
The Role of Steps on Silver Nanoparticles in Electrocatalytic Oxygen Reduction


Centre for Catalysis and Clean Energy, School of Environment and Science, Griffith University, Gold Coast, QLD 4222, Australia
*
Author to whom correspondence should be addressed.
Catalysts 2022, 12(6), 576; https://doi.org/10.3390/catal12060576
Submission received: 28 April 2022
/
Revised: 19 May 2022
/
Accepted: 20 May 2022
/
Published: 24 May 2022
(This article belongs to the Special Issue Heterogeneous Electrocatalysis: Fundamentals and Applications)
Abstract
:Hydrogen fuel cell technology is an essential component of a green economy. However, it is limited in practicality and affordability by the oxygen reduction reaction (ORR). Nanoscale silver particles have been proposed as a cost-effective solution to this problem. However, previous computational studies focused on clean and flat surfaces. High-index surfaces can be used to model active steps presented in nanoparticles. Here, we used the stable stepped Ag(322) surface as a model to understand the ORR performance of steps on Ag nanoparticles. Our density functional theory (DFT) results demonstrate a small dissociation energy barrier for O2 molecules on the Ag(322) surface, which can be ascribed to the existence of low-coordination number surface atoms. Consequently, the adsorption of OOH* led to the associative pathway becoming ineffective. Alternatively, the unusual dissociative mechanism is energetically favored on Ag(322) for ORR. Our findings reveal the importance of the coordination numbers of active sites for catalytic performance, which can further guide electrocatalysts’ design.
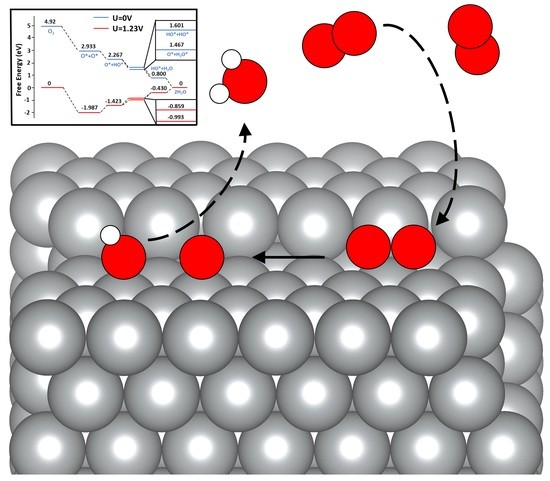
1. Introduction
The looming energy crisis is inevitable unless current energy sources are replaced with sustainable green alternatives. Energy sources have become more diverse and greener in recent years, yet solar and wind power alone are unlikely to overcome our predicament. The electrification of our power sector will meet the challenges of long-distance transport, metallurgy, etc. [1]. Hydrogen is an abundant element that could address the limitations of the current clean energy sector. It can be used for metallurgic purposes and as a source of portable high energy density storage technology [2,3]. However, extracting the energy from hydrogen has proved challenging. The hydrogen fuel cell is the primary technology that converts this gas into electricity and is the pathway to a 100% renewable economy [4,5]. Unfortunately, this technology is held back by the kinetically poor cathodic oxygen reduction reaction (ORR) [6,7,8,9]. Moreover, expensive Pt-based catalysts are necessary for commercial hydrogen fuel cells. Therefore, the low energy conversion efficiency and high cost of the electrocatalysts prevent the hydrogen fuel cell to compete with traditional alternatives. An intuitive approach for this problem is to increase the electrocatalytic performance of a non-expensive material [10]. However, these substitutes are difficult to mass-produce or have poor lifetimes [11,12]. Therefore, an earth-abundant elemental catalyst would be ideal.
Silver is a potential alternative electrocatalyst for hydrogen fuel cells. This is because Ag has similar properties to Pt [13,14,15]. In addition, Ag-based catalysts are 50 times cheaper than the industrial Pt/C standard [14]. Previous studies have revealed that Ag nanowires, nanocubes, and nanoparticles have high catalytic performance due to their unique surface morphology [16]. Since Ag nanomaterials are rich in high-Miller-index surfaces, their outstanding performance may be ascribed to the existence of these stepped surfaces [15]. Ag is facet-dependent and has shown significant variation in its catalytic properties due to the different bonding strengths with the adsorbates [13,17,18,19,20,21,22]. High-Miller-index surfaces often show stronger catalytic activity than their flat counterparts. Vattuone et al. found that Ag (210) showed different absorption properties of oxygen molecules at 105 K. Additionally, when Ag (210) absorbed an oxygen molecule, it became easier for the surface to absorb additional oxygen molecules. Jiang et al. investigated the benzene adsorption on stepped Ag(211), (533), (322), (755), and (544) surfaces. They found that the work function change of the surface was strongly related to the density of the steps [23]. In addition, the adsorption energy of benzene on the Ag(322) surface was found to be the strongest among all the considered systems, which was 9% stronger than that on Ag(111). While the ORR mechanisms of flat, low-Miller-index surfaces are well understood, minimal theoretical work has been conducted to investigate the mechanisms of their ORR pathways on these rough stepped surfaces [24]. Furthermore, a better understanding of the ORR pathways on stepped surfaces can illuminate ways to improve the overall performance of other nanocatalysts. To this end, an atomistic investigation of the ORR mechanisms on stepped surfaces is required.
Previous studies have demonstrated that computational methodology plays a crucial role in accurately understanding the adsorption properties of the O2 on Ag surfaces [21,25,26,27]. The adsorption of the oxygen molecule is also controversial, as it has several adsorption states [28,29,30]. Studying ORR is notorious for being beyond the limits of the generalized gradient approximation (GGA) level studies. Several limitations, such as the self-interaction error and poor descriptions of two-body vdW forces, prevent the broad application of DFT [31,32,33]. Computations with some van der Waals (vdW) correction methods, e.g., D3, still fall short of experimental expectations [34]. Only hybrid or non-local functionals can reliably reproduce experimental values, but they suffer from large computational costs [35,36]. However, the SCAN-rVV10 method overcomes these limitations [37,38]. Our previous study demonstrated its excellent non-local vdW and close-range predictions [8]. Both SCAN and rVV10 have been shown individually to work around the self-interaction error and replicate an accurate oxygen dissociation energy barrier for Ag(111) [8,39]. Therefore, this method could be useful for further studies involving O2 adsorption.
In this study, the ORR mechanism of the Ag(322) surface was investigated using the SCAN-rVV10 method. The Ag(322) surface was selected since it is a stable stepped surface with high reactivity [23,40], which can be reflective of the features of nanoparticles. The results showed that the dissociative pathway was energetically preferred for ORR on Ag(322). Furthermore, the coordination numbers of the surface atoms are essential to their catalytic performance.
2. Results and Discussion
2.1. Surface Properties of Ag(322)
High-Miller-index surfaces have higher catalytic activity because of the differences in layer separation and the greater number of available bonding states [21]. However, the main drawback of high-Miller-index nanocatalysts is their short lifetime. Most high-Miller-index surfaces have large surface energies. For example, the (211) and (210) surfaces studied by Jiang et al. and Vattuone et al. had surface energies of 0.87 and 0.90 J/m2, which is 14% and 18% higher than that of the most stable Ag(111) surfaces, respectively [40]. Such high surface energy leads to reconstruction to a thermodynamically stable configuration, limiting the lifetime of the high-Miller-index surfaces. By comparison, the Ag(322) surface has almost identical surface energy to that of the Ag(111) while still being a high-Miller surface [40]. The Ag(322) surface has noticeable surface relaxation because of its stepped edge. The interlayer distances between the first and second topmost layers (Δ12) and between the second and third layers (Δ23) are reduced by 19.58% and 4.55%, respectively. Conversely, the interlayer relaxation in the Ag(111) surface is negligible [8]. The large relaxation in Ag(322) is influenced by its unique morphology. The Ag(322) surface has three different coordination numbered (CN) Ag atoms (see Figure 1a). The CN of the topmost Ag atoms is 7. As a comparison, all the atoms in the topmost Ag(111) surface have a CN of 9. These differing CN atoms have different electronic configurations. The partial density of states (PDOS) of the Ag 4d states is shown in Figure 1b. It can be seen that the states of the CN7 atoms have sharper peaks, which are closer to the Fermi energy level. The PDOS images suggest that CN7 atoms are more reactive, which means they could have stronger adsorption capabilities in terms of the other surface atoms with larger CNs.
2.2. Adsorption of Atomic and Molecular Oxygen on Ag(322)
Several adsorption sites, including the fcc and hcp sites on the terrace, the bridge site at the step, and the hollow sites next to the step, were investigated in this study. The stable configurations for oxygen and molecular oxygen adsorption on Ag(322) are shown in Figure 2. For atomic O, the step-hollow (SH) and hollow (H) sites were optimal for adsorption (see Figure 2a,b). After the adsorption of the atomic oxygen, the average Ag-O bond length at the SH site was about 5% longer than that at the H site. A structural distortion adjacent to the H site was seen, as shown in Figure 2b. This is because the strong adsorption of the oxygen with stepped Ag atoms weakened the interaction between the CN7 atom and the neighboring CN9 atoms. Consequently, considerable surface relaxation was observed. Furthermore, the adsorbed oxygen atom bonded with two CN7 and two CN9 surface Ag atoms at the SH site. At the H site, the oxygen atom was only associated with two CN7 and one CN10 atom. As such, while a shortened Ag-O bond length was found at the H site, the overall adsorption energy of atomic oxygen at the SH site was lower (see Table 1). For the same reason, the adsorption energy of the atomic oxygen at the SH site on Ag(322) was about 10% lower than that on Ag(111) since the oxygen atom only interacted with three CN9 atoms on the (111) surface.
Molecular oxygen could stably adsorb at the fcc step-hollow (fSH) and bridge step-hollow (bSH) sites (see Figure 2c,d). The SH, fSH, and bSH sites were all between the two terraces and bonded to the step atoms. The difference between fSH and bSH is the orientation of the O-O bond. In bSH, the O-O bond is parallel to the step edge, while that in fSH is almost perpendicular. At the bSH site, two O atoms only interact with one CN7 and one CN10 Ag atom. Meanwhile, at the fSH site, an O2 molecule bonds to two CN7 atoms, two CN10 atoms, and one CN9 Ag atom. The different coordination geometries of adsorbed O2 lead to various adsorption strengths. The adsorption energy of the O2 molecules at the fSH site was 150% stronger than that at the bSH site (see Table 1). Figure 3a,b show the PDOS of the O 2p states and the 4d states of the Ag atoms associated with O2. It can be seen that the O2 2p and Ag 4d states were closer to the Fermi energy level in the energy range of −1 to 0 eV when O2 adsorbed at the bSH site. Additionally, the O2 2p states in the energy range of −4 to −3 eV were sharper at the bSH site in comparison with that at the fSH sites. The subtle difference supports the weaker adsorption strength of O2 at the bSH site. The O-O bonding strength was further analyzed by using the partial crystal orbital Hamilton population (−pCOPH), as shown in Figure 3c,d. Within this range between −6 to −8 eV, the fSH site had a smaller bonding contribution than bSH. The fSH site anti-bonding contribution was larger and more jagged than that of bSH. Quantitatively, the −IpCOHP valued the average O-O interaction of the fSH site as −3.35 eV/bond, which is 0.10 eV/bond smaller than that of bSH. Therefore, both the qualitative and quantitative analyses confirmed the weakened O-O bond when O2 adsorbs at the fSH site, which supports the stronger adsorption strength at this site.
The magnetic moments of all systems were zero, suggesting that only the peroxo species was present (see Table 1). The peroxo species is ideal, as it contributes two e- to the Ag-O bond. The Ag(322) fSH site had the largest O-Ag bond length but the shortest vibrational frequency, thus indicating a weak O-O bond and strong Ag-O interaction. Interestingly, the adsorption properties of O2 on the Ag(322) surface were similar to those on Pt(111). The adsorption energy on Pt(111) is −0.68 eV with an O-O vibrational frequency of 743 cm−1, similar to Ag(322) [34]. This suggests that the Ag(322) surface may have outstanding ORR performance, similar to Pt(111).
2.3. Dissociation of O2 on Ag(322)
The oxygen dissociation at both the fSH and bSH sites was considered here using the climbing image nudged elastic band (CI-NEB) method with eight images between the initial and final states (see Figure 4). Starting from the adsorbed O2 at the fSH site, one oxygen atom can shift to the hollow site of the terrace with another oxygen atom next to the step. The energy barrier of this pathway is 0.66 eV. When O2 adsorbs at the bSH site, both oxygen atoms can shift to the SH site. This pathway has a lower energy barrier of 0.47 eV. The dissociation barriers of both pathways are much lower than on the flat Ag(111) surface, which is 1.22 eV [8]. The significantly lower energy barriers on the stepped Ag(322) surface demonstrated that the surface morphology played a crucial role with its larger reactive surface area and varying CN.
2.4. Electrocatalytic ORR Performance on Ag(322)
The Gibbs free energy changes of the adsorbed oxygen intermediates were used as the descriptor to evaluate the electrocatalytic ORR performance of Ag(322), which were calculated on the basis of the computational hydrogen electrode (CHE) method developed by Norskov and colleagues [41,42]. Under standard conditions, ORR may follow two pathways: the associative mechanism (see Equation (1)) or the dissociative mechanism (see Equations (2)–(3)) [7,8,13,41]. For the associative mechanism, the O2 adsorbs to the catalytic site (*) and then becomes protonated multiple times.
The dissociative mechanism starts with the dissociation of adsorbed O2*.
or
In this study, the Gibbs free energy changes were investigated with the consideration of both the associative and dissociative mechanisms (see Figure 5 and Figure 6). The associative overpotential for Ag(111) was large compared with the Pt(111) overpotential of 0.441 V [8,21,27,28,41]. The Ag(322) surface performed significantly worse, with a value ~85% larger than that of Pt(111). The dissociative overpotential for Pt(111) is 0.457 eV, which is smaller than that found in our Ag(111) results [8,21,27,28,41]. The Ag(322) surface was comparable to that of Pt, with a difference between the dissociative overpotentials of ~20%. This was supported by nanoparticle studies, as silver nanoparticles significantly improve the catalytic performance compared with the (111) surface crystal [43]. The large adsorption energy of the OH* and OOH* species caused large associative overpotentials and differences between computational studies. In comparison with the reported values, our adsorption energies for OH* and OOH* species were larger. This discrepancy could be caused by the different XC functionals. The GGA functional, such as PBE, has been widely used in most previous studies without the correct consideration of vdW forces [13,21,24,27,28,31,41]. Montemore et al. mentioned this drawback in their work and pointed out the trend of current ORR studies relying on outdated methodologies [28]. Methods with vdW correction, e.g., SCAN-rVV10, can predict better the adsorption properties of the OOH* and OH* species [31].
In comparison to Ag(111), the (322) surface showed better dissociative overpotentials but a worse associative overpotential. The associative overpotential of Ag(322) suffered from unideal OH and OOH adsorption energies. Previous studies have demonstrated that low CN atoms decrease the Gibbs free energy via strong interactions with intermediate species [44,45,46]. However, this was not the case for OOH*. The adsorption energies of OH and OOH on Ag(322) were −3.48 eV and −1.07 eV, respectively (see Figure 5). Comparatively, Ag(111) had OH and OOH adsorption energies of −3.12 eV and −1.23 eV, respectively (see Figure 5). The rate-limiting step for Ag(322) was the formation of OOH* species. The adsorption energy of OOH was too weak and required substantial energy to break the O-OH bond. Consequently, the of the reaction increased (see Figure 6). This is because the orientation of the OOH* species was not ideal, as it was difficult for it to fit in the fSH site. Additionally, the dissociative pathway of Ag(322) is promising, as it showed a low dissociation energy barrier and bypassed the energy traps of the associative method, such as H2O2 production [28].
The dissociative Gibbs energy profile of Ag(322) showed a minimal preference for H2O(l) or OH* formation (see Figure 6), which can increase the removal of OH* species [47]. OH* removal is a unique challenge for Ag. The characteristically strong OH* adsorption on Ag surfaces is often the potential-limiting step. Since is larger on Ag(322), the performance improves and prevents an energy well from forming. Compared with Pt(111), the and of Ag(322) were similar [41]. The potential-limiting step for the dissociation mechanism was the protonation of O*. The adsorption energy of O* was greater than that of OH*, which makes the process uphill under a potential of 1.23 V. However, the availability of low-CN Ag atoms played a crucial role in matching Pt(111) performance. The varying CN atoms not only weakened the O-O but also increased the ORR activity.
Some recent experimental studies on Ag nanoparticles have also suggested the existence of the dissociative pathway for ORR [18,43,48]. Since the 4e-associated mechanism for ORR has been experimentally and theoretically demonstrated on Ag(111) [43,48], the existence of steps in Ag nanoparticles may lead to the dissociative pathway, which closely matches our theoretical prediction in this study. However, while our DFT results can provide the insights to show the specific role of steps in the Ag nanoparticles for electrocatalytic ORR applications, it is worth noting that the theoretical data, e.g., the predicted ORR overpotential, may be different from experimental data. This is because the oversimplified models employed here without consideration of the impact of the water solvent, electrolyte, and applied bias potential cannot model the electrocatalysts under the working conditions [24,49,50,51]. Unfortunately, the consideration of working conditions is still computationally unaffordable and deserves further investigation using more advanced simulation methodologies in the future.
3. Computational Method
All DFT computations were performed using the Vienna Ab initio Simulation Package (VASP) [52] and based on the projected augmented wave (PAW) method [53]. It is known that the O2 molecule is in a triplet state in its ground state. As such, spin-polarization was considered in all the computations for O2 adsorption and dissociation. The electron–ion interaction was described using the PAW potentials, with 5s14d10, 1s1, and 2s22p4 treated as valence electrons of Ag, H, and O, respectively. The smooth part of the wave function was expanded by a kinetic energy cut-off value of 520 eV. During geometric optimization, atoms could relax until the Hellmann–Feynman forces were smaller than 1 × 10−3 eV/Å. The convergence criterion for the electronic self-consistent loop was set to 1 × 10−5 eV. The gamma-centered k-point meshes utilized a reciprocal space resolution of 2π × 0.02 Å−1. For the electron exchange and correlation interactions, the SCAN functional at the meta-GGA level was used [37]. To better consider the vdW interactions, -rVV10 was coupled with SCAN [38]. Both were proven to be effective for this system in our previous work on Ag(111) [8].
The Ag(322) surface was modeled by a (2 × 1) unit cell with a vacuum layer of 15 Å to avoid interaction between the surfaces of neighboring slabs in the z-direction. A four-layer slab was used, in which the bottom two layers were fixed at the bulk position, and the other two were relaxed during structural optimization. The Ag(111) surface was constructed in the same way but used a (2√3×3) supercell [8].
The adsorption energy of O*, OH*, and OOH* species (see Equation (4)) is:
where N is the number of adsorbates per unit cell, Ead/surf, Esurf, and Ead are the energies of the Ag(111) surface with the adsorbate, the clean silver surface, and the isolated adsorbate, respectively. For the calculations of the isolated atoms and molecules, a (20 × 20 × 20) Å3 unit cell and a Gamma-only k-point grid were used. Multiple adsorption sites were identified and are discussed within the paper. The strength of the O-O bond when adsorbed was also studied through the partial density of states (PDOS) and crystal orbital Hamilton population (COPH) analysis [54], vibrational frequency, and magnetic moments [52]. The O2 dissociation energy barrier was investigated using the climbing image nudged elastic band (CI-NEB) method with eight images between the initial and final states [55].
The theoretical catalytic performance can be visualized via the Gibbs free energy. The Gibbs free energy is given by the standard definition as:
where , , , and are the DFT-calculated total binding energy, the zero-point energy change, the entropic contributions, and the shift in caused by a change in relative potential with respect to the hydrogen electrode, respectively. Herein the was obtained by Nørskov et al. (2005) [56]. is the pH contribution to the . For our study, pH was set to 0 to negate the effect of and simplify the model. The Gibbs free energy of each adsorbate was found through:
where, E*, EOH*, EO* and EOOH* are the total energies of the bare surface, the surface with adsorbed OH, the surface with adsorbed O, and the surface with the adsorbed OOH, respectively. EH2O and EH2 are the total energy of the isolated H2O and H2, respectively. The associative free energy diagrams were constructed using Equations (6)–(8) and following the chemical Equation (1). Then, the Gibbs free energy of each elementary step based on the associative mechanism could be calculated through Equations (9)–(12):
The Gibbs free energy of each elementary step of the dissociative mechanism (see Equations (13)–(15)) is:
4. Conclusions
Hydrogen fuel cells are held back by their inflated cost from using Pt-based catalysts. Ag nanoparticles are an affordable and potential replacement for the industrial standard. In this study, we used SCAN-rVV10 to reveal the ORR performance of Ag(322), which is considered to be a substitute for silver nanoparticles. This stepped surface outperformed Ag(111) in adsorption energy, dissociation energy barrier, and theoretical overpotential. Specifically, we modeled a novel dissociation pathway on Ag(322) with an energy barrier of 0.471 eV. This pathway was supported by low-CN step atoms and weakened the O-O bond, thus allowing for a dissociation energy barrier comparable to that of Pt(111). The associative pathway was not preferred due to an overpowering OOH* adsorption, which increased the respective overpotential. Our results, therefore, highlight the importance of the effect of surface morphology and CN numbers for catalyst designs.
Author Contributions
Methodology, software, formal analysis, writing-original draft preparation, J.J.H.; formal analysis and methodology, J.L.; methodology, J.J.W.; project administration, supervision, conceptualization, methodology, resources, writing—review and editing, Y.W. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The data that support the findings of this study are available within the article.
Acknowledgments
This research was undertaken on the supercomputers at National Computational Infrastructure (NCI) in Canberra, Australia, which is supported by the Australian Commonwealth Government, and the Pawsey Supercomputing Centre in Perth with funding from the Australian government and the Government of Western Australia.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bakenne, A.; Nuttall, W.; Kazantzis, N. Sankey-Diagram-based insights into the hydrogen economy of today. Int. J. Hydrogen Energy 2016, 41, 7744–7753. [Google Scholar] [CrossRef]
- Hirscher, M.; Yartys, V.A.; Baricco, M.; von Colbe, J.B.; Blanchard, D.; Bowman, R.C.; Broom, D.P.; Buckley, C.E.; Chang, F.; Chen, P.; et al. Materials for hydrogen-based energy storage – past, recent progress and future outlook. J. Alloys Compd. 2020, 827, 153548. [Google Scholar] [CrossRef]
- Kushnir, D.; Hansen, T.; Vogl, V.; Åhman, M. Adopting hydrogen direct reduction for the Swedish steel industry: A technological innovation system (TIS) study. J. Clean. Prod. 2020, 242, 118185. [Google Scholar] [CrossRef]
- Oliveira, A.M.; Beswick, R.R.; Yan, Y. A green hydrogen economy for a renewable energy society. Curr. Opin. Chem. Eng. 2021, 33, 100701. [Google Scholar] [CrossRef]
- McDowall, W.; Eames, M. Towards a sustainable hydrogen economy: A multi-criteria sustainability appraisal of competing hydrogen futures. Int. J. Hydrogen Energy 2007, 32, 4611–4626. [Google Scholar] [CrossRef]
- Cruz-Martínez, H.; Rojas-Chávez, H.; Matadamas-Ortiz, P.T.; Ortiz-Herrera, J.C.; López-Chávez, E.; Solorza-Feria, O.; Medina, D.I. Current progress of Pt-based ORR electrocatalysts for PEMFCs: An integrated view combining theory and experiment. Mater. Today Phys. 2021, 19, 100406. [Google Scholar] [CrossRef]
- Fu, C.; Liu, C.; Li, T.; Zhang, X.; Wang, F.; Yang, J.; Jiang, Y.; Cui, P.; Li, H. DFT calculations: A powerful tool for better understanding of electrocatalytic oxygen reduction reactions on Pt-based metallic catalysts. Comput. Mater. Sci. 2019, 170, 109202. [Google Scholar] [CrossRef]
- Hinsch, J.J.; Liu, J.; Wang, Y. Reinvestigating oxygen adsorption on Ag(111) by using strongly constrained and appropriately normed semi-local density functional with the revised Vydrov van Voorhis van der Waals force correction. J. Chem. Phys. 2021, 155, 234704. [Google Scholar] [CrossRef]
- Zhao, W.; Huang, K.; Zhang, Q.; Wu, H.; Gu, L.; Yao, K.; Shen, Y.; Shao, Y. In-situ synthesis, operation and regeneration of nanoporous silver with high performance toward oxygen reduction reaction. Nano Energy 2019, 58, 69–77. [Google Scholar] [CrossRef]
- Sarapuu, A.; Kibena-Põldsepp, E.; Borghei, M.; Tammeveski, K. Electrocatalysis of oxygen reduction on heteroatom-doped nanocarbons and transition metal–nitrogen–carbon catalysts for alkaline membrane fuel cells. J. Mater. Chem. A 2018, 6, 776–804. [Google Scholar] [CrossRef]
- Ikram, R.; Jan, B.M.; Ahmad, W. An overview of industrial scalable production of graphene oxide and analytical approaches for synthesis and characterization. J. Mater. Res. Technol. 2020, 9, 11587–11610. [Google Scholar] [CrossRef]
- Gewirth, A.A.; Varnell, J.A.; DiAscro, A.M. Nonprecious Metal Catalysts for Oxygen Reduction in Heterogeneous Aqueous Systems. Chem. Rev. 2018, 118, 2313–2339. [Google Scholar] [CrossRef]
- Erikson, H.; Sarapuu, A.; Tammeveski, K. Oxygen Reduction Reaction on Silver Catalysts in Alkaline Media: A Minireview. ChemElectroChem 2019, 6, 73–86. [Google Scholar] [CrossRef]
- Kitayev, A.; Zysler, M.; Hardisty, S.; Page, M.; Tal-Gutelmacher, E.; Zitoun, D. Silver Oxygen Reduction Electrocatalyst in Alkaline Medium: Aging and Protective Coating. Energy Technol. 2021, 9, 2100546. [Google Scholar] [CrossRef]
- Nubla, K.; Sandhyarani, N. Ag nanoparticles anchored Ag2WO4 nanorods: An efficient methanol tolerant and durable Pt free electro-catalyst toward oxygen reduction reaction. Electrochim. Acta 2020, 340, 135942. [Google Scholar] [CrossRef]
- Linge, J.M.; Erikson, H.; Merisalu, M.; Matisen, L.; Käärik, M.; Leis, J.; Sammelselg, V.; Aruväli, J.; Kaljuvee, T.; Tammeveski, K. Oxygen Reduction on Silver Nanoparticles Supported on Carbide-Derived Carbons. J. Electrochem. Soc. 2018, 165, F1199–F1205. [Google Scholar] [CrossRef]
- Shi, Y.M.; Zhou, W.; Lu, A.Y.; Fang, W.J.; Lee, Y.H.; Hsu, A.L.; Kim, S.M.; Kim, K.K.; Yang, H.Y.; Li, L.J.; et al. van der Waals Epitaxy of MoS2 Layers Using Graphene As Growth Templates. Nano Lett. 2012, 12, 2784–2791. [Google Scholar] [CrossRef]
- Wang, Q.; Cui, X.; Guan, W.; Zhang, L.; Fan, X.; Shi, Z.; Zheng, W. Shape-dependent catalytic activity of oxygen reduction reaction (ORR) on silver nanodecahedra and nanocubes. J. Power Sources 2014, 269, 152–157. [Google Scholar] [CrossRef]
- Jones, T.E.; Rocha, T.C.R.; Knop-Gericke, A.; Stampfl, C.; Schlögl, R.; Piccinin, S. Insights into the Electronic Structure of the Oxygen Species Active in Alkene Epoxidation on Silver. ACS Catal. 2015, 5, 5846–5850. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, H.; Wang, J.; Yang, S.; Liu, T.; Tao, K.; Chang, H. A silver wire aerogel promotes hydrogen peroxide reduction for fuel cells and electrochemical sensors. J. Mater. Chem. A 2019, 7, 11497–11505. [Google Scholar] [CrossRef]
- Zhang, N.; Chen, F.; Liu, D.; Xia, Z. Electrochemical Oxygen Reduction Reaction in Alkaline Solution at a Low Overpotential on (220)-Textured Ag Surface. ACS Appl. Energy Mater. 2018, 1, 4385–4394. [Google Scholar] [CrossRef]
- White, J.J.; Liu, J.; Hinsch, J.J.; Wang, Y. Theoretical understanding of the properties of stepped iron surfaces with van der Waals interaction corrections. Phys. Chem. Chem. Phys. 2021, 23, 2649–2657. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, J.; Su, G.; Ferri, N.; Liu, W.; Tkatchenko, A. Tuning the work function of stepped metal surfaces by adsorption of organic molecules. J. Phys. Condens. Matter 2017, 29, 204001. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; White, M.G.; Liu, P. Oxygen Reduction Reaction on Ag(111) in Alkaline Solution: A Combined Density Functional Theory and Kinetic Monte Carlo Study. ChemCatChem 2018, 10, 540–549. [Google Scholar] [CrossRef]
- Andryushechkin, B.V.; Shevlyuga, V.M.; Pavlova, T.V.; Zhidomirov, G.M.; Eltsov, K.N. Adsorption of molecular oxygen on the Ag(111) surface: A combined temperature-programmed desorption and scanning tunneling microscopy study. J. Chem. Phys. 2018, 148, 244702. [Google Scholar] [CrossRef]
- Raukema, A.; Butler, D.A.; Box, F.M.A.; Kleyn, A.W. Dissociative and non-dissociative sticking of O2 at the Ag(111) surface. Surf. Sci. 1995, 347, 151–168. [Google Scholar] [CrossRef]
- Xu, Y.; Greeley, J.; Mavrikakis, M. Effect of Subsurface Oxygen on the Reactivity of the Ag(111) Surface. J. Am. Chem. Soc. 2005, 127, 12823–12827. [Google Scholar] [CrossRef]
- Montemore, M.M.; Spronsen, M.A.V.; Madix, R.J.; Friend, C.M. O2 Activation by Metal Surfaces: Implications for Bonding and Reactivity on Heterogeneous Catalysts. Chem. Rev. 2018, 118, 2816–2862. [Google Scholar] [CrossRef]
- Chen, Z.; Li, J.; Zeng, X.C. Unraveling Oxygen Evolution in Li-Rich Oxides: A Unified Modeling of the Intermediate Peroxo/Superoxo-like Dimers. J. Am. Chem. Soc. 2019, 141, 10751–10759. [Google Scholar] [CrossRef]
- Wasileski, S.A.; Janik, M.J. A first-principles study of molecular oxygen dissociation at an electrode surface: A comparison of potential variation and coadsorption effects. Phys. Chem. Chem. Phys. 2008, 10, 3613–3627. [Google Scholar] [CrossRef]
- Otani, M. Electrocatalysis: Theory and experiment at the interface. Phys. Chem. Chem. Phys. 2008, 10, 3607–3608. [Google Scholar]
- Polo, V.; Kraka, E.; Cremer, D. Electron correlation and the self-interaction error of density functional theory. Mol. Phys. 2002, 100, 1771–1790. [Google Scholar] [CrossRef]
- Bao, J.L.; Gagliardi, L.; Truhlar, D.G. Self-Interaction Error in Density Functional Theory: An Appraisal. J. Phys. Chem. Lett. 2018, 9, 2353–2358. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Huang, Z.-Q.; Zhang, Y.; Chang, C.-R. Trends in water-promoted oxygen dissociation on the transition metal surfaces from first principles. Phys. Chem. Chem. Phys. 2017, 19, 2364–2371. [Google Scholar] [CrossRef]
- Sargeant, E.; Illas, F.; Rodríguez, P.; Calle-Vallejo, F. Importance of the gas-phase error correction for O2 when using DFT to model the oxygen reduction and evolution reactions. J. Electroanal. Chem. 2021, 896, 115178–115184. [Google Scholar] [CrossRef]
- Tesch, R.; Kowalski, P.M.; Eikerling, M.H. Properties of the Pt(111)/electrolyte electrochemical interface studied with a hybrid DFT–solvation approach. J. Phys. Condens. Matter 2021, 33, 1. [Google Scholar] [CrossRef]
- Sun, J.; Ruzsinszky, A.; Perdew, J.P. Strongly Constrained and Appropriately Normed Semilocal Density Functional. Phys. Rev. Lett. 2015, 115, 036402–036408. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Yang, Z.-H.; Perdew, J.P.; Sun, J. Versatile van der Waals Density Functional Based on a Meta-Generalized Gradient Approximation. Phys. Rev. X 2016, 6, 041005. [Google Scholar] [CrossRef] [Green Version]
- Sharkas, K.; Wagle, K.; Santra, B.; Akter, S.; Zope, R.R.; Baruah, T.; Jackson, K.A.; Perdew, J.P.; Peralta, J.E. Self-interaction error overbinds water clusters but cancels in structural energy differences. Proc. Natl. Acad. Sci. USA 2020, 117, 11283–11288. [Google Scholar] [CrossRef]
- Kamrani Moghaddam, L.; Ramezani Paschepari, S.; Zaimy, M.A.; Abdalaian, A.; Jebali, A. The inhibition of epidermal growth factor receptor signaling by hexagonal selenium nanoparticles modified by SiRNA. Cancer Gene Ther. 2016, 23, 321–326. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jónsson, H. Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Na My, O.; Xuan Huynh, N.T.; Thi, P.T.; Chihaia, V.; Ngoc Son, D. Mechanism and activity of the oxygen reduction reaction on WTe2 transition metal dichalcogenide with Te vacancy. RSC Adv. 2020, 10, 8460–8469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linge, J.M.; Erikson, H.; Merisalu, M.; Sammelselg, V.; Tammeveski, K. Oxygen reduction on silver catalysts electrodeposited on various nanocarbon supports. SN Appl. Sci. 2021, 3, 263. [Google Scholar] [CrossRef]
- Li, B.; Gao, W.; Jiang, Q. Electronic and geometric determinants of adsorption: Fundamentals and applications. J. Phys. Conf. Energy 2021, 3, 022001. [Google Scholar] [CrossRef]
- Calle-Vallejo, F.; Tymoczko, J.; Colic, V.; Vu, Q.H.; Pohl, M.D.; Morgenstern, K.; Loffreda, D.; Sautet, P.; Schuhmann, W.; Bandarenka, A.S. Finding optimal surface sites on heterogeneous catalysts by counting nearest neighbors. Science 2015, 350, 185–189. [Google Scholar] [CrossRef]
- Nanba, Y.; Koyama, M. An Element-Based Generalized Coordination Number for Predicting the Oxygen Binding Energy on Pt3M (M = Co, Ni, or Cu) Alloy Nanoparticles. ACS Omega 2021, 6, 3218–3226. [Google Scholar] [CrossRef]
- Ge, X.; Sumboja, A.; Wuu, D.; An, T.; An, B.; Goh, F.W.T.; Hor, T.S.A.; Zong, Y.; Liu, Z. Oxygen Reduction in Alkaline Media: From Mechanisms to Recent Advances of Catalysts. ACS Catal. 2015, 5, 4643–4667. [Google Scholar] [CrossRef]
- Linge, J.M.; Erikson, H.; Kozlova, J.; Aruväli, J.; Sammelselg, V.; Tammeveski, K. Oxygen reduction on electrodeposited silver catalysts in alkaline solution. J. Solid State Electrochem. 2018, 22, 81–89. [Google Scholar] [CrossRef]
- Liu, J.; Wang, Y. Theory-experimental gap. In Multiscale Modeling of Electrochemical Reactions and Processes; Wang, Y., Ed.; AIP Publishing: Melville, NY, USA, 2021; pp. 1-1–1-14. [Google Scholar]
- Wang, Y. Numerical simulation of electrified solid-liquid interface. In Multiscale Modeling of Electrochemical Reactions and Processes; Wang, Y., Ed.; AIP Publishing: Melville, NY, USA, 2021; pp. 3-1–3-18. [Google Scholar]
- Wang, Y.; Liu, X.; Liu, J.; Al-Mamun, M.; Liew, A.W.; Yin, H.; Wen, W.; Zhong, Y.L.; Liu, P.; Zhao, H. Electrolyte Effect on Electrocatalytic Hydrogen Evolution Performance of One-Dimensional Cobalt–Dithiolene Metal–Organic Frameworks: A Theoretical Perspective. ACS Appl. Energy Mater. 2018, 1, 1688–1694. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Blochl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Deringer, V.L.; Tchougréeff, A.L.; Dronskowski, R. Crystal orbital Hamilton population (COHP) analysis as projected from plane-wave basis sets. J. Phys. Chem. A 2011, 115, 5461–5467. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jonsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy path. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef] [Green Version]
- Nørskov, J.K.; Bligaard, T.; Logadottir, A.; Kitchin, J.R.; Chen, J.G.; Pandelov, S.; Stimming, U. Trends in the Exchange Current for Hydrogen Evolution. J. Electrochem. Soc. 2005, 152, J23. [Google Scholar] [CrossRef] [Green Version]
- Suen, N.T.; Hung, S.F.; Quan, Q.; Zhang, N.; Xu, Y.J.; Chen, H.M. Electrocatalysis for the oxygen evolution reaction: Recent development and future perspective. Chem. Soc. Rev. 2017, 46, 337–365. [Google Scholar] [CrossRef]
Figure 1.
(a) Diagram of the (322) surface from the side to visualize the step and differing coordination numbers (CN means coordination number). (b) DOS of the 4d state in differing CN Ag atoms on the Ag(322) surface.
Figure 1.
(a) Diagram of the (322) surface from the side to visualize the step and differing coordination numbers (CN means coordination number). (b) DOS of the 4d state in differing CN Ag atoms on the Ag(322) surface.

Figure 2.
Top and side view of the preferred adsorption sites of molecular and atomic oxygen on the Ag(322) surface. (a,b) The atomic adsorption at the step-hollow (SH) and hollow (H) sites, respectively. (c,d) The molecular adsorption at the bridge step-hollow (bSH) and fcc step-hollow (fSH) sites, respectively. Color code: silver; Ag, red; O. The red dashed line represents the location of the steps in the top view.
Figure 2.
Top and side view of the preferred adsorption sites of molecular and atomic oxygen on the Ag(322) surface. (a,b) The atomic adsorption at the step-hollow (SH) and hollow (H) sites, respectively. (c,d) The molecular adsorption at the bridge step-hollow (bSH) and fcc step-hollow (fSH) sites, respectively. Color code: silver; Ag, red; O. The red dashed line represents the location of the steps in the top view.

Figure 3.
PDOS of O2 adsorption on the Ag(322) surface at the fSH site (a) and bSH site (b). -pCOHP of O-O at fSH (c) and bSH (d) site.
Figure 3.
PDOS of O2 adsorption on the Ag(322) surface at the fSH site (a) and bSH site (b). -pCOHP of O-O at fSH (c) and bSH (d) site.

Figure 4.
O2 dissociation pathways on the (322) surface starting from the O2 adsorption at the fSH site (left) or bSH site (right). Color code: silver; Ag, red; O. The red dashed line represents the location of the steps in the top view.
Figure 4.
O2 dissociation pathways on the (322) surface starting from the O2 adsorption at the fSH site (left) or bSH site (right). Color code: silver; Ag, red; O. The red dashed line represents the location of the steps in the top view.

Figure 5.
Adsorption sites of O, OH, and OOH on Ag(111) and Ag(322). Color code: silver; Ag, red; O, white; H. The red dashed line represents the location of the steps in the top view.
Figure 5.
Adsorption sites of O, OH, and OOH on Ag(111) and Ag(322). Color code: silver; Ag, red; O, white; H. The red dashed line represents the location of the steps in the top view.

Figure 6.
Gibbs free energy diagrams of ORR activity on the Ag(322) and Ag(111) surface. (a,b) show the associative free energy for (322) and (111), respectively. (c,d) show the dissociative free energy for (322) and (111), respectively. η is the overpotential for the respective pathway. Blue is where U = 0 V and red is where U = 1.23 V.
Figure 6.
Gibbs free energy diagrams of ORR activity on the Ag(322) and Ag(111) surface. (a,b) show the associative free energy for (322) and (111), respectively. (c,d) show the dissociative free energy for (322) and (111), respectively. η is the overpotential for the respective pathway. Blue is where U = 0 V and red is where U = 1.23 V.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Comparison of O* and O2* adsorption properties on different silver surfaces. Ads stands for adsorption, Eads is adsorption energy, dO-Ag is the average O-Ag bond length, vO-O is the vibrational frequency of the O-O bond, and m is the magnetic moment.
Table 1.
Comparison of O* and O2* adsorption properties on different silver surfaces. Ads stands for adsorption, Eads is adsorption energy, dO-Ag is the average O-Ag bond length, vO-O is the vibrational frequency of the O-O bond, and m is the magnetic moment.
| Absorbate | Ads Site | Eads (eV) | dO-Ag (Å) | vO-O (cm−1) | REF | ||
|---|---|---|---|---|---|---|---|
| 322 | O | SH | −4.40 | 2.24 | n/a | 0 | This work |
| H | −3.02 | 2.13 | n/a | 0 | |||
| O2 | fSH | −0.70 | 2.37 | 668 | 0 | ||
| bSH | −0.27 | 2.36 | 762 | 0 | |||
| 111 | O | fcc | −4.05 | 2.10 | n/a | 0 | [8] |
| O2 | fH | −0.38 | 2.22 | 827 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hinsch, J.J.; Liu, J.; White, J.J.; Wang, Y. The Role of Steps on Silver Nanoparticles in Electrocatalytic Oxygen Reduction. Catalysts 2022, 12, 576. https://doi.org/10.3390/catal12060576
AMA Style
Hinsch JJ, Liu J, White JJ, Wang Y. The Role of Steps on Silver Nanoparticles in Electrocatalytic Oxygen Reduction. Catalysts. 2022; 12(6):576. https://doi.org/10.3390/catal12060576
Chicago/Turabian StyleHinsch, Jack Jon, Junxian Liu, Jessica Jein White, and Yun Wang. 2022. "The Role of Steps on Silver Nanoparticles in Electrocatalytic Oxygen Reduction" Catalysts 12, no. 6: 576. https://doi.org/10.3390/catal12060576
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.