3.2. Synthesis and Spectral Data
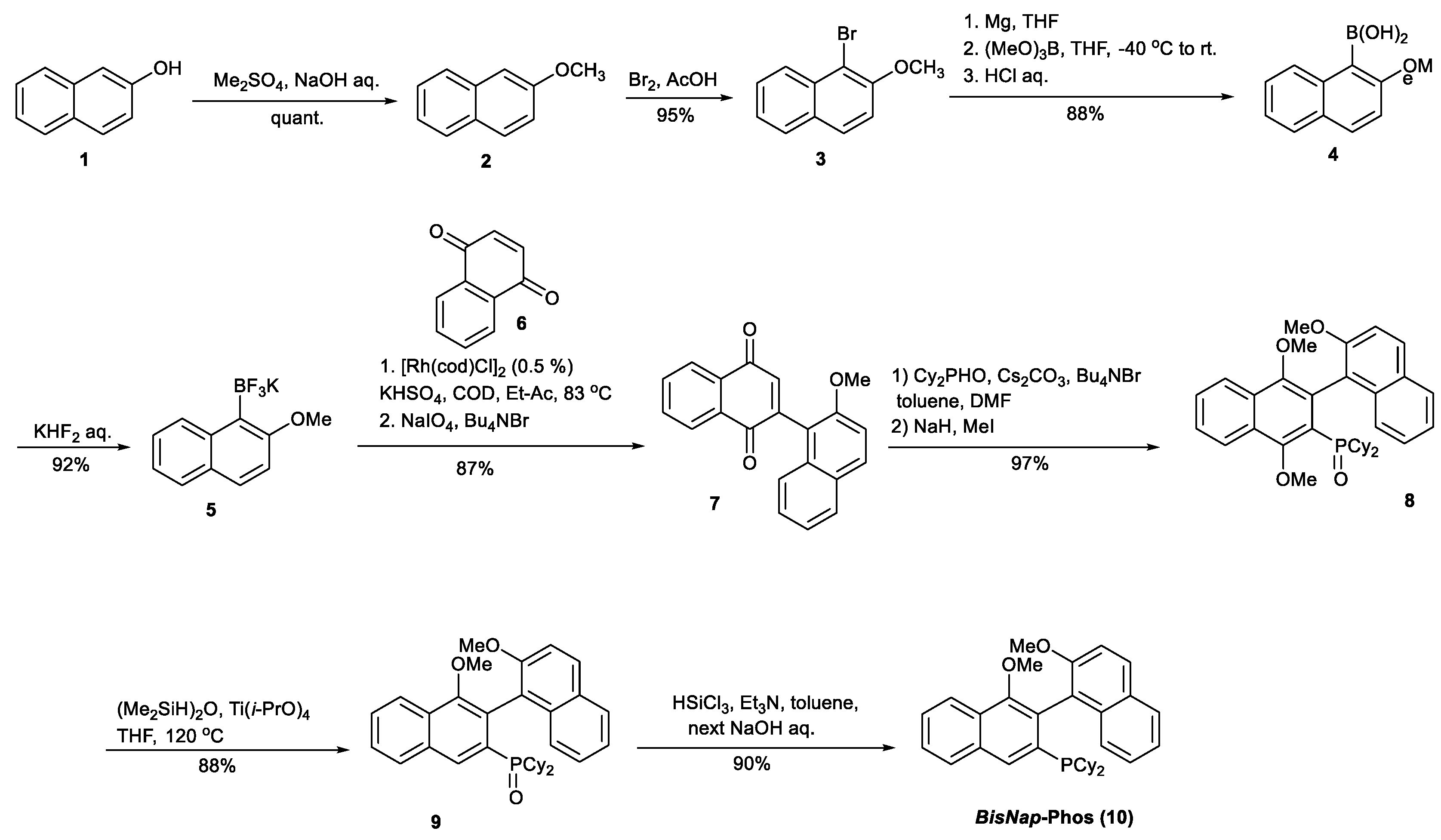
Synthesis of rac-dicyclohexyl(1′,2,4′-trimethoxy-5′,8′-dihydro-1,2′-binaphthalen-3′-yl)phosphane oxide (8).
A reactor equipped with a magnetic stirrer was charged with compound
7 (3 g, 9.6 mmol), obtained as presented [
27], CsCO
3 (3.1 g, 9.6 mmol), Cy
2PHO (3 g, 14 mmol), Bu
4NHSO
4 (0.1g, 0.3 mmol), toluene (15 mL), and DMF (10 mL) Next, the reaction mixture was protected by an argon atmosphere, sealed with a glass stopper, and stirred at 35 °C for 96 h. After that time, the reaction mixture was cooled down to 0 °C, and CH
3I (2.3 mL, 5.2 g, 37 mmol) and K
2CO
3 (5.2 g, 37 mmol) were added. Protected by the argon atmosphere, the mixture was stirred in a closed reactor at 35 °C for 96 h. The obtained mixture was poured on 100 g of ice, and carefully acidified with 1M HCl to an acidic pH. The crude product was extracted with DCM, dried with MgSO
4, and purified on a SiO
2 column eluted by a hexane: acetone (3: 1) mixture to afford 5.2g (97%) of pure compound
8.
Alternatively, 8 could be obtained in a reaction catalyzed by Bi(OTf)3 according to the following procedure. A reactor equipped with a magnetic stirrer was charged with compound 7 (0.5 g), Cy2PHO (0.5 g), Bi(OTf)3 (40 mg), and 10 mL of DMF. Next, the reaction mixture was protected by an argon atmosphere, sealed with a glass stopper, and stirred at 70 °C for 48 h. After that time, the reaction mixture was cooled down to 20 °C, and 10 mL of DMD, 50 mg of Bu4NHSO4, and CsCO3 (0.3 g) were added, followed by the addition of 40% NaH (0.37g) realized in a small portion with respect to foaming H2 gas. Once a liberation of H2 was completed, 0.5 mL of CH3I was added, the reactor sealed with a glass stopper, and the reaction mixture was stirred at 35 °C for 48 h. The obtained mixture was poured on 100 g of ice, and carefully acidified with 1M HCl to an acidic pH. The crude product was extracted with DCM, dried with MgSO4, and purified on a SiO2 column eluted by a hexane: acetone = 3: 1 mixture to afford 0.82 g (91%) of pure compound 8.
1H NMR (400 MHz, CDCl3): δ 0.96–2.16 (m, 22H), 3.45 (s, 3H), 3.86 (s, 3H), 4.14 (s, 3H), 7.17–7.28 (m, 3H), 7.34 (d, J = 9.2 Hz, 1H), 7.60–7.64 (m, 2H), 7.78–7.82 (m, 1H), 7.90 (d, J = 9.0 Hz, 1H), 8.16–8.21 (m, 2H).
13C NMR (100 MHz, CDCl3): δ 25.7, 26.1, 26.2, 26.3, 26.5, 26.6, 26.7, 26.8, 26.8, 26.9, 27.0, 27.1, 39.4 (d, J = 18.0 Hz), 40.1 (d, J = 18.0 Hz), 55.3, 61.4, 62.4, 111.8, 119,8, 121.5, 122.3, 122.4, 123.7, 125.0, 125.3, 126.0, 126.9, 127.4, 128.0, 128.6, 129.1, 130.7, 131.2, 151.2, 154.2, 154.6.
31P NMR (161 MHz, CDCl3): δ 50.17 ppm.
HRMS (ESI): m/z = 557.3340 [C35H41O4P+H]+, m/z (teor.) = 557.3175
Synthesis of dicyclohexyl(1′,2-dimethoxy-5′,8′-dihydro-1,2′-binaphthalen-3′-yl)phosphane oxide (9).
A reactor equipped with a magnetic stirrer and reflux condenser connected to the argon line was charged with compound 8 (1.7 g, 3 mmol), 20 mL of THF, TMDS (1.7 mL, 1.3 g, 9.7 mmol), and Ti(OiPr)4 (1 mL. 1 g, 3.5 mmol). The reaction was heated to afford a gentle reflux condition for 24 h and cooled down to ambient temperature. The solvents were evaporated off under the reduced pressure and the product 9 isolated on a SiO2 column applied the gradient of eluents hexane:acetone = 6-3: 1 to afford 1.4g (88%) of pure compound 9. Chiral chromatographic analysis of 9 was performed on HPLC-MS Column CHIRALPAK® AS-H, 250 × 4, 6 mm, 5 µm eluting with CH3CN: H2O = 50: 50 at flow 0.45 mL/min. which showed two signals at 8.07 min and 9.2 min with the same pseudomolecular ion mass 527 Da, which corresponds to the stereoisomers R- and S- of compound 9.
1H NMR (500 MHz, CDCl3): δ 1.13–1.68 (m, 22H), 3.56 (s, 3H), 3.90 (s, 3H), 7.14 (d, J = 8.4 Hz, 1H), 7.23–7.36 (m, 1H), 7.31–7.36 (m, 1H), 7.42 (d, J = 9.1 Hz, 1H), 7.62–7.66 (m, 2H), 7.86 (d, J = 8.0 Hz, 1H), 8.02 (d, J = 9.0 Hz, 1H), 8.07–8.11 (m, 1H), 8.14–8.18 (m, 1H), 8.58 (d, J = 12.1 Hz, 1H),
13C NMR (126 MHz, CDCl3): δ 25.4, 25.5, 25.6, 25.8, 26.1, 26.2, 26.3, 26.5, 26.6, 26.7, 26.8, 36.7 (d, J = 3.7 Hz), 37.7 (d, J = 3.7 Hz), 55.8, 61.5, 112.3, 119.8, 122.6, 123.7, 126.0, 126.2, 126.7, 126.8, 128.8, 129.2, 130.2, 131.1, 132.2, 132.3, 133.4, 133.8, 134.2, 154.4, 154.6, 154.8.
31P NMR (202 MHz, CDCl3): δ 47.54 ppm.
Synthesis of rac-dicyclohexyl(1′,2-dimethoxy-5′,8′-dihydro-1,2′-binaphthalen-3′-yl)phosphane (rac-10).
A reactor equipped with a magnetic stirrer and filled with argon was charged with compound 9 (0.5 g, 0.95 mmol), 20 mL of toluene, 10 mL of Et3N, and 1 mL of SiHCl3. The reactor was sealed with a glass stoper and the reaction mixture was stirred at 120 °C for 24 h. After that time, the reactor was cooled down to 0 °C, and 20 mL of toluene and 10 mL of 15% NaOH were added, maintaining the intense stirring. The formed organic phase was separated, washed with water, and dried by MgSO4. The MgSO4 was filtered off, and the solvent was completely evaporated off under reduced pressure. To the crude product, 15 mL of methanol was added, the air in the flask was replaced with argon, and the flask was sealed with the glass stoper and heated at 90 °C to dissolve the crude product. The pure compound 10 (435 mg, 90%) was crystallized from the solution after 24 h, storing at 0 °C.
Starting from the enantiomerically pure (S)-9, the enantiomerically pure (S)-10 was obtained [α]20D = +57.7 (c = 0.5, Et2O). The spectral data of (Sa)-10 were identical to those recorded for the racemic compound.
1H NMR (500 MHz, CDCl3): 1.02–1.90 (m, 22H), 3.49 (s, 3H), 3.88 (s, 3H), 7.22 (d, J = 8.5 Hz, 1H), 7.28–7.31 (m, 2H), 7,41 (d, J = 8.8 Hz, 1H), 7.57–7.61 (m, 2H), 7.86 (d, J = 8.0 Hz, 1H), 7.96–8.02 (m, 3H), 8.19–8.23 (m, 1H).
13C NMR (126 MHz, benzene d-6): 26.7, 27.1, 27.5, 27.6, 27.7, 27.8, 27.8, 27.8, 27.9, 29.9, 30.1, 30.2, 30.4, 30.5, 30.6, 30.7, 30.8, 34.9 (d, J = 17.8 Hz), 55.3, 61.0, 112.8, 121.8 (d, J = 6.9 Hz), 123.1, 123.5, 126.2, 126.5, 126.7, 127.8, 127.9, 128.1, 128.3, 128.6 (d, J = 3.4 Hz), 129.4, 129.8, 132.2, 132.6, 134.7, 134.8, 137.6, 137.8, 154.9, 155.0, 155.1.
31P NMR (126 MHz, CDCl3): δ −9.50 ppm.
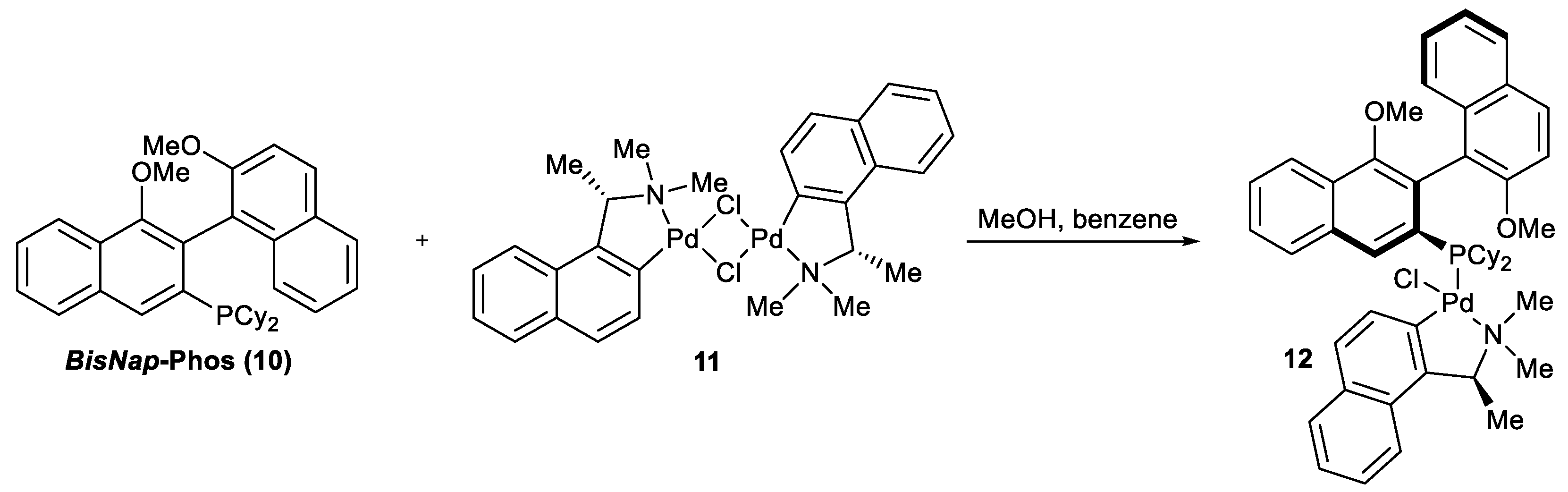
Synthesis of palladium complex 12.
A glass flask equipped with a magnetic stirrer was charged with 266 mg of palladium complex
(S)-11 dissolved in 6 mL of methanol and added to a stirring solution of 346 mg of
rac-10 in 12 mL of a benzene: methanol (1: 1) mixture. The air in the flask was replaced with argon, the flask was closed with a glass stopper, and the reaction mixture was stirred for 16 h at 50 °C. After the reaction completion, methanol was evaporated under reduced pressure and the product was purified by column chromatography eluted with a hexane: Et
2O: MeOH = 2: 1: 0.1 mixture to yield 55 mg of complex
12 as a mixture of diastereomers enriched with the less polar one. Next, the obtained product was crystallized twice with the same solvent mixture to yield 150 mg of a single diastereomer
S,Sa-12. The relative configuration of the complex was determined by X-ray diffraction (
Figure 2a, CCDC 2222975). Due to the restricted rotation of substituents in the complex, the
1H and
13NMR spectra of
12 contained unspecific very wide and not resolved signals:
31P NMR (126 MHz, CDCl3): δ = 73.89 ppm (wide).
MS (ESI): m/z = 850.27 [C48H56ClNO2PPd + H]+, m/z (teor.) = 850.28; and 814.29 [C48H55NO2PPd]+, m/z (teor.) = 814, 30. The isotopic profile of the signals corresponds to that theoretically calculated.
Synthesis of (Sa)-dicyclohexyl(1′,2-dimethoxy-5′,8′-dihydro-1,2′-binaphthalen-3′-yl)phosphane oxide ((Sa)-9).
An amount of 150 mg of (S,Sa)-12 and 150 mg of dppe were dissolved in 30 mL of DCM. The reaction mixture was sealed and stirred at RT for 48 h, then heated at 100 °C for 20 min. 10 mL of 15% H2O2 was added and stirring continued for the next 4 h. The organic phase was separated and dried with MgSO4 and the product was isolated by SiO2 column chromatography eluting with a hexane: acetone (3: 1) mixture to afford 80 mg of (Sa)-9. The chiral HPLC-MS chromatogram recorder, with the application of CHIRALPAK® AS-H, 250 × 4, 6 mm, 5 µm column eluted with CH3CN: water=50: 50 at flow 0.45 mL/min signal of enantiomer (S)- at 9.2 min, while the signal of enantiomer (R)-9- showed up at 8.0 min, was not presented. [α]20D = +67.7 (C = 1, DCM).
Separation of enantiomers of 9 by co-crystallization with TADDOL.
An amount of 4 g of racemic
9 and 3.5 g of (-)-TADDOL were dissolved in 60 mL of EtOH and 9 mL of CHCl
3, in a sealed glass pressure vial at 100 °C. The vial was allowed to cool down to ambient temperature. The crystalline complex was precipitated within 24 h with a yield of 1.6 g and [α]
20D = +13.3 (
c = 1, methanol). The diastereomeric excess of the obtained complex was measured by the NMR technique as reported [
28]: 6 mg of complex and 35 mg of (
S)-naproxen were dissolved in 1 mL of CDCl
3, and
31P,
1H spectra were recorded. The spectrum of racemic complex contains the signals at 52.0 and 51.9 ppm, with the single diastereomer (
S)- at 51.9 ppm. Note that the chemical shifts of the signals corresponding to the enantiomer
9 are dependent on the concentration and the ratio of
9 to naproxen, so the assignment of the absolute configuration only through the NMR technique could not be precise if only a single enantiomer were present in the mixture. Pure
(S)-9 was obtained after a flash column separation of its complex with TADDOL eluting with a hexane: acetone = 3: 1 mixture in a quantitative yield with [α]
20D = + 67.7 (
c = 1, DCM). Chromatographic analysis of
9 was performed on HPLC Column CHIRALPAK
® AS-H, 250 × 4, 6 mm, 5 µm eluting with CH
3CN: water = 50: 50 at flow 0.45 mL/min; the enantiomer (
R)- shows up at 8.07 min, and enantiomer (
S)- at 9.2 min. The spectral data of (
Sa)-
9 were identical to those recorded for the racemic compound. The enantiomer (
Ra)-
9 could be obtained after the crystallization of the filtrate: the solution was evaporated under the reduced pressure and dissolved in acetone in a pressure vial to afford a 10% solution. The crystals of
(Ra)-9 were formed upon cooling down to ambient temperature. In some experiments, complete enantiomer separation requires the recrystallization of the obtained crystals from the same solvent system.
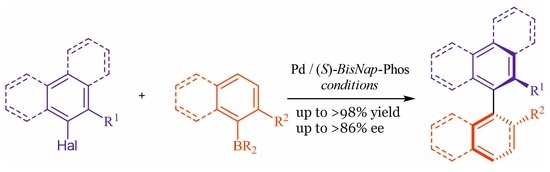
General procedures for asymmetric synthesis of tetra-ortho-substituted biaryl compounds:
General procedure A: A 10 mL round-bottom flask equipped with a stirring bar was charged with 0.3% aqueous solution of SDS and base Na2CO3 (3 mmol). Next, ortho-substituted aryl bromide (1 mmol), ortho-substituted aryl boronic acid or its derivative (1.2 mmol), ligand BisNap-Phos (4 mol%), and the pre-catalyst PdCl2(C6H5CN)2 (2 mol%) were dissolved in a minimum amount of THF and added to the mixture. The reaction was stirred at 60 °C for 18 h, then extracted with DCM (3 × 10 mL), and the combined organic layer was dried over MgSO4 and filtered. The solvent was removed under reduced pressure and the crude product was isolated by column chromatography.
General procedure B: A 10 mL round-bottom flask equipped with a stirring bar was charged with 0.3% aqueous solution of brij 97 and base Na2CO3 (3 mmol). Next, ortho-substituted aryl bromide (1 mmol), ortho-substituted aryl boronic acid or its derivative (1.2 mmol), ligand BisNap-Phos (4 mol%), and the pre-catalyst PdCl2(C6H5CN)2 (2 mol%) were dissolved in a minimum amount of THF and added to the mixture. The reaction was stirred at 60 °C for 18 h, then extracted with DCM (3 × 10 mL), and the combined organic layer was dried over MgSO4 and filtered. The solvent was removed under reduced pressure and the crude product was isolated by column chromatography.
General procedure C: A 10 mL round-bottom flask equipped with a stirring bar was charged with anhydrous DME as a solvent and anhydrous Cs2CO3 (3 mmol). Next, ortho-substituted aryl bromide (1 mmol), ortho-substituted aryl boronic acid or it derivative (1.2 mmol), BisNap-Phos (4 mol%), and pre-catalyst PdCl2(C6H5CN)2 (2 mol%) were dissolved in a minimum amount of THF and added to the reaction mixture. The reaction was stirred at 80 °C for 18 h, then extracted with DCM (3 × 10 mL), and the combined organic layer was dried over MgSO4 and filtered. The solvent was removed under reduced pressure and the crude product was isolated by column chromatography.
2,2′-dimethoxy-1,1′-binaphthyl (13).
The compound was synthesized according to General Procedure A and then purified by column chromatography on silica gel using hexane:acetone (6:1) as an eluent. The compound was obtained as colorless crystals. mp = 218.4–220.4 °C (lit. [
29] mp = 223–227 °C). The enantiomeric excess was determined by LCMS using a reversed phase chiral column: AS-RH; Mobile phase: H
2O: CH
3CN (50: 50); Flow: 0.45; t(
R) = 7.847 min. (73.5%); t(
S) = 8.529 min., λ = 254 nm; and HPLC using a chiral column: OD-H; Mobile phase: hexane: ethanol (99.5: 0.5); Flow: 0.5 mL/min; t
(R) = 18.609; t(
S) = 20.334; λ = 254 nm. The specific rotation of the compound with an 47% ee (
R) was [α]
D = +23.3 (
c 1.0, CHCl
3) (lit. [
29] [α]
D = + 57.54;
c 1.0, CHCl
3). Yield: 96%. Elemental Analysis: found C, 84.05; H, 5.66; theoretical: C, 84.05; H, 5.77.
1H NMR (500 Hz, CDCl3): δ 3.78 (s, 6H, OCH3), 7.10–7.12 (m, 2H), 7.21–7.23 (m, 2H), 7.31–7.34 (m, 2H), 7.47 (d, J = 9.1 Hz, 2HH), 7.87–7.89 (m, 2H), 7.99 (d, J = 8.8 Hz, 2H).
13C NMR (126 Hz, CDCl3): δ 56.9 (CH3), 114.2, 119.6, 123.5, 125.2, 126.3, 127.9, 129.2, 129.4, 134.0, 155.0.
13C NMR (DEPT 135, CDCl3): δ 56.9 (CH3), 114.2, 123.5, 125.2, 126.3, 127.9, 129.4.
2-methoxy-2′-methyl-1,1′-binaphthalene (15).
The compound was synthesized according to General Procedure A and then purified by column chromatography on silica gel using hexane:acetone = 9:1 as an eluent. The compound was obtained as a yellowish solid. mp = 116–118 °C (lit.[
30] mp = 119–120 °C). The enantiomeric excess was determined by LCMS using a reversed phase chiral column: AS-RH; Mobile phase: H
2O: CH
3CN (50: 50); Flow: 0.45; t(
S) = 13.156 min.; t(
R) = 14,070 min. (85%); λ = 254 nm. The specific rotation of the compound with an 70% ee (
R) was [α]
D = +10.3 (
c = 1.03, CHCl
3) (lit. [
30] [α]
D = + 22.3; (
c 1.3, CHCl
3, 92% ee). Yield: 98%. Elemental Analysis: found C, 88.25; H, 5.97; theoretical: C, 88.56; H, 6.08.
1H NMR (500 MHz, CDCl3): δ 2.11 (s, 3H, CH3), 3.77 (s, 3H, OCH3), 7.00–7.02 (m, 1H), 7.13–7.15 (m, 1H), 7.20–7.23 (m, 2H), 7.34 (ddd, J = 8.2, 6.6 and 1.3 Hz, 1H), 7.39 (ddd, J = 8.1, 6.7 and 1.3 Hz, 1H), 7.47 (d, J = 9.1 Hz, 1H), 7.53 (d, J = 8.2 Hz, 1H), 7.89 (dd, J = 8.4 and 3.9 Hz, 2H), 8.00 (d, J = 8.8 Hz, 1H).
13C NMR (126 MHz, CDCl3): δ 20.3 (CH3), 56.6 (OCH3), 113.8, 122.0, 123.6, 124.7, 125.1, 125.8, 126.5, 127.48, 127.9, 127.9, 128.7, 129.2, 129.4, 132.1, 132.3, 133.2, 133.6, 135.0.
13C NMR (DEPT 135, CDCl3): δ 20.3 (CH3), 56.6 (OCH3), 113.8, 123.6, 124.7, 125.1, 125.8, 125.8, 126.5, 127.5, 127.9, 127.9, 128.67, 129.4.
2,6-Dimethoxy-2′,6′-dimethyl-1,1′-biphenyl (21).
The compound was synthesized according to General Procedure A from chloride
17 or bromide
18 and then purified by column chromatography on silica gel using hexane: acetone (99.7: 0.3) as an eluent. The pure compound was isolated as colorless crystals. Mp = 108–110 °C (lit. [
31] m.p. = 110–112 °C, crystallized from MeOH). Yield: 40–61%.
1H NMR (500 MHz, CDCl3): δ 2.01 (s, 6H, CH3), 3.72 (s, 6H, OCH3), 6.67 (d, J = 8.4 Hz, 2H), 7.15 (t, J = 8.42 Hz, 1H), 7.16 (d, J = 8.2 Hz, 2H), 7.35 (t, J = 8.2 Hz, 1H).
13C NMR (126 MHz, DEPT 135, CDCl3): δ 20.1 (CH3), 55.8 (OCH3), 103.9, 126.8, 127.0, 128.6.
5-Cyano-2,3,2′,6′-tetrametoxy-1,1′-biphenyl (22).
The compound was synthesized according to General Procedure A from chloride 19 and then purified by column chromatography on silica gel using hexane: acetone (6: 1) as an eluent. The pure compound was isolated as colorless crystals. Mp = 119–120 °C. Yield: 56%.
1H NMR (1H NMR (300.33 MHz, CDCl3): δ 3.68 (s, 3H, OCH3), 3.74 (s, 6H, OCH3), 3.92 (s, 3H, OCH3), 6.65 (d, J = 8.4 Hz, 2H), 7.13 (s, 2H), 7.31–7.36 (t, J = 8.2 Hz, 1H).
13C NMR (75.52 MHz, CDCl3): δ 55.8, 55.9, 60.5, 103.8, 104.1, 106.4, 113.9, 114.3, 119.2, 129.2, 129.6, 129.8, 151.7, 152.8, 157.7.
HRMS (ESI): m/z = 300.1227 [C17H17NO4+H]+, m/z (teor.) = 300.1230, diff. = −1.00 ppm.
2-Acetyl-2′,6′-dimethoxy-1,1′-biphenyl (23).
The compound was synthesized according to General Procedure A from chloride
20 and then purified by column chromatography on silica gel using hexane:ethyl acetate (9:1) as an eluent. The pure compound was isolated as colorless crystals. Mp = 51–52 °C (lit. [
32] m.p. = 51–52 °C). Yield: 87%.
1H NMR (500 MHz, CDCl3): δ 3.64 (s, 3H, C(O)OCH3), 3.71 (s, 6 H, OCH3), 6.65 (d, J = 8.4 Hz, 2H, CH), 7.29 (t, J = 8.4 Hz, 1H), 7.34 (dd, J = 7.7, and 1.1 Hz, 1H), 7.40 (td, J = 7.7 and 1.5 Hz, 1H), 7.54 (td, J = 7.5 and 1.5 Hz, 1H), 7.96 (dd, J = 7.7 and 1.3 Hz, 1H).
13C NMR (DEPT 135, CDCl3): δ = 51.6 (C(O)OCH3), 55.8 (OCH3), 103.9, 126.9, 128.7, 129.6, 131.2, 132.4.
2,2′-dimethyl-1,1′-binaphthalene (25).
The compound was synthesized according to General Procedure A and then purified by column chromatography on silica gel using hexane as an eluent. The pure compound was a colorless oily liquid. The enantiomeric excess was determined by the specific rotation and then compared with the literature data. The specific rotation of the compound with an 77% ee (
S) was [α]
D = +27.5 (
c 1.025, CHCl
3), (lit. [
30] [α]
D = +32.5 c = 0.8, CHCl
3, 90% ee (
S)). The specific rotation of the compound with an 71% ee (
R) was [α]
D = −24.4 (
c 1.01, CHCl
3) (lit. [
33] [α]
D = −40.0;
c = 1.12; CHCl
3). Yield: 85%. Elemental Analysis: found C, 93.66; H, 5.79; theoretical: C, 93.99; H, 6.01.
1H NMR (500 MHz, CDCl3): δ 2.05 (s, 6H, CH3), 7.05–7.07 (m, 2H), 7.22 (ddd, J = 8.4, 6.8 and 1.3 Hz, 2H), 7.41 (ddd, J = 8.1, 6.9 and 1.1 Hz, 2H), 7.52 (d, J = 8.5 Hz, 2H), 7.88–7.92 (m, 4H).
13C NMR (126 MHz, CDCl3): δ = 20.0 (CH3), 124.9, 125.6, 126.1, 127.4, 127.9, 128.7, 132.2, 132.7, 134.2,135.1.
13C NMR (DEPT 135, CDCl3): δ = 20.0 (CH3), 124.9, 125.6, 127.4, 127.9, 128.7.
2-methyl-1,1′-binaphthalene (27).
The compound was synthesized according to General Procedure C and then purified by column chromatography on silica gel using hexane as an eluent. The pure compound was isolated as colorless crystals. mp = 95.1 °C (lit. [
34] mp = 82.8–87.1 °C). The enantiomeric excess was determined by HPLC using a chiral column: OJ-H, Mobile phase: hexane: isopropanol (80: 20); Flow: 1 mL/min; t(
R) = 9.146 min. (68, 5%); t(
S) = 12.253 min.; λ = 254 nm. The specific rotation of the compound with an 37% ee (
R) was [α]
D = −17.8 (
c 1.0, CHCl
3), (lit. [
35] [α]
D = −15.0;
c = 1.0, CHCl
3, λ = 589 nm, 49% ee). Yield: 80%. Elemental Analysis: found C, 93.66; H, 5.79; theoretical: C, 93.99; H, 6.01.
1H NMR (500 MHz, CDCl3): δ 2.12 (s, 3H, CH3), 7.15–7.17 (m, 1H), 7.22–7.24 (m, 2H), 7.27–7.30 (m, 1H), 7.39 (dd, J = 6.9, and 1.3 Hz, 1H), 7.41 (ddd, J = 8.2, 6.6 and 1.3 Hz, 1H), 7.48 (ddd, J = 8.2, 6.6 and 1.6 Hz, 1H), 7.51 (d, J = 8.8 Hz, 1H), 7.63 (dd, J = 8.2 and 6.9 Hz, 1H), 7.89 (dd, J = 8.4 and 3.3 Hz, 2H), 7.97 (d, J = 8.2 Hz, 2H).
13C NMR (126 MHz, CDCl3): δ 20.5 (CH3), 124.8, 125.6, 125.9, 125.9, 126.0, 126.1, 126.3, 127.5, 127.6, 127.7, 127.8, 128.3, 128.6, 131.9, 132.6, 133.5, 133.7, 134.4, 136.0, 137.5.
13C NMR (DEPT 135, CDCl3): δ 20.5 (CH3), 124.8, 125.6, 125.9, 125.9, 126.0, 126.1, 126.3, 127.5, 127.6, 127.7, 127.7, 128.2, 128.6.
2-methoxy-1,1′-binaphthalene (28).
The compound was synthesized according to General Procedure B and then purified by column chromatography on silica gel using hexane as an eluent. The pure compound was isolated as colorless crystals. Mp = 105.9 °C (lit. [
36] mp = 109–110 °C - crystallized from MeOH). The enantiomeric excess was determined by HPLC using a chiral column: OJ-H, Mobile phase: hexane: isopropanol (95: 5); Flow: 1 mL/min; t(
S) =16.607 min.; t(
R) = 28.830 min. (67%); λ = 254 nm. The specific rotation of the compound with an 34% ee (
R) was [α]
D = −16.5 (
c = 1.02, CHCl
3, λ = 589 nm), (lit. [
35] (
S) [α]
D = +8.0;
c = 1.0, CHCl
3, λ = 589 nm, 80% ee). Yield: 95%. HRMS (ESI): m/z = 285.1275 [C
21H
16O+H]+, m/z (teor.) = 285.1274, diff. = 0.35 ppm.
1H NMR (500 MHz, CDCl3): δ 3.78 (s, 3H, OCH3), 7.16 (dd., J = 5.7 and 1.3 Hz, 2H), 7.23 (ddd, J = 8.2, 6.6 and 1.3 Hz, 1H), 7.29 (ddd, J = 8.1, 5.2 and 2.2 Hz, 1H), 7.34 (d, J = 8.8 Hz, 1H), 7.46 (d, J = 8.8 Hz, 1H), 7.46 (dd, J = 6.9 and 1.3 Hz, 2H), 7.63 (dd, J = 8.2 and 6.9 Hz, 1H), 7.89 (d, J = 8.2 Hz, 1H), 7.94–8.01 (m, 3H).
13C NMR (126 MHz, CDCl3): δ 56.8 (OCH3), 113.8, 123.2, 123.5, 125.51, 125.58, 125.7, 125.9, 126.2, 126.3, 127.7, 127.8, 128.2, 128.4, 129.1, 129.5, 132.9, 133.7, 134.2, 134.5, 154.6.
13C NMR (DEPT 135, CDCl3): δ 56.8 (OCH3), 113.8, 123.67, 125.5, 125.6, 125.7, 125.9, 126.2, 126.4, 127.7, 127.8, 128.2, 128.4, 129.5.
2-(2-methoxynaphthalen-1-yl)—4,6-dimethylaniline (30).
The compound was synthesized according to General Procedure C and then purified by column chromatography on silica gel using hexane as an eluent; when 2-methoxynaphthalene was removed from the column, the system was changed to hexane: acetone (98: 2). The pure compound was isolated as colorless crystals. Mp = 165.1 °C (DME). The enantiomeric excess was determined by HPLC using a chiral column: AS-H, Mobile phase: hexane: isopropanol (95: 5); Flow: 0.3 mL/min; t1 = 24.778 min. (54.5%); t2 = 26.990 min.; λ = 254 nm. Due to the low enantiomeric excess of the compound of 9%, specific rotation was not determined. Yield: 86%. Anal. calcd for C19H19NO: C 82.28; H, 6.90; N, 5.05. Found: C, 81.52; H, 6.80; N, 4.95.
1H NMR (500 MHz, CDCl3): δ 2.27 (s, 3H, CH3), 2.31 (s, 3H, CH3), 3.31 (s, 2 H, NH2), 3.90 (s,3H, OCH3), 6.82 (s, 1H), 7.02 (s, 1H), 7.35–7.39 (m, 2H), 7.42 (d, J = 8.8 Hz, 1H), 7.44–7.46 (m, 1H), 7.84–7.86 (m, 1H), 7.93 (d, J = 8.8 Hz, 1H).
13C NMR (126 MHz, CDCl3): δ 17.9 (CH3), 20.5 (CH3), 56.8 (OCH3), 113.9, 121.6, 121.8, 122.5, 123.7, 125.2, 126.6, 127.0, 127.8, 129.27, 129.4, 129.78, 130.6, 133.5, 140.3, 154.4.
13C NMR (DEPT 135, CDCl3): δ = 17.9 (CH3), 20.5 (CH3), 56.8 (OCH3), 113.8, 123.7, 125.2, 126.6, 127.8, 129.4, 129.7, 130.6.
2,4-dimethyl-6-(2-methylnaphthalen-1-yl)aniline (31).
The compound was synthesized according to General Procedure C and then purified by column chromatography on silica gel using hexane as an eluent; when methylnaphthalene was removed from the column, the system was changed to hexane: acetone (99: 1). The pure compound was isolated as grayish crystals. Mp = 106.5 °C (DME). The enantiomeric excess was determined by HPLC using a chiral column: OD-H, Mobile phase: hexane: isopropanol (95: 5); Flow: 1 mL/min; t1 = 8.259 min.; t2 = 8.788 min. (59.5%); λ = 254 nm. The specific rotation of the compound with an 19% ee was [α]D = −1.2 (c = 1.0, CHCl3, λ = 594 nm, 20 °C). Yield: 92%. Elemental Analysis: found C, 86.72; H, 7.30; N, 5.25; theoretical: C, 87.31; H 7.33; N, 5.36.
1H NMR (500 MHz, CDCl3): δ 2.26 (s, 3H, CH3), 2.28 (s, 3H, CH3), 2.30 (s, 3H, CH3), 3.20 (s, 2H, NH2), 6.74 (s, 1H), 7.00 (s, 1H), 7.36 (ddd, J = 8.2, 6.6 and 1.3 Hz, 1H), 7.43–7.47 (m, 3H), 7.81 (d, J = 8.5 Hz, 1H), 7.85–7.86 (m, 1H).
13C NMR (126 MHz, CDCl3): δ 17.9 (CH3), 20.4 (CH3), 20.5 (CH3), 122.8, 124.8, 125.0, 125.7, 126.1, 127.5, 127.8, 128.8, 128.9, 130.4, 132.3, 132.6, 134.7, 139.3.
13C NMR (DEPT 135, CDCl3): δ 17.9 (CH3), 20.4 (CH3), 20.5 (CH3), 125.0, 125.7, 126.1, 127.5, 127.8, 128.8, 128.9, 130.4.
2,4-dimethyl-6-(naphthalen-1-yl)aniline (32).
The compound was synthesized according to General Procedure C and then purified by column chromatography on silica gel using hexane as an eluent; when naphthalene was removed from the column, the system was changed to hexane: acetone (99: 1). The pure compound was isolated as pinkish crystals. Mp = 127.8 °C (DME). The enantiomeric excess was determined by HPLC using a chiral column: OJ-H, Mobile phase: hexane:isopropanol (80: 20); Flow: 1 mL/min; t1 = 10.971 min (52%).; t2 = 13.418 min.; λ = 254 nm. Due to the low enantiomeric excess of the compound of 4%, specific rotation was not determined. Yield: 93%. Elemental Analysis: found C, 87.01 H, 6.85; N, 5.54; theoretical: C, 87.41; H, 6.93; N, 5.66.
1H NMR (500 MHz, CDCl3): δ 2.26 (s, 3H, CH3), 2.30 (s, 3H, CH3), 3.33 (s, 2H, NH2), 6.88 (m, 1H), 7.01 (m, 1H), 7.43 (ddd, J = 8.2, 6.6 and 1.3 Hz, 1H), 7.46 (dd, J = 6.9 and 1.3 Hz, 1H), 7.51 (ddd, J = 8.2, 6.6 and 1.3 Hz, 1H), 7.56 (dd, J = 8.2 and 6.9 Hz, 1H), 7.66 (d, J = 8.2 Hz, 1H), 7.91 (dd, J = 15.1 and 8.2 Hz, 2H).
13C NMR (126 MHz, CDCl3): δ 17.8 (CH3), 20.4 (CH3), 122.5, 125.8, 125.8, 125.9, 126.2, 126.2, 127.1, 127.6, 127.8, 128.2, 129.4, 130.6, 131.8, 133.8, 137.4, 139.8.
13C NMR (DEPT 135, CDCl3): δ 17.8 (CH3), 20.4 (CH3), 125.8, 125.9, 126.2, 126.2, 127.6, 127.8, 128.2, 129.4, 130.6.
9-(2-methylnaphthalen-1-yl)phenanthrene (34).
The compound was synthesized according to General Procedure C and then purified by column chromatography on silica gel using hexane as an eluent. The pure compound was isolated as colorless crystals. mp = 145.7 °C (lit. [
37] mp = 143–144 °C crystallized from EtOH-acetone). The enantiomeric excess was determined by HPLC using a chiral column: OJ-H, Mobile phase: hexane: isopropanol (95: 5); Flow: 1 mL/min; t1 = 11.078 min.; t2 = 16.962 min. (67%); λ = 254 nm. The specific rotation of the compound with an 35% ee was [α]
D = +55.4 (
c = 1.01, CHCl3, λ = 589 nm, 20 °C). Yield: 80%. Elemental Analysis: found C, 92.80; H, 5.81; theoretical: C, 94.30; H, 5.70.
1H NMR (500 MHz, CDCl3): δ 2.20 (s, 3H, CH3), 7.23 (ddd, J = 7.9, 6.6 and 1.3 Hz, 1H), 7.29–7.32 (m, 2H), 7.39–7.43 (m, 2H), 7.55 (d, J = 8.2 Hz, 1H), 7.66–7.70 (m, 3H), 7.75 (ddd, J = 8.4, 6.9 and 1.4 Hz, 1H), 7.91–7.94 (m, 3H), 8.83–8.86 (m, 2H).
13C NMR (126 MHz, CDCl3): δ 20.5 (CH3), 122.6, 122.9, 124.8, 125.9, 126.2, 126.6, 126.6, 126.7, 126.8, 127.6, 127.8, 128.4, 128.6, 130.1, 131.7, 131.8, 132.0, 134.5, 135.9, 136.0.
13C NMR (DEPT 135, CDCl3): δ 20.5 (CH3), 122.67, 122.9, 124.8, 125.9, 126.2, 126.5, 126.6, 126.7, 126.8, 127.6, 127.8, 128.4, 128.6.
2′-methoxy-[1,1′-binaphthalen]-2-yl diethylcarbamate (36).
The compound was synthesized according to General Procedure B and then purified by column chromatography on silica gel using hexane:acetone (9:1) as an eluent; when 2-methoxy naphthalene was removed from the column, the system was changed to hexane: acetone (6: 1). The pure compound was isolated as yellowish crystals. Mp = 110.7 °C. The enantiomeric excess was determined by HPLC using a chiral column: OJ-H, Mobile phase: hexane:ethanol (98: 2); Flow: 1 mL/min; t1 = 23.080 min.; t2 = 25.855 min. (79.5%); λ = 254 nm. The specific rotation of the compound with an 59% ee was [α]D = +30.8 (c = 1.02, CHCl3, λ = 589 nm). Yield: 72%. HRMS (ESI): m/z = 400.1925 [C26H25NO3+H]+, m/z (theor.) = 400.1907, diff. = 5.00 ppm. Mobile phase: CH3CN: H2O (65: 35), Flow: 0.3 mL/min. Elemental Analysis: found C, 77.82; H, 6.22; N, 3.41 theoretical: C, 78.17; H, 6.31; N, 3.51.
1H NMR (500 MHz, CDCl3): δ 0.43–0.46 (m, 3H, CH3), 0.82–0.85 (m, 3H, CH3), 2.68–2.69 (m, 2H, CH2), 3.03–3.11 (m, 2H, CH2), 3.76 (s, 3H, OCH3), 7.19–7.21 (m, 1H), 7.24 (ddd, J = 8.51, 6.31 and 1.26 Hz, 1H, CH), 7.28 (dd, J = 4.4 and 0.9 Hz, 2H, CH2), 7.31 (ddd, J = 8.1, 6.5 and 1.4 Hz, 1H), 7.42 (d, J = 9.1 Hz, 1H), 7.44 (dd, J = 7.9 and 4.1 Hz, 1H), 7.62 (d, J = 8.8 Hz, 1H), 7.84 (d, J = 8.2 Hz, 1H), 7.93 (d, J = 8.2 Hz, 1H), 7.97 (d, J = 8.8 Hz, 1H), 7.98 (d, J = 8.8 Hz, 1H).
13C NMR (126 MHz, CDCl3): δ 13.0 (CH3), 13.1 (CH3), 41.2 (CH2), 41.7 (CH2), 56.8 (OCH3), 113.8, 118.5, 122.6, 123.6, 124.6, 125.0, 125.6, 126.0, 126.2, 126.5, 127.6, 1281, 128.7, 129.0, 129. 7, 131.4, 133.6, 133.9, 147.4, 153.5, 1554.0.
13C NMR (DEPT 135, CDCl3): δ 13.0 (CH3), 13.1 (CH3), 41.2 (CH2), 41.7 (CH2), 56.8 (OCH3), 113.8, 122.6, 123.6, 125.0, 125.6, 126.0, 126.2, 126.5, 127.6, 128.1, 128.7, 129.7.
2′-methyl-[1,1′-binaphthalen]-2-yl diethylcarbamate (37).
The compound was synthesized according to General Procedure A and then purified by column chromatography on silica gel using hexane: acetone (9: 1) as an eluent. The pure compound was isolated as yellowish crystals. Mp = 100.5 °C. The enantiomeric excess was determined by HPLC using a chiral column: OJ-H, Mobile phase: hexane: isopropanol (95: 5); Flow: 1 mL/min; t1 = 8.204 min.; t2 = 10.755 min. (83.5%); λ = 254 nm. The specific rotation of the compound with an 67% ee was [α]D = +65,4 (c = 1.105, CHCl3, λ = 589 nm). Yield: 82%. HRMS (ESI): m/z = 370.1782 [C26H25NO2+H]+, m/z (theor.) = 370.1802, diff. = −5.40 ppm.
1H NMR (500 MHz, CDCl3): δ 0.35–0.38 (m, 3H, N(CH2CH3)2), 0.81–0.83 (m, 3H, N(CH2CH3)2), 2.11 (s, 3H, CH3), 2.55–2.64 (m, 2H, CH2), 3.01–3.07 (m, 2H, CH2), 7.18–7.25 (m, 3H), 7.27–7.30 (m, 1H), 7.36–7.39 (m, 1H), 7.43–7.48 (m, 1H), 7.47 (d, J =8.4 Hz, 1H), 7.58 (d, J = 8.8 Hz, 1H), 7.85 (d, J = 8.4 Hz, 2H), 7.95 (d, J = 8.2 Hz, 1H), 7.99 (d, J = 8.8 Hz, 1H).
13C NMR (126 MHz, CDCl3): δ 12.9 (N(CH2CH3)2), 12.9 N(CH2CH3)2), 20.3 (CH3), 41.2 (N(CH2CH3)2), 41.7 (N(CH2CH3)2), 122.7, 124.8, 125.2, 125.6, 125.9, 126.0, 126.5, 127.5, 127.5, 127.7, 128.1, 128.5, 128.7, 131.2, 131.5, 132.0, 133.1, 133.1, 135.3, 147.1, 153.4.
13C NMR (DEPT 135, CDCl3): δ 12.9 (N(CH2CH3)2), 12.9 (N(CH2CH3)2), 20.3 (CH3), 41.2 (N(CH2CH3)2), 41.7 (N(CH2CH3)2), 122.7, 124.8, 125.2, 125.6, 125.9, 126.0, 126.5, 127.5, 127.7, 128.1, 128.5, 128.7.
2,2′-bis-[1,1′-binaphthalen]-2-yl N,N-diethylcarbamate (39).
The compound was synthesized according to General Procedure B and then purified by column chromatography on silica gel using hexane: acetone (9: 1) as an eluent. The pure compound was isolated as a yellowish solid. Mp = 66–68 °C (lit. [
38] mp = 67–68 °C). The enantiomeric excess was determined by HPLC using a chiral column: OD-H, Mobile phase: hexane: ethanol (99.5: 0.5); Flow: 0.5 mL/min; t1 = 21.610 min.; t2 = 24.678 min. (75%); λ = 254 nm. The specific rotation of the compound with an 50% ee (
R) was [α]
D = +62.0 (
c = 0.995, CHCl
3, λ = 589 nm), (lit. [
38] [α]
D = +117.0;
c = 2.0, CHCl
3, λ = 589 nm). Yield: 80%. Elemental Analysis: found C, 73.83; H, 6.53; N, 5.61; theoretical: C, 74.36; H, 6.66; N, 5.78.
1H NMR (500 MHz, CDCl3): δ 0.37–0.40 (m, 6H, CH3), 0.84–0.87 (m, 6H, CH3), 2.63–2.71 (m, 4H, CH2), 2.99–3.13 (m, 4H, CH2), 7.29 (ddd, J = 7.9, 6.6 and 1.3 Hz, 1H), 7.33–7.35 (m, 1H), 7.43 (ddd, J = 8.1, 6.7 and 1.3 Hz, 1H), 7.60 (d, J = 8.8 Hz, 2H), 7.90 (d, J = 8.2 Hz, 2H), 7.96 (d, J = 8.8 Hz, 2H).
13C NMR (126 MHz, CDCl3): δ 12.9 (CH3), 13.00 (CH3), 41.3 (CH2), 41.8 (CH2), 122.5, 123.7, 125.2, 126.1, 126.4, 127.7, 128.8, 131.2, 133.4, 147.5, 153.3.
13C NMR (DEPT 135, CDCl3): δ 12.9 (CH3), 13.00 (CH3), 41.3 (CH2), 41.8 (CH2), 122.5, 125.2, 126.1, 126.4, 127.7, 128.8.
2-methoxy-2′-(2-pivaloyloxyethoxy)-1,1′-binaphthyl (41).
The compound was synthesized according to General Procedure B and then purified by column chromatography on silica gel using hexane: acetone (99: 1) as an eluent. The pure compound was isolated as an oily liquid. The enantiomeric excess was determined by HPLC using a chiral column: OD-H, Mobile phase: hexane: ethanol (99.5: 0.5); Flow: 0.5 mL/min; t1 = 20.882 min. (68%); t2 = 22.780 min.; λ = 254 nm. The specific rotation of the compound with an 36% ee was [α]D = +13.7 (c = 1.015, CHCl3, λ = 589 nm). Yield: 72%. HRMS (ESI): m/z = 451.1883 [C28H28O4+Na]+, m/z (teor.) = 451.1880, diff. = 0.66 ppm
1H NMR (500 MHz, CDCl3): δ 0.99 (s, 9H, CH3), 3.78 (s, 3H, OCH3), 4.05–4.07 (m, 2H, CH2), 4.10–4.19 (m, 2 H, CH2), 7.11 (d, J = 16.4 Hz, 1H), 7.13 (d, J = 16.7 Hz, 1H), 7.19–7.25 (m, 2H), 7.30–7.33 (m, 1H), 7.33–7.37 (m, 1H), 7.46 (dd, J = 9.0 and 2.7 Hz, 2H), 7.87 (dd, J = 18.9 and 9.1 Hz, 2H), 7.97 (dd, J = 9.0 and 3.0 Hz, 2H).
13C NMR (126 MHz, CDCl3): δ 26.9 (CH3), 38.5 (C(CH3)3), 56.7 (OCH3), 63.0 (CH2), 67.7 (CH2), 113.9, 116.3, 119.2, 121.0, 123.4, 123.8, 125.1, 125.4, 126.3, 126.3, 127.8, 127.9, 129.1, 129.3, 129.4, 129.6, 133.9, 134,0, 153.9, 154.8, 178.3.
13C NMR (DEPT 135, CDCl3): δ 26.9 (CH3), 56.7 (OCH3), 63.0 (CH2), 67.7 (CH2), 113.9, 116.3, 123.4, 123.9, 125.1, 125.4, 126.3, 126.3, 127.8, 127.9, 129.4, 129.4.
2-((2′-((diethylcarbamoyl)oxy)-[1,1′-binaphthalen]-2-yl)oxy)ethyl pivalate (42).
The compound was synthesized according to General Procedure B and then purified by column chromatography on silica gel using hexane: acetone (95: 5) as an eluent. The pure compound was isolated as a yellowish solid. Mp = 91.8 °C. The enantiomeric excess was determined by HPLC using a chiral column: OJ-H, Mobile phase: hexane: ethanol (98: 2); Flow: 1 mL/min; t1 = 12.668 min.; t2 = 17.935 min. (68%); λ = 254 nm. The specific rotation of the compound with an 36% ee was [α] = + 19.9 (c = 1.005, CHCl3, λ = 589 nm, 20 °C). Yield: 87%. Elemental Analysis: found C, 74.34; H, 6.74; N, 2.73; theoretical: C, 74.83; H, 6.87; N, 2.73.
1H NMR (500 MHz, CDCl3): δ 0.42–0.45 (m, 3H, N(CH2CH3)2), 0.80–0.82 (m, 3H, N(CH2CH3)2), 1.01 (s, 9H, (CH3)3), 2.68–2.69 (m, 2H, N(CH2CH3)2), 2.97–3.09 (m, 2H, N(CH2CH3)2), 4.04–4.12 (m, 4H, OCH2CH2O), 7.23–7.26 (m, 2H), 7.28–7.29 (m, 2H), 7.35 (ddd, J = 7.9, 6.4 and 2.2 Hz, 1H), 7.42 (d, J = 9.0 Hz, 1H), 7.42–7.45 (m, 1H), 7.61 (d, J = 9.1 Hz, 1H), 7.85 (d, J = 8.2 Hz, 1H), 7.93 (d, J = 8.2 Hz, 1H), 7.97 (t, J = 8.8 Hz, 2H).
13C NMR (126 MHz, CDCl3): δ 12.9 N(CH2CH3)2), 13.1 N(CH2CH3)2), 26.9((CH3)3), 38.5 (C(CH3)3), 41.2 N(CH2CH3)2), 41.7 N(CH2CH3)2), 62.9 (OCH2CH2), 67.6 (OCH2CH2), 115.7, 119.9, 122.5, 124.0, 124.4, 125.0, 125.7, 125.9, 126.1, 126.5, 127.5, 128.0, 128.7, 129.4, 129.6, 131.3, 133.5, 133.9, 147.4, 153.4, 153.9, 178.3.
13C NMR (DEPT 135, CDCl3): δ 12.9 N(CH2CH3)2), 13.1 N(CH2CH3)2), 26.9 ((CH3)3), 41.2 N(CH2CH3)2), 41.7 N(CH2CH3)2), 62.9 (OCH2CH2), 67.6 (OCH2CH2), 115.7, 122.5, 124.0, 125.0, 125.7, 125.9, 126.1, 126.5, 127.5, 128.0, 128.7, 129.6.
(2-methoxy-1,1′-binaphthalen-2-yl)methyl 2,2′-dimethylpropanoate (44).
The compound was synthesized according to General Procedure B and then purified by column chromatography on silica gel using hexane: acetone (98: 2) as an eluent. The pure compound was isolated as an oily liquid. The enantiomeric excess was determined by HPLC using a chiral column: OD-H, Mobile phase: hexane: ethanol (98: 2); Flow: 0.5 mL/min; t1 = 7.395 min.; t2 = 7.865 min (93%); λ = 254 nm. The specific rotation of the compound with an 86% ee was [α]D = −10.7 (c = 1.025, CHCl3, λ = 589 nm). Yield: 66%. HRMS (ESI): m/z = 421.1827 [C27H26O3+H]+, m/z (teor.) = 421.1774, diff. = 12.58 ppm.
1H NMR (500 MHz, CDCl3): δ 1.07 (s, 9H, (CH3)3), 3.78 (s, 3H, OCH3), 4.79 (d, J = 12.9 Hz, 1H, CH2), 4.97 (d, J = 12.9 Hz, 1H, CH2), 6.98–7.00 (m, 1H), 7.16–7.18 (m, 1H), 7.20 (ddd, J = 8.5, 6.9 and 1.3 Hz, 1H), 7.25 (ddd, J = 8.4, 6.8 and 1.3Hz, 1H), 7.32 (ddd, J = 8.1, 6.9 and 1.3 Hz, 1H), 7.44–7.48 (m, 2H, CH), 7.69 (d, J = 8.5 Hz, 1H), 7.84–7.88 (m, 1H), 7.93 (d, J = 8.3Hz, 1H), 7.98–8.02 (m, 2H).
13C NMR (126 MHz, CDCl3): δ 27.0 (CH3), 38.7 (C(CH3)3) 56.4 (OCH3), 64.8 (CH2), 113.3, 119.7, 123.6, 125.1, 125.7, 125.9, 126.1, 126.2, 126.3, 126.7, 126.8, 127.2, 127.6, 127.8, 127.9, 128.0, 128.0, 128.1, 129.0, 129.4, 130.0 132.9, 133,0, 133.2, 133.2, 133.9, 154.4, 178.3.
13C NMR (DEPT 135, CDCl3): δ 27.0 (CH3), 56.4 (OCH3), 64.8 (CH2), 113.3, 123.6, 125.1, 125.7, 125.9, 126.2, 126.3, 126.7, 127.9, 128.0, 128.0, 130.0.
2-(2′-((diethylcarbamoyl)oxy)-[1.1′-binaphthalen]-2-yl)methyl pivalate (45).
The compound was synthesized according to General Procedure B and purified by column chromatography on silica gel using hexane: acetone (99: 1) as an eluent. The pure compound was isolated as colorless crystals. Mp = 69–72 °C. The enantiomeric excess was determined by HPLC using a chiral column: OD-H, Mobile phase: hexane: ethanol (98: 2); Flow: 1 mL/min; t1 = 7.861 min.; t2 = 8,513 min. (88%); λ = 254 nm. The specific rotation of the compound with a 76% ee was [α]D = +31.5 (c = 1.0, CHCl3, λ = 589 nm). Yield: 47%. Elemental Analysis: found C, 75.87; H, 6.80; N, 2.73; theoretical: C, 76.99; H, 6.88; N, 2.90.
1H NMR (500 MHz, CDCl3): δ 0.37–0.40 (m, 3H, N(CH2CH3)2), 0.78–0.81 (m, 3 H, N(CH2CH3)2), 1.11 (s, 9H, (CH3)3), 2.65 (q, J = 6.9 Hz, 2H, N(CH2CH3)2), 2.98–3.09 (m, 2H, N(CH2CH3)2), 4.83 (d, J = 12.9 Hz, 1H, CH2), 5.00 (d, J = 12.9 Hz, 1H, CH2), 7.21–7.23 (m, 1H), 7.28–7.32 (m, 3H), 7.45–7.48 (m, 2H), 7.60 (d, J = 8.8 Hz, 1H), 7.64 (d, J = 8.5 Hz, 1H), 7.91 (d, J = 8.2 Hz, 1H), 7.97 (t, J = 7.9 Hz, 2H), 8.02 (d, J = 8.5 Hz, 1H).
13C NMR (126 MHz, CDCl3): δ 12.9 N(CH2CH3)2), 13.0 N(CH2CH3)2), 27.1 (CH3), 41. N(CH2CH3)2), 41.8 N(CH2CH3)2), 64.4 (CH2), 122.5, 125.4, 125.5, 125.7, 125.8, 126.0, 126.4, 126.6, 126.6, 127.6, 128.1, 128.3, 129.3, 131.4, 132.1, 132.7, 133.1, 133.3, 147.3, 178.1.
13C NMR (DEPT 135, CDCl3): δ 12.9 N(CH2CH3)2), 13.0 N(CH2CH3)2), 27.1 (CH3), 41.2 N(CH2CH3)2), 41.8 N(CH2CH3)2), 64.4 (CH2), 122.5, 125.4, 125.5, 125.8, 126.1, 126.4, 126.6, 126.6, 127.6, 128.1, 128.4, 129.3.
9-(2-methoxynaphthalen-1-yl)phenanthrene (46).
The compound was synthesized according to General Procedure C and then purified by column chromatography on silica gel using hexane as an eluent. The pure compound was isolated as colorless crystals. Mp = 184.9–187.7 °C. The enantiomeric excess was determined by HPLC using a chiral column: OJ-H, Mobile phase: hexane: ethanol (98: 2); Flow: 1 mL/min; t1 = 17.219 min. (52.5%); t2 = 26.766 min.; λ = 254 nm. Due to the low enantiomeric excess of the compound of 5%, specific rotation was not determined. Yield: 91%. HRMS (ESI): m/z = 335.1454 [C25H18O+H]+, m/z (teor.) = 335.1430, diff. = 7.16 ppm.
1H NMR (500 MHz, CDCl3): δ 3.79 (s, 3H, CH3), 7.21–7.24 (m, 1H), 7.28–7.30 (m, 1H), 7.34 (ddd, J = 8.2, 6.6 and 1.3 Hz, 1H), 7.39–7.40 (m, 2H), 7.49 (d, J = 9.1 Hz, 1H), 7.62–7.67 (m, 2H), 7.72 (ddd, J = 8.2, 6.9 and 1.3 Hz, 1H), 7.73 (s, 1H), 7.89–7.91 (m, 2H), 8.02 (d, J = 8.8 Hz, 1H), 8.80–8.83 (m, 2H).
13C NMR (126 MHz, CDCl3): δ 56.8 (OCH3), 113.8, 122.6, 122.8, 123.6, 125.5, 126.4, 126.5, 126.5, 126.6, 126.6, 126.8, 127.8, 128.7, 129.1, 129.6, 130.3, 131.9, 132.1, 134.3, 154.8.
13C NMR (DEPT 135, CDCl3): δ 56.8 (OCH3), 113.8, 122.6, 122.8, 123.6, 125.5, 126.4, 126.5, 126.5, 126.6, 126.6, 126.8, 127.8, 128.7, 129.1, 129.6.
2-(2-methoxynaphthalen-1-yl)phenyl 2,2-dimethylpropanoate (48).
The compound was synthesized according to General Procedure A and then purified by column chromatography on silica gel using hexane:acetone (9:1) as an eluent. The pure compound was isolated as an oily liquid. The enantiomeric excess was determined using the europium tris [3-(heptafluoropropylhydroxymethylene)-(+)-camphorate] as an optically active NMR shift reagent. Signals from which it was found that the compound exhibited an enantiomeric excess of 3% were 0.84 ppm and 0.86 ppm derived from hydrogen in the pivaloyl group. These signals were derived from split and shifted towards higher chemical shifts signal at 0.72 ppm. Yield: 35%. HRMS (ESI): m/z = 357.1439 [C22 H22O3+Na]+, m/z (teor.) = 357.1461, diff. = −6.16 ppm.
1H NMR (500 MHz, CDCl3): δ = 0.73 (s, 9H, (CH3)3), 3.84 (s, 3H, OCH3), 7.25–7.27 (m, 1H, CH), 7.32–7.36 (m, 3H), 7.37–7.38 (m, 3H), 7.47–7.49 (m, 1H), 7.79–7.81 (m, 1H), 7.88 (d, J = 8.8 Hz, 1H).
13C NMR (126 MHz, CDCl3): δ = 26.5 (CH3), 38.6, (C(CH3)3), 56.7 (OCH3), 113.4, 120.4, 122.7, 123.5, 125.2, 125.6, 126.4, 127.6, 128.6, 128.8, 129.4, 129.5, 132.5, 133.5, 149.5, 154.2, 176.1.
13C NMR (DEPT 135, CDCl3): δ = 26.5 (CH3), 56.7 (OCH3), 113.4, 122.7, 123.5, 125.2, 125.6, 126.4, 127.6, 128.6, 129.4, 132.5.
2-(2-methoxynaphthalen-1-yl)phenyl diethylcarbamate (50).
The compound was synthesized according to General Procedure A and then purified by column chromatography on silica gel using hexane: acetone (9: 1) as an eluent. The pure compound was isolated as a colorless oily liquid. Using the HPLC method or chemical shift reagent europium tris[3-(heptafluoropropylhydroxymethylene)-(+)-camphorate], the enantiomers were not distinguished. Yield: 85%. HRMS (ESI): m/z = 350.1750 [C22 H23NO3+H]+, m/z (teor.) = 350.1751, diff. = −0.29 ppm
1H NMR (500 MHz, CDCl3): δ 0.46–0.49 (m, 3H, N(CH2CH3)2), 0.80–0.84 (m, 3H, N(CH2CH3)2), 2.67–2.71 (m, 2H, N(CH2CH3)2), 3.00–3.12 (m, 2H, N(CH2CH3)2), 3.83 (s, 3H, OCH3), 7.30–7.36 (m, 5H), 7.42–7.49 (m, 3H, CH), 7.78–7.80 (m, 1H), 7.88 (d, J = 8.9 Hz, 1H).
13C NMR (126 MHz, CDCl3): δ 12.9 N(CH2CH3)2), 13.2 N(CH2CH3)2), 41.1 N(CH2CH3)2), 41.7 N(CH2CH3)2), 56.7 (OCH3), 113.5, 120.9, 122.5, 123.0, 123.4, 124.8, 125.4, 126.3, 127.5, 128.4, 128.8, 128.9, 129.3, 131.03, 132.2, 133.5, 149.9, 153.3, 154.2.
13C NMR (DEPT 135, 125.77 MHz, CDCl3): δ 12.9 N(CH2CH3)2), 13.2 N(CH2CH3)2), 41.1 N(CH2CH3)2), 41.6 N(CH2CH3)2), 56.7 (OCH3), 113.5, 123.0, 123.4, 124.8, 125.4, 126.3, 127.5, 128.4, 129.3, 132.2.
9-(naphthalen-1-yl)phenanthrene (51).
The compound was synthesized according to General Procedure C and then purified by column chromatography on silica gel using hexane as an eluent. The pure compound was isolated as colorless crystals. Mp = 115.9–117.2 °C (DME) (lit. [
39] mp = 126–127 °C). The enantiomeric excess was determined by HPLC using a chiral column: AS-H, Mobile phase: hexane: isopropanol (90: 10); Flow: 0.2 mL/min; t1 = 26.394 min. (51%); t2 = 28.445 min.; λ = 254 nm. Due to the low enantiomeric excess of the compound of 3%, specific rotation was not determined. Yield: 98%. Elemental Analysis: found C, 93.74; H, 5.12 theoretical: C, 94.70; H, 5.30.
1H NMR (500 MHz, CDCl3): δ 7.28–7.31 (m, 1H, CH), 7.39–7.44 (m, 2H), 7.45–7.51 (m, 2H), 7.57–7.59 (m, 1H), 7.62–7.68 (m, 3H), 7.72–7.75 (m, 1H), 7.80 (s, 1H), 7.91–7.93 (m, 1H), 7.97–8.00 (m, 2H, CH), 8.82 (t, J = 8.8Hz, 2H).
13C NMR (126 MHz, CDCl3): δ 122.6, 122.7, 125.4, 125.8, 126.0, 126.5, 126.5, 126.6, 126.7, 126.8, 127.4, 127.8, 128.0, 128.19, 128.4, 128.7 130.2, 130.3, 131.6, 132.1, 132.9, 133.5, 137.5, 137.1, 138.4.
13C NMR (DEPT 135, CDCl3): δ 122.6, 122.7, 125.4, 125.8, 126.0, 126.5, 126.5, 126.6, 126.7, 126.9, 127.4, 127.8, 128.0, 128.1, 128.4, 128.7.
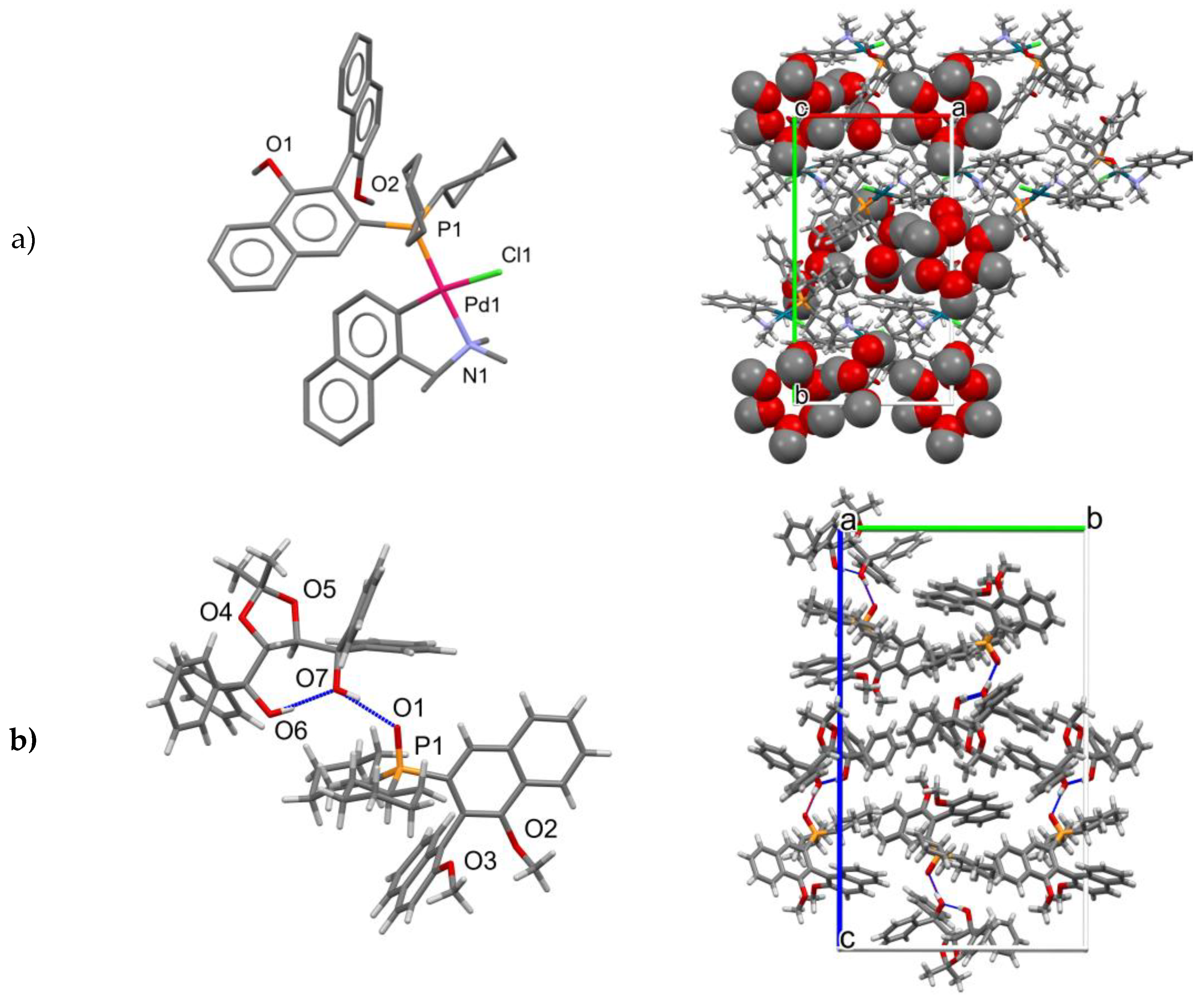
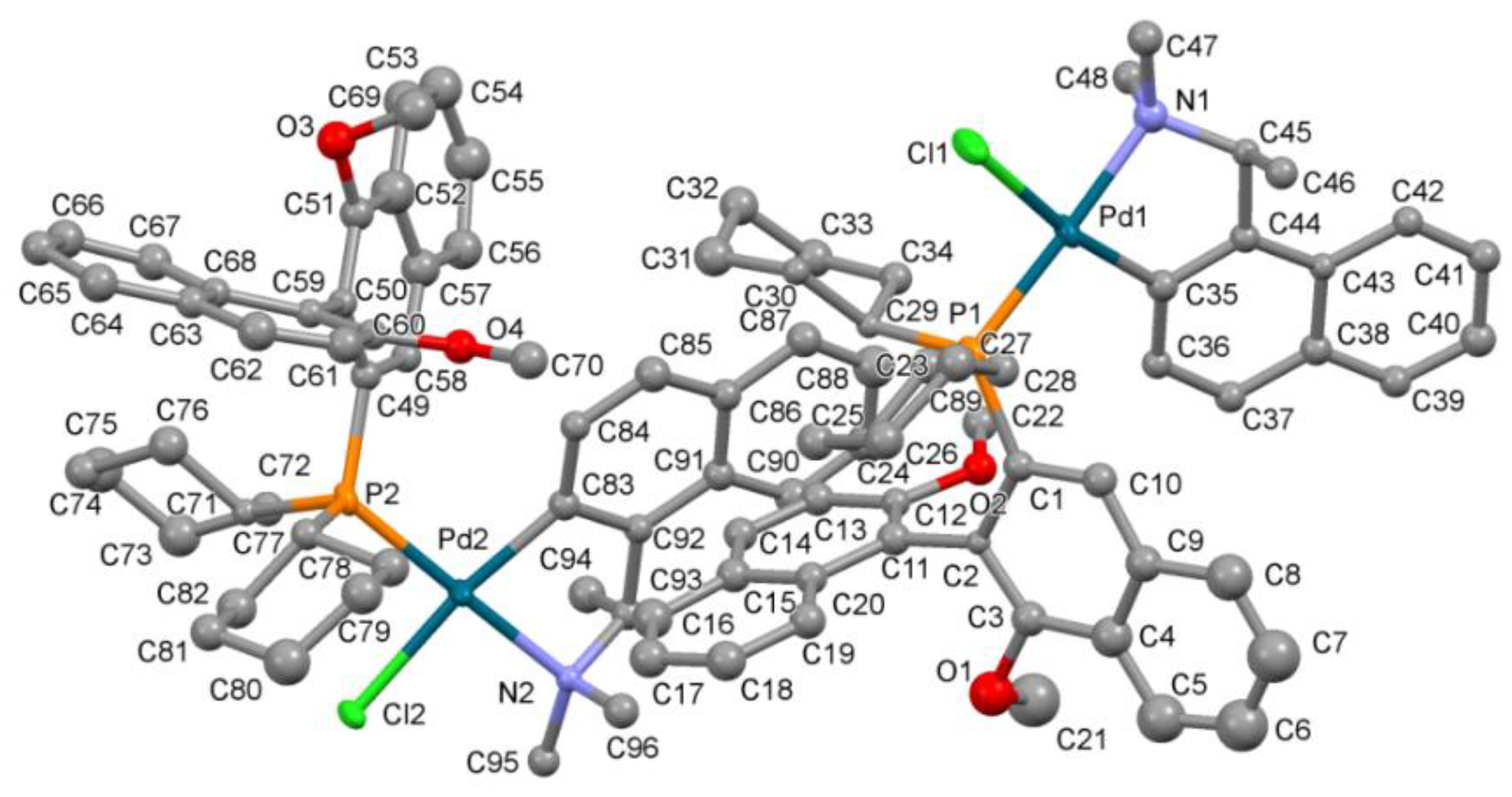
X-ray crystal structure determination of Pd complex (S,Sa)-12 and cocrystal of compound 9 with TADDOL.
The labeling scheme of atoms in coordination units of
(S,Sa)-12 is shown in
Figure 3.
Crystal data for the Pd complex
(S,Sa)-12 and a cocrystal of compound
9 with TADDOL are provided in
Table 18,
Table 19 and
Table 20.
Diffraction reflections were collected on Bruker Nonius Kappa CCD and on Xcalibur Sapphire2 diffractometers with Mo Kα radiation (0.71073 Å). The absorption corrections were applied by the multi-scan method from Blessing [
40]. Crystal structure for compound
9 was refined with anisotropic non-hydrogen atoms. The proper configuration was established on the base of the synthesis route and the presence of the TADDOL molecule in the cocrystal structure.
The compound
12 has the formula 2(C
48H
55ClNO
2PPd) 9(CH
3OH) 2(H
2O) and crystalizes in the monoclinic
P2
1 space group as a solvate of a pure
(S,
Sa)-diastereoisomer with two neutral coordination units, nine methanol molecules, and two water molecules in the asymmetric part. Due to the very poor crystals of the Pd complex, only the Pd, P, and Cl atoms were refined anisotropically. Further calculations with all non-hydrogen atoms refined anisotropically presented no better solution. The hydrogen atoms in the coordination units were introduced at calculated positions and refined riding on their carrier atoms. The hydrogen atoms from solvent molecules were omitted due to the low quality of the experimental data. The absolute configuration of the chiral complex was confirmed by using the Flack parameter [
41]. The supplementary crystallographic data for Pd complex
(S,Sa)-12 CCDC No. 2222975 and for the cocrystal of compound
(Ra)-9 with TADDOL CCDC No. 2232483 can be obtained free of charge from the Cambridge Crystallographic Data Centre via
www.ccdc.cam.ac.uk/data_request/cif, accessed on 1 December 2022.
Crystal data for Pd complex (S,Sa)-12 C105H130Cl2N2O15P2Pd2 (M = 2005.75 g/mol): monoclinic, space group P21, a = 14.0761(1) Å, b = 25.813(1) Å, c = 14.586(1) Å, β = 90.26(1)°, V = 5299.7(4) Å3, Z = 2, T = 100(2) K, μ(MoKα) = 0.480 mm−1, Dcalc = 1.257 g/cm3, 15122 reflections measured (2.8° ≤ 2Θ ≤ 51.92°), 10364 unique (Rint = 0.0458, Rsigma = 0.0526) which were used in all calculations. The final R1 was 0.0585 (>2σ(I)) and wR2 was 0.1553 (all data).
Crystal data for the cocrystal of compound (Ra)-9 with TADDOL C65H69O7P (M =993.17 g/mol): orthorhombic, space group P212121 (no. 19), a = 9.8002(6) Å, b = 18.1101(10) Å, c = 30.8874(18) Å, V = 5482.0(6) Å3, Z = 4, T = 293(2) K, μ(MoKα) = 0.104 mm−1, Dcalc = 1.203 g/cm3, 46268 reflections measured (4.688° ≤ 2Θ ≤ 50.482°), 9878 unique (Rint = 0.0849, Rsigma = 0.0747), which were used in all calculations. The final R1 was 0.0608 (I > 2σ(I)) and wR2 was 0.1529 (all data).



 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





























































