprosium(III) Complexes. Inorg. Chem. 2022, 61, 1264–1269. [Google Scholar] [CrossRef]
- Shimazaki, Y.; Stack, T.D.P.; Storr, T. Detailed Evaluation of the Geometric and Electronic Structures of One-Electron Oxidized Group 10 (Ni, Pd, and Pt) Metal(II)-(Disalicylidene) Diamine Complexes. Inorg. Chem. 2009, 48, 8383–8392. [Google Scholar] [CrossRef]
- Rotthaus, O.; Thomas, F.; Jarjayes, O.; Philouze, C.; Saint-Aman, E.; Pierre, J.L. Valence Tautomerism in Octahedral and Square-Planar Phenoxyl-Nickel(II) Complexes: Are Imino Nitrogen Atoms Good Friends? Chem.—A Eur. J. 2006, 12, 6953–6962. [Google Scholar] [CrossRef]
- Pistner, A.J.; Moon, H.W.; Silakov, A.; Yennawar, H.P.; Radosevich, A.T. Stable Open-Shell Phosphorane Based on a Redox-Active Amidodiphenoxide Scaffold. Inorg. Chem. 2017, 56, 8661–8668. [Google Scholar] [CrossRef]
- Poyatos, M.; Maisse-François, A.; Bellemin-Laponnaz, S.; Gade, L.H. Coordination Chemistry of a Modular N,C-Chelating Oxazole-Carbene Ligand and Its Applications in Hydrosilylation Catalysis. Organometallics 2006, 25, 2634–2641. [Google Scholar] [CrossRef]
- Schneider, N.; Bellemin-Laponnaz, S.; Wadepohl, H.; Gade, L.H. A New Class of Modular Oxazoline-NHC Ligands and Their Coordination Chemistry with Platinum Metals. Eur. J. Inorg. Chem. 2008, 2008, 5587–5598. [Google Scholar] [CrossRef]
- Jahnke, M.C.; Pape, T.; Hahn, F.E. Platinum Complexes with Picoline-Functionalized Benzimidazolin-2-Ylidene Ligands. Z. Naturforsch.—Sect. B J. Chem. Sci. 2010, 65, 341–346. [Google Scholar] [CrossRef]
- Meyer, D.; Zeller, A.; Strassner, T. Platinum Complexes with Pyrimidine-Functionalized N-Heterocyclic Carbene Ligands-Synthesis and Solid State Structures. J. Organomet. Chem. 2012, 701, 56–61. [Google Scholar] [CrossRef]
- Cao, P.; Cabrera, J.; Padilla, R.; Serra, D.; Rominger, F.; Limbach, M. Hydroamination of Unactivated Alkenes Catalyzed by Novel Platinum(II) N-Heterocyclic Carbene Complexes. Organometallics 2012, 31, 921–929. [Google Scholar] [CrossRef]
- Wei, C.H.; Hingerty, B.E.; Busing, W.R. Structure of Tetrakis(Pyridine)Platinum(II) Chloride Trihydrate: Unconstrained Anisotropic Least-Squares Refinement of Hydrogen and Non-Hydrogen Atoms from Combined X-Ray–Neutron Diffraction Data. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1989, 45, 26–30. [Google Scholar] [CrossRef]
- Thomas, F.; Jarjayes, O.; Duboc, C.; Philouze, C.; Saint-Aman, E.; Pierre, J.-L. Intramolecularly Hydrogen-Bonded versus Copper(II) Coordinated Mono- and Bis-Phenoxyl Radicals. Dalt. Trans. 2004, 17, 2662–2669. [Google Scholar] [CrossRef]
- Chiang, L.; Kochem, A.; Jarjayes, O.; Dunn, T.J.; Vezin, H.; Sakaguchi, M.; Ogura, T.; Orio, M.; Shimazaki, Y.; Thomas, F.; et al. Radical Localization in a Series of Symmetric NiII Complexes with Oxidized Salen Ligands. Chem.—A Eur. J. 2012, 18, 14117–14127. [Google Scholar] [CrossRef]
- Storr, T.; Wasinger, E.C.; Pratt, R.C.; Stack, T.D.P. The Geometric and Electronic Structure of a One-Electron-Oxidized Nickel(II) Bis(Salicylidene)Diamine Complex. Angew. Chem. 2007, 119, 5290–5293. [Google Scholar] [CrossRef]
- Mruthunjaya, A.K.V.; Torriero, A.A.J. Mechanistic Aspects of the Electrochemical Oxidation of Aliphatic Amines and Aniline Derivatives. Molecules 2023, 28, 471. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, J.; Chen, Z.; Zhang, A.; Ma, C. Synthesis of Nitrocarbazole Compounds and Their Electrocatalytic Oxidation of Alcohol. Cuihua Xuebao/Chin. J. Catal. 2016, 37, 533–538. [Google Scholar] [CrossRef]
- Steckhan, E. Indirect Electroorganic Syntheses—A Modern Chapter of Organic Electrochemistry [New Synthetic Methods (59)]. Angew. Chem. Int. Ed. Engl. 1986, 25, 683–701. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An Update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Spek, A.L. Structure Validation in Chemical Crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; Van De Streek, J. Mercury: Visualization and Analysis of Crystal Structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Dunning, T.H.; Hay, P.J. Gaussian Basis Sets for Molecular Calculations. In Methods of Electronic Structure Theory; Springer: Boston, MA, USA, 1977; pp. 1–27. [Google Scholar]
- Hay, P.J.; Wadt, W.R. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for the Transition Metal Atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Wadt, W.R.; Hay, P.J. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for Main Group Elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for K to Au Including the Outermost Core Orbitale. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-Fitting Basis Sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Peterson, K.A.; Figgen, D.; Goll, E.; Stoll, H.; Dolg, M. Systematically Convergent Basis Sets with Relativistic Pseudopotentials. II. Small-Core Pseudopotentials and Correlation Consistent Basis Sets for the Post-d Group 16-18 Elements. J. Chem. Phys. 2003, 119, 11113–11123. [Google Scholar] [CrossRef]
- Neese, F. Software Update: The ORCA Program System, Version 4.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2018, 8, e1327–e1332. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Moiety Formula Sum Formula | 2(C39H55N3O2Pt), 2(BF4), 3(CH2Cl2) C81H116B2Cl6F8N6O4Pt2 |
|---|---|
| formula weight | 2014.28 |
| temperature [K] | 100(2) |
| wavelength [Å] | 0.71073 |
| crystal system, space group | (No. 2) |
| a [Å] | 9.7661(12) |
| b [Å] | 13.8600(15) |
| c [Å] | 16.8963(19) |
| α [deg] | 102.107(3) |
| β [deg] | 91.972(4) |
| γ [deg] | 99.446(3) |
| V [Å3] | 2200.2(4) |
| Z, Dc [g cm−3] | 1, 1.520 |
| absorption coefficient [mm−1] | 3.424 |
| F(000) | 1016 |
| crystal size [mm] | 0.30 × 0.10 × 0.02 |
| Θ range for data collection [deg] | 2.4–32.0 |
| limiting indices | −14 ≤ h ≤ 14, −20 ≤ k ≤ 20, −25 ≤ l ≤ 25 |
| reflections measured reflections unique observed reflections [I > 2σ(I)] | 113,538 15,266 14,441 |
| GOF on F2 | 1.093 |
| data/restraints/parameters | 15266/2/516 |
| final R indices [I > 2σ(I)] | R1 = 0.0241, wR2 = 0.0588 |
| R indices (all data) | R1 = 0.0262, wR2 = 0.0593 |
| largest diff. peak and hole [e Å−3] | 2.02 and −1.47 |
| CCDC number | 2,277,791 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikhailov, I.K.; Gafurov, Z.N.; Kagilev, A.A.; Morozov, V.I.; Kantyukov, A.O.; Zueva, E.M.; Ganeev, G.R.; Sakhapov, I.F.; Toropchina, A.V.; Litvinov, I.A.; et al. Redox Chemistry of Pt(II) Complex with Non-Innocent NHC Bis(Phenolate) Pincer Ligand: Electrochemical, Spectroscopic, and Computational Aspects. Catalysts 2023, 13, 1291. https://doi.org/10.3390/catal13091291
Mikhailov IK, Gafurov ZN, Kagilev AA, Morozov VI, Kantyukov AO, Zueva EM, Ganeev GR, Sakhapov IF, Toropchina AV, Litvinov IA, et al. Redox Chemistry of Pt(II) Complex with Non-Innocent NHC Bis(Phenolate) Pincer Ligand: Electrochemical, Spectroscopic, and Computational Aspects. Catalysts. 2023; 13(9):1291. https://doi.org/10.3390/catal13091291
Chicago/Turabian StyleMikhailov, Ilya K., Zufar N. Gafurov, Alexey A. Kagilev, Vladimir I. Morozov, Artyom O. Kantyukov, Ekaterina M. Zueva, Gumar R. Ganeev, Ilyas F. Sakhapov, Asiya V. Toropchina, Igor A. Litvinov, and et al. 2023. "Redox Chemistry of Pt(II) Complex with Non-Innocent NHC Bis(Phenolate) Pincer Ligand: Electrochemical, Spectroscopic, and Computational Aspects" Catalysts 13, no. 9: 1291. https://doi.org/10.3390/catal13091291