

Hydrogen, Ammonia and Symbiotic/Smart Fertilizer Production Using Renewable Feedstock and CO2 Utilization through Catalytic Processes and Nonthermal Plasma with Novel Catalysts and In Situ Reactive Separation: A Roadmap for Sustainable and Innovation-Based Technology

Abstract
:
1. Introduction
1.1. Motivation for a Sustainable Distributed Process-Intensified Ammonia Technology
1.2. Critical Materials
1.2.1. Nano-Structured Microporous PolyHIPE Polymers (PHPs)
1.2.2. Need for Symbiotic/Smart Fertilizers
1.2.3. PolyHIPE Polymer for In Situ Ammonia Sequestration and Symbiotic/Smart Fertilizer Generation
1.2.4. Natural Materials for In Situ Ammonia Sequestration and Soil Additives
1.2.5. Heterogeneous Catalysts
1.2.6. Heterogeneous Catalysts for Plasma Processes
1.2.7. Plasma–Catalyst Interactions
1.2.8. Supported Nano-Catalysts with Semi-Conductor Characteristics and Crystal Defects
1.2.9. Microwave-Induced Plasma Generating Catalysts
1.3. Critical Processes
1.3.1. Gasification
1.3.2. Syngas Cleaning
1.3.3. Gasifier Ash as Micronutrient-Rich Soil Additive
1.3.4. Tars as a Natural Herbicide and Pesticide
1.4. Critical Process-Intensified Reactors
1.4.1. Process Intensification (PI)
1.4.2. Multi-Reaction Zone Reactors (M-RZR) for Ammonia and Symbiotic Fertilizer Production and In Situ Sequestration/Separation
2. Globally Integrated Process-Intensified Sustainable Hydrogen, Ammonia and Fertilizer Production with CO2 Sequestration: A Roadmap
3. Hydrogen Generation and Biomass-to-Electricity Conversion by Gasification
3.1. Performance of a Large-Scale Down-Draft Fixed-Bed Biomass Gasifier
3.2. Multi-Oxidant and Up-Down-Draft Fixed-Bed Gasifier Design
- (a)
- Feed Zone (68): Biomass is fed into the Feed Zone (68) using the feed screw (70) via an airlock.
- (b)
- Drying Zone (72) and Combustion Zone (80): As the biomass fuel moves downwards, hot combustion gases from the Combustion Zone (80) heat the biomass fuel and remove moisture from the fuel. The oxidant for the Combustion Zone (80) is air-fed from the air inlet (78). This facility is primarily used if bridging takes place. This method ensures that syngas quality is not compromised and the bridge is broken without having to use physical methods.
- (c)
- Pyrolysis Zone (82): Below the localized Combustion Zone (80), the Pyrolysis Zone (82) is located for which heat is provided by the oxidation zone below.
- (d)
- Oxidation Zone-1 (84): The partial oxidation of biomass is initially provided by supplying either air or externally oxygen-enriched air and the flue gases are removed from the exhaust (76) through an up-draft gasification mode. Once the temperature reaches ca. 800 °C, the oxygen-selective membranes become operational. Then the flue gas valve is closed and the syngas is withdrawn from the top of the outer reactor as illustrated in Figure 3a,b. When the steady state is established and the temperature in the oxidation Zone-1 reaches ca. 1200 °C, CO2 or H2O is injected into this zone (84a) through the inlet (100). A similar facility is present to inject CO2 or H2O into the reduction Zone (90) as shown in Figure 3c.
- (e)
- Reduction Zone (90): The Reduction Zone (90) is in the form of a catalytic vortex flow reactor, illustrated in Figure 3c. Gases from the Oxidation Zone (84) enter into the Reduction Zone (90) through the perforated grid (88). Zone (90) is in the form of a ring-main to which four vortex arms (94) are attached. The number of vortex arms can be increased with increasing syngas generation capacity. These tubular stainless steel vortex reactors are lined with porous nickel mesh and coated [34,35] using a silica-supported nickel catalyst (denoted as Ni/Si = 1/4 Mw-A) [82,83] for in situ thermal tar cracking. Syngas is accelerated through this reactor while mixing with either carbon dioxide or steam as oxidant. The injection of water into the ring-main (90) results in a rapid pressure rise and steam surge through the vortex arms and it results in the removal of particles from the catalyst surface. At the outlet, gases from the vortex arms impinge onto the wall of the outer reactor to separate the gases from the particulates in a cyclone process. These particles are small (<0.5 mm) as larger and denser particles move from the oxidation Zone-1 (84) directly into the second oxidation Zone-2 (103) under gravity. In order to reduce the load on the syngas suction fan, water injection is carried out at 3 bar through a non-return valve. The evaporation of water increases the pressure in the gasifier Reduction Zone (90).
- (f)
- Oxidation Zone-2 (103): Separated gases from the Reduction Zone (90) are sucked upwards in the annular space between the walls of the inner and outer reactors and leave the gasifier. Under steady-state operations, the gasifier operates in down-draft mode. However, in order to strip the bio-ash from its carbon, the surviving biomass particles from gasification are subjected to oxidation in the Oxidation Zone-2. The supply of the oxidant to this zone is identical to that in Oxidation Zone-1. The resulting gases from this Zone (103) flow vertically upwards into the Reduction Zone (90) under the action of the suction fan. In Zone (90), they mix with the gases from the oxidation Zone-1 (84) while the carbon-stripped bio-ash is collected at the entrance of the ash-removal auger (30) and removed gradually as they also act as a seal for the gasifier. Due to the high concentration of solids in this zone, a surface scraper (106) operates in this zone where the operation mode of the gasifier is up-draft.
- (g)
- Air Separation/Oxygen Enrichment Zone (20): There are two air separation/oxygen enrichment Zones both represented as (20) in Figure 3a and Figure 3d. They provide oxygen-enriched air to oxidation Zones 1 and 2. They are identical in structure but the oxygen enrichment level in Zone-2 (84b) is lower depending on the desired level of carbon-stripping. The structure of these zones is shown in Figure 3d. Air intake (16) into the air-space (108) allows the establishment of thermal equilibrium so that the oxygen-selective dense membrane (15) is at ca. 800 °C although these membranes can operate at a lower temperature (ca. 600 °C) [3]. Therefore, the temperatures in the oxidation Zones 1 and 2 are >800 °C and 600 °C, respectively, commensurate with the temperatures of these zones. However, the surface wall temperatures in these zones are very high (ca. 1000–1300 °C) due to syngas combustion on oxygen-rich surfaces. The oxygen-selective membrane (15) is supported by a microporous Ni-B metal to protect the membrane against thermal spikes. Oxygen atoms permeate through the membrane and recombine to form oxygen molecules on the permeate side in the O2 space (110) before entering into the oxidation Zone (84) through the perforated high-temperature stainless steel wall (111) and another perforated ceramic lining (112). Examples of oxygen-selective membranes include perovskites such as BaCo(0.4)Fe(0.6−x)Zr(x)O(3−δ) where x = 0–0.4 and δ is the oxygen vacancy. Oxygen permeation is enhanced by the presence of an exothermic reaction on the permeate side as this creates a very large entropy-based driving force for diffusion as shown previously [3,138]. Membranes are sealed in their position using commercially available high-temperature ceramic sealants [3,138]. The retentate air stream leaves the oxygen separation zone at the exit (18) with enhanced nitrogen content.
- (h)
- Other Facilities: Syngas from the gasifier outlet is subject to syngas cleaning, which results in tar recovery. Tars and bio-ash can either be utilized in various applications or returned to the gasifier from the inlet port (37). As seen from Figure 3b, in addition to the main syngas outlet and outlet (76) there are four gas sampling ports, which are located at the Drying Zone (72), Pyrolysis (82) Oxidation Zone-1 (84) and Reduction Zone (90). The temperatures in these zones as well as at the oxidation Zone-2 (103) and syngas outlet are also monitored. The data collected are used to control the syngas quality as well as to ensure a continuous operation. Under normal operating conditions, the gasifier is started by using air- or oxygen-enriched air so that excessive heating of the gasifier is avoided. Alternatively, a mixture of oxygen and CO2 can be used for this purpose. When the equilibrium is reached with temperature in the oxidation zone ca. 1200 °C, intermitted water injection can start for steam gasification. However, water injection lowers the temperature and hence water injection is terminated until the next cycle when the temperature of the oxidation zone is reduced to 1000 °C. In this way, the average temperature in the oxidation zone is kept at ca. 1100 ± 100 °C. However, this temperature can be increased or reduced at will through primary changes in the equivalence ratio and water injection frequency and its amount.
3.3. Multi-Oxidant Gasification of Biomass for Hydrogen Generation
4. Syngas Cleaning
4.1. Sulfonated PolyHIPE Polymer (s-PHP) in Tar Removal from Syngas
4.2. Syngas Cleaning Equipment
4.3. Model Syngas and Tar
4.4. Syngas Tar Removal Equipment Performance
5. Catalytic Plasma Splitting of Carbon Dioxide with Oxygen Sequestration
5.1. Background
- (a)
- In addition to ammonium carbamate formation when NH3 is sequestrated, do nitrated ammonium derivatives such as ammonium nitrate, urea, etc. form in the catalytic syngas conversion to fertilizers?
- (b)
- Can the level of oxygen in the reaction mixture be controlled to enhance conversion and selectivity towards a target product through oxygen sequestration by in situ reactive separation?
- (c)
- Can any such sequestration process yield a sustainable and useful product so as to lower the energy cost of CO2 capture and utilization?
- (d)
- In addition to the optimization of process variables such as the type and strength of plasma and plasma reactor, can plasma catalysts be designed to achieve the desired product and to achieve cost benefits?
- (e)
- Finally, can biochar as well as ash from the gasification of biomass (i.e., bio-ash) be used as a catalyst in plasma-induced CO2 splitting?
5.2. Catalytic Dielectric Discharge Barrier (DBD) Nonthermal Plasma Equipment
- EC-1a: Both electrodes (2 and 6 in Figure 7b) are outside the plasma zone and isolated through the inner and outer walls of the quartz reactor.
- EC-3: Both electrodes (2 and 6) are within the plasma zone and hence both electrodes are not isolated.
- EC-4: Ground electrode (6) is behind the perforated outer wall. High voltage electrode (2) is behind the inner wall which is not perforated. This applies to the M-RZR system shown in Figure 7b.
5.3. Carbon Dioxide-to-Carbon Monoxide Conversion in a Single Reaction Zone DBD Reactor
5.4. Effect of Nitrogen Concentration in CO2 Conversion
5.5. Catalytic Plasma Carbon Dioxide Splitting in the Presence of Biochar as a Catalyst and In Situ Oxygen Scavenger
5.6. Biochar as Plasma Catalyst and Oxygen Scavenger
6. Direct Conversion of Hydrogen Enriched Model Syngas to Ammonia via Ammonium Carbamate with CO2 Sequestration and Utilization
6.1. Background
6.2. Experimental Results
7. Piezoelectric Catalyst Development
7.1. Microwave Radiation Induced Ferroelectric-to-Piezoelectric Phase Transition under Plasma and Its Application to Synthesis of Piezoelectric Black Barium Titanate Catalyst
- (a)
- Clear reflection intensity change at 44.90°, (002) and 45.37°, (200) for the black-colored BaTiO3 and equalization of the corresponding intensities for the cream-colored BaTiO3.
- (b)
- The broadening of the peaks in the microwave-irradiated samples indicates that, as expected, the microwave-irradiated samples have higher amorphous fractions than the fresh BaTiO3. However, the cream-colored BaTiO3 is more amorphous than the black-colored BaTiO3.
- (c)
- Due to the amorphization process, a number of weak peaks disappear in the microwave-irradiated samples.
- (d)
- In the microwave-irradiated samples, in addition to the broadening of the peaks, peak positions shift towards higher Bragg angles.
7.2. Defect Engineering in Catalysts and Perovskites
7.3. Anion Vacancies
7.4. Cation Vacancies
7.5. Amorphization Process in Ferroelectric-Piezoelectric Catalysts
7.6. Chemical and Morphological Heterogeneity of the Microwave Irradiated BaTiO3
7.7. Morphological Heterogeneity and Amorphization of the Microwave Irradiated Barium Titanate with Plasma Generation
7.8. BaTiO3 Crystal Lattice Structure, Domain Walls and Grain Boundaries
7.9. Experimental Results on Microwave Radiation and Plasma-Induced Morphological Transformations in BaTiO3
7.10. Mechanism of Lattice-Exudate Formation and Amorphization of BaTiO3
8. Preparation of Supported High-Entropy Catalysts for Nonthermal Plasma Facilitated Reactions
8.1. Background
8.2. Significance of Supported High-Entropy Catalysts
8.3. Experimental Results
8.4. Compositional and Morphological Heterogeneity
9. Quantum Effects in Catalytic Plasma Processes, Catalyst Porogenesis and Generation of Negatively Charged Plasma Dust Particles as Collider Catalysts
9.1. Background
9.2. Analytical Techniques and In Situ Observation of Catalyst/CO2 Interactions
9.3. Quantum Effects in Catalytic Plasma Reactions: Catalyst—CO2 Interactions
9.4. Quantum Effects in Catalytic Plasma Reactions: Plasma-Induced Catalyst Erosion and Dusty (Complex) Plasma Characteristics
- (a)
- Formation of dust particles by plasma-induced catalyst erosion, which is likely to be acute due to the extensive morphological defects which are present in photo- and plasma-catalysts.
- (b)
- Nano-dust particles are formed either as quantum dots with semi-conduction characteristics or nano-particles from the reactive precursors.
- (c)
- (d)
- (e)
- The ion density and electric potential profile are almost independent of both, dust size as well as dust density [340].
- (f)
- Results available from the RF-generated plasma suggest that the plasma and dust cloud appear to coexist and coupling of both is weak [340].
- (g)
- Many-particle quantum mechanics calculations by Balzer et al. [341] predict that energetic ion-particle collision can excite the electrons in the solid leading to the occupation of a single lattice position by two electrons thus forming a doubloon. Doubloons have homogeneous distribution in this non-equilibrium state [341].
- (h)
- In the investigation of dust density waves (propagation of dust particles in the subsonic ion flow direction or at an oblique angle), it was found that the impact between electrons and neutral gas atoms (which results in plasma glow as a signature of energetic electrons) was enhanced in the presence of dust and the maximum in glow brightness was followed by dust density maximum [342].
- (i)
- If the interparticle spacing is on the order of or less than the Debye length, the dust particles will interact strongly through their mutual Coulomb fields [337]. When the plasma-generating supported perovskite or spinel catalysts, (including BaTiO3) are used as packed beds in a plasma reactor, this condition is satisfied.
9.5. Experimental Demonstration of Microwave Plasma-Induced Erosion of BaTiO3 Particles
9.6. Mechanism of Particle Generation and Porogenesis
9.7. Third Body Impact in Catalysis
10. Mechanism of Catalytic Plasma CO2 Reduction Using BaTiO3 and M/Si = X Catalysts
10.1. Background
10.2. Simplified Modelling of TiO2-Terminated BaTiO3 (100) Surface and Reaction Mechanism
- (a)
- Reaction-1. CO2 reduction at oxygen vacancy [Ti3+ {#} Ti3+] and restoration of {O2−} site. We can assume that microwave plasma processed BaTiO3 already has a range of oxygen vacancies as depicted in Figure 23 (Site-A). Reaction-1 illustrates the restoration of the {O2−} site through electron transfer from two Ti3+ cations bordering the oxygen vacancy to the CO2 molecule with the release of CO.
- (b)
- Reaction-2. CO2+ cation generation at the [Ti3+ {#} Ti4+] sites. In nonthermal plasma reactions, CO2 cations are known to be generated and attributed to high-energy electron impact with CO2 molecules [91]. These ions can also be generated when CO2 molecules are adsorbed at the [Ti3+ {#} Ti4+] oxygen vacancy sites as in Figure 23 (Site-C). As a result, the Ti4+ cations are converted into a more active Ti3+ state as in [Ti3+ {#} Ti3+]. The reaction is shown in Reaction-2.
- (c)
- Reaction-3. O2 generation at [Ti3+ {O2−} Ti4+] and restoration of [Ti3+ {#} Ti3+] sites. CO2+ cations generated in Reaction-2 are consumed at the active Site-B leading to CO and O2 generation and the restoration of the oxygen vacancy [Ti3+ {#} Ti3+] as shown in Reaction-3. When combined, Reactions 1 and 2 restore the existing oxygen vacancies, leading to CO2 dissociation into CO and O2.
- (d)
- Reaction-4. Charged catalyst plasma dust particles as an electron source and a third-body, M(n)−. The above mechanisms take place on the catalyst surface. Several other reactions occur in the plasma space based on electron impact. It is often proposed that CO2 decomposition can take place in the plasma space through electron impact. The electron-rich plasma dust particles can be represented as M(n)−, where n is the number of electrons per dusty plasma particle. We can assume that the impact of CO2 with them results in the reduction of CO2 and the formation of oxygen atoms according to Reaction-4.
- (e)
- Reaction-5. Reaction of oxygen radicals at [Ti4+ {O2−} Ti4+] sites. Oxygen radicals are highly soluble and can diffuse within the crystal lattice [323] to react with the oxygen anions and cause the formation of oxygen vacancies.
11. Preparation of Composite Supported Metal Oxide Catalysts and Piezoelectric Barium Titanate
11.1. Background
11.2. Experimental Results
11.3. Implications of Piezoelectric Phase Formation during Plasma Reactions
12. Conclusions and Recommendations
12.1. Processes for Hydrogen and Syngas Generation for Ammonia and Symbiotic-Smart Fertilizers
- (a)
- The production is carried out in small-scale, distributed platforms with an energy handling capacity of ca. 10–50 MWe, rather than at GWe levels as in centralized facilities such as the Haber–Bosch ammonia plants [3,39,366]. Distributed plants have short start-up and shut-down times, and are able to operate with highly variable feedstock, sourced locally. The fully integrated process-intensified plants do not have the burden of “economies of scale”, which are the characteristics of centralized production plants [135].
- (b)
- In order to provide low capital and operating costs, IPI-based reactors should perform more than one unit operation, such as those in Multi-Reaction Zone Reactors [3,39] and those proposed by Hessel et al. [148,151]. In this respect, non-thermal, atmospheric, low-temperature, catalytic plasma is ideally suited as the primary reaction zone. The examples provided here include ammonia synthesis and ammonia sequestration using either CO2 to obtain ammonium carbamate/urea or acidic porous materials to obtain symbiotic-smart fertilizers. Other examples include: (i) plasma CO2 splitting using biochar or bio-ash from gasifiers as catalysts and in situ biochar upgrading, (ii) use of hydrogen-enriched syngas from a multi-oxidant biomass gasifier to obtain ammonium carbamate as a precursor for urea.
- (c)
- As shown, by using the multi-oxidant (oxygen-enriched air and water) gasification, the hydrogen content of syngas can be brought to a level for direct ammonium carbamate production. However, the resulting syngas still requires cleaning/tar elimination before it can be used for ammonia/ammonium carbamate production. As hydrogen is the limiting component in most applications, if needed, the whole syngas can be converted to hydrogen through a water–gas shift reaction (CO + H2O = H2 + CO2) [367].
- (d)
- The pilot-plant size 50 kWe multi-oxidant up-down-draft fixed-bed gasifier benefits from in situ air separation using oxygen-selective membranes operating in the oxidation zone at the mean temperature of 1100 °C. The function of this type of membrane is not to completely separate air but to enrich the air oxygen concentration in order to increase the temperature of the oxidation zone in the catalyst for water injection and also to lower the nitrogen concentration in syngas so that it is suitable for direct ammonium carbamate and subsequently its conversion to anhydrous ammonia. The permeation flux of oxygen can be further increased (by nearly 100-fold) through the activation of the oxygen-selective membranes at ca. 1000 °C by using non-Faradaic electrochemical catalysis (NEMCA) [368,369,370].
- (e)
- Throughout this study, the catalytic plasma technology has been promoted in various conversion processes as the Reaction Zone-1 (RZ-1) in the Multi-Reaction Zone Reactor (M-RZR). Therefore, there are issues to be considered: (1) Plasma reactors, and (2) Catalysts for use in plasma reactors.
- (1)
- Dielectric Barrier Discharge (DBD) reactors are routinely used in catalytic plasma synthesis. Although some engineering knowledge is available for such reactors (see for example [39,170] and references therein), they are not satisfactory for industrial-scale operations due to the presence of high electric fields, precision engineering, limited capacity and explosive gases. Therefore, the use of microwave-induced plasma reactors should be considered in industrial applications as industrial-scale microwave reactors are already in use.
- (2)
- In this study, the nature of efficient radiation-induced catalyst activation and their performance is investigated. It is clear that the attributes of efficient catalysts for plasma and UV-radiation applications are significantly different than the thermochemical catalysts. However, plasma and UV-radiation catalysts themselves have also differences. UV-radiation is necessary for the catalyst excitation whereas no such requirement is needed for plasma catalysts, except for piezoelectric plasma catalysts, which can be activated through external stressors such as thermal fluctuations or microwave/ultrasound excitations which create transient polarization in such catalysts.
12.2. Black-Barium Titanate (BaTi1−rMrO3−x−y{#}xNz) as a Unique Piezoelectric Plasma Catalyst with Quantum Effects: Synthesis, Structure Evaluation and Performance in CO2 Splitting
- (a)
- Following the review of recent developments in catalysts for UV-radiation-induced reactions, in particular for water-splitting reactions, catalysts for plasma applications were considered. A novel process is presented, which is based on microwave radiation-induced conversion of paraelectric-ferroelectric BaTiO3 to black piezoelectric barium titanate with disordered lattice containing N- and Ti -substitutions and oxygen vacancies {#}. The non-stoichiometric formula of the resulting black-barium titanate is BaTi1−rMrO3−x−y{#}xNz, where M is a transition metal catalyst (optional).
- (b)
- The radiation-induced chemical heterogeneity leading to the formation of BaTi1−rMrO3−x−y{#}xNz also results in structural heterogeneity which is evaluated by extensive SEM and EDS studies. These studies reveal the mechanism of chemical and morphological transformations. It is shown that during the microwave irradiation of HNO3 (or H2SO4), doped BaTiO3 underwent rapid solid-state transformation, which was accompanied by color change (white-to-black) and extensive amorphization as detected by XRD. The resulting material had a large number of morphological features including nano- and micro-plates decorated and separated by nano-sized domains which are described as crystal lattice exudates.
- (c)
- Another important microscopic morphological transition is porogenesis when particles in the size range of ca. 10 nm–1000 nm are ejected from the surface of the BaTiO3 particles during the microwave radiation-induced phase change. This porogenetic process also penetrates the bulk making the chemically heterogenetic barium titanate (BaTi1−rMrO3−x−y{#}xNz) porous which is also useful in plasma catalysis for surface area enhancement and plasma propagation.
- (d)
- The porogenetic particles with a chemical structure of BaTi1−rMrO3−x−y{#}xNz, can be considered plasma dust particles, which are catalytically more active than the bulk material due to their nano-size. They are essentially negatively charged BaTi1−rMr O3−x−y{#}xNz assemblies but do not have the electron holes. The implications of the nano-structure in the BaTi1−rMrO3−x−y{#}xNz catalyst and in the porogenetic particles are considered in terms of quantum effects which can provide new catalytic pathways in plasma reactions.
- (e)
- The occurrence of dusty (complex) plasma ranges from natural to artificial environments, covering a host of scientific disciplines, from astrophysics to magnetic fusion devices (tokamaks) to semiconductor processing. This range also indicates the size range of the plasma environment. It will be interesting to investigate the quantum effects within the pores of catalysts where the plasma environment is confined to μm-size domains with nano-size catalytic particles.
12.3. Supported High Entropy Catalysts
- (a)
- An example of a supported high-entropy catalyst synthesis is provided for the first time. The synthesis was performed using the microwave shock as described previously [82,83]. This catalyst can be represented as (Cr0.1Fe0.1Co0.1Cu0.1Zn0.1)O3−x {#}x/SiO2 = 1. The catalytic activity of such a catalyst is based on the cocktail effect of the components and their alloys, which is further enhanced by its highly amorphous morphology. The SEM and EDS studies show that the structure is devoid of nano-structured decorations. The surface composition is dictated by Co but Zn is almost absent.
- (b)
- In order to understand the structure formation in the supported high-entropy catalysts, silica-supported medium entropy catalysts were also investigated. It was shown that Cu- and Co-containing supported catalysts are most likely to form surface decorations. However, in high-entropy catalysts formed from components with similar atomic size and multi-valent states, the probability of alloying increases, and hence the surface decorations (which form as a result of phase separation) are absent.
12.4. Biochar/Bio-Ash as a Natural Co-Catalyst in CO2 Splitting and Biochar Upgrading
- (a)
- CO2 splitting to CO and O2 was studied using a nonthermal catalytic plasma reactor. As catalyst, BaTi1−rMrO3−x−y{#}xNz and Co/Si = 1/4 catalyst were used. The results were interpreted in terms of phase transition in BaTiO3 to the defect-rich piezoelectric barium titanate. The presence of a silica-supported Co-catalyst enhanced CO2 conversion provided that BaTiO3 was in the piezoelectric state. In addition, biochar was used in order to sequestrate the evolved O2 which enhanced conversion and at the same time upgraded biochar (for use as a soil additive) by oxygenation.
- (b)
- A mechanism of catalytic plasma-induced CO2 is proposed based on TiO2-terminated BaTiO3−x{#}x (100) surface. The overall reaction mechanism is based on oxygen vacancy generation – restoration reactions involving Ti3+, Ti4+, O2− and dusty-plasma particles. It is argued that this type of modelling is more informative in the understanding of heterogeneous catalytic reactions.
12.5. Ammonia Synthesis and Efficiency Evaluation
- (a)
- We consider the effect of combined BaTiO3−x−y{#}xNz and Ni/Si = 1/4 catalysts in ammonia synthesis in order to demonstrate the importance of defect-rich piezoelectric black-BaTiO3 catalysts and negatively charged plasma dust particles. It is important to note that, as some 85% of anhydrous ammonia is used in fertilizer production, direct ammonia-based fertilizer production should be the priority, which can make the current catalytic plasma production economically feasible. Therefore, the comparison included various data on catalytic plasma NH3 synthesis and it’s in situ conversion to highly effective symbiotic-smart ammonia fertilizers.
- (b)
- A comprehensive review of unconventional catalysis by Bogaerts et al. [367] indicated that in catalytic plasma ammonia synthesis, the lowest energy cost was 1.5 MJ/mol [371] and the outlet NH3 concentration of 9.0 mol% (yield 17.1%) [69]. It is important to note that the corresponding values for Haber–Bosch ammonia are: 0.5 MJ/mol and 8–15 mol%. Very high outlet ammonia concentrations can be obtained under special conditions. Shah et al. [372] obtained an ammonia concentration of 19.1 mol% by using RF-plasma under reduced pressure whereas, 19.4% ammonia concentration per pass (nitrogen conversion 40.4%) was achieved with in situ ammonia sequestration using multi-reaction zone reactor catalytic plasma with BaTiO3−x−y{#}xNz and Ni/Si = 1/4 catalyst [3,67]. Wang et al. [373] also used a membrane-based in situ separation via catalyst protection in a DBD reactor to obtain an ammonia yield of 1.5 g/kWh with 5.3% outlet NH3 concentration. The effects of reactor type, electrode configuration and catalyst on NH3 outlet concentration and energy yield are shown in Table 15.
- (c)
- The effect of catalyst and co-catalyst is also illustrated using DBD-1 and DBD-2 reactors. It was found that Ni/Si = X catalyst performed best compared with Co/Si = X and Fe/Si = X catalyst [67] and hence subsequently, Ni/Si = X catalyst was used in all the ammonia synthesis experiments, including when these catalysts were used with BaTiO3 as a co-catalyst/plasma catalysis promoter. The efficiency of Ni catalyst in [M/Si = X]/BaTiO3 = Y (M = Ni, Co, Fe) over Co and Fe was confirmed recently [93]. It can be seen from Table 15 that, the Ni/Si = 1/4 catalyst performs better than BaTiO3 with respect to outlet ammonia concentration and energy yield. When Ni/Si = 1/4 catalyst and BaTiO3 are used together, efficiency increases although the outlet ammonia concentration remains unchanged at 6.4%. The outlet concentration increases with increasing power input; at the power input of 195 W, the outlet concentration reaches 9.0%, which is comparable to that of the Haber–Bosch process.
- (d)
- It can be seen from Table 15 that when ammonia was sequestrated using acidic PolyHIPE Polymer using the Multi-Reaction Zone Reactor (Number- 8; M-RZR in Table 15), the maximum outlet concentration increased to 19.4% and the energy cost reduced 13.2 MJ/mol-NH3. It must be pointed out that these results were obtained under non-optimized conditions both for Ni/Si = X (X could be increased to 2) and processing conditions, including plasma power and frequency and total gas flow rate, etc. Optimization enhancements of energy yield efficiency by a factor of 3 [359].
- (e)
- The combination of a metal catalyst with a ferroelectric perovskite plasma catalytic promoter, lead zirconate titanate (PZT) was used by Navascues et al. [303] as the Curie temperature of PZT is significantly higher than that of BaTiO3. These catalyst systems were (1) PZT; (2) Al2O3 coated PZT and (3) Ru/Al2O3 coated PZT. It was concluded that, unlike several previous reports, the yield and energy efficiency for the ammonia synthesis did not significantly improve when including Ru particles, even at temperatures at which an incipient catalytic activity could be inferred. However, under some operating conditions, Al2O3-coated PZT gave a higher yield and energy efficiency. It is not clear that the structural modifications described in the current study for BaTiO3 apply to PZT and therefore should be investigated.
- (f)
- Although it is possible that these results are specific to the PZT and Ru/Al2O3 combination, they indicate the importance of both the plasma catalysis promotor (moderator) and the supported catalyst system. BaTiO3 represents a unique position as a ferroelectric/piezoelectric catalyst with both anion and cation substitutions and oxygen vacancies which renders its relatively low Curie Temperature irrelevant for use as a catalyst above 120 °C. Furthermore, as demonstrated previously [3,67,69,82,83] (see also Table 10), the supported single catalysts (M/Si = X) or binary catalysts (M(1)/M(2)/Si = X/Y/Z) or the composite catalysts with BaTi1−rMrO3−x−y{#}xNz appear to yield higher outlet ammonia concentration as well as higher energy efficiency.
- (g)
- High ammonia outlet temperature is important because it affects the cost incursion in the downstream processes, notably the ammonia recovery and the recycling of the unreacted gases. The recent ammonia production cost analyses by Wang et al. [374] and Rouwenhorst et al. [40,149] indicate that low outlet NH3, the recycling cost constitutes more than that of the ammonia synthesis cost for a distributed production platform. Therefore, several techniques have been adopted to enhance ammonia output concentration [3,39,40,148,149,375,376]. Evidently, (see for example Table 15), this level of conversion (0.2%) used in the model is too low. Therefore, the cost prediction should substantially improve when using conversions of ca. 12% or more. In order to enhance the conversion, in addition to the optimization of various processing parameters (plasma pulsation, temperature, flow rate, etc.), the main enhancement will come from plasma-promoting design and optimization, microwave plasma processing, integrated intensified processes, and in situ ammonia sequestration [2].
- (h)
- We have already discussed the enhancement expectations from plasma promoting/catalyst design and integrated-intensified microwave plasma processing using plasma generating-chemical looping catalysts [3,39,67,82,83,170]. In situ ammonia sequestration using either acids or CO2 [3,39,67] resulted in further enhancement in conversion (ca. 20% NH3 outlet concentration and ca. 40% nitrogen conversion) and energy cost reduction (13.2 MJ/mol) with energy yield of 4.64 g/kWh. Recently, Rouwenhorst et al. [149] added another technique of sequestration to enhance energy efficiency to 2.3 g NH3/kWh. These improvements are insufficient to get to the energy efficiency level of Haber–Bosch (≈155 g NH3/kWh) for the production of anhydrous NH3. However, it is likely that the implementation of the proposed measures to produce symbiotic/smart ammonia-based fertilizers through the use of plasma-generating catalysts in microwave reactors with in situ sequestration using acid-containing, water absorbents will create a new generation of fertilizers which will help to reduce ammonia/fertilizer cost and generate symbiotic-smart fertilizer systems which can enhance crop yield, fertilizer, and water efficiencies.
- (i)
- The main energy cost in ammonia synthesis without in situ recovery consists of energy consumption associated with synthesis (i.e., Energy Cost, EC), ammonia recovery and unreacted gas recycle. Ammonia recovery and gas recycle costs are inversely proportional to ammonia yield. Hence, for a given ammonia plant, the total energy cost (ET) of ammonia production can be assumed to be inversely proportional to the outlet ammonia concentration, Cout and given by:where n ≥ 1 and the value of the power index, n, is a function of recovery and recycle operations. High values of (n) indicate low efficiency gas separation and recycle.ET = EC/[Cout]n
- (j)
- The catalyst and reactor/process efficiency comparisons can be misleading when they are only based on Energy Cost (EC), Energy Yield, (EY) or outlet concentration Cout, all of which depend on the Specific Input Energy (SIE). A better comparison can be based on the Energy Cost per unit mol% conversion (EU); i.e., EU = ET = EC/[Cout]n (n = 1). This will ensure that the cost of ammonia recovery is incorporated in the overall assessment of a given catalyst and process. As no data are available for the energy cost of the post-synthesis recovery-recycle, we assume that n = 1 as a comparator. In Table 15, the variation of ET (n = 1) is shown as a single comparator data for various plasma reactors, processes and catalysts some of which have sufficient attributes for commercialization. ET (n = 1) for the Haber–Bosch process, ranges from 6.25 to 3.33 MJ/mol2 whereas the best result for the DBD reactor with in situ NH3 sequestration is 68 MJ/mol2. Nevertheless, this result is not optimized and only reflects the effects of NH3 sequestration and the type of catalyst used. However, both of these variables themselves have not been optimized and the resulting product is not NH3 but a fertilizer with no NH3 recovery and N2 + H2 recycle does not require cyclic decompression-compression and cooling as in HB-process. Clearly, the use of EC/Cout as a measure of ammonia cost is a more accurate differentiator than Energy Cost, which does not take into account the NH3 separation cost.
12.6. The Significance of Porogenetic Plasma Dust as Catalyst
- (a)
- The recent theoretical study by Perillo, et al. [377] and van’t Veer et al. [378] and experimental investigations by Navascues et al. [379,380] on the apparent very low conversion in ammonia synthesis indicate that, after formation, NH3 undergoes dissociation (reverse reaction) in the plasma space due to electron impact. Another reason for the low NH3 concentration is the recombination processes termed N-MAR (nitrogen molecular-activated recombination) [377]. This theoretical study concludes that two nitrogen-including recombination reaction paths are dominant; the ion conversion of NH species followed by dissociative recombination and the proton transfer between H2+ and N2, producing N2H+ which then undergoes dissociation into N2 and H upon electron impact [381]. These findings led to the inevitable question if catalysts are in fact useful in plasma synthesis and the necessity of optimizing the feed H2/N2 ratio according to the catalyst [303,382,383]. Alternative methods of preventing NH3 dissociation include product removal from the plasma zone as discussed previously or pulsate plasma [384,385], which also promotes the enhancement of CO2+ cations leading to CO2 splitting [386].
- (b)
- As plasma-generated dust particles remove the electrons from the plasma space, NH3 dissociation due to electron impact should be reduced while the recombination of reactive N species should be prevented. This process thus explains why the use of BaTiO3−x−y{#}xNz type of piezoelectric catalysts [82,83,170], which are formed as a result of plasma and microwave irradiation, yield high NH3 in the range of ca. 9–20% [3,39,67,69].
- (c)
- As concluded by Rouwenhorst et al. [387], in order to prevent reverse reactions from taking place in the plasma space, the dissociation of N2 and H2 should take place on the catalyst surface. However, this is only possible in low-power DBD plasmas [367]. Therefore, porogenetically created plasma dust particles can generate low-power plasma space but have high catalytic activity on their surface.
- (d)
- In piezoelectric catalysts, an external force is required for the generation of electrons and holes. However, in dusty plasma particles, lattice excitation results from the emission of phonons, and hence such particles do not require an external energy field to acquire electrons, and their large size ensures the retention of their large electron concentration.
12.7. A Roadmap for Sustainable Production of Hydrogen, Ammonia and Symbiotic-Smart Fertilizers
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tilman, D.; Cassman, K.G.; Matson, P.A.; Naylor, R.; Polasky, S. Agricultural sustainability and intensive production practices. Nature 2002, 418, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Akay, G. Bioprocess and chemical process intensification. In Encyclopaedia of Chemicals Processing; Lee, S., Ed.; Taylor & Francis: New York, NY, USA, 2006; Volume 1, pp. 183–199. [Google Scholar]
- Akay, G. Sustainable ammonia and advanced symbiotic fertilizer production using catalytic multi-reaction-zone reactors with nonthermal plasma and simultaneous reactive separation. ACS Sustain. Chem. Eng. 2017, 5, 11588–11606. [Google Scholar] [CrossRef]
- Akay, G.; Burke, D.R. AgroProcess intensification through synthetic rhizosphere media for nitrogen fixation and yield enhancement. Am. J. Agric. Biol. Sci. 2012, 7, 150–172. [Google Scholar]
- Akay, G.; Fleming, S. AgroProcess Intensification: Microbioreactors as soil additives with nitrogen fixing bacterium Azospirillum brasilense to enhance its potential as self-sustaining biofertiliser. Green Process. Synth. 2012, 1, 427–437. [Google Scholar]
- Akay, G.; Burke, D.R. Synthetic Symbiotic System as Soil Additives to Deliver Active Ingredients through Plant Roots for Enhanced Plant and Crop Yield. US Patent US8898955, 2 December 2014. [Google Scholar]
- Dunbabin, V.M.; Postma, J.A.; Schnepf, A.; Pagès, L.; Javaux, M.; Wu, L.; Leitner, D.; Chen, Y.L.; Rengel, Z.; Diggle, A.J. Modelling root–soil interactions using three–dimensional models of root growth, architecture and function. Plant Soil 2013, 372, 93–124. [Google Scholar] [CrossRef]
- Chen, J.; Lü, S.; Zhang, Z.; Zhao, X.; Li, X.; Ning, P.; Liu, M. Environmentally friendly fertilizers: A review of materials used and their effects on the environment. Sci. Total Environ. 2018, 614, 829–839. [Google Scholar] [CrossRef]
- Gutierrez, C.F.; Sanabria, J.; Raaijmakers, J.M.; Oyserman, B.O. Restoring degraded microbiome function with self-assembled communities. FEMS Microbiol. Ecol. 2020, 96, fiaa225. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, J.; Cowie, A.; Masiello, C.A.; Kammann, C.; Woolf, D.; Amonetta, J.E.; Cayuela, M.L.; Camps-Arbestain, M.; Whitman, T. Biochar in climate mitigation. Nat. Geosci. 2021, 14, 883–892. [Google Scholar] [CrossRef]
- Chen, J.; Wu, J.; Raffa, P.; Picchioni, F.; Koning, C.E. Superabsorbent Polymers: From long-established, microplastics generating systems, to sustainable, biodegradable and future proof alternatives. Prog. Polym. Sci. 2022, 125, 101475. [Google Scholar] [CrossRef]
- Bi, R.; Zhang, Q.; Zhan, L.; Xu, X.; Zhang, X.; Dong, Y.; Yan, X.; Xiong, Z. Biochar and organic substitution improved net ecosystem economic benefit in intensive vegetable production. Biochar 2022, 4, 46. [Google Scholar] [CrossRef]
- Venugopalan, V.K.; Nath, R.; Chandran, S. Smart fertilizers-a way ahead for sustainable agriculture. J. Plant Nutr. 2022, 45, 2068–2076. [Google Scholar] [CrossRef]
- Akay, G. Flow-induced phase inversion in the intensive processing of concentrated emulsions. Chem. Eng. Sci. 1998, 53, 203–223. [Google Scholar] [CrossRef]
- Akay, G.; Dawnes, S.; Price, V.J. Microcellular Polymers as Cell Growth Media and Novel Polymers. European Patent EP1183328, 8 August 2012. (Priority GB19980026701 (1998) and represents the first ever paper on the use of POLYHIPE Polymer in tissue engineering and bioprocess intensification). [Google Scholar]
- Akay, G.; Noor, Z.Z.; Calkan, O.F.; Ndlovu, T.M.; Burke, D.B. Process for Preparing a Functionalized PolyHIPE Polymer. US Patent US 07820729, 26 October 2010. [Google Scholar]
- Akay, G.; Bokhari, M.A.; Byron, V.J.; Dogru, M. Development of nano-structured materials and their application in bioprocess-chemical process intensification and tissue engineering. In Chemical Engineering Trends and Developments; Galan, M.A., Del Valle, E.M., Eds.; Wiley: London, UK, 2005; pp. 171–196. [Google Scholar]
- Akay, G.; Birch, M.A.; Bokhari, M.A. Microcellular polyHIPE polymer supports osteoblast growth and bone formation in vitro. Biomaterials 2004, 25, 3991–4000. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Sanguramath, R.A.; Israel, S.; Silverstein, M.S. Emulsion templating: Porous polymers and beyond. Macromolecules 2019, 52, 5445–5479. [Google Scholar] [CrossRef]
- Dikici, B.A.; Malayeri, A.; Sherborne, C.; Dikici, S.; Paterson, T.; Dew, L.; Hatton, P.; Asencio, I.O.; MacNeil, S.; Langford, C.; et al. Thiolene- and polycaprolactone methacrylate-based polymerized High Internal Phase Emulsion (PolyHIPE) scaffolds for tissue engineering. Biomacromolecules 2022, 23, 720–730. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Liu, C.; Jiang, M.; Yuan, H. Design and development of fertilizers point-applied device in root-zone. Appl. Eng. Agric. 2022, 38, 559–571. [Google Scholar] [CrossRef]
- Barby, D.; Haq, Z. Low Density Porous Cross-Linked Polymeric Materials and Their Preparation. European Patent EP0060138, 3 September 1986. [Google Scholar]
- Akay, G. Theoretical Bases of “Confinement Phenomenon” and the Use of PolyHIPE Polymer in Its Proof and Applications; Progress Report for Unilever Research Port Sunlight Laboratory (Document Number: E ENG 0656); Exeter University: Exeter, UK, September 1992. [Google Scholar]
- Akay, G.; Fleming, S. Engineered ecosystem development for AgroProcess Intensification. WIT Trans. Ecol. Environ. 2012, 114, 485–495. [Google Scholar]
- Akay, G.; Erhan, E.; Keskinler, B. Bioprocess intensification in flow-through monolithic microbioreactors with immobilized bacteria. Biotechnol. Bioeng. 2005, 90, 180–190. [Google Scholar] [CrossRef]
- Bokhari, M.A.; Akay, G.; Zhang, S.G.; Birch, M.A. Enhancement of osteoblast growth and differentiation in vitro on a peptide hydrogel-polyHIPE polymer hybrid material. Biomaterials 2005, 26, 5198–5208. [Google Scholar] [CrossRef]
- Akay, G.; Noor, Z.Z.; Dogru, M. Process intensification in water-in-crude oil emulsion separation by simultaneous application of electric field and novel demulsifier adsorbers based on PolyHIPE Polymers. In Microreactor Technology and Process Intensification; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2005; Volume 914, pp. 378–392. [Google Scholar]
- Akay, G.; Vickers, J. Methods for Separating Oil and Water Using Polyhipe Polymers. US Patent US8177985, 15 May 2012. [Google Scholar]
- Akay, G.; Pekdemir, T.; Shakorfow, A.M.; Vickers, V. Intensified demulsification and separation of thermal oxide reprocessing interfacial crud (THORP-IFC) simulants. Green Synth. Process. 2012, 1, 109–127. [Google Scholar] [CrossRef]
- Akay, G. Stable Oil in Water Emulsions and a Process for Preparing Same. European Patent EP0649867, 17 September 2001. (Priority EP93308521, 13 December 1993). [Google Scholar]
- Akay, G. Flow induced phase inversion in powder structuring by polymers. In Polymer Powder Technology; Narkis, M., Rosenzweig, N., Eds.; Wiley: New York, NY, USA, 1995; Chapter 20; pp. 542–587. [Google Scholar]
- Kumar, A.; Li, S.; Cheng, C.-M.; Lee, D. Recent developments in phase inversion emulsification. Ind. Eng. Chem. Res. 2015, 54, 8375–8396. [Google Scholar] [CrossRef]
- Kumar, A.; Li, S.; Cheng, C.-M.; Lee, D. Flow-induced phase inversion emulsification in tapered microchannels. Lab Chip. 2016, 21, 4173–4180. [Google Scholar] [CrossRef]
- Akay, G.; Calkan, B.; Hasni, H.; Mohamed, R. Preparation of Nanostructured Microporous Composite Foams. European Patent EP2342272, 25 September 2013. [Google Scholar]
- Akay, G.; Calkan, B. Preparation of nanostructured microporous metal foams through flow induced electroless deposition. J. Nanomat. 2015, 2015, 275705. [Google Scholar] [CrossRef]
- Kovačič, J.M.; Terezija Ciringer, T.; Ambrožič-Dolinšek, J.; Kovačič, S. Use of emulsion-templated, highly porous polyelectrolytes for in vitro germination of chickpea embryos: A new substrate for soilless cultivation. Biomacromolecules 2022, 23, 3452–3457. [Google Scholar] [CrossRef]
- Barlık, N.; Keskinler, B.; Kocakerim, M.M.; Akay, G. Surface modification of monolithic PolyHIPE Polymers for anionic functionality and their ion exchange behaviour. J. Appl. Polym. Sci. 2015, 132, 42286. [Google Scholar] [CrossRef]
- Barlik, N.; Keskinler, B.; Kocakerim, M.M.; Akay, G. Functionalized PolyHIPE Polymer monoliths as an anion-exchange media for removal of nitrate ions from aqueous solutions. Desalination Water Treat. 2016, 57, 26440–26447. [Google Scholar] [CrossRef]
- Akay, A. Catalytic Multi-Reaction Zone Reactor System. US Patent US11498845, 15 November 2022. [Google Scholar]
- Rouwenhorst, K.H.R.; Lefferts, L. Feasibility study of plasma-catalytic ammonia synthesis for energy storage applications. Catalysts 2020, 10, 999. [Google Scholar] [CrossRef]
- Mosley, L.M.; Willson, P.; Hamilton, B.; Butler, G. The capacity of biochar made from common reeds to neutralise pH and remove dissolved metals in acid drainage. Environ. Sci. Pullut. Res. 2015, 22, 15113–15122. [Google Scholar] [CrossRef] [PubMed]
- Sahin, O.; Taskin, M.B.; Kaya, E.C.; Atakol, O.; Emir, E.; Inali, A.; Gunes, A. Effect of acid modification of biochar on nutrient availability and maize growth in a calcareous soil. Soil Use Manag. 2017, 33, 447–456. [Google Scholar] [CrossRef]
- Vaughn, S.F.; Kenar, J.A.; Tisserat, B.; Jackson, M.J.; Joshee, N.; Vaidya, B.N.; Peterson, S.C. Chemical and physical properties of Paulownia elongata biochar modified with oxidants for horticultural applications. Ind. Crops Prod. 2017, 97, 260–267. [Google Scholar] [CrossRef]
- Shibaoka, M. Carbon content of fly ash and size distribution of unburnt char particles in fly ash. Fuel 1986, 65, 449–450. [Google Scholar] [CrossRef]
- Jordan, C.A.; Akay, G. Speciation and distribution of alkali, alkali earth metals and major ash forming elements during gasification of fuel cane bagasse. Fuel 2012, 91, 253–263. [Google Scholar] [CrossRef]
- Ukwattage, N.L.; Ranjith, P.G.; Bouazza, M. The use of coal combustion fly ash as a soil amendment in agricultural lands (with comments on its potential to improve food security and sequester carbon). Fuel 2013, 109, 400–408. [Google Scholar] [CrossRef]
- Dzantor, E.K.; Adeleke, E.; Kankarla, V. Using coal fly ash in agriculture: Combination of fly ash and poultly litter as soil amendments for bioenergy feedstock production. Coal Combust. Gasif. Prod. 2015, 7, 33–39. [Google Scholar]
- Yao, X.; Zhao, Z.; Chen, S.; Zhou, H.; Xu, K. Migration and transformation behaviours of ash residues from a typical fixed-bed gasification station for biomass syngas production in China. Energy 2020, 201, 117646. [Google Scholar] [CrossRef]
- Mu, L.; Li, T.; Zuo, S.; Yin, H.; Dong, M. Effect of leaching pretreatment on the inhibition of slagging/sintering of aquatic biomass: Ash transformation behavior based on experimental and equilibrium evaluation. Fuel 2022, 323, 124391. [Google Scholar] [CrossRef]
- Li, X.; Bai, C.; Qiao, Y.; Wang, X.; Yang, K.; Colombo, P. Preparation, properties and applications of fly ash-based porous geopolymers: A review. J. Clean. Production 2022, 359, 132043. [Google Scholar] [CrossRef]
- Arrobas, M.; de Almeida, S.F.; Raimundo, S.; da Silva Domingues, L.; Rodrigues, M.Â. Leonardites rich in humic and fulvic acids had little effect on tissue elemental composition and dry matter yield in pot-grown olive cuttings. Soil Syst. 2022, 6, 7. [Google Scholar] [CrossRef]
- Kaya, C.; Şenbayram, M.; Akram, N.A.; Ashraf, M.; Alyemeni, M.N.; Ahmad, P. Sulfur-enriched leonardite and humic acid soil amendments enhance tolerance to drought and phosphorus deficiency stress in maize (Zea mays L.). Sci. Rep. 2020, 10, 6432. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Ding, W.; Yang, Y.; Sun, J.; Ding, Q. Humic acids derived from leonardite-affected growth and nutrient uptake of corn seedlings. Commun. Soil Sci. Plant Anal. 2016, 47, 1275–1282. [Google Scholar]
- Bhattacharya, S.S.; Kim, K.H.; Das, S.; Uchimiya, M.; Jeon, B.H.; Kwon, E.; Szulejko, J.E. A review on the role of organic inputs in maintaining the soil carbon pool of the terrestrial ecosystem. J. Environ. Manag. 2016, 167, 214–227. [Google Scholar] [CrossRef]
- Yolcu, H.; Seker, H.; Gullap, M.K.; Lithourgidis, A.; Gunes, A. Application of cattle manure, zeolite and leonardite improves hay yield and quality of annual ryegrass (Lolium multiflorum Lam.) under semiarid conditions. Aust. J. Crop. Sci. 2011, 5, 926–931. [Google Scholar]
- Ece, A.; Saltali, K.; Eryigit, N.; Uysal, F. The effects of leonardite applications on climbing bean (Phaseolus vulgaris L.) yield and some soil properties. J. Agronomy. 2007, 6, 480–483. [Google Scholar]
- Akinremi, O.O.; Janzen, R.L.; Lemke, R.L.; Larney, F.J. Response of canola, wheat and green beans to Leonardite additions. Can. J. For. Res. 2000, 80, 437–443. [Google Scholar] [CrossRef]
- Rouwenhorst, K.H.R.; Engelmann, Y.; van ‘t Veer, K.; Postma, R.S.; Bogaerts, A.; Lefferts, L. Plasma-driven catalysis: Green ammonia synthesis with intermittent electricity. Green Chem. 2020, 22, 6258–6287. [Google Scholar] [CrossRef]
- Yan, C.; Waitt, C.; Akintola, I.; Lee, G.; Easa, J.; Clarke, R.; Geng, F.; Poirier, D.; Otor, H.O.; Rivera-Castro, G.; et al. Recent advances in plasma catalysis. J. Phys. Chem. C 2022, 126, 9611–9614. [Google Scholar] [CrossRef]
- Neyts, E.C.; Bogaerts, A. Understanding plasma catalysis through modelling and simulation. J. Phys. D Appl. Phys. 2014, 47, 1–18. [Google Scholar] [CrossRef]
- Neyts, E.C.; Ostrikov, K.; Sunkara, M.K.; Bogaerts, A. Plasma catalysis: Synergistic effects at the nanoscale. Chem. Rev. 2015, 115, 13408–13446. [Google Scholar] [CrossRef]
- Mehta, P.; Barboun, P.; Herrera, F.A.; Kim, J.; Rumbach, P.; Go, D.B.; Hicks, J.C.; Schneider, W.F. Overcoming ammonia synthesis scaling relations with plasma-enabled catalysis. Nat. Catal. 2018, 1, 269–275. [Google Scholar] [CrossRef]
- Chen, J.G.; Crooks, R.M.; Seefeldt, L.C.; Bren, K.L.; Bullock, R.M.; Darensbourg, M.Y.; Holland, P.L.; Hoffman, B.; Janik, M.J.; Jones, A.K.; et al. Beyond fossil fuel-Driven nitrogen transformations. Science 2018, 360, eaar6611. [Google Scholar] [CrossRef]
- Mehta, P.; Barboun, P.M.; Engelmann, Y.; Go, D.B.; Bogaerts, A.; Schneider, W.F.; Hicks, J.C. Plasma-catalytic ammonia synthesis beyond the equilibrium limit. ACS Catal. 2020, 10, 6726–6734. [Google Scholar] [CrossRef]
- Vervloessem, E.; Gorbanev, Y.; Nikiforov, A.; De Geyter, N.; Bogaerts, A. Sustainable NOx production from air in pulsed plasma: Elucidating the chemistry behind the low energy consumption. Green Chem. 2022, 24, 916–929. [Google Scholar] [CrossRef]
- Bogaerts, A.; Zhang, Q.; Zhang, Y.; Van Laer, K.; Wang, W. Burning questions of plasma catalysis: Answers by modelling. Catal. Today 2019, 337, 3–14. [Google Scholar] [CrossRef]
- Akay, G. Ammonia Production by Integrated Intensified Processes. US Patent US9416019, 16 August 2016. (Priority: GB Patent Application, 1014304.8A, 27 August 2010). [Google Scholar]
- Kim, H.-H.; Teramoto, Y.; Ogata, A.A.; Takagi, H.; Nanba, T. Atmospheric-pressure nonthermal plasma synthesis of ammonia over ruthenium catalysts. Plasma Process. Poly. 2017, 14, 1–9. [Google Scholar] [CrossRef]
- Akay, G.; Zhang, K. Process intensification in ammonia synthesis using novel co-assembled supported micro-porous catalysts promoted by non-thermal plasma. Ind. Eng. Chem. Res. 2017, 56, 457–468. [Google Scholar] [CrossRef]
- Chiremba, E.; Zhang, K.; Kazak, C.; Akay, G. Direct nonoxidative conversion of methane to hydrogen and higher hydrocarbons by dielectric barrier discharge plasma with plasma catalysis promotors. AIChE J. 2017, 63, 4418–4429. [Google Scholar] [CrossRef]
- Barboun, P.; Mehta, P.; Herrera, F.A.; Go, D.B.; Schneider, W.F.; Hicks, J.C. Distinguishing plasma contributions to catalyst performance in plasma-assisted ammonia synthesis. ACS Sustain. Chem. Eng. 2019, 7, 8621–8630. [Google Scholar] [CrossRef]
- Wang, Y.; Craven, M.; Yu, X.; Ding, J.; Bryant, P.; Huang, J.; Tu, X. Plasma-enhanced catalytic synthesis of ammonia over a Ni/Al2O3 catalyst at near-room temperature: Insights into the importance of the catalyst surface on the reaction mechanism. ACS Catal. 2019, 9, 10780–10793. [Google Scholar] [CrossRef]
- Shah, J.; Wu, T.; Lucero, J.; Carreon, M.A.; Carreon, M.L. Nonthermal plasma synthesis of ammonia over Ni-MOF-74. ACS Sustain. Chem. Eng. 2019, 7, 377–383. [Google Scholar] [CrossRef]
- Akay, G.; Zhang, K.; Al-Harrasi, W.S.S.; Sankaran, R.M. Catalytic plasma Fischer–Tropsch synthesis using hierarchically connected porous Co/SiO2 catalysts prepared by microwave-induced co-assembly. Ind. Eng. Chem. Res. 2020, 59, 12013–12027. [Google Scholar] [CrossRef]
- Barboun, P.M.; Hicks, J.C. Unconventional catalytic approaches to ammonia synthesis. Annu. Rev. Chem. Biomol. Eng. 2020, 11, 503–521. [Google Scholar] [CrossRef] [PubMed]
- Patil, B.S.; van Kaathoven, A.S.R.; Peeters, F.; Cherkasov, N.; Wang, Q.; Lang, J.; Hessel, V. Deciphering the synergy between plasma and catalyst support for ammonia synthesis in a packed DBD reactor, J. Phys. D Appl. Phys. 2020, 53, 144003. [Google Scholar] [CrossRef]
- Gorbanev, Y.; Engelmann, Y.; van’t Veer, K.; Vlasov, E.; Ndayirinde, C.; Yi, Y.; Bals, S.; Bogaerts, A. Al2O3-supported transition metals for plasma-catalytic NH3 synthesis in a DBD plasma: Metal activity and insights into mechanisms. Catalysts 2021, 11, 1230. [Google Scholar] [CrossRef]
- Ma, H.; Sharma, R.K.; Welzel, S.; van de Sanden, M.C.M.; Tsampas, M.N.; Schneider, W.F. Observation and rationalization of nitrogen oxidation enabled only by coupled plasma and catalyst. Nat. Commun. 2022, 13, 402. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhu, X.; Hu, X.; Tu, X. Plasma-assisted ammonia synthesis in a packed-bed dielectric barrier discharge reactor: Roles of dielectric constant and thermal conductivity of packing materials. Plasma Sci. Technol. 2022, 24, 025503. [Google Scholar] [CrossRef]
- Palma, V.; Cortese, M.; Renda, S.; Ruocco, C.; Martino, M.; Meloni, E. A Review about the recent advances in selected nonthermal plasma assisted solid–gas phase chemical processes. Nanomaterials 2020, 10, 1596. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Zhou, R.; Zhou, R.; Liu, B.; Zhang, T.; Xian, Y.; Cullen, P.J.; Lu, X.; Ostrikov, K. Sustainable ammonia production by non-thermal plasmas: Status, mechanisms, and opportunities. Chem. Eng. J. 2021, 421, 129544. [Google Scholar] [CrossRef]
- Akay, G. Co-assembled supported catalysts: Synthesis of nano-structured supported catalysts with hierarchic pores through combined flow and radiation induced co-assembled nano-reactors. Catalysts 2016, 6, 80. [Google Scholar] [CrossRef]
- Akay, G. Plasma generating—Chemical looping catalyst synthesis by microwave plasma shock for nitrogen fixation from air and hydrogen production from water for agriculture and energy technologies in global warming prevention. Catalysts 2020, 10, 152. [Google Scholar] [CrossRef]
- Argyle, M.D.; Bartholomew, C.H. Heterogeneous catalyst deactivation and regeneration: A review. Catalysts 2015, 5, 145–269. [Google Scholar] [CrossRef]
- Xu, S.; Chansai, S.; Xu, S.; Stere, C.E.; Jiao, Y.; Yang, S.; Hardacre, C.; Fan, X. CO poisoning of Ru catalysts in CO2 hydrogenation under thermal and plasma conditions: A combined kinetic and diffuse reflectance infrared Fourier Transform Spectroscopy–Mass Spectrometry study. ACS Catal. 2020, 10, 12828–12840. [Google Scholar] [CrossRef]
- Al-Harrasi, W.S.S.; Zhang, K.; Akay, G. Process intensification in gas-to-liquid reactions: Plasma promoted Fischer-Tropsch synthesis for hydrocarbons at low temperatures and ambient pressure. Green Process. Synth. 2013, 2, 479–490. [Google Scholar] [CrossRef]
- Zhang, K.; Zhang, G.; Liu, X.; Phan, A.N.; Luo, K.; Akay, G. Correction to “A study on CO2 decomposition to CO and O2 by the combination of catalysis and dielectric-barrier discharges at low temperatures and ambient pressure”. Ind. Eng. Chem. Res. 2020, 59, 502. [Google Scholar] [CrossRef]
- Gómez-Ramírez, A.; Cotrino, J.; Lambert, R.M.; González-Elipe, A.R. Efficient synthesis of ammonia from N2 and H2 alone in a ferroelectric packed-bed DBD reactor. Plasma Sour. Sci. Technol. 2015, 24, 065011. [Google Scholar] [CrossRef]
- Gómez-Ramírez, A.; Montoro-Damas, A.M.; Cotrino, J.; Lambert, R.M.; González-Elipe, A.R. About the enhancement of chemical yield during the atmospheric plasma synthesis of ammonia in a ferroelectric packed bed reactor. Plasma Process. Polym. 2017, 14, 1600081. [Google Scholar] [CrossRef]
- Gómez-Ramírez, A.; Álvarez, R.; Navascués, P.; García-García, F.J.; Palmero, A.; Cotrino, J.; González-Elipe, A.R. Electrical and reaction performances of packed-bed plasma reactors moderated with ferroelectric or dielectric materials. Plasma Process. Polym. 2021, 18, 2000193. [Google Scholar] [CrossRef]
- Navascués, P.; Cotrino, J.; González-Elipe, A.R.; Gómez-Ramírez, A. Plasma assisted CO2 dissociation in pure and gas mixture streams with a ferroelectric packed-bed reactor in ambient conditions. Chem. Eng. J. 2022, 430, 133066. [Google Scholar] [CrossRef]
- Ma, Y.; Tian, Y.; Zeng, Y.; Tu, X. Plasma synthesis of ammonia in a tangled wire dielectric barrier discharge reactor: Effect of electrode materials. J. Energy Inst. 2021, 99, 137–144. [Google Scholar] [CrossRef]
- Zhu, X.; Liu, J.; Hu, X.; Zhou, Z.; Li, X.; Wang, W.; Wu, R.; Tu, X. Plasma-catalytic synthesis of ammonia over Ru-based catalysts: Insights into the support effect. J. Energy Inst. 2022, 102, 240–246. [Google Scholar] [CrossRef]
- Flury, M.; Aramrak, S. Role of air-water interfaces in colloid transport in porous media: A review. Water Resour. Res. 2017, 53, 5247–5275. [Google Scholar] [CrossRef]
- Martins-Costa, M.T.C.; Anglada, J.M.; Franciscoc, J.S.; Ruiz-Lopez, M.F. Photosensitization mechanisms at the air–water interface of aqueous aerosols. Chem. Sci. 2022, 13, 2624. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, M.P.; Faria, J.A. Catalysis at the solid-liquid-liquid interface of water-oil Pickering emulsions: A tutorial review. ACS Eng. Au 2022, 2, 295–319. [Google Scholar] [CrossRef]
- Akay, G.; Zhang, K.; Chiremba, E. CO2 reforming of CH4 over nano-porous catalysts promoted by dielectric-barrier discharges. In Proceedings of the EU COPIRIDE Integrated Project Review Meeting Presentation, Mainz, Germany, 24 July 2012. Full report: COPIRIDE Project Output (17-02-2013). [Google Scholar]
- Zhang, K.; Mukhriza, T.; Liu, X.; Greco, P.P.; Chiremba, E. A study on CO2 and CH4 conversion to synthesis gas and higher hydrocarbons by the combination of catalyst and dielectric-barrier discharges. Appl. Catal. A Gen. 2015, 502, 138–149. [Google Scholar] [CrossRef]
- Akay, G.; Jordan, A.C. Gasification of fuel cane bagasse in a downdraft gasifier: Influence of lignocellulosic composition and fuel particle size on syngas composition and yield. Energy Fuel 2011, 25, 2274–2283. [Google Scholar] [CrossRef]
- Wu, N.; Lan, K.; Yao, Y. An integrated techno-economic and environmental assessment for carbon capture in hydrogen production by biomass gasification, Resources. Conserv. Recycl. 2023, 188, 106693. [Google Scholar] [CrossRef]
- Lan, K.; Zhang, B.; Yao, Y. Circular utilization of urban tree waste contributes to the mitigation of climate change and eutrophication. One Earth 2022, 5, 944–957. [Google Scholar] [CrossRef]
- Maitlo, G.; Ali, I.; Mangi, K.H.; Ali, S.; Maitlo, H.A.; Unar, I.N.; Pirzada, A.M. Thermochemical conversion of biomass for syngas production: Current status and future trends. Sustainability 2022, 14, 2596. [Google Scholar] [CrossRef]
- Nunes, L.J.R. Biomass gasification as an industrial process with effective proof-of-concept: A comprehensive review on technologies, processes and future developments. Results Eng. 2022, 14, 100408. [Google Scholar] [CrossRef]
- Tezer, Ö.; Karabağ, N.; Öngen, A.; Çolpan, C.Ö.; Ayol, A. Biomass gasification for sustainable energy production: A review. Int. J. Hydrogen Energy 2022, 47, 15419–15433. [Google Scholar] [CrossRef]
- Piazzi, S.; Patuzzi, F.; Baratieri, M. Energy and exergy analysis of different biomass gasification coupled to Fischer-Tropsch synthesis configurations. Energy 2022, 249, 123642. [Google Scholar] [CrossRef]
- Antolini, D.; Piazzi, S.; Menin, L.; Baratieri, M.; Patuzzi, F. High hydrogen content syngas for biofuels production from biomass air gasification: Experimental evaluation of a char-catalyzed steam reforming unit. Int. J. Hydrogen Energy 2022, 64, 27421–27436. [Google Scholar] [CrossRef]
- Ruiz, M.; Schnitzer, A.; Courson, C.; Mauviel, M. Fe-doped olivine and char for in-bed elimination of gasification tars in an air-blown fluidised bed reactor coupled with oxidative hot gas filtration. Carbon Resour. Convers. 2022, 5, 271–288. [Google Scholar] [CrossRef]
- Shahbaz, M.; AlNouss, A.; Ghiat, I.; Mckay, G.; Mackey, H.; Elkhalifa, S.; Al-Ansari, T. A comprehensive review of biomass based thermochemical conversion technologies integrated with CO2 capture and utilisation within BECCS networks. Resour. Conserv. Recycl. 2021, 173, 105734. [Google Scholar] [CrossRef]
- Donnison, C.; Holland, R.A.; Hastings, A.; Armstrong, L.-M.; Eigenbrod, F.; Taylor, G. Bioenergy with carbon capture and storage (BECCS): Finding the win–wins for energy, negative emissions and ecosystem services—Size matters. GCB Bioenergy 2020, 12, 586–604. [Google Scholar] [CrossRef]
- Centi, G.; Iaquaniello, G.; Perathoner, S. Chemical engineering role in the use of renewable energy and alternative carbon sources in chemical production. BMC Chem. Eng. 2019, 1, 5. [Google Scholar] [CrossRef]
- Sansaniwala, S.K.; Rosen, M.A.; Tyagic, S.K. Global challenges in the sustainable development of biomass gasification: An overview. Renew. Sust. Energy Rev. 2017, 80, 23–43. [Google Scholar] [CrossRef]
- Yao, Y.; Graziano, D.; Riddle, M.; Cresko, J.; Masanet, E. Greener pathways for energy-intensive commodity chemicals: Opportunities and challenges. Curr. Opin. Chem. Eng. 2014, 6, 90–98. [Google Scholar] [CrossRef]
- Gilbert, P.; Alexander, S.; Thurnley, P.; Brammer, J. Assessing economically viable carbon reductions for the production of ammonia from biomass gasification. J. Clean. Prod. 2014, 64, 581–589. [Google Scholar] [CrossRef]
- Dogru, M.; Akay, G. Catalytic Gasification. US Patent US08252072, 28 August 2012. [Google Scholar]
- Saidur, R.; Boroumandjazi, G.; Mekhilef, S.; Mohammed, H.A. A review on exergy analysis of biomass based fuels. Renew. Sust. Energy Rev. 2012, 16, 1217–1222. [Google Scholar] [CrossRef]
- Liu, Q.; Zhong, W.; Zhou, J.; Yu, Z. Effects of S and Al on K migration and transformation during coal and biomass co-combustion. ACS Omega 2022, 7, 15880–15891. [Google Scholar] [CrossRef]
- Zhang, Z.; Xiang, X.; Shen, Z.; Wang, Y.; Li, X.; Zou, X.; Xu, H.; Cao, Y.; Chen, H.; Wang, Y. Construction of high-performance NiCe-MOF derived structured catalyst for steam reforming of biomass tar model compound. Int. J. Hydrogen Energy 2022, 47, 32004–32014. [Google Scholar] [CrossRef]
- Míguez, J.L.; Porteiro, J.; Behrendt, F.; Blanco, D.; Patiño, D.; Dieguez-Alonso, A. Review of the use of additives to mitigate operational problems associated with the combustion of biomass with high content in ash-forming species. Renew. Sust. Energy Rev. 2021, 141, 110502. [Google Scholar] [CrossRef]
- Jordan, C.A.; Akay, G. Effect of CaO on Tar Production and Dew Point Depression during Gasification of Fuel Cane Bagasse in a Novel Downdraft Gasifier. Fuel Process. Technol. 2013, 106, 654–660. [Google Scholar] [CrossRef]
- Li, B.; Mbeugang, C.F.M.; Huang, Y.; Liu, D.; Wang, Q.; Zhang, S. A review of CaO based catalysts for tar removal during biomass gasification. Energy 2022, 244, 123172. [Google Scholar] [CrossRef]
- Zhang, Q.; Bu, T.; Lu, J.; Jin, Z.; Chen, D.; Qiu, F.; Feng, Y.; Hu, W. Effects of quicklime conditioning on the volatile reforming and tar elimination performance of sewage sludge pyrochar. J. Anal. Appl. Pyrolysis 2022, 168, 105732. [Google Scholar] [CrossRef]
- Woolcock, P.J.; Brown, R.C. A review of cleaning technologies for biomass-derived syngas. Biomass Bioenergy 2013, 52, 54–84. [Google Scholar] [CrossRef]
- Akay, G.; Jordan, C.A.; Mohamed, A.H. Syngas cleaning with nano-structured micro-porous ion exchange polymers in biomass gasification using a novel downdraft gasifier. J. Energy Chem. 2013, 22, 426–435. [Google Scholar] [CrossRef]
- Jordan, C.A.; Akay, G. Occurrence, composition and dew point of tars produced during gasification of fuel cane bagasse in a downdraft gasifier. Biomass Bioenergy. 2012, 42, 51–58. [Google Scholar] [CrossRef]
- Rueda, Y.G.; Helsen, L. The role of plasma in syngas tar cracking. Biomass Convers. Biorefinery 2020, 10, 857–871. [Google Scholar] [CrossRef]
- Soedarsono, H.P.; Faizal, F.; Panatarani, C.; Joni, I.M. Carbon functionalized material derived from byproduct of plasma tar-cracking unit on biomass gasifier collected using standard impinger method. Processes 2022, 10, 1733. [Google Scholar] [CrossRef]
- Okati, A.; Khani, M.R.; Shokri, B.; Monteiro, E.; Rouboa, A. Parametric studies over a plasma co-gasification process of biomass and coal through a restricted model in Aspen plus. Fuel 2023, 331, 125952. [Google Scholar] [CrossRef]
- Hagner, M.; Pasanen, T.; Penttinen, O.-P.; Lindqvist, B.; Lindqvist, I.; Tiilikkala, K.; Setälä, H. Effects of birch tar oil on soil organisms and plants. Agric. Food Sci. 2010, 19, 13–23. [Google Scholar] [CrossRef]
- Hagner, M.; Penttinen, O.-P.; Pasanen, T. Acute toxicity of birch tar oil on aquatic organisms. Agric. Food Sci. 2010, 19, 24–33. [Google Scholar] [CrossRef]
- Tiilikkala, K.; Fagernäs, L.; Tiilikkala, J. History and use of wood pyrolysis liquids as biocide and plant protection product. Open Agric. J. 2010, 4, 111–118. [Google Scholar] [CrossRef]
- Richert, A.; Kalwasinska, A.; Brzezinska, M.S.; Dabrowska, G.B. Biodegradability of novel polylactide and polycaprolactone materials with bacteriostatic properties due to embedded birch tar in different environments. Int. J. Mol. Sci. 2021, 22, 10228. [Google Scholar] [CrossRef] [PubMed]
- Richert, A.; Olewnik-Kruszkowska, E.; Dabrowska, G.B.; Dabrowski, H.P. The role of birch tar in changing the physicochemical and biocidal properties of polylactide-Based films. Int. J. Mol. Sci. 2022, 23, 268. [Google Scholar] [CrossRef]
- Stacey, R.J.; Dunne, J.; Brunning, S.; Devièse, T.; Mortimer, R.; Ladd, S.; Parfitt, K.; Evershed, R.; Bull, I. Birch bark tar in early Medieval England—Continuity of tradition or technological revival? J. Archaeol. Sci. Rep. 2020, 29, 102118. [Google Scholar] [CrossRef]
- Tintner, J.; Leibrecht, F.; Pfeifer, C.; Konuk, M.; Srebotnik, E.; Woitsch, J. Pitch oil production–An intangible cultural heritage in Central Europe. J. Anal. Appl. Pyrolysis 2021, 159, 105309. [Google Scholar] [CrossRef]
- Akay, G. Renewable resources come together. Chem. Eng. 2005, 784, 27–30. [Google Scholar]
- Kawahara, A.; Takahashi, Y.; Hirano, Y.; Hirano, M.; Ishihara, T. Importance of pore structure control in porous substrate for high oxygen penetration in La0.6Sr0.4Ti0.3Fe0.7O3 thin film for CH4 partial oxidation. Solid State Ion. 2011, 190, 53–59. [Google Scholar] [CrossRef]
- Hashim, S.S.; Mohamed, A.R.; Bhatia, S. Oxygen separation from air using ceramic-based membrane technology for sustainable fuel production and power generation. Renew. Sust. Energy Rev. 2011, 15, 1284–1293. [Google Scholar] [CrossRef]
- El Naggar, A.M.A.; Akay, G. Novel intensified catalytic nan-structured nickel-zirconia supported palladium based membrane for high temperature hydrogen production from biomass generated syngas. Int. J. Hydrogen Energy 2013, 38, 6618–6632. [Google Scholar] [CrossRef]
- Shin, D.; Kang, S. Optimization of an ion transport membrane reactor system for syngas production. Energy Rep. 2022, 8, 3767–3779. [Google Scholar] [CrossRef]
- Micari, M.; Agrawal, K.A. Oxygen enrichment of air: Performance guidelines for membranes based on techno-economic assessment. J. Membr. Sci. 2022, 641, 119883. [Google Scholar] [CrossRef]
- Pandiyan, A.; Kyriakou, V.; Neagu, D.; Welzel, S.; Goede, A.; van de Sanden, M.C.M.; Tsampas, M.N. CO2 conversion via coupled plasma-electrolysis process. J. CO2 Util. 2022, 57, 101904. [Google Scholar] [CrossRef]
- Amhamed, A.I.; Qarnain, S.; Hewlett, S.; Sodiq, A.; Abdellatif, Y.; Isaifan, R.J.; Alrebei, O.F. Ammonia production plants—A review. Fuels 2022, 3, 408–435. [Google Scholar] [CrossRef]
- Das, A.; Peu, S.D. A Comprehensive Review on recent advancements in thermochemical processes for clean hydrogen production to decarbonize the energy sector. Sustainability 2022, 14, 11206. [Google Scholar] [CrossRef]
- Yang, X.; Kan, T.; Kheradmand, A.; Xu, H.; Strezov, V.; Yu, A.; Jiang, Y. Tunable syngas production from two-stage sorption-enhanced steam gasification of sewage sludge. Chem. Eng. J. 2021, 404, 126069. [Google Scholar] [CrossRef]
- Santos, M.P.S.; Hanak, D.P. Techno-economic feasibility assessment of sorption enhanced gasification of municipal solid waste for hydrogen production. Int. J. Hydrogen Energy 2022, 47, 6586–6604. [Google Scholar] [CrossRef]
- Peng, P.; Cheng, Y.; Hatzenbeller, R.; Addy, M.; Zhou, N.; Schiappacasse, C.; Chen, D.; Zhang, Y.; Anderson, E.; Liu, Y.; et al. Ru-based multifunctional mesoporous catalyst for low-pressure and non-thermal plasma synthesis of ammonia. Int. J. Hydrogen Energy 2017, 42, 19056–19066. [Google Scholar] [CrossRef]
- Peng, P.; Chen, P.; Addy, M.; Cheng, Y.; Anderson, E.; Zhou, N.; Schiappacasse, C.; Zhang, Y.; Chen, D.; Hatzenbeller, R.; et al. Atmospheric plasma-assisted ammonia synthesis enhanced via synergistic catalytic absorption. ACS Sustain. Chem. Eng. 2019, 7, 100–104. [Google Scholar]
- Sarafraz, M.M.; Tran, N.N.; Pourali, N.; Rebrov, E.V.; Hessel, V. Thermodynamic potential of a novel plasma-assisted sustainable process for co-production of ammonia and hydrogen with liquid metals. Energy Convers. Manag. 2020, 210, 112709. [Google Scholar] [CrossRef]
- Rouwenhorst, K.H.R.; Mani, S.; Lefferts, L. Improving the Energy Yield of Plasma-Based Ammonia Synthesis with In Situ Adsorption. ACS Sustain. Chem. Eng. 2022, 10, 1994. [Google Scholar] [CrossRef]
- Zhang, S.; Gao, Y.; Sun, H.; Fan, Z.; Shao, T. Dry reforming of methane by microsecond pulsed dielectric barrier discharge plasma: Optimizing the reactor structures. High Volt. 2022, 7, 718–729. [Google Scholar] [CrossRef]
- Sarafraz, M.M.; Christo, F.C.; Tran, N.N.; Fulcheri, L.; Hessel, V. Conversion of greenhouse gases to synthetic fuel using a sustainable cyclic plasma process. Int. J. Hydrogen Energy 2023, 48, 6174–6191. [Google Scholar] [CrossRef]
- Hong, J.; Prawer, S.; Murphy, A.B. Plasma catalysis as an alternative route for ammonia production: Status, mechanism, and Prospects for Progress. ACS Sustain. Chem. Eng. 2018, 6, 15–31. [Google Scholar] [CrossRef]
- Akay, E.M.; Okumus, Z.; Yildirim, O.S.; Bokhari, M.A.; Akay, G. Synthetic organs for transplant and bio-mimic reactors for process intensification using nano-structured micro-porous materials. WIT Trans. Biomed. Health 2010, 15, 383–394. [Google Scholar]
- Dogru, M.; Howarth, C.R.; Akay, G.; Keskinler, B.; Malik, A.A. Gasification of hazelnut shells in a downdraft gasifier. Energy 2002, 27, 415–427. [Google Scholar] [CrossRef]
- Akay, G.; Dogru, M.; Calkan, O.F.; Calkan, B. Biomass processing in biofuel applications. In Biofuels for Fuel Cells; Lens, P., Westermann, P., Haberbauer, M., Moreno, A., Eds.; IWA Publishing: London, UK, 2005; Chapter 4; pp. 51–75. [Google Scholar]
- Akay, G.; Dogru, M.; Calkan, O.F. Biomass to rescue. Chem. Eng. 2006, 787, 55–57. [Google Scholar]
- Akay, G.; Calkan, O.F. Performance of an intensified downdraft gasifier at 50 kWe and 1 MWe. In Proceedings of the AIChE Spring Meeting, New Orleans, LA, USA, 6–10 April 2008. ACS Symposium Series, Paper No: 171a. [Google Scholar]
- Balas, M.; Lisy, M.; Kubicek, J.; Pospisil, J. Syngas cleaning by wet scrubber. WSEAS Trans. Heat Mass Transf. 2014, 9, 195–204. [Google Scholar]
- Min, J.; Nam, S.B.; Kim, N.R.; Kim, D.-J.; Yoon, Y.-S.; Park, S.-N.; Gu, J.-H. A study on the characteristics of particulate matter in the syngas produced from the waste gasification with cleaning systems for energy utilization. J. Mater. Cycles Waste Manag. 2017, 19, 1155–1165. [Google Scholar] [CrossRef]
- Naaz, Z.; Ravi, M.R.; Kohli, S. Modelling and simulation of downdraft biomass gasifier: Issues and challenges. Biomass Bioenergy 2022, 162, 106483. [Google Scholar] [CrossRef]
- Ibrahim, A.; Veremieiev, S.; Gaskell, P.H. An advanced, comprehensive thermo-chemical equilibrium model of a downdraft biomass gasifier. Renew. Energy 2022, 194, 912–925. [Google Scholar] [CrossRef]
- Wang, Y.; Khan, T.S.; Wildfire, C.; Shekhawat, D.; Hu, J. Microwave-enhanced catalytic ammonia synthesis under moderate pressure and temperature. Catal. Commun. 2021, 159, 106344. [Google Scholar] [CrossRef]
- Akay, G.; Wakeman, R.J. Mechanisms of permeate flux decay, solute rejection and concentration polarisation in crossflow filtration of a double chain ionic surfactant dispersion. J. Membr. Sci. 1994, 88, 177–195. [Google Scholar] [CrossRef]
- Akay, G.; Wakeman, R.J. Electric field enhanced crossflow microfiltration of hydrophobically modified water soluble polymers. J. Membr. Sci. 1996, 131, 229–236. [Google Scholar] [CrossRef]
- Giddings, J.C. A new separation concept based on a coupling of concentration and flow nonuniformities. Sep. Sci. 1966, 1, 123–125. [Google Scholar] [CrossRef]
- Akay, G. Stress-Induced diffusion and chemical reaction in nonhomogeneous velocity gradient fields. Polym. Eng. Sci. 1982, 22, 798–804. [Google Scholar] [CrossRef]
- Solymosi, F. The bonding, structure and reactions of CO2 adsorbed on clean and promoted metal surfaces. J. Mol. Catal. 1991, 65, 337–358. [Google Scholar] [CrossRef]
- Snoeckx, R.; Heijkers, S.; Van Wesenbeeck, K.; Lenaerts, S.; Bogaerts, A. CO2 conversion in a dielectric barrier discharge plasma: N2 in the mix as a helping hand or problematic impurity? Energy Environ. Sci. 2016, 9, 30–39. [Google Scholar] [CrossRef]
- Bogaerts, A.; Tu, X.; Whitehead, J.C.; Centi, G.; Lefferts, L.; Guaitella, O.; Azzolina-Jury, F.; Kim, H.-H.; Murphy, A.B.; Schneider, W.F.; et al. The 2020 plasma catalysis roadmap. J. Phys. D Appl. Phys. 2020, 53, 443001. [Google Scholar] [CrossRef]
- Akay, G. Synthesis of Plasma Generating-Chemical Looping Catalysts. US Patent Application Publication US2023/0064758, 2 March 2023. [Google Scholar]
- Rashid, M.S.; Liu, G.; Yousaf, B.; Hamid, Y.; Rehman, A.; Arif, M.; Ahmed, R.; Song, Y.; Ashraf, A. Role of biochar-based free radicals in immobilization and speciation of metals in the contaminated soil-plant environment. J. Environ. Manag. 2023, 325, 116620. [Google Scholar] [CrossRef]
- Nishimiya, K.; Hata, T.; Imamura, Y. Analysis of chemical structure of wood charcoal by X-ray photoelectron spectroscopy. J. Wood Sci. 1998, 44, 56–61. [Google Scholar] [CrossRef]
- Cohen-Ofri, I.; Weiner, L.; Boaretto, E.; Mintz, G.; Weiner, S. Modern and fossil charcoal: Aspects of structure and diagenesis. J. Archaeol. Sci. 2006, 33, 428–439. [Google Scholar] [CrossRef]
- Cha, J.S.; Park, S.H.; Jung, S.-C.; Ryu, C.; Jeon, J.-K.; Shin, M.-C.; Park, Y.-K. Production and utilization of biochar: A review. J. Ind. Eng. Chem. 2016, 40, 1–15. [Google Scholar] [CrossRef]
- Li, S.; Huang, A.; Chen, Y.-J.; Li, D.; Turng, L.-S. Highly filled biochar/ultra-high molecular weight polyethylene/linear low density polyethylene composites for high-performance electromagnetic interference shielding. Compos. Part B Eng. 2018, 153, 277–284. [Google Scholar] [CrossRef]
- Li, L.; Zhang, Y.; Cai, D.; Yu, M.; Cao, K.; Sun, J.; Zhang, L.; Zhang, Q.; Zou, G.; Song, Z. Characteristics and mechanism of microwave-induced discharge of spherical bio-char with graphite addition. Fuel 2023, 331, 125771. [Google Scholar] [CrossRef]
- Liu, Q.; Meki, K.; Zheng, H.; Yuan, Y.; Shao, M.; Luo, X.; Li, X.; Jiang, Z.; Li, F.; Xing, B. Biochar application in remediating salt-affected soil to achieve carbon neutrality and abate climate change. Biochar 2023, 5, 45. [Google Scholar] [CrossRef]
- Mood, S.H.; Pelaez-Samaniego, G.-P. Perspectives of engineered biochar for environmental applications: A review. Energy Fuels 2022, 36, 7940–7986. [Google Scholar] [CrossRef]
- Zhao, J.J.; Shen, X.-J.; Domene, X.; Alcaniz, J.-M.; Liao, X.; Palet, C. Comparison of biochars derived from different types of feedstock and their potential for heavy metal removal in multiple-metal solutions. Sci. Rep. 2019, 9, 9869. [Google Scholar] [CrossRef] [PubMed]
- de Mello, M.D.; Ahmad, M.; Lee, D.T.; Dimitrakellis, P.; Miao, Y.; Zheng, W.; Nykypanchuk, D.; Vlachos, D.G.; Tsapatsis, M.; Boscoboinik, J.A. In situ tracking of nonthermal plasma etching of ZIF-8 films. ACS Appl. Mater. Interfaces 2022, 14, 19023–19030. [Google Scholar] [CrossRef]
- O’Connor, D.; Peng, T.; Zhang, J.; Tsang, D.C.W.; Alessi, D.S.; Shen, Z.; Bolan, N.S.; Huo, D. Biochar application for the remediation of heavy metal polluted land: A review of in situ field trials. Sci. Total Environ. 2018, 619, 815–826. [Google Scholar] [CrossRef]
- Bakshi, S.; Banik, C.; Laird, D.A. Estimating the organic oxygen content of biochar. Sci. Rep. 2020, 10, 13082. [Google Scholar] [CrossRef] [PubMed]
- Shao, S.; Zhang, J.; Zhang, Z.; Zheng, P.; Zhao, M.; Li, J.; Wang, C. High piezoelectric properties and domain configuration in BaTiO3 ceramics obtained through the solid-state reaction route. J. Phys. D Appl. Phys. 2008, 41, 125408. [Google Scholar] [CrossRef]
- Huang, S.; Zeng, J.; Zheng, L.; Man, Z.; Ruan, X.; Shi, X.; Li, G. A novel piezoelectric ceramic with high Curie temperature and high piezoelectric coefficient. Ceram. Int. 2020, 46, 6212–6216. [Google Scholar] [CrossRef]
- Meng, Y.; Chen, G.; Huang, M. Piezoelectric materials: Properties, advancements, and design strategies for high-temperature applications. Nanomaterials 2022, 12, 1171. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, J.; Liu, C.; Hou, X.; Wang, J. Phase field modelling of coupling evolution of fracture and dielectric breakdown in ferroelectric materials. Int. J. Mech. Sci. 2022, 236, 107747. [Google Scholar] [CrossRef]
- Starr, M.; Wang, X. Fundamental analysis of piezocatalysis process on the surfaces of strained piezoelectric materials. Sci. Rep. 2013, 3, 2160. [Google Scholar] [CrossRef]
- Wu, J.; Qin, N.; Bao, D. Effective enhancement of piezocatalytic activity of BaTiO3 nanowires under ultrasonic vibration. Nano Energy 2018, 45, 44–51. [Google Scholar] [CrossRef]
- Xiong, F.; Tan, S.; Yao, X.; An, Q.; Mai, L. Crystal defect modulation in cathode materials for non-lithium ion batteries: Progress and challenges. Mater. Today 2021, 45, 169–190. [Google Scholar] [CrossRef]
- Wei, X.-K.; Domingo, N.; Sun, Y.; Balke, N.; Dunin-Borkowski, R.E.; Mayer, J. Progress on emerging ferroelectric materials for energy harvesting, storage and conversion. Adv. Energy Mater. 2022, 12, 2201199. [Google Scholar] [CrossRef]
- Ji, M.; Kim, J.H.; Ryu, C.-H.; Lee, Y.-I. Synthesis of self-modified black BaTiO3–x nanoparticles and effect of oxygen vacancy for the expansion of piezocatalytic application. Nano Energy 2022, 95, 106993. [Google Scholar] [CrossRef]
- Bai, S.; Zhang, N.; Gao, C.; Xiong, Y. Defect engineering in photocatalytic materials. Nano Energy 2018, 53, 296–336. [Google Scholar] [CrossRef]
- Lin, L.; Huang, J.; Li, X.; Abass, M.A.; Hang, S. Effective surface disorder engineering of metal oxide nanocrystals for improved photocatalysis. Appl. Catal. B Environ. 2017, 203, 615–624. [Google Scholar] [CrossRef]
- Hoshina, T.; Sase, R.; Nishiyama, J.; Takeda, H.; Tsurumi, T. Effect of oxygen vacancies on intrinsic dielectric permittivity of strontium titanate ceramics. J. Ceram. Soc. Japan. 2018, 126, 263–268. [Google Scholar] [CrossRef]
- Yamada, T.; Takeda, H.; Tsurumi, T.; Hoshina, T. Possibility of ferroelectric bismuth and nitrogen co-doped barium titanate. J. Ceram. Soc. Jpn. 2020, 128, 486–491. [Google Scholar] [CrossRef]
- Liu, Q.; Zhai, D.; Xiao, Z.; Tang, C.; Sun, Q.; Bowen, C.R.; Luo, H.; Zhang, D. Piezo-photoelectronic coupling effect of BaTiO3@TiO2 nanowires for highly concentrated dye degradation. Nano Energy 2022, 92, 106702. [Google Scholar] [CrossRef]
- Sickafus, K.E.; Larson, A.C.; Yu, N.; Nastasi, M.; Hollenberg, G.W.; Garner, F.A.; Bradt, R.C. Cation disorder in high dose, neutron-irradiated spinel. J. Nucl. Mater. 1995, 219, 128–134. [Google Scholar] [CrossRef]
- Kang, Y.; Wang, L.; Xu, X.; Chen, X.-Q.; Wang, L.; Liu, G. Homogeneously nitrogen doped BaLa4Ti4O15 extending visible light responsive range for photocatalytic oxygen evolution. ACS Mater. Lett. 2023, 5, 275–281. [Google Scholar] [CrossRef]
- Wang, J.; Huang, J.; Meng, J.; Li, Q.; Yang, J. Double-hole codoped huge-gap semiconductor ZrO2 for visible-light photocatalysis. Phys. Chem. Chem. Phys. 2016, 18, 17517–17524. [Google Scholar] [CrossRef]
- Fo, Y.; Zhou, X. A theoretical study on tetragonal BaTiO3 modified by surface co-doping for photocatalytic overall water splitting. Int. J. Hydrogen Energy 2022, 47, 19073–19085. [Google Scholar] [CrossRef]
- Goumri-Said, S.; Kanoun, M.B. Insight into the effect of anionic–anionic co-doping on BaTiO3 for visible light photocatalytic water splitting: A first-principles hybrid computational study. Catalysts 2022, 12, 1672. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Tang, Y.; Kageyama, T.; Yamashita, H.; Masuda, N.; Hosokawa, S.; Kageyama, H. Titanium-based hydrides as heterogeneous catalysts for ammonia synthesis. J. Am. Chem. Soc. 2017, 139, 18240–18246. [Google Scholar] [CrossRef] [PubMed]
- Nedumkandathil, R.; Jaworski, A.; Grins, J.; Bernin, D.; Karlsson, M.; Eklof-Osterberg, C.; Neagu, A.; Tai, C.-W.; Pell, A.J.; Haussermann, U. Hydride reduction of BaTiO3-oxyhydride versus O vacancy formation. ACS Omega 2018, 3, 11426–11438. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Jaworski, A.; Chen, Z.; Lu, C.; Slabon, A.; Häussermann, U. Barium titanium oxynitride from ammonia-free nitridation of reduced BaTiO3. Inorganics 2021, 9, 62. [Google Scholar] [CrossRef]
- Jiang, W.; Loh, H.; Low, B.Q.L.; Zhu, H.; Low, J.; Heng, J.Z.X.; Tang, K.Y.; Li, Z.; Loh, X.J.; Ye, E.; et al. Role of oxygen vacancy in metal oxides for photocatalytic CO2 reduction. Appl. Catal. B Environ. 2023, 321, 122079. [Google Scholar] [CrossRef]
- Manjunatha, H.C.; Seenappa, L.; Chandrika, B.M.; Chikka, H. A study of photon interaction parameters in barium compounds. Ann. Nucl. Energy 2017, 109, 310–317. [Google Scholar] [CrossRef]
- Mudiyanselage, K.; Szanyi, J. NO2 uptake under practically relevant conditions on BaO/Pt(111). Catal. Today 2012, 181, 116–123. [Google Scholar] [CrossRef]
- Rached, A.; Wederni, M.A.; Khirouni, K.; Alaya, S.; Martín-Palma, R.J.; Dhahri, J. Structural, optical and electrical properties of barium titanate. Mater. Chem. Phys. 2021, 267, 124600. [Google Scholar] [CrossRef]
- Borkar, S.N.; Aggarwal, P.; Deshpande, V.K. Effect of calcium substitution on structural, dielectric, ferroelectric, piezoelectric, and energy storage properties of BaTiO3. Curr. Appl. Phys. 2022, 39, 205–213. [Google Scholar] [CrossRef]
- Ertekin, N.; Rezaee, S. Effect of anion and cation vacancies pairs in conduct of the Ba(1–3x) TiO3(1–x) and BaTi(1–3x/2)O3(1–x) (x = 0.0033) as a memristor. Mater. Today Commun. 2022, 31, 103333. [Google Scholar] [CrossRef]
- Qiao, Z.; Wang, C.; Zou, Y.; Wu, X.; Liu, Z. Oxygen vacancy and pyroelectric polarization collaboratively enhancing PEC performance in BaTiO3 photoelectrodes. Colloids Surf. A Physicochem. Eng. Asp. 2022, 647, 129073. [Google Scholar] [CrossRef]
- Chen, X.; Liu, L.; Yu, P.Y.; Mao, S.S. Increasing solar absorption for photocatalysis with black hydrogenated titanium dioxide nanocrystals. Science 2011, 331, 745–750. [Google Scholar] [CrossRef]
- Wang, B.; Shen, S.; Mao, S.S. Black TiO2 for solar hydrogen conversion. J. Mater. 2017, 3, 96–111. [Google Scholar]
- Rajaraman, T.S.; Parikh, S.P.; Gandhi, V.G. Black TiO2: A review of its properties and conflicting trends. Chem. Eng. J. 2020, 389, 123918. [Google Scholar] [CrossRef]
- Lu, G.M.; Feng, X.T.; Li, Y.H.; Zhang, X. The Microwave-induced fracturing of hard rock. Rock Mech. Rock. Eng. 2019, 52, 3017–3032. [Google Scholar] [CrossRef]
- Samir, M.; Deyab, H.R.; Hassani, F.; Kermani, M.; Agus, P.; Sasmito, A.P. Experimental investigation on the effects of microwave irradiation on kimberlite and granite rocks. J. Rock Mech. Geotech. Eng. 2021, 13, 267–274. [Google Scholar]
- Peinsitt, T.; Kuchar, F.; Hartlieb, P.; Moser, P.; Kargl, H.; Restner, U.; Sifferlinge, N.A. Microwave heating of dry and water saturated basalt, granite and sandstone. Int. J. Min. Miner. Eng. 2010, 2, 18–29. [Google Scholar] [CrossRef]
- Ma, E. Amorphization in mechanically driven material systems. Scr. Mater. 2003, 49, 941–946. [Google Scholar] [CrossRef]
- Akay, G.; Tincer, T.; Aydin, E. The effect of orientation on the radiation induced degradation of polymers. Eur. Polym. J. 1980, 16, 597–600. [Google Scholar] [CrossRef]
- Akay, G.; Tincer, T. The effect of orientation on radiation-induced degradation in high density polyethylene. Polym. Eng. Sci. 1982, 21, 8–17. [Google Scholar] [CrossRef]
- Liu, Q.; Yang, H.; Zhao, J.; Liu, S.; Xia, L.; Hu, P.; Lv, Y.; Huang, Y.; Kong, M.; Li, G. Acceleratory and inhibitory effects of uniaxial tensile stress on the photo-oxidation of polyethylene: Dependence of stress, time duration and temperature. Polymer 2018, 148, 316–329. [Google Scholar] [CrossRef]
- Yeh, J.-W.; Chen, S.-K.; Lin, S.-J.; Gan, J.-Y.; Chin, T.-S.; Shun, T.-T.; Tsau, C.-H.; Chang, S.-Y. Nanostructured high-entropy alloys with multiple principal elements: Novel alloy design concepts and outcomes. Adv. Eng. Mater. 2004, 6, 299–303. [Google Scholar] [CrossRef]
- Cantor, B.; Chang, I.; Knight, P.; Vincent, A. Microstructural development in equiatomic multicomponent alloys. Mater. Sci. Eng. A 2004, 375, 213–218. [Google Scholar] [CrossRef]
- Jian, W.-R.; Xie, Z.; Xu, S.; Yao, X.; Beyerlein, I.J. Shock-induced amorphization in medium entropy alloy CoCrNi. Scr. Mater. 2022, 209, 114379. [Google Scholar] [CrossRef]
- Pacchioni, G. High-entropy materials go nano. Nat. Rev. Mater. 2022, 7, 156. [Google Scholar] [CrossRef]
- Li, K.; Chen, W. Recent progress in high-entropy alloys for catalysts: Synthesis, applications, and prospects. Mater. Today Energy 2021, 20, 100638. [Google Scholar] [CrossRef]
- Chen, L.; Deng, S.; Liu, H.; Wu, J.; Qi, H.; Chen, J. Giant energy-storage density with ultrahigh efficiency in lead-free relaxors via high-entropy design. Nat. Commun. 2022, 13, 3089. [Google Scholar] [CrossRef]
- Akay, G. Process Intensification in the Catalytic Synthesis of Supported Catalysts with Hierarchic Pore Structure. International Patent Publication PCT WO/2013/108045, 25 July 2013. [Google Scholar]
- Fan, C.; Li, L.; Wen, W.; Li, H.; Fu, J.; Ruan, W.; Ren, S.; Sohrabi, S.; Zhang, Z.; Liang, X.; et al. Rapid amorphization of CrMnFeCoNi high-entropy alloy under ultrasonic vibrations. Mater. Des. 2023, 225, 111575. [Google Scholar] [CrossRef]
- Li, L.; Lyu, G.-J.; Li, H.; Fan, C.; Wen, W.; Lin, H.; Huang, B.; Sohrabi, S.; Ren, S.; Liang, X.; et al. Ultra-fast amorphization of crystalline alloys by ultrasonic vibrations. J. Mater. Sci. Technol. 2023, 142, 76–88. [Google Scholar] [CrossRef]
- Wang, B.; Wang, C.; Yu, X.; Cao, Y.; Gao, L.; Wu, C.; Yao, Y.; Lin, S.; Zou, Z. General synthesis of high-entropy alloy and ceramic nanoparticles in nanoseconds. Nat. Synth. 2022, 1, 138–146. [Google Scholar] [CrossRef]
- Potnis, P.R.; Tsou, N.-T.; Huber, J.E. A Review of domain modelling and domain imaging techniques in ferroelectric crystals. Materials 2011, 4, 417–447. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.C.; Bhattacharya, K. Domain patterns and macroscopic behaviour of ferroelectric materials. Philos. Mag. B 2001, 81, 2021–2054. [Google Scholar] [CrossRef]
- Arlt, G.; Sasko, P. Domain configuration and equilibrium size of domains in BaTiO3 ceramics. J. Appl. Phys. 1980, 51, 4956–4960. [Google Scholar] [CrossRef]
- Trolier-McKinstry, S.; Gharb, N.B.; Damjanovic, D. Piezoelectric nonlinearity due to motion of 180° domain walls in ferroelectric materials at subcoercive fields: A dynamic poling model. Appl. Phys. Lett. 2006, 88, 202901. [Google Scholar] [CrossRef]
- Kannan, V.; Trassin, M.; Kochmann, D.M. Kinetics of ferroelectric switching in poled barium titanate ceramics: Effects of electrical cycling rate. Materialia 2022, 25, 101553. [Google Scholar] [CrossRef]
- Hoshina, T.; Kigoshi, Y.; Hatta, S.; Teranishi, T.; Takeda, H.; Tsurumi, T. Size effect and domain-wall contribution of barium titanate ceramics. Ferroelectrics 2010, 402, 29–36. [Google Scholar] [CrossRef]
- Ghosh, D.; Sakata, S.; Carter, J.C.; Thomas, P.A.; Han, H.; Nino, J.C.; Jones, J.L. Domain wall displacement is the origin of superior permittivity and piezoelectricity in BaTiO3 at intermediate grain sizes. Adv. Funct. Mater. 2014, 24, 885–896. [Google Scholar] [CrossRef]
- Bencan, A.; Oveisi, E.; Hashemizadeh, S.; Veerapandiyan, V.K.; Hoshina, T.; Rojac, T.; Deluca, M.; Drazic, G.; Damjanovic, D. Atomic scale symmetry and polar nanoclusters in the paraelectric phase of ferroelectric materials. Nat. Commun. 2021, 12, 3509. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Rodríguez-Córdoba, W.E.; Liu, Z.; Zhu, H.; Lian, T. Beyond band alignment: Hole localization driven formation of three spatially separated long-lived exciton states in CdSe/CdS nanorods. ACS Nano 2013, 7, 7173–7185. [Google Scholar] [CrossRef]
- Gao, D.; Xu, J.; Wang, L.; Zhu, B.; Yu, H.; Yu, J. Optimizing atomic hydrogen desorption of sulfur-rich NiS1+x cocatalyst for boosting photocatalytic H2 evolution. Adv. Mater. 2022, 34, 2108475. [Google Scholar] [CrossRef]
- Law, V.J.; Dowlin, D.P. Converting a microwave oven into a plasma reactor: A review. Int. J. Chem. Eng. 2018, 3, 2957194. [Google Scholar] [CrossRef]
- Zamri, A.A.; Ong, M.Y.; Nomanbhay, S.; Show, P.L. Microwave plasma technology for sustainable energy production and the electromagnetic interaction within the plasma system: A review. Environ. Res. 2021, 197, 111204. [Google Scholar] [CrossRef]
- Wirth, B.D. How does radiation damage materials? Science 2007, 318, 923–924. [Google Scholar] [CrossRef] [PubMed]
- Uberuaga, B.P.; Hoagland, R.G.; Voter, A.F.; Valone, S.M. Direct transformation of vacancy voids to stacking fault tetrahedra. Phys. Rev. Lett. 2007, 99, 135501. [Google Scholar] [CrossRef]
- Matsukawa, Y.; Zinkle, S.J. One-dimensional fast migration of vacancy clusters in metals. Science 2007, 318, 959–962. [Google Scholar] [CrossRef]
- Wei, J.; Feng, B.; Tochigi, E.; Shibata, N.; Ikuhara, Y. Direct imaging of the disconnection climb mediated point defects absorption by a grain boundary. Nat. Commun. 2022, 13, 1455. [Google Scholar] [CrossRef]
- Odette, G.R.; Alinger, M.J.; Wirth, B.D. Recent developments in irradiation-resistant steels. Annu. Rev. Mater. Res. 2008, 38, 471–503. [Google Scholar] [CrossRef]
- Sickafus, K.E.; Grimes, R.; Valdez, J.; Cleave, A.; Tang, M.; Ishimaru, M.; Corish, S.M.; Stanek, C.R.; Uberuaga, B.P. Radiation-induced amorphization resistance and radiation tolerance in structurally related oxides. Nat. Mater. 2007, 6, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.; Balogh, A.G.; Hahn, H. Instability of irradiation induced defects in nanostructured materials. Nucl. Instrum. Methods Phys. Res. Sect. B 1997, 127–128, 119–122. [Google Scholar] [CrossRef]
- Nita, N.; Schaeublin, R.; Victoria, M. Impact of irradiation on the microstructure of nanocrystalline materials. J. Nucl. Mater. 2004, 329–333, 953–957. [Google Scholar] [CrossRef]
- Fu, C.-C.; Torre, J.D.; Willaime, F.; Bocquet, J.-L.; Barbu, A. Multiscale modelling of defect kinetics in irradiated iron. Nat. Mater. 2005, 4, 68–74. [Google Scholar] [CrossRef]
- Bai, X.-M.; Voter, A.F.; Hoagland, R.G.; Nastasi, M.; Uberuaga, B.P. Efficient annealing of radiation damage near grain boundaries via interstitial emission. Science 2010, 327, 1631–1634. [Google Scholar] [CrossRef]
- Frolov, T.; Olmsted, D.L.; Asta, M.; Mishin, Y. Structural phase transformations in metallic grain boundaries. Nat. Commun. 2013, 4, 1899. [Google Scholar] [CrossRef]
- Chugunov, A.I.; Horowitz, C.J. Breaking stress of neutron star crus. Mon. Not. R. Astron. Soc. 2010, 407, L54–L58. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Baba, T.; Jiang, H.; Liu, C.; Guan, Y.; Elleuch, O.; Kuech, T.; Morgan, D.; Idrobo, J.-C.; et al. Radiation-induced segregation in a ceramic. Nat. Mater. 2020, 19, 992–998. [Google Scholar] [CrossRef]
- Albedwawi, S.H.; AlJaberi, A.; Haidemenopoulos, G.N.; Polychronopoulou, K. High entropy oxides-exploring a paradigm of promising catalysts: A review. Mater. Des. 2021, 202, 109534. [Google Scholar] [CrossRef]
- Bao, H.; Zhu, S.; Zhou, L.; Fu, H.; Zhang, H.; Cai, W. Mars–van-Krevelen mechanism-based blackening of nano-sized white semiconducting oxides for synergetic solar photo-thermocatalytic degradation of dye pullutants. Nanoscale 2020, 12, 4030–4039. [Google Scholar] [CrossRef]
- Botros, M.; Janek, J. Embracing disorder in solid state batteries. Science 2022, 378, 1273–1274. [Google Scholar] [CrossRef]
- Zhang, F.; Cheng, F.; Cheng, C.; Guo, M.; Liu, Y.; Miao, Y.; Gao, F.; Wang, X. Preparation and electrical conductivity of (Zr, Hf, Pr, Y, La) O high entropy fluorite oxides. J. Mater. Sci. Technol. 2022, 105, 122–130. [Google Scholar] [CrossRef]
- Zeng, Y.; Ouyang, B.; Liu, J.; Byeon, Y.-W.; Cai, Z.; Miara, L.J.; Wang, Y.; Ceder, G. High-entropy mechanism to boost ionic conductivity. Science 2022, 378, 1320–1324. [Google Scholar] [CrossRef]
- Liu, H.; Cui, Z.; Chen, M.; Zhao, B. Phase equilibrium study of ZnO-‘FeO’-SiO2 system at fixed Po2 10−8 atm. Metall. Mater. Trans. B 2016, 47, 164–173. [Google Scholar] [CrossRef]
- Lee, C.S.; Matori, K.A.; Ab Aziz, S.H.; Kamari, H.M.; Ismail, I.; Zaid, M.H. Fabrication and characterization of glass and glass-ceramic from rice husk ash as a potent material for opto-electronic applications. J. Mater. Sci. Mater. Electron. 2017, 28, 17611–17621. [Google Scholar] [CrossRef]
- Liu, H.; Cui, Z.; Chen, M.; Zhao, B. Phase equilibria study of the ZnO–‘FeO’–SiO2–MgO system at Po2 10−8 atm. Miner. Process. Extr. Metall. 2018, 127, 242–249. [Google Scholar] [CrossRef]
- Ma, S.; Huang, S.D.; Liu, Z.P. Dynamic coordination of cations and catalytic selectivity on zinc–chromium oxide alloys during syngas conversion. Nat. Catal. 2019, 2, 671–677. [Google Scholar] [CrossRef]
- Mostafa, M.M.M.; Bajafar, W.; Gu, L.; Narasimharao, K.; Abdel Salam, M.; Alshehri, A.; Khdary, N.H.; Al-Faifi, S.; Chowdhury, A.D. Electrochemical characteristics of nanosized Cu, Ni, and Zn cobaltite spinel materials. Catalysts 2022, 12, 893. [Google Scholar] [CrossRef]
- Noormohammadi, E.; Sanjabi, S.; Soavi, F.; Poli, F. Electrodeposited Cobalt–Copper mixed oxides for supercapacitor electrodes and investigation of the Co/Cu ratio on the electrochemical performance. Mater. Renew. Sustain. Energy 2023, 12, 53–61. [Google Scholar] [CrossRef]
- Zhang, Q.; Hu, Y.; Wu, H.; Zhao, X.; Wang, M.; Wang, S.; Feng, R.; Chen, Q.; Song, F.; Chen, M.; et al. Entropy-stabilized multicomponent porous spinel nanowires of NiFeXO4 (X = Fe, Ni, Al, Mo, Co, Cr) for efficient and durable electrocatalytic oxygen evolution reaction in alkaline medium. ACS Nano 2023, 17, 1485–1494. [Google Scholar] [CrossRef]
- Aydinyan, S.; Kirakosyan, H.; Sargsyan, A.; Volobujeva, O.; Kharatyan, S. Solution combustion synthesis of MnFeCoNiCu and (MnFeCoNiCu)3O4 high entropy materials and sintering thereof. Ceram. Int. 2022, 48, 20294–20305. [Google Scholar] [CrossRef]
- Wang, Y.; Li, H.; Liu, H.; Yang, L.; Zeng, C. Preparation and formation mechanism of Cr-free spinel-structured high entropy oxide (MnFeCoNiCu)3O4. Ceram. Int. 2023, 49, 1940–1946. [Google Scholar] [CrossRef]
- Sarkar, A.; Breitung, B.; Hahn, H. High entropy oxides: The role of entropy, enthalpy and synergy. Scr. Mater. 2020, 187, 43–48. [Google Scholar] [CrossRef]
- Anandkumar, M.; Trofimov, E. Synthesis, properties, and applications of high-entropy oxide ceramics: Current progress and future perspectives. J. Alloys Compd. 2023, 960, 170690. [Google Scholar] [CrossRef]
- Ye, Y.F.; Wang, Q.; Lu, J.; Liu, C.T.; Yang, Y. High-entropy alloy: Challenges and prospects. Mater. Today 2016, 19, 349–362. [Google Scholar] [CrossRef]
- Pan, Y.; Liu, J.-X.; Tu, T.-Z.; Wang, W.; Zhang, G.-J. High-entropy oxides for catalysis: A diamond in the rough. Chem. Eng. J. 2023, 451, 138659. [Google Scholar] [CrossRef]
- Ortega-Zarzosa, G.; Araujo-Andrade, C.; Compean-Jasso, M.E.; Mart, J.R.; Ruiz, F. Cobalt oxide/silica xerogels powder: X-ray diffraction, infrared and visible absorption studies. J. Sol-Gel Sci. Technol. 2002, 24, 23–29. [Google Scholar] [CrossRef]
- Cheng, W.; Rechberger, F.; Ilari, G.; Ma, H.; Lin, W.-I.; Niederberger, M. Amorphous cobalt silicate nanobelts@carbon composites as stable anode material for lithium ion batteries. Chem. Sci. 2015, 6, 6908. [Google Scholar] [CrossRef]
- Guragain, D.; Zequine, C.; Gupta, R.K.; Mishra, S.R. Facile synthesis of bio-template tubular MCo2O4 (M = Cr, Mn, Ni) microstructure and its electrochemical performance in aqueous electrolyte. Processes 2020, 8, 343. [Google Scholar] [CrossRef]
- Saaid, F.I.; Arsyad, A.; Azman, N.S.H.; Kumar, A.; Yang, C.-C.; Tseng, T.-T.; Winie, T. The synergistic effect of iron cobaltite compare to its single oxides as cathode in supercapacitor. J. Electroceram. 2020, 44, 183–194. [Google Scholar] [CrossRef]
- Li, J.; Jiang, N.; Liao, J.; Feng, Y.; Liu, Q.; Li, H. Nonstoichiometric Cu0.6Ni0.4Co2O4 nanowires as an anode material for high performance lithium storge. Nanomaterials 2020, 10, 191. [Google Scholar]
- Tian, G.; Liu, X.; Zhang, C.; Fan, X.; Xiong, H.; Li, Z.; Yan, B.; Zhang, L.; Wang, N.; Peng, H.-J.; et al. Accelerating syngas-to-aromatic conversion via spontaneously monodispersed Fe in ZnCr2O4 spinel. Nat. Commun. 2022, 13, 5567. [Google Scholar] [CrossRef]
- Rost, C.M.; Sachet, E.; Borman, T.; Moballegh, A.; Dickey, E.C.; Hou, D.; Jones, J.L.; Curtarolo, S.; Maria, J.-P. Entropy-stabilized oxides. Nat. Commun. 2015, 6, 8485. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Gong, J.; Cai, D.; Li, Y.; Sun, Y.; Wang, W.; Feng, C. Flexible synthesis of CuCo2O4 hexagonal nanocrystal by melting salt modified combustion method as high-performance anode materials for lithium-ion batteries. J. Electroceram. 2023, 50, 57–66. [Google Scholar] [CrossRef]
- Navrotsky, A.; Kleppa, O. The thermodynamics of cation distributions in simple spinels. J. Inorg. Nucl. Chem. 1967, 29, 2701–2714. [Google Scholar] [CrossRef]
- Fang, Y.; Zhang, S.; Ohodnicki, P.R.; Wang, G. Relation between cation distribution and chemical bonds in spinel NiFe2O4. Mater. Today Commun. 2022, 33, 104436. [Google Scholar] [CrossRef]
- Cheng, Z.; Yang, H.; Xu, Y.; Jiang, J.; Xu, Q. Active bimetallic Fe–O–Co sites built on 2D trimetallic complex spinel oxides for industrially oxygen evolution electrocatalyst. Mater. Today Chem. 2022, 26, 101214. [Google Scholar] [CrossRef]
- Lin, C.-C.; Chang, C.-W.; Kaun, C.-C.; Su, Y.-H. Stepwise evolution of photocatalytic spinel-structured (Co, Cr, Fe, Mn, Ni)3O4 high entropy oxides from first principles calculations to machine learning. Crystals 2021, 11, 1035. [Google Scholar] [CrossRef]
- Cieslak, J.; Reissner, M.; Berent, K.; Dabrowa, J.; Stygar, M.; Mozdzierz, M.; Zajusz, M. Magnetic properties and ionic distribution in high entropy spinels studied by mossbauer and ab initio methods. Acta Mater. 2021, 206, 116600. [Google Scholar] [CrossRef]
- Sarkar, A.; Eggert, B.; Witte, R.; Lill, J.; Velasco, L.; Wang, Q.; Sonar, J.; Ollefs, K.; Bhattacharya, S.S.; Brand, R.A.; et al. Comprehensive investigation of crystallographic, spin-electronic and magnetic structure of (Co0.2Cr0.2Fe0.2Mn0.2Ni0.2)3O4: Unraveling the suppression of configuration entropy in high entropy oxides. Acta Mater. 2022, 226, 117581. [Google Scholar] [CrossRef]
- Johnstone, G.H.J.; González-Rivas, M.U.; Taddei, K.M.; Sutarto, R.; Sawatzky, G.A.; Green, R.J.; Oudah, M.; Hallas, A.M. Entropy engineering and tunable magnetic order in the spinel high-entropy oxide. J. Am. Chem. Soc. 2022, 144, 20590–20600. [Google Scholar] [CrossRef]
- Poleti, D.; Vasović, D.; Karanović, L.; Branković, Z. Synthesis and characterization of ternary zinc-antimony-transition metal spinels. J. Solid State Chem. 1994, 112, 39–44. [Google Scholar] [CrossRef]
- Wang, Z.L. Splendid one-dimensional nanostructures of zinc oxide: A new nanomaterial family for nanotechnology. ACS Nano 2008, 2, 1987–1992. [Google Scholar] [CrossRef] [PubMed]
- Morkoç, H.; Özgür, Ü. Zinc Oxide: Fundamentals, Materials and Device Technology; Wiley-VCH: Weinheim, Germany, 2009; ISBN 978-3-527-40813-9. [Google Scholar]
- Fortunato, E.; Gonçalves, A.; Pimentel, A.; Barquinha, P.; Gonçalves, G.; Pereira, L.; Ferreira, I.; Martins, R. Zinc oxide, a multifunctional material: From material to device applications. Appl. Phys. A 2009, 96, 197–205. [Google Scholar] [CrossRef]
- Wojnarowicz, J.; Chudoba, T.; Lojkowski, W. A review of microwave synthesis of zinc oxide nanomaterials: Reactants, process parameters and morphologies. Nanomaterials 2020, 10, 1086. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, H.; Nakamura, N.; Watanabe, S.; Kim, J.; Kitano, M.; Hosono, H. Electronic promotion of methanol synthesis over Cu-loaded ZnO-based catalysts. J. Phys. Chem. Lett. 2023, 14, 1259–1264. [Google Scholar] [CrossRef] [PubMed]
- Flores-Lasluisa, J.X.; Quílez-Bermejo, J.; Ramírez-Pérez, A.C.; Huerta, F.; Cazorla-Amorós, D.; Morallón, E. Copper-doped cobalt spinel electrocatalysts supported on activated carbon for hydrogen evolution reaction. Materials 2019, 12, 1302. [Google Scholar] [CrossRef]
- Wang, L.; Yi, Y.; Guo, H.; Du, X.; Zhu, B.; Zhu, Y. Highly dispersed Co nanoparticles prepared by an improved method for plasma-driven NH3 decomposition to produce H2. Catalysts 2019, 9, 107. [Google Scholar] [CrossRef]
- Xu, C.; Tan, J.; Zhang, X.; Huang, Y. Petal-like CuCo2O4 spinel nanocatalyst with rich oxygen vacancies for efficient PMS activation to rapidly degrade pefloxacin. Sep. Purif. Technol. 2022, 291, 120933. [Google Scholar] [CrossRef]
- Ferreira, T.A.S.; Waerenborgh, J.C.; Mendonça, M.H.R.M.; Nunes, M.R.; Costa, F.M. Structural and morphological characterization of FeCo2O4 and CoFe2O4 spinels prepared by a coprecipitation method. Solid State Sci. 2003, 5, 383–392. [Google Scholar] [CrossRef]
- Kurnaz Yetim, N.; Kurşun Baysak, F.; Koç, M.M.; Nartop, D. Characterization of magnetic Fe3O4@SiO2 nanoparticles with fluorescent properties for potential multipurpose imaging and theranostic applications. J. Mater. Sci. Mater. Electron. 2020, 31, 18278–18288. [Google Scholar] [CrossRef]
- Favors, Z.; Wang, W.; Bay, H.H.; George, A.; Ozkan, M.; Ozkan, C.S. Stable cycling of SiO₂ nanotubes as high-performance anodes for lithium-ion batteries. Sci. Rep. 2014, 4, 4605. [Google Scholar] [CrossRef]
- George, A.; Shen, B.; Craven, M.; Wang, Y.; Kang, D.; Wu, C.; Tu, X. A review of non-thermal plasma technology: A novel solution for CO2 conversion and utilization. Renew. Sustain. Energy Rev. 2021, 135, 109702. [Google Scholar] [CrossRef]
- Navascués, P.; Garrido-García, J.; Cotrino, J.; González-Elipe, A.R. Gómez-Ramírez, Incorporation of a metal catalyst for the ammonia synthesis in a ferroelectric packed-bed plasma reactor: Does it really matter? ACS Sustain. Chem. Eng. 2023, 11, 3621–3632. [Google Scholar] [CrossRef] [PubMed]
- Ou, Y.; Li, S.; Wang, F.; Duan, X.; Yuan, W.; Yang, H.; Zhang, Z.; Wang, Y. Reversible transformation between terrace and step sites of Pt nanoparticles on titanium under CO and O2 environments. Chin. J. Catal. 2022, 43, 2026–2033. [Google Scholar] [CrossRef]
- Pantzas, K.; Patriarche, G. Experimental quantification of atomically-resolved HAADF-STEM images using EDX. Ultramicroscopy 2021, 220, 113152. [Google Scholar] [CrossRef] [PubMed]
- Cha, E.; Chung, H.; Jang, J.; Lee, J.; Lee, E.; Ye, J.C. Low-dose sparse-view HAADF-STEM-EDX tomography of nanocrystals using unsupervised deep learning. ACS Nano 2022, 16, 10314–10326. [Google Scholar] [CrossRef]
- Diebold, U.; Li, S.-C.; Schmid, M. Oxide surface science. Annu. Rev. Phys. Chem. 2010, 61, 129–148. [Google Scholar] [CrossRef]
- Setvin, M.; Hulva, J.; Parkinson, G.S.; Schmid, M.; Diebold, U. Electron transfer between anatase TiO2 and an O2 molecule directly observed by atomic force microscopy. Proc. Natl. Acad. Sci. USA 2017, 114, E2556–E2562. [Google Scholar] [CrossRef] [PubMed]
- Sokolovic, I.; Reticcioli, M.; Calkovsky, M.; Wagner, M.; Schmid, M.; Franchini, C.; Diebold, U.; Setvin, M. Resolving the absorption of molecular O2 on the rutile TiO2(110) surface by noncontact atomic force microscopy. Proc. Natl. Acad. Sci. USA 2020, 117, 14827–14837. [Google Scholar] [CrossRef] [PubMed]
- Katsube, D.; Shimizu, R.; Sugimoto, Y.; Hitosugi, T.; Abe, M. Identification of OH groups on SrTiO3(100)-()-R33.7° reconstructed surface by non-contact atomic force microscopy and scanning tunnelling microscopy. Appl. Phys. Lett. 2023, 122, 017602. [Google Scholar] [CrossRef]
- Lakhera, S.K.; Rugma, T.P.; Rishi Krishna, B.S.; Rabiee, N.; Bernaurdshaw, N. Pivotal role of oxygen during the synthesis of Cu(OH)2/TiO2 and its effect on photocatalytic hydrogen production activity. Catal. Today 2023, 43, 114007. [Google Scholar] [CrossRef]
- Li, S.; Czap, G.; Li, J.; Zhang, Y.; Yu, A.; Yuan, D.; Kimura, H.; Wu, R.; Ho, W. Confinement-induced catalytic dissociation of hydrogen molecules in a scanning tunnelling microscope. J. Am. Chem. Soc. 2022, 144, 9618–9623. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.D. Cations play an essential role in CO2 reduction. Nat. Catal. 2021, 4, 641–642. [Google Scholar] [CrossRef]
- Ridge, S.; Resasco, J.; Walton, A.; Seger, B.; Bell, A.T.; Chan, K. Understanding cation effects in electrochemical CO2 reduction. Energy Environ. Sci. 2019, 12, 3001–3014, Erratum in Energy Environ. Sci. 2020, 13, 646–647. [Google Scholar]
- Holland, H.D. The oxygenation of the atmosphere and oceans. Philos. Trans. R. Soc. Lond. Ser. B 2006, 361, 903–915. [Google Scholar] [CrossRef]
- Lu, Z.; Chang, Y.C.; Yin, Q.-Z.; Ng, C.Y.; Jackson, W.M. Evidence for direct molecular oxygen production in CO2 photodissociation. Science 2014, 346, 61–64. [Google Scholar] [CrossRef]
- Schlegel, H.B. Exploring potential energy surfaces for chemical reactions: An overview of some practical methods. J. Comput. Chem. 2003, 24, 1514–1527. [Google Scholar] [CrossRef]
- Szabó, I.; Császár, A.G.; Czakó, G. Dynamics of the F− + CH3Cl → Cl− + CH3F SN2 reaction on a chemically accurate potential energy surface. Chem. Sci. 2013, 4, 4362. [Google Scholar] [CrossRef]
- Wang, Y.; Song, H.; Szabó, I.; Czakó, G.; Guo, H.; Yang, M. Mode specific SN2 reaction dynamics. J. Phys. Chem. Lett. 2016, 7, 3322. [Google Scholar] [CrossRef]
- Maeda, S.; Harabuchi, Y.; Takagi, M.; Taketsugu, T.; Morokuma, K. Artificial force induced reaction (AFIR) method for exploring quantum chemical potential energy surfaces. Chem. Rec. 2016, 16, 2232–2248. [Google Scholar] [CrossRef]
- Rousseau, R.; Glezakou, V.A.; Selloni, A. Theoretical insights into the surface physics and chemistry of redox-active oxides. Nat. Rev. Mater. 2020, 5, 460–475. [Google Scholar] [CrossRef]
- Tasi, D.A.; Győri, T.; Czakó, G. On the development of a gold-standard potential energy surface for the OH− + CH3I reaction. Phys. Chem. Chem. Phys. 2020, 22, 3775–3778. [Google Scholar] [CrossRef]
- Hussain, A.; Nabi, A.G.; Javaid, S. Remarkable reactivity of Fe modified Cu(100) surface towards CO2 decomposition: A DFT study. Mater. Today Commun. 2023, 34, 105395. [Google Scholar] [CrossRef]
- Zhao, C.; Xu, H. Activation of CO2 by Direct Cleavage Triggered by Photoelectrons on Rutile TiO2(110). J. Phys. Chem. Lett. 2023, 14, 1928–1933. [Google Scholar] [CrossRef] [PubMed]
- Ganyecz, A.; Mezei, P.D.; Kállay, M. Oxygen reduction reaction on TiO2 rutile (110) surface in the presence of bridging hydroxyl groups. Comput. Theor. Chem. 2019, 1168, 112607. [Google Scholar] [CrossRef]
- Song, H.; Xie, W.; Zhang, C.; Yang, M. Toward a comprehensive understanding of mode-specific dynamics of polyatomic reactions: A Full-Dimensional Quantum Dynamics Study of the H + NH3 Reaction. J. Phys. Chem. A 2022, 126, 663–669. [Google Scholar] [CrossRef]
- Fernando, A.; Weerawardene, K.L.D.M.; Karimova, N.V.; Aikens, C.M. Quantum mechanical studies of large metal, metal oxide, and metal chalcogenide nanoparticles and clusters. Chem. Rev. 2015, 115, 6112–6216. [Google Scholar]
- Taifan, W.; Boily, J.-F.; Baltrusaitis, J. Surface chemistry of carbon dioxide revisited. Surf. Sci. Rep. 2016, 71, 595–671. [Google Scholar] [CrossRef]
- Kitamura, J.; Mizuno, H.; Kato, N.; Aoki, I. Plasma-Erosion Properties of Ceramic Coating Prepared by Plasma Spraying. Mater. Trans. 2006, 47, 1677–1683. [Google Scholar] [CrossRef]
- Kasashima, Y.; Tabaru, T.; Matsuda, O.; Motomura, T. Investigation of the relationship between plasma etching characteristics and microstructures of alumina ceramics for chamber parts. Jpn. J. Appl. Phys. 2019, 58, 041001. [Google Scholar] [CrossRef]
- Bouchoule, A.; Boufendi, L. Particulate formation and dusty plasma behaviour in argon-silane RF discharge. Plasma Sources Sci. Technol. 1993, 2, 204. [Google Scholar] [CrossRef]
- Deka, B.C.T.; Bailung, Y.; Sharma, D.; Sharma, S.K.; Bailung, H. Spatiotemporal evolution of a self-excited dust density wave in a nanodusty plasma under strong Havnes effect. Phys. Plasmas 2021, 28, 123702. [Google Scholar]
- Petersen, A.; Asnaz, O.H.; Tadsen, B.; Greiner, F. Decoupling of dust cloud and embedding plasma for high electron depletion in nanodusty plasmas. Commun. Phys. 2022, 5, 308. [Google Scholar] [CrossRef]
- Chen, X.; Hogan, C.J. Nanoparticle dynamics in the spatial afterglows of nonthermal plasma synthesis reactors. Chem. Eng. J. 2021, 411, 128383. [Google Scholar] [CrossRef]
- Fortova, V.E.; Ivlevb, A.V.; Khrapakb, S.A.; Khrapaka, A.G.; Morfill, G.E. Complex (dusty) plasmas: Current status, open issues, perspectives. Phys. Rep. 2005, 421, 1–103. [Google Scholar] [CrossRef]
- Morfill, G.E.; Ivlev, A. Complex plasmas: An interdisciplinary research field. Rev. Mod. Phys. 2009, 81, 1353–1404. [Google Scholar] [CrossRef]
- Merlino, R. Dusty plasmas: From Saturn’s rings to semiconductor processing devices. Adv. Phys. X 2021, 6, 1. [Google Scholar] [CrossRef]
- Haves, O.; Morfill, G.E.; Goertz, C.K. Plasma potential and grain charges in a dust cloud embedded in a plasma. J. Geophys. Res. Space Phys. 1984, 89, 10999–11003. [Google Scholar] [CrossRef]
- Staps, A.T.J.; Donders, T.J.M.; Platier, B.; Beckers, J. In-situ measurement of dust charge density in nanodusty plasma. J. Phys. D Appl. Phys. 2022, 55, 08LT01. [Google Scholar] [CrossRef]
- Davari, H.; Farokhi, B.; Asgarian, M.A. Particle simulation of the strong magnetic field effect on dust particle charging process. Sci. Rep. 2023, 13, 1111. [Google Scholar] [CrossRef]
- Balzer, K.; Rasmussen, M.R.; Schlünzen, N.; Joost, J.-P.; Bonitz, M. Doublon formation by ions impacting strongly correlated finite lattice system. Phys. Rev. Lett. 2018, 121, 267602. [Google Scholar] [CrossRef]
- Arp, O.; Caliebe, D.; Menzel, K.O.; Piel, A.; Goree, J.A. Experimental investigation of dust density waves and plasma glow. IEEE Trans. Plasma Sci. 2010, 38, 842–846. [Google Scholar] [CrossRef]
- Mondol, M.M.H.; Kim, C.-U.; Jhung, S.H. Titanium nitride@nitrogen-enriched porous carbon derived from metal–organic frameworks and melamine: A remarkable oxidative catalyst to remove indoles from fuel. Chem. Eng. J. 2022, 450, 138411. [Google Scholar] [CrossRef]
- Kong, L.; Liang, X.; Wang, M.; Wu, C.-M.L. Theoretical screening of transition metal-embedded Ti2n for high-efficiency hydrogen evolution reaction. ACS Sustain. Chem. Eng. 2022, 10, 4152–4160. [Google Scholar] [CrossRef]
- Bai, X.; Guan, J. Applications of MXene-based single atom catalysts. Small Struct. 2023, 4, 2200354. [Google Scholar] [CrossRef]
- Valentin, C.D.; Finazzi, E.; Pacchioni, G.; Selloni, A.; Livraghi, S.; Paganini, M.C.; Giamello, E. N-doped TiO2: Theory and experiment. Chem. Phys. 2007, 339, 44–56. [Google Scholar] [CrossRef]
- Foo, C.; Li, Y.; Lebedev, K.; Chen, T.; Day, S.; Tang, C.; Tsang, S.C.E. Characterisation of oxygen defects and nitrogen impurities in TiO2 photocatalysts using variable-temperature X-ray powder diffraction. Nat. Commun. 2021, 12, 661. [Google Scholar] [CrossRef]
- Eichler, J.E.; Burrow, J.N.; Wang, Y.; Calabro, D.C.; Mullins, C.B. Unraveling porogenesis in nitrogen rich K+-activated carbons. Carbon 2022, 186, 711–723. [Google Scholar] [CrossRef]
- Schlögl, R.; Abd Hamid, S.B. Nanocatalysis: Mature science revisited or something really new? Angew. Chem. Int. Ed. 2004, 43, 1628–1637. [Google Scholar] [CrossRef]
- Fu, Q.; Yang, F.; Bao, X. Interface-confined oxide nanostructures for catalytic oxidation reactions. Acc. Chem. Res. 2013, 46, 1692–1701. [Google Scholar] [CrossRef]
- Lu, L.; Zou, S.; Fang, B. The critical impact of ligands on heterogeneous nanocatalysis: A review. ACS Catal. 2021, 11, 6020–6058. [Google Scholar] [CrossRef]
- Yang, R.; Bao, Z.; Sun, Y. Probing and leveraging the structural heterogeneity of nano-materials for enhanced catalysis. ACS Nanosci. Au 2023, 3, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Jing, W.; Shen, H.; Qin, R.; Wu, Q.; Liu, K.; Zheng, N. Surface and interface coordination chemistry learned from model heterogeneous metal nanocatalysts: From atomically dispersed catalysts to atomically precise clusters. Chem. Rev. 2023, 123, 5948–6002. [Google Scholar] [CrossRef]
- Matsugi, A. Modeling third-body effects in the thermal decomposition of H2O2. Combust. Flame 2021, 225, 444–452. [Google Scholar] [CrossRef]
- Barbet, M.C.; Burke, M.P. Impact of “missing” third-body efficiencies on kinetic model predictions of combustion properties. Proc. Combust. Inst. 2021, 38, 425–432. [Google Scholar] [CrossRef]
- Oladeji, A.V.; Courtney, J.M.; Rees, N.V. Copper deposition on metallic and non-metallic single particles via impact electrochemistry. Electrochim. Acta 2022, 405, 139838. [Google Scholar] [CrossRef]
- Culbertson, C.M.; Flak, A.T.; Yatskin, M.; Cheong, P.H.-Y.; Cann, D.P.; Dolgos, M.R. Neutron total scattering studies of Group-II titanates (ATiO3, A2+=Mg, Ca, Sr, Ba). Sci. Rep. 2020, 10, 3729. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Huyan, H.; Sarkar, A.; Gao, W.; Yan, X.; Addiego, C.; Kruk, R.; Hanh, H.; Pan, X. Direct observation of elemental fluctuation and oxygen octahedral distortion-dependent charge distribution in high entropy oxides. Nat. Commun. 2022, 13, 2358. [Google Scholar] [CrossRef]
- Liu, J.; Zhu, X.; Zhou, C.; Du, J.; Gan, Y.; Chen, G.; Tu, X. Plasma-catalytic ammonia synthesis over BaTiO3 supported metal catalysts: Process optimization using response surface methodology. Vacuum 2022, 203, 111205. [Google Scholar] [CrossRef]
- Yang, K.; Dai, Y.; Huang, B. Review of first-principles studies of TiO2: Nanocluster, bulk, and material interface. Catalysts 2020, 10, 972. [Google Scholar] [CrossRef]
- Fo, Y.; Ma, Y.; Dong, H.; Zhou, X. Tuning the electronic structure of BaTiO3 for an enhanced photocatalytic performance using cation-anion cooping: A first principles study. New J. Chem. 2021, 45, 8228–8239. [Google Scholar] [CrossRef]
- Rakotovelo, S.P.; Moussounda, G.; Haroun, F.M.; Legare, P.; Rakotomahevitra, A.; Parlebas, C.J. DFT study of BaTiO3 (001) surface with O and O2 adsorption. Eur. Phys. J. B 2007, 57, 291–297. [Google Scholar] [CrossRef]
- Chun, H.-J.; Youngju Lee, Y.; Kim, S.; Yoon, Y.; Kim, Y.; Park, S.-C. Surface termination of BaTiO3(111) single crystal: A combined DFT and XPS study. Appl. Surf. Sci. 2022, 578, 152018. [Google Scholar] [CrossRef]
- Wang, Q.; Domen, K. Particulate photocatalysts for light-driven water splitting: Mechanisms, challenges, and design strategies. Chem. Rev. 2020, 120, 919–985. [Google Scholar] [CrossRef] [PubMed]
- Landau, M.V.; Vidruk, R.; Vingurt, D.; Fuks, D.; Herskowitz, M. Grain boundaries in nanocrystalline catalytic materials as a source of surface chemical functionality. Rev. Chem. Eng. 2014, 30, 379–401. [Google Scholar] [CrossRef]
- Pal, D.B.; Chand, R.; Upadhyay, S.N.; Mishra, P.K. Performance of water gas shift reaction catalysts: A review. Renew. Sust. Energy Rev. 2018, 93, 549–565. [Google Scholar] [CrossRef]
- Bogaerts, A.; Centi, G.; Hessel, V.; Rebrov, E. Challenges in unconventional catalysis. Catal. Today 2023, 420, 114180. [Google Scholar] [CrossRef]
- Stoukides, M.; Vayenas, C.G. The effect of electrochemical oxygen pumping on the rate and selectivity of ethylene oxidation on polycrystalline silver. J. Catal. 1981, 70, 137–146. [Google Scholar] [CrossRef]
- Vernoux, P.; Lizarraga, L.; Tsampas, M.N.; Sapountzi, F.M.; De Lucas-Consuegra, A.; Valverde, J.L.; Souentie, S.; Vayenas, C.G.; Tsiplakides, D.; Balomenou, S.; et al. Ionically conducting ceramics as active catalyst supports. Chem. Rev. 2013, 113, 8192–8260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, C.; Du, X.; Zhu, Y.; Huang, L.; Yang, K.; Zhao, J.; Liang, C.; Yu, Q.; Li, S.; et al. Deciphering exogenous electric field promoting catalysis from the perspectives of electric energy and electron transfer: A review. Chem. Eng. J. 2023, 452, 139098. [Google Scholar] [CrossRef]
- Hollevoet, L.; Jardali, F.; Gorbanev, Y.; Creel, J.; Bogaerts, A.; Martens, J.A. Towards green ammonia synthesis through plasma-driven nitrogen oxidation and catalytic reduction. Angew. Chem. Int. Ed. 2020, 59, 23825–23829. [Google Scholar] [CrossRef]
- Shah, J.; Wang, W.; Bogaerts, A.; Carreon, M.L. Ammonia synthesis by radio frequency plasma catalysis: Revealing the underlying mechanisms. ACS Appl. Energy Mater. 2018, 1, 4824–4839. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, W.; Xu, S.; Zhao, S.; Chen, G.; Weidenkaff, A.; Hardacre, C.; Fan, X.; Huang, J.; Tu, X. Shielding protection by mesoporous catalysts for improving plasma-catalytic ambient ammonia synthesis. J. Am. Chem. Soc. 2022, 144, 12020–12031. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Du, X.; Chen, K.; Zheng, Z.; Liu, Y.; Shen, X.; Hu, C. Predicting the ammonia synthesis performance of plasma catalysis using an artificial neural network model. ACS Sustain. Chem. Eng. 2023, 11, 4543–4554. [Google Scholar] [CrossRef]
- Zhang, J.; Li, X.; Zheng, J.; Du, M.; Wu, X.; Song, J.; Cheng, C.; Li, T.; Yang, W. Non-thermal plasma-assisted ammonia production: A review. Energy Convers. Manag. 2023, 293, 117482. [Google Scholar] [CrossRef]
- Osorio-Tejada, J.; van’t Veer, K.; Long, N.V.D.; Tran, N.N.; Fulcheri, L.; Patil, B.S.; Bogaerts, A.; Hessel, V. Sustainability analysis of methane-to-hydrogen-to-ammonia conversion by integration of high-temperature plasma and non-thermal plasma processes. Energy Convers. Manag. 2022, 269, 116095. [Google Scholar] [CrossRef]
- Perillo, R.; Chandra, R.; Akkermans, G.; Vijvers, W.; Graef, W.; Classen, I.; Van Dijk, J.; De Baar, M. Studying the influence of nitrogen seeding in a detached-like hydrogen plasma by means of numerical simulation. Plasma Phys. Control. Fusion 2018, 60, 105004. [Google Scholar] [CrossRef]
- Van’t Veer, K.; Engelmann, Y.; Reniers, F.; Bogaerts, A. Plasma Catalytic Ammonia Synthesis in a DBD Plasma: Role of Micro-discharges and Their Afterglows. J. Phys. Chem. C 2020, 124, 22871–22883. [Google Scholar] [CrossRef]
- Navascués, P.; Obrero-Pérez, J.M.; Cotrino, J.; González-Elipe, A.R.; Gómez-Ramírez, A. Isotope Labelling for Reaction Mechanism Analysis in DBD Plasma Processes. Catalysts 2019, 9, 45. [Google Scholar] [CrossRef]
- Navascués, P.; Obrero-Pérez, J.M.; Cotrino, J.; González-Elipe, A.R.; Gómez-Ramírez, A. Unraveling Discharge and Surface Mechanisms in Plasma-Assisted Ammonia Reactions. ACS Sustain. Chem. Eng. 2020, 8, 14855–14866. [Google Scholar] [CrossRef]
- Yaala, M.B.; Saeedi, A.; Scherrer, D.-F.; Moser, L.; Steiner, R.; Zutter, M.; Oberkofler, M.; De Temmerman, G.; Marot, L.; Meyer, E. Plasma-assisted catalytic formation of ammonia in N2–H2 plasma on a tungsten surface. Phys. Chem. Chem. Phys. 2019, 21, 16623–16633. [Google Scholar] [CrossRef]
- Andersen, J.A.; Christensen, J.M.; Østberg, M.; Bogaerts, A.; Jensen, A.D. Plasma-catalytic dry reforming of methane: Screening of catalytic materials in a coaxial packed-bed DBD reactor. Chem. Eng. J. 2020, 397, 125519. [Google Scholar] [CrossRef]
- Andersen, J.A.; Holm, M.C.; van’t Veer, K.; Christensen, J.M.; Østberg, M.; Bogaerts, A.; Jensen, A.D. Plasma-catalytic ammonia synthesis in a dielectric barrier discharge reactor: A combined experimental study and kinetic modelling. Chem. Eng. J. 2023, 457, 141294. [Google Scholar] [CrossRef]
- Zeng, X.; Zhang, S.; Liu, Y.; Hu, X.; Ostrikov, K.K.; Shao, T. Energy-efficient pathways for pulsed-plasma-activated sustainable ammonia synthesis. ACS Sustain. Chem. Eng. 2023, 11, 1110–1120. [Google Scholar] [CrossRef]
- Kim, M.; Biswas, S.; Nava, G.; Wong, B.M.; Mangolini, L. Reduced energy cost of ammonia synthesis via RF plasma pulsing. ACS Sustain. Chem. Eng. 2022, 10, 15135–15147. [Google Scholar] [CrossRef]
- Fu, Q.; Wang, Y.; Chang, Z. Study on the conversion mechanism of CO2 to O2 in pulse voltage dielectric barrier discharge at Martian pressure. J. CO2 Util. 2023, 70, 102430. [Google Scholar] [CrossRef]
- Rouwenhorst, K.H.R.; Kim, H.-H.; Lefferts, L. Vibrationally excited activation of N2 in plasma-enhanced catalytic ammonia synthesis: A kinetic analysis. ACS Sustain. Chem. Eng. 2019, 7, 17515–17522. [Google Scholar] [CrossRef]
- Rouwenhorst, K.H.R.; Van der Ham, A.G.J.; Mul, G.; Kersten, S.R.A. Islanded ammonia power systems: Technology review & conceptual process design. Renew. Sustain. Energy Rev. 2019, 114, 109339. [Google Scholar]
- Fasihi, M.; Weiss, R.; Savolainen, J.; Breyer, C. Global potential of green ammonia based on hybrid PV-wind power plants. Appl. Energy 2021, 294, 116170. [Google Scholar] [CrossRef]
- Valera-Medina, A.; Amer-Hatem, F.; Azad, A.K.; Dedoussi, I.C.; de Joannon, M.; Fernandes, R.X.; Glarborg, P.; Hashemi, H.; He, X.; Mashruk, S.; et al. Review on Ammonia as a Potential Fuel: From Synthesis to Economics. Energy Fuels 2021, 35, 6964–7029. [Google Scholar] [CrossRef]
- Bouaboula, H.; Ouikhalfan, M.; Saadoune, I.; Chaouki, J.; Zaabout, A.; Belmabkhout, Y. Addressing sustainable energy intermittence for green ammonia production. Energy Rep. 2023, 9, 4507–4517. [Google Scholar] [CrossRef]
- Campion, N.; Nami, H.; Swisher, P.R.; Hendriksen, P.V.; Münster, M. Techno-economic assessment of green ammonia production with different wind and solar potentials. Renew. Sustain. Energy Rev. 2023, 173, 113057. [Google Scholar] [CrossRef]
- Osorio-Tejada, J.; Tran, N.N.; Hessel, V. Techno-environmental assessment of small-scale Haber-Bosch and plasma-assisted ammonia supply chains. Sci. Total Environ. 2022, 826, 154162. [Google Scholar] [CrossRef] [PubMed]
- Antonini, C.; Treyer, K.; Moioli, E.; Bauer, C.; Schildhauer, T.J.; Mazzotti, M. Hydrogen from wood gasification with CCS—A techno-environmental analysis of production and use as transport fuel. Sustain. Energy Fuels 2021, 5, 2602–2621. [Google Scholar] [CrossRef]
- Terlouw, T.; Bauer, C.; Rosa, L.; Mazzotti, M. Large-scale hydrogen production via water electrolysis: A techno-economic and environmental assessment. Energy Environ. Sci. 2022, 15, 3583–3602. [Google Scholar] [CrossRef]
- Zhu, X.; Xie, W.; Wu, J.; Miao, Y.; Xiang, C.; Chen, C.; Ge, B.; Gan, Z.; Yang, F.; Zhang, M.; et al. Recent advances in direct air capture by adsorption. Chem. Soc. Rev. 2022, 51, 6574–6651. [Google Scholar]
- Castro-Amoedo, R.; Granacher, J.; Daher, M.A.; Maréchal, F. On the role of system integration of carbon capture and mineralization in achieving net-negative emissions in industrial sectors. Energy Environ. Sci. 2023, 16. [Google Scholar] [CrossRef]
- Terlouw, T.; Bauer, C.; Rosa, L.; Mazzotti, M. Life cycle assessment of carbon dioxide removal technologies: A critical review. Energy Environ. Sci. 2021, 14, 1701–1721. [Google Scholar] [CrossRef]
- Witte, C.-P. Urea metabolism in plants. Plant Sci. 2011, 180, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Verma, S.; Gupta, A.; Paul, A.R.; Jain, A.; Haque, N. Life Cycle Analysis for The Production of Urea Through Syngas. IOP Conf. Ser. Earth Environ. Sci. 2021, 795, 012031. [Google Scholar] [CrossRef]
- Khalidy, R.; Arnaud, E.; Santos, R.M. Natural and human-induced factors on the accumulation and migration of pedogenic carbonate in soil: A Review. Land 2022, 11, 1448. [Google Scholar] [CrossRef]
- Dia Milani, D.; Ali Kiani, A.; Nawshad Haque, N.; Sarabjit Giddey, S.; Paul Feron, P. Green pathways for urea synthesis: A review from Australia’s perspective. Sustain. Chem. Clim. Action 2022, 1, 100008. [Google Scholar] [CrossRef]
- Sun, X.; Ding, R. Recent progress with electrocatalysts for urea electrolysis in alkaline media for energy-saving hydrogen production. Catal. Sci. Technol. 2020, 10, 1567–1581. [Google Scholar] [CrossRef]
- Tong, Y.; Chen, L.; Dyson, P.J.; Fei, Z. Boosting hydrogen production via urea electrolysis on an amorphous nickel phosphide/graphene hybrid structure. J. Mater. Sci. 2021, 56, 17709–17720. [Google Scholar] [CrossRef]
- Anuratha, K.S.; Rinawati, M.; Wu, T.H.; Yeh, M.H.; Lin, J.Y. Recent development of nickel-based electrocatalysts for urea electrolysis in alkaline solution. Nanomaterials 2022, 12, 2970. [Google Scholar] [CrossRef] [PubMed]
- Mora, J.C.; Singh, K.; Hill, J.M.; Thangadurai, V.; Ponnurangam, S. Surface Basicity Controlled Degradation and Recoverability of Proton Conducting Perovskites, BaZr0.8Ce0.1Y0.1O3–δ and Ba0.5Sr0.5Ce0.6Zr0.2Gd0.1Y0.1O3–δ, in the Presence of CO2. J. Phys. Chem. C 2023, 127, 8529–8538. [Google Scholar] [CrossRef]
- Kitchen, H.J.; Vallance, S.R.; Kennedy, J.L.; Tapia-Ruiz, N.; Carassiti, L.; Harrison, A.; Whittaker, A.G.; Drysdale, T.D.; Kingman, S.W.; Gregory, D.H. Modern microwave methods in solid-state inorganic materials chemistry: From fundamentals to manufacturing. Chem. Rev. 2014, 114, 1170–1206. [Google Scholar] [CrossRef]
- Horikoshi, S.; Schiffmann, R.F.; Fukushima, J.; Serpone, N. Microwave Chemical and Materials Processing; Springer Nature: Singapore, 2017; ISBN 978-981-10-6465-4. [Google Scholar]
- Ramirez, A.; Hueso, J.L.; Abian, M.; Alzueta, M.U.; Mallada, R.; Santamaria, J. Escaping undesired gas-phase chemistry: Microwave-driven selectivity enhancement in heterogeneous catalytic reactors. Sci. Adv. 2019, 5, eaau9000. [Google Scholar] [CrossRef]
- Priecel, P.; Lopez-Sanchez, J.A. Advantages and limitations of microwave reactors: From chemical synthesis to the catalytic valorization of biobased chemicals. ACS Sustain. Chem. Eng. 2019, 7, 3–21. [Google Scholar] [CrossRef]
- Baldry, M.; Gurieff, N.; Keogh, D. Imagining sustainable human ecosystems with power-to-x in-situ resource utilisation technology. Acta Astronaut. 2022, 192, 190–198. [Google Scholar] [CrossRef]
- Nguyen, M.T.P.; Knowling, M.; Tran, N.N.; Burgess, A.; Fisk, I.; Watt, M.; Escribà-Gelonch, M.; This, H.; Culton, J.; Hessel, V. Space farming: Horticulture systems on spacecraft and outlook to planetary space exploration. Plant Physiol. Biochem. 2023, 194, 708–721. [Google Scholar] [CrossRef]
- Pathak, H.S.; Brown, P.; Best, T. A systematic literature review of the factors affecting the precision agriculture adoption process. Precis. Agric. 2019, 20, 1292–1316. [Google Scholar] [CrossRef]
- Butler, C.; Fan, Y.; Grewal, S.; Winter, L.R. At-field and on-demand nitrogenous fertilizer synthesis. ACS Sust. Chem. Eng. 2023, 11, 5803–5818. [Google Scholar] [CrossRef]



































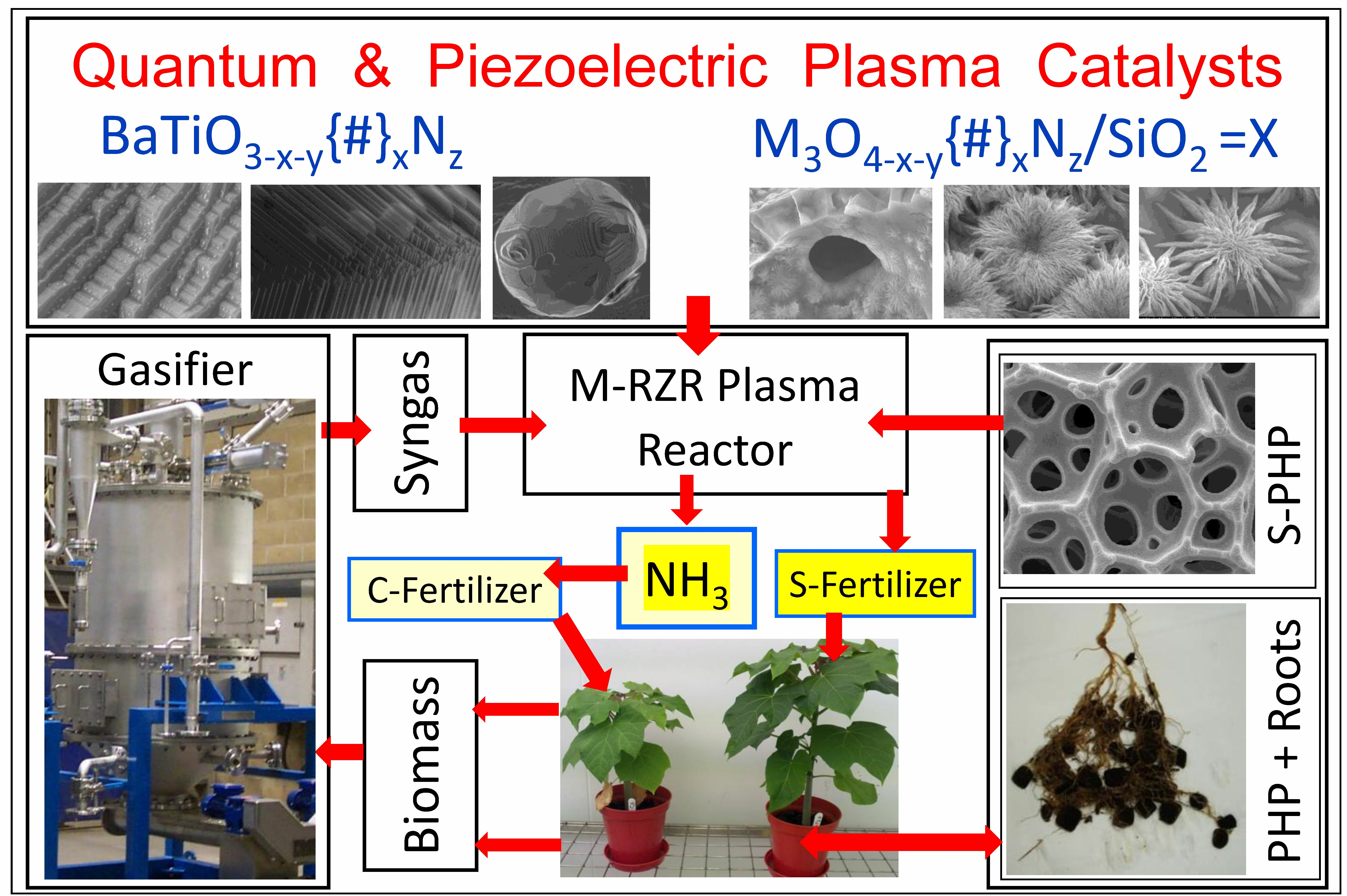{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proximate Analysis & Physical Property | Ultimate Analysis & Heating Values | ||
|---|---|---|---|
| Moisture Content (wt%) | 12.8 | Carbon (wt%) | 45.1 |
| Volatile matter (wt% db *) | 63.8 | Hydrogen (wt%) | 6.1 |
| Fixed carbon (wt% db *) | 18.7 | Oxygen (wt%) | 44.5 |
| Absolute density (kg/m3) | 841 | Nitrogen (wt%) | 0.6 |
| Bulk density (kg/m3) | 283 | Sulfur (wt%) | 0.4 |
| Approximate wood chips size (cm) | 10 | Ash content (wt% db) | 3.3 |
| High Heating Value (HHV) MJ/kg | 17.9 | ||
| Low Heating Value (LHV) MJ/kg | 16.6 | ||
| Syngas Composition before Water Scrubbing | Effluent Gas Composition from Gas Engine Generator | |||
|---|---|---|---|---|
| Parameter | (Vol %) | Parameter | Measured | EWID Limits |
| H2 | 13.7 | Particulates mg/Nm3 | <5 | 10 |
| CO | 16.2 | Total Organic Carbon mg/Nm3 | <3 | 10 |
| CH4 | 2.0 | HCl mg/Nm3 | <5.6 | 10 |
| C2+ | 0.2 | HF mg/Nm3 | <0.45 | 1 |
| O2 | 2.6 | SO2 mg/Nm3 | <38 | 50 |
| H2O | 3.1 | NOx mg/Nm3 | <240 | 400 |
| CO2 | 12.4 | CO mg/Nm3 | <100 | Site specific |
| N2 | 52.8 | Total Heavy Metals mg/Nm3 | <0.25 | 0.5 |
| Tar | 5.0 g/Nm3 | Hg mg/Nm3 | <0.01 | 0.05 |
| LHV | 5.2 MJ/Nm3 | Cd +TI mg/Nm3 | <0.007 | 0.05 |
| Particulates | 10 g/Nm3 | Dioxins & Furans ng/Nm3 | <0.002 | 0.1 |
| Oxidant Status | Range of Dry Gas Composition (Vol %) | Average H2/CO Ratio | Average H2/N2 Ratio | Average Cold Gas Efficiency (%) | Average LHV (MJ/Nm3) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| H2 | CO | CH4 | CO2 | C2Hn | N2 | |||||
| Without Water | 10–14 | 12–17 | 2–4 | 8–12 | 0–1 | 58–47 | 0.83 | 0.22 | 78 | 6.0 |
| With Water | 35–37 | 27–30 | 5–7 | 12–15 | 0–1 | 10–21 | 1.3 | 2.3 | 89 | 12.4 |
| Electrode Type → | Full Electrode Insulation | Partial Electrode Insulation | ||||
|---|---|---|---|---|---|---|
| Applied Voltage (kV) | 0 | 10 | 25 | 0 | 10 | 25 |
| Efficiency without s-PHP (%) | 19.1 ± 1.2 | 21.8 ± 1.3 | 30.1 ± 1.8 | 19.6 ± 1.2 | 80.1 ± 1.6 | 97.5 ± 1.3 |
| Efficiency with s-PHP (%) | 61.8 ± 3.8 | 67.3 ± 4.2 | 78.2 ± 4.5 | 62.1 ± 3.8 | 86.6 ± 2.6 | 98.7 ± 0.9 |
| Plasma Power (W) | CO2 Conversion under Different Electrode Configurations | ||||
|---|---|---|---|---|---|
| EC-1a from Ref [87] | EC-2 | EC-3 | EC-4 | ||
| BaTiO3 + Catalysts | BaTiO3 Only | BaTiO3 Only | |||
| 46 | 12.7 | 10.0 | 16.3 | 15.6 | 10.6 |
| 66 | 16.8 | 12.9 | 19.8 | 19.8 | 16.0 |
| 86 | 19.7 | 16.1 | 22.9 | 24.3 | 19.6 |
| 106 | 23.0 | 18.8 | 26.4 | - | 23.6 |
| Flow Rate (mL/min) | 25 | 50 | 75 | 100 |
| CO2 Conversion (mol%) | 27.2 | 16.1 | 13.1 | 10.3 |
| Total Gas Flow Rate (mL/min) → | 50 | 50 | 75 | 100 | 50 | 50 | 50 | 50 | 50 | 50 | 50 | 50 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N2 Concentration, (mol%) → | 0 | 2.5 | 2.5 | 2.5 | 5 | 10 | 20 | 25 | 50 | 75 | 90 | 93 |
| EA—Apparent CO2 conversion (mol%) | 18.8 | 29.9 | 21.0 | 16.2 | 31.0 | 33.0 | 34.6 | 35.7 | 41.6 | 47.0 | 47.4 | 47.6 |
| EE—Effective CO2 conversion (mol%) | 18.8 | 29.2 | 20.5 | 15.8 | 29.5 | 29.7 | 27.7 | 26.8 | 20.8 | 11.8 | 4.74 | 3.33 |
| Plasma power (W) | 25 | 46 | 50 | 66 | 75 | 86 | 100 | 106 |
| EA—Apparent CO2 conversion (mol%) | 13.3 | 21.7 | 22.4 | 29.4 | 31.8 | 36.8 | 39.2 | 41.6 |
| EE—Effective CO2 conversion (mol%) | 6.65 | 10.9 | 11.2 | 14.7 | 15.9 | 16.4 | 16.9 | 20.8 |
| Before Reaction | After Reaction | |
|---|---|---|
| Total gas flow rate = 50 mL/min | Total gas flow rate = 35.8 mL/min | |
| Inlet composition | Product composition (measured) | |
| CO2 = 10%; | CO2 =11.3% | C2 = 0.6% |
| H2 = 40%; | H2 = 24.9% | C3 = 0.03% |
| CH4 = 4% | CH4 =18.3% | C4 = 0.21% |
| CO = 10% | CO = 2.0% | C5 = 0.01% |
| N2 = 36% | N2 = 42.7% | |
| [H2]/[N2] = 1.11 | [H2]/[N2] = 0.58 | Solid product: (NH2)COO(NH4) |
| Domain Type | [O] | [Ba] | [Ti] | [N] | [Ba]/[Ti] | RO |
| (0) Fresh BaTiO3 | 0.611 | 0.196 | 0.193 | 0.00 | 1.02 | 1.57 |
| (1) O-depleted; N-free | 0.333 | 0.343 | 0.324 | 0.00 | 1.06 | 0.50 |
| (2) O-rich; N-free | 0.676 | 0.165 | 0.159 | 0.00 | 1.04 | 2.09 |
| (3) Dusty surface: O-poor; N-rich | 0.581 | 0.128 | 0.140 | 0.151 | 0.914 | 2.17 |
| (4) O-rich; N-poor | 0.678 | 0.136 | 0.116 | 0.070 | 1.17 | 2.69 |
| (5) O-rich; N-rich; Ti-depleted | 0.680 | 0.094 | 0.007 | 0.219 | 13.4 | 6.73 |
| Elements | Si | Cr | Fe | Co | Cu | Zn |
|---|---|---|---|---|---|---|
| At % | 34.1 | 13.1 | 13.4 | 23.8 | 14.5 | 1.1 |
| Sample | Crystallite Size at Dominant Peak * (nm) | EDS Analysis: Atomic Fractions at Decorative and Plain Surfaces + | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Decorations (Flowers) | Plain | |||||||||
| Co3O4 | CuO | Co | Cu | Si | Co + Cu Si | Co | Cu | Si | Co + Cu Si | |
| Co/Si = 1/4 A (1 kW) | 9.20 | 0.0601 | 0.0 | 0.193 | 0.311 | 0.0477 | 0.0 | 0.208 | 0.229 | |
| Cu/Si = 1/4 A (1.8 kW) | 11.7 | 0.0 | 0.172 | 0.204 | 0.843 | 0.0 | 0.108 | 0.252 | 0.429 | |
| Co/Cu/Si=1/1/8(AB) ** Thermal@300 °C | 11.8 | 25.3 | 0.0899 | 0.121 | 0.102 | 2.06 | 0.0451 | 0.0356 | 0.191 | 0.424 |
| Co/Cu/Si = 1/1/8 A (1 kW) | 6.48 | 11.6 | 0.0361 | 0.0521 | 0.191 | 0.462 | 0.0252 | 0.0241 | 0.182 | 0.271 |
| Co/Cu/Si = 1/1/8 A (1.8 kW) | 5.46 | 7.93 | 0.095 | 0.235 | 0.103 | 3.24 | 0.048 | 0.045 | 0.323 | 0.288 |
| Co/Cu/Si = 1/1/8 AB (1 kW + 600 °C) | 11.7 | 35.1 | 0.0251 | 0.0263 | 0.209 | 0.245 | 0.0245 | 0.0270 | 0.195 | 0.264 |
| EDS results for nanoplates (Spot-1) | 0.0302 | 0.137 | 0.178 | 0.939 | See Figure 19c SEM for identification of spot analysis | |||||
| Catalyst | Crystallite Size at Dominant Peak * (nm) | Surface Composition + (At. Fraction) | ||||
|---|---|---|---|---|---|---|
| Co3O4 | Fe3O4 | Co | Fe | Si | Co + Fe Si | |
| Fe/Si = 1/4 AB | - | 3.86 | 0.0 | 0.078 | 0.175 | 0.446 |
| Co/Fe/Si = 1/1/8 A | 8.74 | 3.93 | 0.026 | 0.035 | 0.195 | 0.313 |
| Co/Fe/Si = 1/1/8 AB | 11.0 | 6.58 | 0.024 | 0.025 | 0.176 | 0.278 |
| Location (See SEM in Figure 21a) | Concentration (Atomic Fraction) | [Ba]/[Ti] | RO | |||
|---|---|---|---|---|---|---|
| [Ba] | [Ti] | [O] | [N] | |||
| Location-1 (Particles in nm size range) | 0.217 | 0.230 | 0.480 | 0.073 | 0.943 | 1.07 |
| Location-2 (Particles in μm size range) | 0.127 | 0.140 | 0.582 | 0.151 | 0.907 | 2.18 |
| Number- Plasma & Process Type | Catalyst | H2 N2 | Flow Rate (mL min) | Outlet Conc. Cout (mol%) | Power (W) | EC Energy Cost (MJ/mol-NH3) | Ref | ET (n = 1) EC/Cout (MJ/mol2) |
|---|---|---|---|---|---|---|---|---|
| 1-RF (0.01 Atm) | Au | 19.1 | 300 | 264 | [372] | 1381 | ||
| 2-DBD-1 | BaTiO3 | 3 | 25 | 3.8 | 93 | 143 | [69] | 3763 |
| 3-DBD-2 | BaTiO3 | 3 | 25 | 3.8 | 77 | 112 | [69] | 2947 |
| 4-DBD-1 | Ni/Si = 1/4 | 3 | 25 | 6.4 | 115 | 132 | [69] | 2063 |
| 5-DBD-2 | Ni/Si = 1/4 BaTiO3 | 3 | 25 | 6.4 | 87 | 81 | [69] | 1266 |
| 6-DBD-1 | Ni/S = 1/4 BaTiO3 | 3 | 25 | 9.0 | 195 | 137 | [69] | 1522 |
| 7-DBD-2 | Ni/Si = 1/4 BaTiO3 | 4 | 50 | 5.4 | 114 | 42.2 | [3] | 781 |
| 8-DBD-M-RZR | Ni/Si = 1/4 BaTiO3 | 4 | 50 | 19.4 | 75 | 13.2 | [3] | 68.0 |
| 9-DBD-Catalyst shield | Ni/MCM-1 | 3 | 40 | 5.3 | 40 | 51 | [373] | 962 |
| 10-Haber-Bosch | Fe | 3 | - | 8–15 | - | 0.5 | [3] | 6.25–3.33 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akay, G. Hydrogen, Ammonia and Symbiotic/Smart Fertilizer Production Using Renewable Feedstock and CO2 Utilization through Catalytic Processes and Nonthermal Plasma with Novel Catalysts and In Situ Reactive Separation: A Roadmap for Sustainable and Innovation-Based Technology. Catalysts 2023, 13, 1287. https://doi.org/10.3390/catal13091287
Akay G. Hydrogen, Ammonia and Symbiotic/Smart Fertilizer Production Using Renewable Feedstock and CO2 Utilization through Catalytic Processes and Nonthermal Plasma with Novel Catalysts and In Situ Reactive Separation: A Roadmap for Sustainable and Innovation-Based Technology. Catalysts. 2023; 13(9):1287. https://doi.org/10.3390/catal13091287
Chicago/Turabian StyleAkay, Galip. 2023. "Hydrogen, Ammonia and Symbiotic/Smart Fertilizer Production Using Renewable Feedstock and CO2 Utilization through Catalytic Processes and Nonthermal Plasma with Novel Catalysts and In Situ Reactive Separation: A Roadmap for Sustainable and Innovation-Based Technology" Catalysts 13, no. 9: 1287. https://doi.org/10.3390/catal13091287