
Quo Vadis Dry Reforming of Methane?—A Review on Its Chemical, Environmental, and Industrial Prospects


1
Department of Inorganic Chemistry, Fritz-Haber-Institut der Max-Planck-Gesellschaft, 14195 Berlin, Germany
2
Department of Heterogeneous Reactions, Max Planck Institute for Chemical Energy Conversion, 45470 Mülheim an der Ruhr, Germany
*
Authors to whom correspondence should be addressed.
Catalysts 2022, 12(5), 465; https://doi.org/10.3390/catal12050465
Submission received: 10 February 2022
/
Revised: 13 April 2022
/
Accepted: 14 April 2022
/
Published: 22 April 2022
(This article belongs to the Special Issue Current Trends in Dry (CO2) Reforming Catalysis)
Abstract
:In recent years, the catalytic dry reforming of methane (DRM) has increasingly come into academic focus. The interesting aspect of this reaction is seemingly the conversion of CO2 and methane, two greenhouse gases, into a valuable synthesis gas (syngas) mixture with an otherwise unachievable but industrially relevant H2/CO ratio of one. In a possible scenario, the chemical conversion of CO2 and CH4 to syngas could be used in consecutive reactions to produce synthetic fuels, with combustion to harness the stored energy. Although the educts of DRM suggest a superior impact of this reaction to mitigate global warming, its potential as a chemical energy converter and greenhouse gas absorber has still to be elucidated. In this review article, we will provide insights into the industrial maturity of this reaction and critically discuss its applicability as a cornerstone in the energy transition. We derive these insights from assessing the current state of research and knowledge on DRM. We conclude that the entire industrial process of syngas production from two greenhouse gases, including heating with current technologies, releases at least 1.23 moles of CO2 per mol of CO2 converted in the catalytic reaction. Furthermore, we show that synthetic fuels derived from this reaction exhibit a negative carbon dioxide capturing efficiency which is similar to burning methane directly in the air. We also outline potential applications and introduce prospective technologies toward a net-zero CO2 strategy based on DRM.
1. Introduction
The combination of two potent greenhouse gases in a chemical transformation, converting them into the universal feedstock synthesis gas (syngas), is a highly attractive motivation for studying the performance and function of processes enabling the dry reforming of methane (DRM). The resulting syngas, a mixture of H2 and CO, can be refined in consecutive catalytic downstream processes into energy storage molecules, i.e., synthetic fuels [1,2], or valuable everyday products, such as fine chemicals or plastics [3]. In the past 25 years, the number of annual publications on DRM that can be found in Scopus and Web of Science has increased from 2 to over 500 [4]. There are enormous estimates in the literature of the potential of this reaction to contribute to the challenge of climate change mitigation and stabilize the feedstock for the future chemical industry. Therefore, we consider some comments on the potential appropriate to manage the expectations of DRM, which represents an option with yet limited, but increasing industrial practice.
The collection of CO2 from unavoidable sources and its conversion into chemicals is part of the technology portfolio called carbon capture and utilization (CCU) [5]. The savings potential in greenhouse gas emissions is not in the lifetime of the product made from this carbon but only in the fossil carbon saved. It is, thus, replacing a fossil-based chemical with one that uses carbon atoms from the collected CO2. Only if fully renewable energy sources are used for the entire process chain, including collection, purification, conversion, and heating, of the initial CO2, the savings potential is 50%. The actual CCU savings potential is likely to be less than 50%, as it will be almost impossible to realize energy utilization strictly based on renewable technologies in the coming decades. This includes the production of H2 likely to be used for chemical reduction, all process energies, and all materials used in the process chain. The situation is different when CO2 and/or methane source is of biological origin (biogas) or when the CO2 is extracted from ambient air (Direct Air Capture, DAC) [6]. It is important to distinguish here between stationary sources (power plants, factories, etc.) and mobile sources (cars, airplanes, etc.) of CO2. At present, there are no practical solutions for on-board capture directly from mobile sources; therefore, the current focus aims at capturing CO2 from stationary sources [7].
It is crucial to critically assess the state of research as well as the feasibility of DRM in the context of other catalytic CO2 processing technologies in order to understand how mature this reaction is and to what extent it will make a significant contribution to the energy transition and CCU on a global scale.
As useful as DRM may look in the family of CO2 utilization reactions, its realistic greenhouse gas savings potential is limited, especially given the high energy requirements for the overall endothermic reaction. Nonetheless, DRM is a viable option for the production of carbon-rich syngas. Such a gas can be used as a feedstock for chemical conversions. The syngas obtained could also serve as a working gas for future carbothermal reduction of iron oxide instead of coal and stoichiometric natural gas if low-carbon hydrogen is used as an external reducing agent. This would allow effective conversion of iron oxide to iron metal and overcome the limitations of the iron–oxygen–hydrogen phase diagram [8].
In order to attain an idea of the maximum tonnage that would be generated by such applications based on potential DRM processes, it is instructive to study some key figures. The chemical industry of the European Union is number two after China with a 15% world market share and would require a total of 300 million ta−1 of CO2 in 2050 for all chemical processes except the synthesis of green fuels. In order to sustain such a CO2-based chemical industry, an additional 250 million ta−1 of biomass and a total of 4900 TWha−1 of green energy are needed. These values come from the DECHEMA projection of a greenhouse gas neutral chemical industry in 2017 [9]. According to a very rough estimate, a total of 2 gta−1 greenhouse gas (CO2 plus methane) could be absorbed by the global chemical industry as an absolute maximum. This figure would increase by a factor if synthetic fuels were used for transportation, which is currently unclear.
To relate these numbers to the challenge of addressing global greenhouse gas emissions, Table 1 lists some values up to 2016. Total greenhouse gas emissions on Earth were 49.4 gt [10]. The table includes relative and absolute values for emission sources that are distributed regionally in weights quite different from this global average [11]. The report from UN Environment Program has shown an increase to 58 gt for 2019 [12].
The listed values show that the maximum use of DRM-derived syngas by the chemical industry is in the range of landfill emissions and emissions from cement production, two sources of unavoidable greenhouse gas emissions [13]. The contribution of DRM to the global greenhouse gas emissions problem is in the range of 5%. However, its relevance is far greater, as any future chemical industry will require a stable carbon supply independent of fossil feedstocks, and here DRM with biogenic waste origins may have a future if combined with CO2-lean hydrogen and/or DAC. From this point of view, it seems appropriate and justified to explore the scientific evidence as a basis for future and current process considerations. DRM is an option if, in future energy systems, the supply of the chemical industry with non-fossil raw materials has to be ensured. The fact that this reaction is a high-temperature process is advantageous because the resulting syngas is purified of many impurities, including biogenic provenance, and can be integrated into chemical complexes with favorable thermal management structures. Since DRM is only feasible with enormous amounts of CO2-free energy, its strategic but quantitatively limited relevance in the energy issue is only realistic in the distant future, when the application fields for green energy are saturated.
In the meantime, technology development can be driven by hybrid processes using partly fossil, partly non-fossil feedstocks and increasingly non-fossil energy sources. One can define two development goals: (i) small-scale and decentralized units with robust operations for biogenic feedstocks and waste sources providing mainly CO, and (ii) large-scale efficiency-optimized processes combined with large-scale CO2 or methane sources. DRM will always target competing processes of CO2 hydrogenation with either pure CO or oxygenated molecules. Thus, DRM is a member of the family of CO2 upgrading reactions. This also becomes clear when we discuss the C1-chemistry reaction network in which DRM is embedded.
Here, we aim at stimulating the debate on DRM and at understanding its relatively slow integration compared to well-established reforming processes such as partial oxidation or steam reforming. As such, we review the current reactor systems designed to achieve high DRM efficiency and outline its impact on removing CO2 from the atmosphere when applied for energy-related technologies based on stoichiometric equations. In this review, we summarize the abundant information on DRM, which has been addressed more comprehensively in the past [3,14,15,16,17], with the aim to rate the potential of this reaction in large-scale applications for a climate-change scenario. This has been suggested as the future niche of DRM by a variety of scientific publications on this topic. Our literature assessment demonstrates that DRM technologies have seemingly reached the level of maturity which is necessary for large-scale applications. To underlie our hypothesis with scientific arguments and to fuel the discussion, we have structured the article hierarchically:
First, we will discuss the educts and introduce the C1 chemistry in order to provide information on how different reactions are interconnected. Second, we present two different catalytic CO2 valorization technologies. Third, we will focus on the complexity of methane reforming and outline important thermodynamic and kinetic aspects of DRM. Forth, we discuss the deactivation of DRM reaction in thermocatalytic systems. Fifth, we highlight the current state of research in the reaction process and the need for operando studies in order to disclose the complex C1 network, which can be assessed by DRM. Sixth, the current state of industrial implementation is discussed. We conclude with a critical assessment of the capabilities of DRM.
2. The Educts and C1 Chemistry
2.1. CO2
The mitigation of anthropogenic greenhouse gas emissions is a central topic in many scientific and societal discussions. With annually increasing emissions of over 49.4 gt [10], CO2 is the main gas responsible for the greenhouse effect in the atmosphere. Moreover, CO2 is also related to the acidification of the oceans, which directly affects the biochemistry of marine organisms. The high levels of CO2 emission may pose a high risk for any aerobic life, including mammals, on our planet.
CO2 emission originates from various anthropogenic and natural sources [18]. In this context, flaring of natural gas waste in oil extraction, the production of cement clinker from limestones, non-energy applications of fuels, for instance, in the form of solvents, and several small processes have been identified as important sources of waste CO2 emissions [18,19]. By far, the largest anthropogenic contributors of CO2 emissions to the atmosphere are the combustion of fossil fuels for power generation [18] and the traditional production of steel from the refinements of ore with coal [20,21].
The capture and reduction of atmospheric and emitting CO2 have been developed into a scientific challenge and fueled various discussions on what to do with the large volumes of available CO2 since its industrial applications are limited [22,23,24,25]. One strategy is to extend the lifetime of carbon in CO2 by chemical transformation into value-added compounds, which can be used to synthesize daily life products, including synthetic fuels. This transformation involves molecules with one C atom (C1), such as formic acid, methanol, methane, and carbon monoxide. Moreover, chemicals of two or more C atoms (C2, C3, etc.) have recently been targeted as the products of CO2 valorization due to their higher market price and higher energy densities compared to C1 compounds [26,27].
Within the vast family of organic chemicals, CO2 reflects the most oxidized and stable form of C1 molecules (Figure 1). CO2 is at the lowest end of the free energy scale and can be considered a thermodynamic sink for carbon compounds. Hence, its activation requires high energy input. The entries in Figure 1 exemplify the Gibbs free energies (ΔGf) for the formation of C1 compounds and their relation to hydrogen, water, and ammonia as representatives of important inorganic molecules relevant to energy conversion [3]. A good analogy of CO2 valorization is the photosynthesis of carbohydrates in photoautotroph organisms [28,29]. The carbon of CO2 is stored in complex chemicals in a reaction cycle that can be interpreted as the inverse of combustion. The energy itself extracted from sunlight by the organism is stored as chemical energy in the form of sugars and starches. However, photosynthesis is a comparatively slow process regarding the large and global scale capture, cycling, storage, and valorization of CO2.
The general functioning of any CO2 valorization strategy relies on transiting upward the energy scales represented in Figure 1, where the amount of involved CO2 is a good indicator of the efficiency of the involved processes. For large-scale applications, the transit through the energy scale is achieved by any chemical reaction of CO2 with partners located at the highest steps of the energy scale (Figure 1, Table 2) and may be accelerated by the presence of heterogeneous catalysts. Suitable reaction partners, such as H2 and CH4, can convert CO2 from the lower end of the energy scale into C1 compounds with intermediate values of ΔGf, such as methanol, formic acid, and carbon monoxide, or even enable Cx chemistry. Overall, energy is stored, which can be released after burning in an oxidative environment or utilized in organic synthesis.
2.2. CH4
Natural gas and associated methane (CH4) is currently one of the cheapest energy sources available on the planet [14,30]. Since most of the world’s natural gas is used to generate heat and electricity by combustion, it is unavoidable to produce large amounts of CO2 when CH4 releases the stored energy. Clearly, finding new technologies for power generation apart from burning fossil fuels would help to remedy current trends of gas emissions. However, because CO2 and natural gas are currently largely available, the conditions are thus favorable for the emergence of technologies that integrate these gases as feedstocks in production chains that have the potential to store and recycle the carbon which would otherwise escape into the atmosphere.
2.3. Industrial C1 Chemistry
CO2 and CH4 form the basis of chemistry that deals with the conversion of compounds with a carbon atom (C1). A complex reaction network allows the catalytic production of valuable basic chemicals or synthetic fuels in direct or downstream processes via CO and methanol (Figure 2). C1 chemistry is intended as an alternative approach to achieving fuels other than refining fossil matter. In the following, this C1 chemistry and its interdependencies will be discussed in the context of methane reforming. Detailed insights into the complexity of the C1 reaction network are presented in various review articles [31,32,33]. The salient feature of Figure 2 is the cyclic nature of the carbon stream. Although a large number of chemicals may be produced and cycled in the production chain, whenever the molecules are burnt for power generation, the carbon content will be transformed into CO2. It remains, for instance, questionable whether the generation of synthetic liquid fuels from CO2, even with an arbitrary efficiency, will solve the problem of greenhouse gas emission. Furthermore, the figure also exemplifies that the other major sink of the carbon stream is plastics, which constitute the second largest environmental pollutant of concern. In general, decarbonisation of the power generation sector seems to be a relevant action to deal with the current pollution.
As shown in Figure 1, the involved chemicals have different equilibrium positions on the energy hypersurface and thus different reactivities and stabilities. These different stabilities significantly complicate coordinated reaction control and management in view of further catalytic reactions in downstream processes. In addition to the different stabilities, different dissociation energies of the chemical bonds also influence the activation of C1 molecules and thus the reaction control, as shown in Table 3. For example, methane has one of the strongest aliphatic C-H bonds (439.3 kJmol−1) [34], so overoxidation to CO2 (for oxidative activation) or deep dehydrogenation to C (for reductive activation) often cannot be avoided [31]. This significantly affects the selectivity distribution of the products.
2.3.1. CH4 Activation
Methane can be activated in two different ways: (i) oxidative or (ii) non-oxidative [31]. Oxidative activation of methane is obtained via the partial oxidation of methane (POM) and the steam reforming of methane (SRM), which lead to oxygenates and syngas [35,36], and the oxidative coupling of methane (OCM) which results in ethane or ethylene [37]. However, both oxidation reactions are affected by overoxidation due to the higher oxidation potential of the products to CO2. Non-oxidative activation processes of methane, such as methane dehydroaromatization (MDA) or methane to olefins, aromatics, and hydrogen (MTOAH), require high temperatures and are deactivated by carbon deposition, which is a common drawback in dehydrogenation reactions of alkanes [38,39,40]. Non-oxidative coupling of methane (NOCM) can proceed selectively at low temperatures but is limited at these temperatures by the position of the reaction equilibrium to very small methane conversions [41].
Within the complex C1 network, CO and its conversion to methanol play a central role. CO is produced from the reforming of methane, where carbon formation can be (partially) suppressed. Using the example of DRM, we will see later that there are indeed catalysts that have already managed the balancing act of high activity and little to no deactivation. In addition to a gaseous and activated C1 product, the same reaction also has the advantage that, with suitable reaction control, no hydrogen is converted into water and can be directly used in downstream processes.
These advantages of DRM, together with the possibility to activate CO2, further motivate a deeper consideration of this reaction.
2.3.2. CO2 Activation
There is a limited number of ways of activating CO2, including urea synthesis by reaction with ammonia (Bosch-Meiser process), DRM to syngas, and direct hydrogenation [3,14,42,43,44,45]. DRM belongs to the reforming technologies intended for the production of syngas, i.e., mixtures of H2, H2O, CO, CO2, and CH4 of variable proportions [3,46]. We refer to syngas as any mixture of H2 and CO. The H2/CO ratio is simply referred to as the syngas ratio and, in reality, takes on any positive value. Syngas itself is the building block for many world-scale industrial processes (Figure 2), such as Fischer–Tropsch, carbonylation, hydroformylation and the synthesis of olefins [47,48,49]. DRM may ideally deliver a product of higher CO contents than is the case for other reforming reactions [3].
On the other hand, CO2 hydrogenation refers to a family of reactions of CO2 with H2 leading directly to a family of functionalized hydrocarbons, including C1 (methanol, CO, formic acid, formaldehyde) and C2+ products such as dimethylether (DME), olefins, liquid fuels and higher alcohols [22,50]. On closer inspection, CO2 hydrogenation can be discussed as process variants of the conversion of a particular syngas mixture. This fact links several CO2 hydrogenation reactions, such as the reverse-water gas shift and the direct formation of formic acid and methanol synthesis, to a common chemical origin where the H2 stoichiometry used to activate the CO2 molecules largely determines the nature of the dominant process.
3. The Reaction Network of C1
3.1. Reforming of Methane
During reforming (Equations (1)–(3)), CH4 is oxidized to carbon monoxide (CO), producing hydrogen (H2) at the same time [14,49]. The syngas ratio varies depending on the type of oxidant used. DRM (Equation (1)) uses CO2 as the oxidant and generates syngas with a ratio of one. Steam reforming of methane (SRM Equation (2)) uses H2O and produces syngas with a ratio of three. For the partial oxidation of methane (POM Equation (3)), O2 is used, and syngas with a ratio of two is obtained. Simultaneously with the syngas production, several side reactions (Equations (4)–(10)) occur, which influence the product ratio or may lead to coking and subsequent catalytic deactivation. Note, the reactions of CO and CO2 hydrogenation (Equations (4), (7)–(10)) reflect variants and intermediate steps of the syngas chemical network (Figure 2) [3]. From Equations (1)–(10), it can be concluded that hydrogenation reactions are mainly exothermic, while, with the exception of Equation (3), the activation of CH4 is endothermic.
CO2 + CH4 → 2CO + 2H2 ΔH298 = 247 kJ mol−1 Dry reforming
H2O + CH4 → CO + 3H2 ΔH298 = 206 kJ mol−1 Steam reforming
½ O2 + CH4 → CO + 2H2 ΔH298 = −44.2 kJ mol−1 Partial oxidation
2H2O + CH4 → CO2 + 4H2 ΔH298 = 169 kJ mol−1 Oxidation by steam/reverse methanation of CO2
2CO → CO2 + C ΔH298 = −172 kJ mol−1 Boudouard reaction
CH4 → C + 2H2 ΔH298 = 75 kJ mol−1 Methane pyrolysis
CO2 + H2 → CO + H2O ΔH298 = 41 kJ mol−1 CO2 Hydrogeneation: RWGS
CO2 + 2H2 → C + 2H2O ΔH298 = −90 kJ mol−1 CO2 Hydrogenation: decarbonation
CO + H2 → C + H2O ΔH298 = −131 kJ mol−1 CO Hydrogenation: decarbonation
CO2 + 3H2 → CH3OH + H2O ΔH298 = −49 kJ mol−1 CO2 Hydrogenation: methanol synthesis
The syngas which is generated by the different reforming routes is targeted to specific applications depending on the H2/CO ratio [14,51]. For instance, SRM (Equation (2)) favors syngas processes with high H2/CO ratios and particularly the H2 production. In addition, the syngas ratio obtained by SRM can be further increased by consecutive WGS (reverse Equation (7)) stages to obtain a hydrogen feedstock of relatively high purity (99.95 %) [52,53]. About 95% of the world’s hydrogen production is achieved by steam reforming of fossil feedstocks [54]. SRM is operated at 800–950 °C and 15–40 bar over Ni-based catalysts [55]. The steam/carbon ratio of the feed is held at above two to ensure that coke-free operation conditions are achieved through the gasification of carbon deposits (reverse of Equation (9)) and the mitigation of coke precursors in the gas phase [56]. This is perhaps the key difference between SRM and DRM processes. Apart from their endothermicities, the constant supply of steam in SRM inhibits the catalytic deactivation by coking.
POM (Equation (3)), on the other hand, is exothermic and can be executed without a catalyst [3]. Industrially, a syngas of ratio two is achieved at 1150–1500 °C and 25–80 bar in oxygen-lean conditions using high purity O2 as a feed [3]. By applying a catalyst (Rh, Ru, Pt, or Ni) the reaction temperature can be lowered to 900–1000 °C. A further decrease in the process temperatures to 500–600 °C at pressures below 5 bar seems possible [57], although this area is still under development. The produced syngas ratio is suitable for many downstream processes such as methanol synthesis [2,58] and Fischer–Tropsch for the preparation of liquid fuels.
Autothermal reforming (ATR) is another technology that aims at the exothermic oxidation of methane with oxygen [59]. However, the utilization of pure oxygen comes with a share of technical and economic problems, so industrial ATR systems are usually operated with a mixture of oxygen, steam, and/or CO2 with methane [14]. Hence, the endothermic and exothermic paths of syngas generation are combined [59,60,61]. Even though the production of syngas is thermodynamically favored for a system that would use only oxygen, the addition of CO2 or H2O to the feed is a common practice that also impacts the H2/CO ratio and allows adaptation of the reformer into many downstream processes such as ammonia and methanol synthesis. Nevertheless, ATR systems are unable to meet the benefit of DRM regarding its unique ability to produce syngas with a ratio close to or below one [14].
Although DRM shares common features with POM and SRM, it poses important challenges on its own. In this sense, DRM has traditionally been considered in its technological infancy compared to the other reforming reactions. Conceptually, the reaction is based on the idea of substituting water in SRM or oxygen in POM with CO2. The potentialities of integrating CO2 into the production chain and cycle the carbon contents are attractive from academic, industrial, and environmental perspectives. The immediate result of DRM compared to POM and SRM is the increase in the CO content in the produced syngas. This unique feature could also promote the implementation of DRM in large-scale processes since the production of syngas with a low H2/CO ratio would be much more difficult to achieve energetically and economically, taking into account the other reforming reactions. Some processes would exclusively require or would largely benefit from the availability of syngas with an elevated CO content ideally composed of a product ratio of one [3,62]. Examples are the direct production of acetic acid (Equation (11)), the synthesis of dimethylether (Equation (12)), the hydroformylation of olefins (Equation (13)), and the Fischer–Tropsch reaction to olefins.
2CO + 2H2 → CH3COOH ΔH298 = −435.0 kJ mol−1 Acetic acid synthesis
3CO+ 3H2 → CH3OCH3 + CO2 Dimethylether synthesis
R-CH=CH2 + CO + H2 → R-CH2-CH2-CHO Hydroformylation
Since the invention of steam reforming in 1912 [63,64], similarities of the three main reforming reactions in terms of mechanisms, deactivation pathways, and catalytic materials have been established [65]. Perhaps the most remarkable aspect of DRM compared to the other reforming reactions is its high tendency to deactivate due to the formation of coke. Coking is the direct consequence of the high C contents of the reaction feed and is often regarded as the main reason that DRM has not been developed until now on a broad front [3,42]. Another feature of DRM is the high energy investment which is the consequence of using the thermodynamic sink CO2 as a reactant [14]. Hence, the true potential of DRM has only recently been considered in-depth for associated catalytic technologies despite being under the radar for around 30 years [49]. Of course, the thermodynamic barrier is of high relevance because DRM is a highly energy-consuming technology and is unfavorable at low temperatures. Efficient large-scale DRM would involve temperatures above 800 °C for stoichiometric transformation in a single-step reactor. Moreover, the reaction should run at 10–40 bar to generate the syngas at pressures that would facilitate integration into conventional downstream processes. In addition, DRM requires a pure and steady source of CO2, which may not necessarily be available in all industrial facilities [3]. These drawbacks of DRM may explain its slow development into a large-scale technology comparable to SRM and POM.
A good point of comparison would be the SRM processes which are industrially mature. Efficient reformers for endothermic SRM consume about 25.5% of the energy value stored in the used methane [14]. At first glance, DRM should be more energy-efficient than SRM since it does not require the generation of steam. However, the high barrier to activating CO2 requires high reaction temperatures and may thus require a larger energy input compared to SRM. Additionally, the different routes of CO2 hydrogenation leading to CO, methanol, and hydrocarbons such as methane can be used for economic comparisons [66,67]. A study in 2016 assessed that the cost of methanol production in Germany by electrical power approaches varied between €608 and €1453 per ton. A pure wind-park-supplied scenario resulted in €1028–1067 per ton. These values may be compared with the production of methanol from syngas processes, which according to estimates of economic sensitivity factors, may cost €307 to €566 per ton [68,69] as the production and storage of H2 itself is the most costly step. The direct path of methanol synthesis from methane reforming is much cheaper due to the low cost of natural gas. Hitting the range of viable energy and economic investment could become a reality in the near horizon of DRM as new products and markets become available for the unique syngas achieved by the process. In addition, the implementation of cheap renewable energy sources [70] and taxation benefits for reducing CO2 emissions will encourage more companies to utilize DRM and associated processes for energy and CO2 storage [67]. Still, the energy investment remains one of the largest constrains of current DRM development due to its intrinsic thermodynamic needs.
3.2. Thermodynamics of DRM
It is of interest to understand the fundamental characteristics of any reaction process in terms of the relevant indicators of chemical performance. Prediction of the equilibrium product composition is useful for ascertaining the extent to which the reaction is complete, as well as its selectivity for a target product and its yield. In general, the thermodynamic information is required to decide when a given process is efficient or when it may be further improved. Note, thermodynamic descriptions of a catalytic reaction consider only the ideal case, in which poisoning or deactivation processes are absent. In reality, and in particular in reforming reactions, coking and sintering of the catalysts lead to deactivation, which limits their performances. Industrial SRM can be used as an example which is operated under somewhat thermodynamic unfavorable conditions in order to avoid coking.
Due to its high endothermicity (Equation (1)), high operating temperatures are needed for efficient catalytic DRM. High temperatures would also favor endothermic side reactions, such as the reverse water-gas shift reaction (Equation (7)), the decomposition of methane (Equation (6)), and the oxidation of methane by steam (Equation (4)). Exothermic reactions, such as the Boudouard reaction (Equation (5)) and the hydrogenation of CO (Equation (9)), would be more favorable at mild temperatures in the range between 400–600 °C. The complex chemical reaction network described in Equations (1)–(10) for methane reforming often involves the simultaneous and dominant occurrence of side reactions for operation temperatures below 600 °C. In this temperature regime, the product selectivity, catalytic activity, and long-term stability may be considered more representative of other reactions rather than of DRM itself. To simplify the description, we refer to the operational reaction temperature interval between 200–600 °C as “mild” and “high” for higher temperature values.
The contributions of the different reactions of the chemical network may be approached by the trends of the equilibrium compositions of the chemical species in the reaction environment. The equilibrium compositions may be retrieved by minimization of the chemical potentials (or Gibbs free energies) of the system. The calculation requires that the molar fractions of the components are positive and that the atomic species balance, as well as the cubic equation of state, are satisfied [71]. The results of this analysis exemplify some important characteristics of DRM for a selection of the most important thermodynamic conditions. Figure 3 shows, for instance, that the conversion of CH4 and CO2 occurs already in the mild temperature regime. For the stoichiometric feed mixture of CH4/CO2= 1, conversions in the range of 90% are thermodynamically predicted at 200 °C. Furthermore, thermodynamic calculations suggest that mainly water and carbon are formed at temperatures below 600 °C rather than syngas. It is apparent that the gas mixture of the educts is reactive even at this low temperature, but the question arises of whether this conversion should be considered true DRM activity. The yield of valuable syngas is still very low in this regime. With the increase in the reaction temperature from 200 to 400 °C, the selectivities towards water and carbon decrease while H2 production is evinced. CO is still only marginally formed in this regime, which leads to an overall syngas ratio much higher than the stoichiometric value of one. The dominance of carbon formation and small CO production indicates the fast occurrence of the Boudouard reaction. Hence, the “mild” temperature interval indicates the occurrence of multiple reactions characterized by low syngas production.
The transition into true DRM activity, i.e., the realization of a product with a syngas composition of one, occurs in the “high” temperature regime close to 800 °C. Coke and water formations are notably reduced, although not entirely suppressed above 800 °C. Some content of H2O is always expected in the products, even with true “dry” feeds. The fact that small amounts of water would always form during the reaction opened the possibility of co-feeding water for the gasification of part of the coke without altering the syngas ratio (Equation (9)). In general, Figure 3 shows that the value of the syngas ratio stabilizes above one, and more carbon is formed in the high-temperature regime when a more reducing feed ratio (CH4 > CO2) is applied. This result indicates that methane transformation may be the source of coking at this temperature interval, contrary to the Boudouard reaction characteristic for “milder” conditions.
Further important correlations have been described [73] to ascertain the influence of pressure variations on the reaction at values between 1–30 bar for a CO2/CH4 ratio of one. With increasing pressure (Figure 4a), carbon formation increases even at high temperatures. In addition, the syngas ratios approach one as the temperature increases for the entire pressure range (Figure 4b). As DRM ideally produces more gaseous molecules than initially present in the reaction, a reduction in pressure should favor conversion to the products at milder temperatures. Between 650 °C and 750 °C, an H2/CO ratio of two, which is representative of POM, can be achieved. CO formation is unfavorable at low temperatures compared to H2, which leads to higher H2/CO ratios. H2O and H2 formation follow opposed trends (Figure 4c,d), with increasing hydrogen yields at lower pressures. Further equilibrium calculations show that the conversion of methane with regard to different CO2/CH4 ratios at temperatures varying between 300 °C and 1100 °C should be almost complete for oxidizing feed ratios (CO2/CH4) of two and three at around 800 °C. However, high conversions cannot be obtained below 1000 °C when stoichiometric mixtures are used in the calculation. High operation pressures lead to a decrease in the reactant’s conversion and favor coking.
Further studies that vary the oxidant feed ratio complement this information. Conversion of methane is favored for oxidizing CO2/CH4 ratios of five at 750 °C. Increasing the temperature above 1000 °C allows for maintaining high conversions of CH4 even for stoichiometric CO2/CH4 ratios [74,75]. In general, these trends suggest that high conversions to syngas and representative H2/CO ratios are expected at the industrially relevant pressure corridor of 10–40 bar only at temperatures above 900 °C. Moreover, the production of syngas by DRM may be understood as the result of two competing processes, i.e., CO2 and CH4 activation. Coking may be considered unavoidable for a large set of operation conditions [56,76,77]. The effects on the reaction of increasing an external parameter while keeping constant the other ones are summarized in Table 4.
The thermodynamic analysis is an important step in the understanding of DRM and its consecutive associated reactions. Nevertheless, its scope is limited compared to the functioning of actual processes in the chemical industry. Minimizing the free energies of the system involves that the trends of Table 4 should be understood as the change of equilibrium compositions expected from an idealized reaction free of kinetic limitations. In reality, reactors may exhibit kinetic limitations at any length scale. Furthermore, reactions usually occur in the presence of a catalyst which is perhaps the most relevant element of current chemical technology. The traditional thermodynamic analysis does not inform about the catalyst, its evolution, its required structure, or its ability to achieve the reaction [78,79,80,81]. For industrial implementation, effective reaction design should also consider the stability of the catalyst, the tendency to sintering of the metallic nanoparticles, poisoning, diffusion effects, redox dynamics at the active sites, and gas flow conditions, just to name a few.
Note, this information pertains mostly to the dynamic functioning of catalysis, and hence its influence is difficult to predict from static models of the reaction conducted under equilibrium conditions. Nevertheless, the thermodynamic information, for instance, of the gaseous compositions, is vital to understanding the basic aspects of the reaction.
For instance, one of the most commonly used types of catalysts for DRM processes may be understood as a phase of a transition metal (Ni, Pt, Co, Fe, Ru, etc.) or its oxide supported on a temperature-resistant material (alumina, silica, zirconia, etc.) [14]. For DRM, the size of the supported particle may lead to results unforeseeable in Table 4. In a study tackling this question, catalysts of differing particle sizes were simulated [82]. In agreement with Table 4, the results showed that CH4 and CO2 conversions are maximized at low pressure and high temperature, while H2/CO ratios of one were achieved with increasing temperatures in the interval between 800–900 °C and 5–10 bar for a large range of particle sizes. In addition, carbon formation was found to increase with nanoparticle diameter in the range between 1 and 20 nm. In the temperature regime below 650 °C the main source of carbon formation was found to be due to Boudouard reaction (Equation (5)) while the decomposition of CH4 (Equation (6)) was more relevant at elevated temperatures. In both cases, carbon formation was severely hindered for particle sizes below 5 nm, which suggests some degree of cooperative effects between adjacent surface metal sites or even the need of a “bulk” metallic phase for the pathways of carbon formation.
3.3. Kinetics of Dry Reforming
The efficiency of DRM is a function of the catalyst architecture, including the type of active metal, size of the nanoparticles, the chemistry of the support, stability of the active phases, and operation conditions. As a consequence, there is not a simple way to describe its mechanism, and aspects such as the rate-determining step and dominant surface species may change among models and experimental approaches. We refer to the most published models, which may fit a large number of observations. One of these is built on the assumption that both CO2 and CH4 activate and recombine on the same surface (surface model). First, CO2 and CH4 decompose catalytically into CO*, O*, CHx*, C*, and H* species. The surface intermediaries further recombine as products of H2, CO, coke, and H2O. Kinetic models based on this kind of Langmuir–Hinshelwood mechanism [83,84] normally assume that the first dissociation of CH4 is the rate-determining step of DRM. Subsequent dehydrogenation reactions of CHx* would rapidly cascade into C* and H* species. Carbon formation would be the result of fast decarbonation rates of CHx* combined with slow oxidation rates of these species by surface O* [14,85,86,87] or by the reverse Boudouard reaction at mild temperatures.
From the product distribution of SRM, POM, and DRM measured with isotopically labeled molecules, it has been concluded that these reactions should share similar elementary steps in a large set of conditions [65,88]. Other approaches [89,90,91] have led to similar conclusions about the proposed reaction steps, which are summarized in the following equations:
CH4 + 2* → CH3* + H* dissociative adsorption
CHx* + * → CHx−1* + H* decarbonation/ deprotonation
CO2 + 2* → CO* + O* dissociative adsorption
H* + O* → OH* + * H* oxidation
CHx* + O* → COHx* + * CHx* oxidation
C* + O* → CO* + * C* oxidation
2H* → H2 + 2* H2 evolution
CO* → CO + * CO evolution
OH* + H* → H2O + 2* H2O evolution
SRM or POM reaction mechanisms could be derived by replacing CO2 in Equation (16) with H2O or O2, respectively. The mechanism implicitly assumes that the activation of reactants and further surface reactions occur at the same surface. However, particular catalyst architectures may favor the activation of CO2 and CH4 on distinct catalytic components. For instance, on oxidic supports exhibiting lattice oxygen vacancies, CO2 is thought to activate by the abstraction of oxygen by the vacancies (Equation (23)) while CH4 would dissociate preferentially on the metal nanoparticle. CO2 would thus refill the lattice vacancies, and the stored oxygen would later oxidize CHx* species which are generated at the nanoparticle. The oxidation of carbon (Equation (24)) would occur close to the metal–support interface. These Mars-van–Krevelen-like kinetic models have been found to hold in catalysts of Ni/CeO2-ZrO2, but they do not seem to apply for Ni/SiO2 catalysts where oxygen vacancies may not be prevalent.
CO2 + Ovacancy → Olattice + CO vacancy filling
C* + Olattice → CO + * + Ovacancy carbon oxidation/vacancy regeneration
The third type of mechanism considers that CO2 does not dissociate or that it can only dissociate with the assistance of other surface species such as H* or O* [92]. It was proposed that CO2 could react directly from the gas phase with surface methyl species in an Eley-Rideal-like mechanism. Furthermore, it has been shown [76] that dissociation of CO2 seems to be the preferred path on Ni-based catalysts, while Pt-based materials favor the adsorption of CO2 without dissociation. The reason for this behavior is the higher tendency of metallic Ni to form oxides compared to Pt and other noble metals such as Rh and Ir. On the latter, adsorbed CO2* would only dissociate when assisted by already existing C* or H* without forming O*. In a similar way, the presence of alkaline, alkaline earth, or rare-earth additives may favor the chemisorption of CO2 to form highly reactive carbonate-like species which subsequently react with carbon deposits originated from methane decomposition:
CO2 + * → CO2*
CO2* + C* → 2CO*
CO2* + H* → CO* +HO*
CO2 + MO → MCO3
There is currently no general agreement in the literature on crucial aspects of the DRM mechanism, especially in the identity of the rate-determining step. Although there is no reason that the rate-determining step would hold the same under the wide range of catalyst architectures and operation conditions of DRM execution, most studies point to the activation of CH4 by hydrogen abstraction (Equation (14)) as the most feasible candidate [65,88,93]. In a theoretical study about the mechanism of DRM and the identity of the active sites [94], it was suggested that the metallic atom (Ni) required at least two adjacent metallic sites to accommodate the intermediaries of CH4 decomposition. Afterward, produced C would give rise to carbide-like species, which may be oxidized in the presence of mobile O*, OH* or by interactions with gaseous oxidants such as steam or O2. The oxidation step of active carbon to CO would be rate-limiting in such a case. In a different study using transient isotopic methods, [95] the results suggested that there was no single rate-determining step in methane reforming over Ni catalysts and that the availability of surface oxygen may play a key role in determining the rate. In contrast, the activation of the first C–H bond has been suggested as the sole kinetically-relevant step for DRM on nickel catalysts [88]. Numerical studies based, for instance, on density functional theory calculations [96] suggested that a three-center C/H/Ni species forms during methyl adsorptions on Ni. In another study [97], the authors addressed the adsorption and dissociation of CH4 and CO2 on Ni(1 1 1), finding that the dissociative adsorption of CH4 is the rate-determining step and that the key intermediate is surface adsorbed CHO. The same authors also investigated the CO2 dissociation with both direct and H-mediated dissociation mechanisms and found that the dissociation of HCOO into CHO and O is unfavorable, while the direct dissociation into CO and O is much easier.
We summarize some of the features of DRM activation kinetics with the chemical scheme presented in Figure 5. Here, gaseous and surface species are linked by chemical transformations. For instance, the presence of CHx* and H* species is caused by the decomposition of CH4. Analogously, the formation of O* species relates to CO2. The relative position of the surface species in the scheme describes their average chemical compositions in a ternary C-O-H diagram. For instance, CO* is placed halfway between C* and O*. Langmuir–Hinshelwood-like kinetics are represented by black arrows with reaction steps as representatives of the surface model, such as dissociative adsorption of the reactants or C* oxidation by O* to give CO. The scheme suggests that inefficient rates of this step would lead to coke formation which may compromise the longevity of the catalyst. Hence, the required effect of the catalyst to produce syngas has to provoke methane cracking while promoting the oxidation of coke. Coking could also result from CO disproportionation as in the Boudouard reaction through CO* intermediaries that decompose into C* and O*.
In addition, reaction steps originated from the direct oxidation of C* species with gaseous H2O and CO2 are represented by red arrows. C* gasification with H2O would not alter the syngas ratio. Contrarily, the path of CH4 oxidation by surface O* (yellow arrow) increases the value of H2/CO above one and decreases the contents of oxides at the catalyst. In agreement with the observations about reforming kinetics, the scheme shows that DRM, SRM, and POM share common elementary steps (Equations (14)–(27)).
The relative contributions of the reaction steps of the scheme on syngas production depend on the operation conditions and catalyst design. A sign that the reaction is efficient would indicate that high conversions of methane and CO2 are reached as well as an H2/CO ratio of one. If the syngas ratio is larger than one, proportionally, more hydrogen is produced with respect to carbon monoxide. Since pure DRM only involves small water formation and the water-gas shift reaction does not occur in significant quantities at high temperatures, the missing CO is essentially related to the formation of soot or further surface species that diffuse into the catalyst bulk and remain stored in its structure. H2/CO ratios below one would as well be linked to carbon formation via CO2 hydrogenation. The occurrence of this family of reactions could be confirmed by unexpected high water formation [22,49].
4. Deactivation of DRM
The goal of current DRM catalyst design strategies is to achieve materials that sustain high conversion levels for long operation periods. The relatively low durability of catalysts has been claimed by several studies as the major impediment of DRM towards its transition into a large-scale technology. The catalyst may deactivate for several reasons. Coking has largely been discussed as the main source of deactivation, but other factors such as nanoparticle agglomeration (sintering), chemical transformation of the active phase, formation of inhibitors, migration of the nanoparticle into the support, support instabilities, or poisoning, are also important factors that need to be considered. In practice, the catalyst may deactivate by the occurrence of several of these processes simultaneously. We address the most significant sources of deactivation that have been reported in the literature: coking, sintering, and poisoning. For more detailed information about these topics, some comprehensive reviews have recently been published and should be consulted [98,99].
4.1. Coking
Coking is considered one of the most important challenges of catalytic DRM in its transition into a mature large-scale process. Coking refers to the formation of carbon on the catalytic material, which may lead to encapsulation of active metal centers or the chemical transformation of the catalyst, thus inhibiting the interaction of the active metal centers to the reactants [100]. In a study about the reforming process of tars on supported Ni-based catalysts [101], the authors found that two types of coke may be generated: type-I, which deposits on the metal particle, and type-II, which grows on the support. A more severe deactivation effect is attributed to coke formed by type-I. The coking behavior depends on several factors, such as the purity of the feed, the size, load, and dispersion of the metallic nanoparticles, the reaction temperature, space-velocity, and chemical effects of the support such as the acid/base character, porosity and redox dynamics.
Apart from the relative location of the carbonaceous deposit, the generated coke during DRM is also classified according to its adopted morphologies (Figure 6) [102]: adsorbed atomic carbon, which may form dispersed layers on the catalyst, or surface and subsurface metallic carbide-like structures, amorphous films and filaments, vermicular whiskers/fibers/filaments and graphitic platelets and films [103]. In many DRM studies, it has been reported that carbon deposits exhibit the whisker morphology independent of the temperature regime. Carbon whiskers normally bear a nanoparticle on the top, are tubular in shape, and are about the same diameter as the nanoparticle they hold. Their geometry suggests that fast decarbonation reactions occur at the nanoparticle until a high concentration gradient is reached through the metallic crystal and the whisker growths. This type of carbon does not deactivate the metal nanoparticle because the decarbonation reaction clearly proceeds, but the physical separation between the active centers and the support may induce inefficient recombination and oxidation rates of carbon. Dramatic formation of whiskers may break down the integrity of the supported catalyst and cause reactor blockage.
Graphitic films, on the other hand, have been observed especially at high temperatures. These morphologies are also generated by carbon diffusion in the metallic nanoparticle, but the process occurs at a slower rate compared to the previous scenario. The longer residence time of C* allows the development of ordered layers parallel to the interface [85]. Graphitic deposits are more easily achieved on nanoparticles above 20 nm in size and induce a dramatic loss in the intrinsic activity due to the decrease in the total active surface area that is packed inside the encapsulating shell [104]. The graphitic deposit is difficult to oxidize by combustion or to gasify with water due to the reduced sticking probabilities of oxygen on graphene/graphite compared to the amorphous carbon found on other types of coke [97]. It is known that spent reforming catalysts may exhibit a mixture of nickel carbides, which are less stable, and the toxic whisker and graphitic deposits, which are stable and will block the active sites. There may be some transformations among the metallic nickel, nickel carbide, and coked surface, giving rise to a combination of several effects [105,106].
There is a variety of catalytic and non-catalytic reactions resulting in coke formation under the conditions of DRM. It has been mentioned that the catalytic Boudouard reaction (Equation (5)) is responsible for carbon formation in mild or moderate temperature regimes and for high syngas production. The Boudouard reaction may be thermodynamically mitigated at high operating temperatures. However, the decomposition of CH4 (Equation (6)) is favorable at high temperatures. Furthermore, non-catalytic radical processes in the gas phase significantly contribute to coking. At low temperatures (below 500 °C), the formation of gas-phase radicals is expected to be of low relevance. Instead, C formation arises mainly by decarbonation of CO, and the formed species may polymerize into an encapsulating amorphous film around the nanoparticle [85,102]. It was found, for instance, that coke produced on activated carbon at mild temperature could be gasified by oxidation with CO2 in a subsequent operation unit [107]. A study with the same concept in a Catformer [108] under conditions such that coking was mitigated but not suppressed revealed that coke selectivity depended on the time of contact of CO with the catalyst. In the Catformer, the first section of the reactor contributes mainly to NiO reduction and to the initial development of the methane reforming reaction far from equilibrium. The catalyst requires periodic regeneration, and this regeneration should lead to the oxidation of coke. As a result, the metallic component of the catalyst is also oxidized. The specific nature of two circulating fluid beds, one for the dry reformer and the other for the catalyst regeneration, already suggests the possibility of reactor designs based on the cyclic operation. The results obtained in the Catformer [108,109] indicate that CO disproportionation is responsible for a large part of coking in DRM at moderate temperatures. Moreover, further kinetic analysis suggests that adjacent free metallic sites are required for coking under allowable thermodynamic conditions. This would be in line with the observation that large metal nanoparticles tend to favor catalyst coking and that high space velocities are preferable in an attempt to “purge” coke precursors away from the reactor.
At high temperatures (above 600 °C), both pyrolytic and catalytic pathways of coking are relevant. In general, the main reaction leading to gas-phase coke precursors can be understood as the homolytic decomposition of CH4. Observations at conditions of industrial applications of DRM (10–20 bar) have recently shown that coke precursors may form before the gas feed contacts the catalytic bed [56]. Complementary carbon deposits are found upstream of the catalytic bed rather than at the catalyst itself or beyond. This behavior strongly suggests the presence of non-catalytic reactions in the gas phase, which ultimately lead to potent coking precursors [110,111,112]. Starting with the pyrolysis of CH4, radical intermediaries lead to heavier coke precursors such as acetylene, ethylene, propylene and aromatics via recombination, dehydrogenation, and addition reactions [113,114]. The polymerization of olefins and the condensation of cyclic fragments would be responsible for the increasing accumulation of heavy aromatics, coke, and soot not only on the catalyst but along the whole reactor. This finding also suggests that gas streams, even with the smallest presence of olefins and aromatics, are unsuitable for DRM [3].
The co-feeding of H2 or H2O in the catalytic reaction as a way to mitigate coking without a relevant change in the produced syngas ratio has also been explored. The beneficial influence of H2 and H2O relates to their ability to buffer out the concentration of C-containing radicals by inhibition of CH4 pyrolysis and subsequent radical propagation reactions [115]. Furthermore, it has been suggested that COH* intermediaries are more easily oxidized than C*, and hence, the use of H2 or steam, which favors the occurrence of this intermediary, would be justified as a strategy to regenerate the catalyst from coking. The presence of water could minimize the formation of methyl radicals produced by non-catalytic reaction paths [56]:
CH4 → CH3· + H
CH3· + H2O → + CH4 + HO
Equation (30) shows the reaction of water with CH3· radicals, thus lowering the amounts of gas-phase coke precursors. Furthermore, H2O is used as a co-feed in DRM processes which paradoxically would turn “dry” reformers into “wet” reactors, yet with a minor sacrifice in terms of reaction yields and product ratios. The benefit of H2O as a co-feed is also evident from the gasification of existing carbon deposits:
which is the reverse of Equation (9) and keeps the syngas ratio at one.
C + H2O → H2 + CO
Equation (32) exemplifies the similar influence of H2 co-feed in coking control.
CH3· + H2 → CH4 + H
One of the key aspects of coke mitigation relies on carefully tuning the relative rates of the reaction steps represented in Figure 5. In these circumstances, coke control involves the enhancement of carbon oxidation compared to its formation [87]. The anti-coking strategies basically rely on two principles; namely, the direct inhibition of coke precursors and the enhanced removal of whichever deposits are formed. Preventing the nanoparticles from sintering, after which coking is favored, is, therefore, a direct anti-coking strategy. Complementary, a larger fraction of the oxidant (CO2, H2O, or O2) in the reaction feed should be applied. In the CALCOR process [116], high CO2 partial pressures are used to enhance the oxidation of C* via the reverse Boudouard reaction. In addition, low total pressures close to the atmospheric one are utilized, which precludes the fast formation of coke. Alkaline, alkaline-earth, and rare-earth additives may have a similar effect to applying an increased oxidant ratio. For instance, the addition of CaO or MgO to the reforming catalyst is known to increase the longevity and suppresses or reduces the coke formation compared to acidic or neutral supports without the additive [86,117]. The chemical effect of these additives has long been discussed in terms of the increased basicity of the catalyst, which in turn increases the uptake rate of CO2 and shifts the Boudouard equilibrium to CO [118]. The rate of CH4 activation and subsequent decarbonation may alternatively be decreased, as is the case in the sulfur passivated reforming (SPARG) process [1,119]. Here, the metal phase of the catalyst is partially poisoned by sulfur, thus reducing its CH4 decomposition ability and overall activity. One sulfur atom quenches four neighboring nickel atoms. Thus, sulfur more effectively inhibits carbon formation than the DRM activity [119] since coking requires a larger ensemble of adjacent nickel atoms.
Maintaining a certain level of surface oxygen or improving O* mobility can also optimize the utilization of CH4 and increase the coking resistance. The catalytic improvement from transition metal catalysts such as Pt [120], Pd [121,122], Ni [78,123], and Fe [124] and the bimetallic effect could be the consequence of dynamic oxygen originating from either the atmosphere, the metal center or the reactive support. Many current coking prevention methods, such as nanoparticle size reduction [125], alkaline metal dosing [126], and alloying of noble metals, may also increase the surface oxygen concentration of the active metal in essence [87]. The presence of surface O in the catalyst may be stimulated by several strategies. One of these involves the utilization of materials with a non-stoichiometric ratio that exhibit lattice oxygen or oxygen vacancies. Among this family of materials, perovskites [127], calcium aluminates [128], and cerium-zirconium oxides have been widely utilized [129] for reforming tars and methane in wet and dry conditions. In principle, the oxygen imbalance and lattice distortion generate or promote the mobility of lattice oxygen or oxygen vacancies that are highly efficient for carbon removal [99].
4.2. Sintering and Poisoning
Sintering refers to the agglomeration or growth of initially small nanoparticles provoking a reduction of the total active area. In DRM, sintering has the additional effect of favoring coking since carbon formation is favorable on larger nanoparticles. Agglomeration or “coalescence” of the metallic phase is a thermochemical effect that affects the whole nanoparticle. If equipped with sufficient thermal energy due to excessively high operation temperatures, the nanoparticles may detach and diffuse on the surface of the support. Eventual collisions among nanoparticles usually lead to their agglomeration and the formation of larger particles with reduced total surface area. Clearly, the adjustment of the metal–support interaction is critical to stabilizing the size of the nanoparticles, for instance, through chemical “anchoring” on the support or by their immobilization inside surrounding porous layers. However, some change in the nanoparticle size can still be expected even in the presence of strong anchoring effects. Individual atoms may be thermally emitted from a standing particle and captured in another portion of the catalyst. The atomic exchange may continue until the nanoparticles reach an equilibrium size which usually results in poorer dispersion compared to the pristine material. Mechanistically, the atomic exchange has a similar effect to the particle size distribution as observed in recrystallizing systems described by Ostwald ripening [130]. Usually, sintering is regarded as an activated process in which a given threshold has to be surpassed in order to induce the transformation of the nanoparticle. In the agglomeration path, the activation energy is related to the metal–support interaction and the average weight of the nanoparticle, while in the atomic migration, the barrier relates to the surface energy. The dominance of each effect is the result of several factors, including operation and preparation temperatures, metal–suppport interactions, nanoarchitecture of the catalyst, and chemical dynamics.
Obviously, preparation and operation temperatures are of major relevance to control sintering. The thermal nature of sintering most likely involves that too high preparation or operation temperatures lead to high rates of particle growth [101]. In a comparative study about the effect of calcination temperature on a NiO-MgO catalyst [131], the authors found that when the catalyst was calcined at 700 °C, the formation of a hard framework between NiO–MgO precluded the reducibility of NiO in the subsequent step, and favored the growth of particles due to thermal effects. Calcination at 500 °C formed an incomplete solid solution of weak NiO–MgO interaction, resulting in a higher degree of nickel reduction. However, facile sintering occurred during reduction due to weak metal–support interactions and the catalyst noticeably deactivated after 50 h of reaction at 750 °C. A mild calcination temperature of 600 °C was found to be optimal, as it enabled the generation of a homogeneous and complete NiO–MgO solid solution which was easy to reduce and resistant to sintering during reduction and reaction.
Another aspect of importance is the loading of the metallic phase, as it is expected that excessively high contents will favor the occurrence of large particles. Moreover, a highly loaded catalyst system exhibits short travel distances among the metallic particles, thus enabling fast agglomeration and sintering. Finally, we mention that a short contact time or high space velocity may help deal with sintering not only during the reaction but also during the prereduction or activation of the catalyst. In a study about the influence of the reduction conditions on the stability of biomass gasification catalysts based on Ni/Al2O3 [132], the authors found that higher reduction gas flow rates had a beneficial effect on the stability of the catalyst. The authors argued that high flows could effectively remove the heat generated during the treatment, thus minimizing the sintering tendency as the energy is dissipated faster and the nanoparticle is kept locally “colder”.
We briefly mention in this context the poisoning effects which have been observed, particularly in reforming systems aimed at the treatment of biomass and biogas. These feedstock sources normally contain small amounts of sulfur (H2S) and nitrogen (NH3), which are known as potent poisons not only for Ni-based but also for precious metal catalysts such as Rh [133]. In particular, sulfur may react with the metal to form inactive sulfides or can reshape the morphology of the nanoparticles and thus promote side reactions that limit the O* mobility of the support. All of these scenarios impact the product selectivities [134]. A desulfurization pre-unit may be necessary for excessive sulfur contents. Other strategies involve the introduction of S traps (Co-Mo or ZnO) in the catalyst design [135] or the addition of reactive elements (K, Ru, Mg, W, Sn, B), which exhibit a higher affinity to sulfur than the active phase [136,137,138]. These additives may also affect the electronic structure of S-metal compounds, thus inducing instabilities and facilitating the desorption of sulfurized molecules. Apart from sulfur, halogen sources such as chlorides, bromides, etc., should be avoided in the catalyst or the reactor walls. Chloride contaminations may form corrosive HCl under reforming conditions, thus destroying the catalyst support, neutralizing important basic sites, and compromising the whole reactor [139]. Finally, oxidant impurities such as NOx may deactivate the active metal by the formation of inactive oxides.
5. Reaction Systems for DRM
Scientific and technological development in DRM has traditionally been assumed to be operated in a fixed bed thermocatalytic reactor. Hence, the main focus for process improvement and its large-scale applicability has been placed on the preparation of active and durable catalysts that can sustain high conversion levels and produce a syngas of steady ratio. Apart from deactivation, for instance, by coking, it is important to consider the large energy investment necessary for this endothermic reaction. Furthermore, it would be ideal to understand which steps of the mechanism are more energy-demanding in order to maximize the reaction potential with a balance of catalyst properties and the control of process parameters.
When a reaction occurs, electrons and atoms are transferred among chemical species. The driving energy for these motions may be supplied by several means. Non-traditional reaction systems different from thermocatalysis have been introduced, which aim at improved energy management as it is currently achieved by classical heat. The energy may be more efficiently invested, for instance, by stimulating the generation of reaction intermediaries at low temperatures or by directly exciting the reaction site. In this context, we also refer to two important families of technologies of great DRM potential, namely, plasma and photochemical systems. We anticipate that these technologies are still in their infancy compared to classical systems due to their higher complexities. Their potential in a green energy transition, however, motivates their study as they are very attractive in this case.
We discuss the operation principles of the most relevant reaction systems and their current limitations, possible improvements, and perspectives. However, our main focus is on the large-scale implementation of DRM for near-future scenarios.
5.1. Thermocatalytic Dry Reforming
In thermocatalysis, the energy necessary for activations and reactions is supplied in the form of heat. It is well established that the utilization of a catalyst is needed to lower the activation energy, tune the selectivity of the reaction, and reduce operation times to values of economic viability. The DRM catalyst intended for classical chemical reactors may come in a variety of forms, such as supported nanoparticles, unsupported phases, metallic alloys, and nano-architectured materials such as porous overlayers, membranes, or thin films. Due to a large number of combinations of active metal phases, supports, dopants, promoters, and architectures, controversial and even contradictory findings are not uncommon in the literature regarding the optimal catalyst and its functioning principles.
The most popular type of thermocatalytic DRM system features a fixed-bed reactor, in which a tube is filled with the catalyst (powder or pellet), and a flow of the mixture of reactants traversing the bed is converted into the mixture of products. The most common catalyst features metallic nanoparticles on an oxide support. Noble metals such as platinum and rhodium are known for their high activity and resilience to coking. However, transition metals such as nickel and cobalt are preferred for commercial applications due to their low cost, notwithstanding their lower activities and durabilities compared with precious metals. The usage of precious metals in DRM reaction has been further reviewed [16] and mostly relates to their effects on a fundamental understanding of the kinetics, mechanisms, and deactivation of these catalysts. Here, we mainly focus on systems with a high potential for large-scale applications. Hence, catalysts in which the precious metals are the main active phase have not been considered.
Several support materials have been applied successfully, including silica, alumina, zeolites, lanthanum oxide, magnesia, carbon, and metallic carbides. Perovskite-based catalysts have recently become a prominent member of the family of reforming catalysts. In general acidic catalysts should be avoided as they enhance hydrocarbon cracking and may favor coking. The role of promoters, among which alkaline, alkaline-earth, and rare-earth elements are widely used, is more usually linked to a diminished coking tendency and increased nanoparticle stabilization than to increased activity compared to the non-promoted analog. Furthermore, the role of the different catalyst modifications is extensively viewed as improving the catalyst durability rather than as increasing the intrinsic activity of the metallic center. Usually, the best performing catalysts will feature the smallest particles of the metallic phase and high oxygen mobility.
A complication to identify particular cross-correlations among different studies relates to the diversity of synthetic strategies, operation conditions, and analytical tools utilized in the literature. This circumstance may be avoided, for instance, with the introduction of unified procedures to achieve particular materials and testing protocols for the reaction. However, important observations have been performed in the past years, which we briefly summarize in the following sections.
5.1.1. The Metallic Phase
Supported Ni-based catalysts are considered as optimal for DRM thanks to their compromise between low cost and high activity. However, they are prone to severe deactivation. It has been shown that keeping the Ni nanoparticles size below 5 nm [140,141] is exceptionally efficient to preclude coke generation. Coking rapidly increases with the nanoparticle size. The coking activity may increase with time on stream as nanoparticle sintering is initiated. For example, the authors of a recent study investigated the coking propensity of supported Ni cluster catalysts down to atomic dispersion. They were prepared by an impregnation method atomically dispersed nickel on hydroxyapatite supports with and without the addition of cerium [142]. The catalysts were found to be formed by clusters of a few atoms and atomically dispersed atoms over the support (Figure 7). The single atoms were highly resistant to coking during DRM but prone to deactivation by sintering. When single atom particles coalesced into large nanoparticles, the coking activity increased. Ce doping on the modified support induced strong metal–support interactions which stabilized Ni single-atom sites and favored the selective activation of only the first C–H bond in methane. As coking occurs mainly by overcracking of methane at high temperatures, the selective activation of only the first C-H bond was found to be key to precluding the formation of coking precursors such as C*. This finding is in line with the mechanism of coke generation at the high temperatures of DRM, which originates from methane cracking and requires the presence of adjacent metallic sites, as discussed above. In addition, support chemistry and metal–support interactions needed to be adjusted to impart durability to the atomically dispersed metallic phase. The metal–support interactions can be tuned by a delicate balance of support material, nanoparticle sizes, type and number of promoters, and nanoarchitecture of the catalyst structure [3,14,143].
In a different study based on the same assumption [144], the authors prepared single-site Ni/MgO and investigated their kinetic behavior by experimental catalysis, DFT, and KMC simulations. Contrary to the previous study, the authors found that although only Ni provided the active site of the reaction, the single-atom Ni site was found to be inactive due to the weak binding to reaction intermediaries and limited neighboring active sites compared to the support MgO. Ni2 and Ni3 sites were classified as presumably inactive. However, clusters of Ni4 atoms increased the catalytic activity. The authors argued that these clusters offered enough active Ni sites that are sufficiently isolated from each other and able to work cooperatively in order to activate CH4 and CO2. This enabled the production of CO, H2, and H2O and completely eliminated carbon deposition. The differences with the previous study may originate from the distinct chemical characters of hydroxyapatite and MgO supports. The latter, a more alkaline material that does not interact strongly with Ni, may have introduced a weaker synergistic action within this system.
Achieving active and durable metallic phases not only involves controlling their structures down to the smallest scales, but pure unsupported metallic nickel has also been utilized successfully. The properties of a “bulk” metallic structure could optionally be tailored at micrometer or millimeter level to achieve an efficient reforming catalyst of low cost. For instance, a catalytic nickel membrane was prepared by the sintered metal method [145]. After compressing alumina-modified nickel powder (particle sizes about 3 μm) without a binder in a cylindrical metal mold, the pellet was then sintered at 900 °C in a hydrogen atmosphere for 2 h. The prepared catalytic nickel membrane was utilized in a combined SRM/DRM reactor. The authors found that CH4 conversion decreased with the increase in the CO2/H2O feed ratio at 700 °C, while the influence of the CO2/H2O feed ratio on CH4 conversion was not significant at higher temperatures. This result may indicate that DRM is intrinsically slower than SRM until a sufficiently high temperature is reached. The authors found that the H2/CO molar ratio could be adjusted by the CO2/H2O feed ratio. No carbon deposition on the catalytic nickel membrane was observed after the combined SRM/DRM reactions. Notably, apart from the huge metallic particles of this study, the catalyst did not deactivate. One of the reasons could be the co-feeding of steam to achieve the SRM reaction while gasifying coke deposits. An additional effect could arise from the specific structure of the membrane reactor. The porous membrane could favor the selective diffusion of H2, thus enabling its separation from the reaction mixture. As the product is continually retrieved, the position of chemical equilibrium could be further shifted according to Le Chatelier’s principle as schematically represented in Figure 8. Contrary to the fixed-bed approach, the membrane could combine two unit operations simultaneously, i.e., reaction and purification. This would increase the overall efficiency of the catalyst.
Apart from nickel, cobalt is another metal of interest that has been applied either alone or in combination with nickel. Co itself is a good reforming catalyst, but in general, its intrinsic activity is lower than the one of Ni. Its most extended use relates to its applications as a modifier that adjusts the propensity to coking of Ni catalysts. In a similar way, precious metals such as Pt, Ru, and Rh exhibiting activities and durabilities superior to that of Ni-based materials may be pragmatically utilized in minute amounts over the nickel catalyst to modify its relevant catalytic properties. Metals are reactive and may oxidize, segregate or migrate under the reaction stimulus. Hence, the chemical influence of a metallic additive may be difficult to elucidate during the reaction. For instance, a study showed that coke deposited on supported monometallic nickel and cobalt catalysts, provoked deactivation. However, catalysts in which both metals were loaded led to little coke formation [146]. Although, in this case, the catalysts did not deactivate by coking, deposition of carbon altered the H2/CO ratio. The low H2/CO ratio reported in this study for the monometallic catalysts could be a sign that soot was being produced by the reaction of H2 with CO2 [14]. This effect was suppressed by the presence of the two metals. In a similar study [147], the effect of Co loading on alumina-supported Ni catalysts with fixed total metallic contents (9 wt.%) was investigated. The results of DRM at 700 °C indicated that catalysts with the highest cobalt contents (9% and 8%) were the most active and stable but produced a large amount of carbon. The higher activity of cobalt-rich catalysts was attributed to its higher intrinsic activity for methane decomposition compared to nickel. Moreover, the enhanced stability of the cobalt-rich catalyst may be related to the selective formation of non-deactivating carbon deposits. In another study, the authors found fundamentally opposite results [148], which suggested that when Ni retained its reduced state, its activity for CH4 activation was preserved, while oxidized Ni2+ was a somewhat less active form of the metal. In the bimetallic catalyst, a comparably higher propensity of the Co phase to form oxides would prevent the oxidation of Ni. The formation of cobalt oxides would favor the oxidation and subsequent removal of carbon deposits, while Ni would maintain its reduced state and high activity for CH4 decomposition (Figure 9). The bimetallic effect would introduce a superior catalytic performance by the combination of the high activity of Ni towards CH4 conversion and the high carbon-removal efficiency of Co.
Furthermore, Ni–Co/SiO2 bimetallic catalysts have been prepared by the incipient wetness impregnation method [146]. Catalysts with small Ni–Co particle size (3.1 nm), high dispersion, metal surface area, and strong interaction with the support could be obtained, yielding high CH4 and CO2 conversions at 700 °C over 30 h on stream and negligible carbon formation. In addition, by forming an alloy with Co, the electron density of Ni became higher due to the donation from neighboring Co, which prevented the oxidation and sintering of Ni [143]. In a related approach, the Ni–Co/SiO2 catalyst was achieved with the formation of a phyllosilicate phase [149]. The authors found that the Ni/Co ratio greatly affected the catalytic performance as it could balance the Co oxidation sites and could facilitate coke removal on Ni sites. With the optimum metallic ratio, catalysts showed a stable performance over 50 h. The addition of Co suppressed carbon formation and Ni sintering, but a disproportionate amount of Co was shown to lead to the oxidation of Ni sites and the subsequent deactivation of the catalysts.
Bimetallic Ni–Fe/SiO2 catalysts have also been demonstrated to have high carbon resistance for the DRM reaction [150,151]. Bimetallic Ni-Fe showed increased activity and stability, with a maximum at a Ni/(Ni + Fe) ratio of 0.8. The authors [151] suggested that Ni remained at the oxidation state Ni0 independently, whether it was alloyed with Fe or physically mixed. While Fe is inactive for DRM, it improved the stability of Ni-based catalysts. Fe in Ni-Fe alloys was partially oxidized to FeO, leading to dealloying and the formation of a Ni-richer alloy. Because FeO is located preferentially at the surface as small domains of a few atoms layer thickness covering a fraction of the Ni-rich particles, it may react with carbon deposits, leading in turn to a reduced coke formation and a local protecting effect.
Alloying Ni with Cu is another strategy to improve the longevity of the Ni/SiO2 catalyst. The addition of copper into the nickel system can stabilize the structure of the site active for methane cracking reaction and for the regeneration process [152], preventing the deactivation caused by sintering or by loss of metallic crystallites. In a study, the addition of Cu into the Ni catalyst was found to produce a Cu-Ni species that inhibited the formation of inactive carbon during the DRM [152]. Hierarchical nanocomposite structures have also been prepared and utilized [153], showing superior catalytic performance and thermostability at high temperatures (>700 °C) compared to the impregnated one. The study suggested that the high surface area, well dispersion of fine alloy nanoparticles, and strong metal–support interactions led to superior catalytic activity and coke and sintering resistance of the hierarchical composite structures during the isothermal testing of 30 h of the bimetallic catalyst.
Doping nickel with platinum is also an option to modify the DRM catalyst. A comparison between monometallic (Ni and Pt) and bimetallic (Pt-Ni) nanoparticles deposited by the reverse microemulsion method on nanofibers of alumina showed a strong bimetallic effect in which Pt-doping favored the formation of Ni and NiO domains rather than the occurrence of NiAl2O4 spinel [154]. The presence of Pt facilitated the redox transition between Ni0 and Ni2+, thus stabilizing its reduced state in the reductive environment of the syngas atmosphere. The bimetallic catalyst was superior in terms of activity, high selectivity to generate a syngas of ratio one, low coking tendency, and overall stability compared to the monometallic analogs.
Ni has also been alloyed with post-transition elements to improve its performance [143]. The presence of 2 wt.-% indium on the surface of a 3 wt.-% Ni/SiO2 catalyst prevented coke formation [155]. The authors found that indium was unstable against sintering without nickel on the silica surface. The bimetallic catalyst remained in the metallic state after reduction at 700 °C. The presence of indium profoundly changed the adsorption, structural and electronic properties of nickel, with indium enrichment on the metallic surface. In addition, TEM imaging of the spent catalysts after 24 h of DRM revealed that the average particle size of the bimetallic catalyst was slightly smaller than that of the monometallic analog.
We would like to indicate that the fundamental reason for these empirical observations of a bimetallic effect is still a topic of scientific interest. We summarize from this overview that the metallic dopant:
- may stabilize the reduced state of active Ni0 by electronic effects,
- may improve the dispersion of Ni inside the metallic nanoparticle, thus preventing the occurrence of large ensembles of adjacent Ni atoms favorable to coking,
- may form in situ oxide phases highly reactive to oxidation of poisonous carbon,
- may tune the strength of the metal–support interaction, or
- may induce synergistic reactions by intrinsic catalysis or co-catalysis.
The exact reason depends on how the two metals interact with each other (i.e., alloy formation or physically separated), with the support, and with the chemical atmosphere (i.e., oxidation states). For instance, the chemical influence of a bimetallic catalyst in which both metals are in the same nanoparticle may largely differ from an otherwise identical catalyst except for the two metals forming physically separated phases. Even in systems in which some degree of homogeneity is expected, extrapolations and generalizations may become inaccurate with the complexity of the underlying chemical processes. Ni and Co form alloys in a large interval of atomic compositions [156]. However, the alloy undergoes a phase transition from hcp to fcc structures with increasing temperatures. Little is known about the behavior of this phase transition under a reactive atmosphere or about its influence on the catalytic activity, which may be critical to achieving the long-term stability of DRM catalysts for large-scale applications. These questions are hard to predict from pristine, static models of the reaction and may further indicate the need for operando investigations.
5.1.2. Promotion Effects
Enhancement in efficiency and longevity of a nickel catalyst may also be realized by the addition of elements that are most likely not incorporated into the metallic phase. These elements are largely viewed as promoters in the sense that they do not have an intrinsic activity but stabilize and facilitate the operation of the metallic phase. For instance, the effect of introducing Ca and K in the DRM catalyst has been thoroughly studied [157]. Below 750 °C, the Ni/SiO2 catalyst promoted by the two additives achieved a conversion of methane of 46%, and optimal conversion of 99.5% was reached at 850 °C and 1 bar. Increasing the pressure in the reactor led to a drop in both methane and CO2 conversions. At 800 °C, the conversion of methane reached 98.7% at 1 bar, 80% at 9 bar, and 70% at 14 bar, closely resembling the trends of thermodynamic calculations. At 860 °C, the conversion of both methane and carbon dioxide were 99.5% and 96.3%. At 14 bar, the conversion increased proportionally to the temperature, and the conversion of CO2 was more important than the conversion of methane at higher temperatures. The authors of this study argued that the addition of Ca and K promoters play a significant role in oxidizing and removing carbon species from the catalyst surface. The alkaline additives could increase the conversion of CO2, thus inducing the presence of surface oxygen species that preclude the growth of coke for long-term operations of 360 h time on stream.
The effects on catalytic durability caused by adding other elements to the nickel-based system have been further investigated. For instance, the effect of adding K, Ca, Mn, and Sn at 0.5 wt.-% over a classical Ni/Al2O3 catalyst was revealed for separate loadings of the promoter [158]. The pristine catalyst without additive achieved 84.7% for methane conversion at 750 °C, which was maintained for a total of 30 h on stream and produced a steady syngas ratio of 0.97. However, severe carbon formation was observed (60 mg/g). Doping caused lower conversion levels and produced syngas that was characterized by H2/CO ratios much lower than one. The effect on the production of coke depended on the loaded metal. The addition of Mn introduced an enormous increase in the production of coke (250 mg/g). At the other end, the loading of K featured a notable decrease in coke generation (10 mg/g) but produced syngas of a very low ratio (0.4) [14,158]. The addition of the other promoters introduced effects that were found to be in-between these two ends.
Further investigations using K in catalyst supported on MgO-ZrO2 showed that the addition of 0.5 wt.-% led to conversion levels closely following the values obtained from equilibrium calculations [159]. Higher concentrations of K, however, showed a decrease in methane conversion. The catalyst that did not have any potassium showed strong initial and long-term deactivation during the experiment, while the deactivation was much slower in the K-doped materials. The catalyst with 0.5 wt.-% of potassium was notably stable with a steady conversion of 90% of methane during the whole period of 14 h. Investigations [160] with the addition of cobalt, lanthanum, cerium, barium, manganese, potassium, and calcium using MgO-ZrO2 as support have revealed some important trends. Co may enhance the catalytic conversion of both methane and CO2, while Ca has the opposite effect. K, Ca, and Co helped to maintain the long-term activity of the catalyst. The addition of Mn to the catalyst boosted carbon formation and catalytic deactivation. In general, the addition of K prevented coking, but a high concentration of this element led to a severe decrease in conversion and product ratio. The effect of K-loading on the performance of the Ni catalyst can hardly be attributed to the formation of an alloy or a separate oxide phase. Its effect, summarized in reduced conversions to syngas, low coking, and generation of syngas of low ratio compared to the stoichiometric value, may be a consequence of an increased metal–support interaction. At low K loadings, the increased interaction prevents the sintering of the metallic phase, which stabilizes the nanoparticle size and favors long-term operation. Too high concentrations of K would provoke an increased metal–support interaction, and, eventually, the support material would start to encapsulate the metallic nanoparticle, leading to a decreased conversion. On average, the surface area of the metal phase would start to decrease. In addition, the increased basicity of the catalyst may increase the affinity for CO2. The combined action of these two effects could explain the observed decrease in the H2/CO ratios.
The promoter effect could be summarized by its influencing effect on the acidity/basicity and on the metal–support interactions. Acidic additives are usually anticipated as non-ideal due to their intrinsic activity for methane cracking [161]. Complementarily, basicity has been treated as a desirable property, as stated in several investigations that highlight the beneficial influence of introducing alkaline materials into the catalyst [140]. These studies claim that basicity would enhance either CO2 adsorption and subsequently enhance its activation [162] or stabilize the dispersion of the metallic phase.
In a study that investigated the role of acidity and basicity on catalysts of Ru supported on Al2O3 and SiO2 [163], the authors concluded that surface hydroxyl groups and the capability of carbon dioxide dissociation, or alternatively the capability of forming formate intermediates, may play a role in the accumulation of coke forming species and the resilience of the resulting material. The authors speculated that catalytic coke formation would result from a bifunctional mechanism, which is either Boudouard or formate mediated and controlled by surface hydroxyl groups. In another study, which was performed on Ni supported by modified silica and alumina, the authors argued that a moderate acidity-basicity is best for a stable and active DRM catalyst [164].
Promoters are usually regarded as enhancers of the metal dispersion on silica, also for noble metals. For instance, moderate amounts of CaO led to a higher dispersion of Rh on SiO2 catalysts [165]. High amounts of CaO decreased Rh dispersion, possibly due to the coverage by the promoter. These catalysts have little application in large-scale scenarios due to economic prohibitions.
Again, the question remains about the fundamental reasons that drive these empirical observations. It is hard to predict where the doping element is located during the reaction, which phases it adopts, or what are its structural and electronic effects on reaction activity, indicating some knowledge gap about the working principles of a performing catalyst which could be tackled, for instance, with further operando investigations.
5.1.3. Support Chemistry
On the other hand, the support is the most abundant component of the catalyst and hence largely determines its large-scale properties such as thermal conductivity and capacity, mechanical resistance, thermal stability, and average acid/base character. Activity, size, and stability of the nanoparticle are the direct consequence of interactions among the components of the catalytic particle in which the support chemistry is critical.
Several supports have been applied successfully to DRM catalysts. Desirable characteristics of the support material include high thermal, chemical and mechanical resistances, high heat capacity and conductivity, small particle size or porous structure, little acidity, and tuned metal–support interactions to stabilize the nanoparticle dispersion [166,167]. Silica and alumina are the most popular support phases for DRM catalysts. Oxides of magnesium, zirconium, and titanium and their mixtures with and without the addition of other elements have also been applied.
In a recent study [140] about how these supports may intrinsically affect the catalyst activity, highly uniform Ni nanoparticles with sizes of 2.6, 5.2, 9.0, and 17.3 nm were synthesized, and overlayers of various metal oxides, including SiO2, Al2O3, MgO, ZrO2 and TiO2, were deposited separately onto the nanoparticles of 5.2 nm. The study revealed that the catalytic activity decreased with nanoparticle size, with 2.6 nm nanoparticles showing CH4 turnover frequencies 4.1 times higher than that of the larger particles of 17.3 nm. Al2O3 exhibited a 4.3 times higher methane turnover frequency than SiO2 with the same 5.2 nm nanoparticles. The other oxides exhibited activities that have been found to be intermediate between those of alumina and silica, corroborating to some degree the high popularity of alumina in DRM catalyst recipes. The study allowed separating the influences of the Ni nanoparticle from the support chemistry, and the results indicated the utmost relevance of designing durable Ni nanoparticles of decreasing sizes, probably to the level of atomic dispersion.
In a similar way to the metallic phase, the support material of commercial DRM catalytic systems has to be easily preparable, non-toxic, and should be made out of cheap precursors. Below, we briefly examine the most popular support materials utilized for DRM applications.
Silica Supports
Silica supports have been extensively applied in DRM studies thanks to their availability, thermal stability, and the possibility to be manufactured with a high specific surface area. In addition, its micro-mesoporosity and acid-base properties can be finely tuned. Several synthesis strategies, mostly based on the stabilization of nickel by an alkaline oxide or rare-earth additive, have been developed in order to overcome sintering effects. A recent review [168] summarized the progress made between 2015 and 2018 in the design of Ni/silica-based catalysts for the dry reforming of methane with special care on the effect of alkaline additives. The different synthetic strategies in this area aim to increase the confinement and control of the nickel particles size. Promoters are usually regarded as enhancers of the metal dispersion on silica not only for Ni-based but also for noble metals catalysts [165]. In general, the alkaline additive will stabilize the silica phase by the formation of silicates which may increase the interactions with the metal particle. A recent study [169] investigated how the synthetic route altered the performance of Ni/SiO2 catalysts. The authors synthesized the catalysts by direct impregnation followed by calcination, or alternatively by a citrate-mediated protocol in which the Ni precursor was forming a chelate intermediary. The citrate addition enabled the reduction of the particle size from 10 nm to 3.3 nm after simple calcination, followed by a reduction in H2. Further materials were achieved from the citrate-modified mixture by either reducing the material directly in hydrogen or by pyrolytic decomposition in a He atmosphere followed by oxidation and a final reduction step. The classical Ni/SiO2 deactivated after only 2 h of operation, and reactor blocking was observed due to extreme coke generation. The citrate-mediated catalysts showed improved performance, reaching stable operations for 100 h on stream at 750 °C and syngas ratios between 0.8 and 1.0. Notably, the catalyst prepared by inert decomposition followed by the redox treatment operated stably without any indications of deactivation, exhibited the highest activities of the set for CH4 and CO2 reaction, and produced syngas with a ratio of 1.0. The authors argued that the pyrolytic decomposition would lead to the formation of a carbonaceous interlayer separating the Ni nanoparticles from the silica support. The interaction of Ni with this layer would prevent particle agglomeration during the calcination/reduction steps, thus stabilizing the metallic dispersion and precluding the formation of coke.
Apart from the possibility of adding an alkaline promoter or altering the synthetic protocol, as shown in the previous study, another interesting approach to outperform the classical Ni/SiO2 catalyst was recently put forward. It is based on the idea of a core-shell structure, i.e., catalyst particles at which the metallic phases are surrounded by porous silica overlayers. The applications of nanoporous silica in DRM processes is a topic of its own which has recently been reviewed [143], including developments such as the utilization of promoters, bimetallic catalysts, stabilization of highly dispersed catalysts, and core-shell architectures (Figure 10).
In a study of high-performance DRM catalysts, monodisperse Ni nanoparticles (5.2 nm) were achieved onto spherical functionalized silica supports (Figure 11) [141]. The Ni/SiO2 catalyst was later covered by porous silica overlayers. The silica-coated Ni catalyst was stable for 170 h under operation conditions and did not show any signs of deactivation. However, the uncoated catalyst deactivated significantly during the experiment. Moreover, the silica-coated catalyst produced syngas with a stable ratio of 0.70, while the uncoated analog gave rise to a syngas ratio that decreased along with deactivation and thoroughly stabilized at 0.55. A closer look at the catalyst morphology attained by (S)TEM imaging revealed whisker-like coke formation and agglomeration of the Ni phase at the uncoated catalyst, which was not observed at the coated analog. The authors suggested that the porosity of silica in the overlayers enabled the transfer of reactants and products while the rigid edifice prevented aggregation of the Ni nanoparticles.
Similar findings were presented in another report [170], in which the Ni nanoparticles were not loaded on spherical silica but on a porous mixed matrix containing the metallic particles embedded in the silica phase. The Ni-in-SiO2 core-shell nanoarchitecture catalyst had reforming activities similar to the commercial analog but introduced a drastic decrease in coking tendency and particle agglomeration. Notably, a syngas of ratio 0.70 was also achieved in this study [169].
Alumina Supports
Alumina is another popular support in classic DRM catalyst recipes and is promising support in industrial applications owing to its high surface area, excellent thermostability, specific surface functional mechanisms, intermediate metal–support interactions, and low cost [171,172,173]. Further studies with tailored alumina architectures have shown that alumina supports may be superior to silica-based catalysts in combined operations of DRM-SRM reactors. For instance, the authors of a study [173] achieved incorporation of the active component into either porous silica (SBA-15 and CeliteS) and porous alumina by either impregnating these materials with a Ni precursor or by direct synthesis of a mixed Ni-alumina ordered porous material which was achieved via a “one-pot” evaporation-induced self-assembly method. The porous silica catalysts deactivated after 10 h on stream at 800 °C under a gas mixture containing CO2, CH4, and steam. The CeliteS catalyst exhibited very poor catalytic conversions, and the spent catalysts of both silica-supported materials were found to produce poisonous carbon whiskers. The alumina-based catalyst was much more durable, notwithstanding their different morphologies (ordered or disordered, Figure 12), which may indicate an intrinsic superior performance of alumina as support compared to silica. The catalyst achieved by the one-pot method exhibited an ordered mesoporous structure in which the Ni phase was rigidly immobilized. The authors found that the syngas ratio, catalytic activities, and stability of the ordered alumina catalyst followed the thermodynamic values for 40 h on stream, while the disordered catalyst had much lower activities and product selectivity according to the expected values. The authors concluded that significantly enhanced Ni–alumina interaction, as well as much smaller and well-confined Ni nanoparticles, were responsible for an improved sintering resistance and for the excellent catalytic stability.
Other authors explored the influence of the alumina morphology on the catalytic performance [171]. Here, the authors prepared alumina with nanosheet, nanofiber, or nanoparticle morphologies and subsequently impregnated the supports with the Ni precursor. All morphologies exhibited high DRM performance in the temperature range of 500–800 °C. The nanosheet structure was the most active and most stable system, while the nanoparticle architecture gave rise to somewhat lower conversion values. A syngas ratio of 0.9 was achieved for all catalysts, but only the nanofiber material was resilient to deactivation. The nanofiber was characterized by constant conversion values of CH4 and CO2 during 50 h on stream at 750 °C. The activity in the other catalysts declined since the beginning of the test. It was further found that the nanosheet Al2O3 phase was defined predominantly by (1 1 0) planes, which favored the metal–support interaction, promoting the initial catalytic activities. Meanwhile, owing to its abundant confined spaces and steady chemisorption behavior, the nanofiber Al2O3 exhibited superior catalytic stability.
Further studies remark on the importance of the 3D ordering of the alumina phase for maintaining the stability of the catalyst [174]. It was shown, for instance, that Ni/alumina catalysts in which the support was an aerogel were active and stable to DRM for 30 h. However, when the alumina support was dosed in the form of a xerogel, the catalyst deactivated after 1 h. The higher specific surface area, lower bulk density, and Ni crystallite sizes of the aerogel-supported catalyst compared to the xerogel support may have facilitated the dispersion of the metallic phase. Furthermore, the structural effect of adding MgO to the catalyst also revealed that the nanoparticles stabilized at sizes of 17 nm, well below the 38 nm of the crystallites in the unpromoted material [175]. Coking was severely reduced for the smallest particles. As catalytic coking requires lateral interactions among carbon surface species, the nanoparticles with a high surface area may favor the coupling of C species and coke build-up. In addition, the increased volume of the nanoparticle subsurface compared to its size may induce subsurface dynamics that are relevant for side reactions [78].
The immobilization of Ni atoms precluding coke formation may also be imparted on the alumina-supported catalyst by thermal methods (Figure 13) [176]. In a study, calcination of Ni/Al2O3 catalysts at elevated temperatures (900 °C) prompted the transformation of NiO and Al2O3 in a NiAl2O4 spinel. The Ni atoms were highly dispersed and more stable when pinned to the spinel structure compared to the merely supported nanoparticles. This resulted in superior resistance to sintering and coking.
The addition of alkaline-earth metal oxides onto the alumina-supported nickel catalysts has also been largely investigated. Usually, the promoted catalyst generates conversion levels of methane that are similar to the values of the unpromoted material. The concentration of the promoter has to be carefully adjusted. A study showed [177] that the addition of 0.9% of MgO improved CO2 conversion by 10% compared to the catalyst without a promoter. The addition of CaO and BaO did not improve the conversion. Furthermore, the H2/CO ratio increased from 0.70 at 500 °C to 0.93 at 860 °C in the catalyst with MgO but reached stoichiometric syngas ratios in the unpromoted catalyst. The production of syngas of a low ratio may originate from CO2 reactions with the catalyst or with hydrogen to give soot. The reaction could as well be a partial decomposition of CO2 into CO catalyzed by the higher affinities of the MgO additive. Overall, the addition of CaO, MgO, and BaO helped maintain high catalytic activities and low deactivation rates. Structurally, the addition of these promoters may stabilize small Ni crystallites efficient for the reaction and with a low propensity to coking.
Rare earth elements modification of the Ni/alumina catalyst is another popular strategy that may lead to several effects [178]. The promoting effects of these additives are termed on the basis of increased surface basicity, the introduction of oxygen vacancies, facilitation of active metal redox transition, and improvement of dispersion of Ni particles [16,179]. In a recent study [179], the authors screened the effects of various rare-earth promoters (Y, Ce, and Pr) and of Sc on Ni supported by MgAl oxide catalysts. A significant improvement in CH4 conversion and catalyst stability was found in the promoted samples, for which Ce and Pr have been identified to be the most promising additives for stable high conversion after 20 h on stream at 750 °C. The authors explained that the addition of rare earth elements enhanced the surface basicity of the catalysts. Therefore, the CO2 adsorption capacity was improved. The absorbed CO2 formed in situ two kinds of carbonate species on the catalyst surface: bidentate and monodentate carbonates, which were especially favorable for CHx conversion. The formed carbonates could effectively react with the deposited carbon, leading to an overall enhanced catalytic stability. The authors further explored the oxygen vacancy and redox properties, which could be enhanced in the presence of promoters exhibiting redox pairs. The enhanced redox properties facilitated the electron transport, thus the rate of CH4/CO2 conversion as well as the elimination of deposited carbon.
A significant part of the promotion effect relates to the specific location of the dopant in the catalytically relevant structure. When dosed in low amounts, La2O3 and CeO2 tend to disperse onto the catalyst matrix, improving Ni dispersions and enhancing CO2 adsorption. However, the effect could be drastically different when the element is directed onto the metallic phase [93,180]. In a study [180], a series of Ni/Al/La catalysts was prepared, consisting of Ni/alumina, Ni-metallic La/alumina, and Ni/lanthanum oxide. The catalyst with metallic lanthanum addition was found to be deactivating the slowest at 700 °C and generated an H2/CO ratio close to one. The other two catalysts had lower CO2 and CH4 conversions and produced syngas of ratios closer to 0.9. The nickel on lanthanum oxide was the least active of the three investigated catalysts. Increasing the temperature to 800 °C led to an increase in conversion for the Ni-alumina and for the catalyst achieved by the addition of metallic lanthanum. However, the latter maintained steady conversion for 35 h, while the unpromoted catalyst deactivated after 5 h of reaction. The authors showed that the particles of the metallic Ni for the most durable catalyst were significantly smaller than the particles of the classical Ni/alumina. This effect was attributed to the enhanced lattice stability imparted by the formation of an inverse spinel structure and LaAlO3 nanocrystallites on the alumina support, both of which aided in minimizing the agglomeration of Ni particles [180].
The effect of adding cerium oxide at concentrations varying between 0 to 15% onto the classical Ni/alumina catalyst was shown to also impact coke generation [181]. It was found that a catalyst without cerium was slightly more active for both methane and CO2 conversions at 800 °C than the Ce-loaded analog. In general, Ce concentration did not affect the overall activity and only reduced it marginally. However, the carbon produced during the reaction varied inversely to the amount of cerium in the catalyst. It was found that, similar to lanthanum, cerium formed a stable CeAlO3 phase during pre-treatment of the catalyst, which may prevail under DRM conditions. This phase could be responsible for carbon deposition mitigation without deteriorating the activity. By nature, the CeAlO3 is active in the catalytic decomposition of CO2 to form active surface oxygen [181]. Therefore, the addition of cerium could improve catalyst longevity for relevant industrial applications [14].
Magnesia and Zirconia Supports
ZrO2, MgO, and rare earth elements such as lanthanum and cerium are not only viewed as popular additives on alumina- and silica-supported catalysts. In some scenarios, they have also been tested as the main support material with and without further additions. A ZrO2-supported nickel catalyst could increase the sulfur resistance of the catalyst. In addition, ZrO2-supported promoted by silica (Ni-Si/ZrO2) and a SiO2-supported analog promoted by zirconia addition (Ni-Zr/SiO2) were found to be active in the DRM reaction for temperatures as low as 400–450 °C [182]. However, the conversion levels were only moderate (below 10%). The Ni-Si/ZrO2 material had a higher initial conversion of both reactants (about twice) and was more stable during the 15 h of DRM reaction in comparison to the Ni-Zr/SiO2. The activity of the latter declined very fast after only 5 h on stream. For both materials, syngas of a low ratio (0.75 on average) was generated. The characterization of the catalyst reducibility revealed that the Ni-Si/ZrO2 could be efficiently reduced at temperatures of 343 °C, while the silica-supported catalyst produced Ni species that survived the reduction treatment still at 447 °C. In addition, the Ni-Si/ZrO2 catalyst featured small nanoparticles of 6–9 nm and a slight electron donor ability, which stabilized the reduced nickel state and favored the formation of easily oxidized carbon species. In a complementary study, a Ni/ZrO2 catalyst was doped with yttrium oxide [183]. In general, the addition of yttrium oxide prompted the formation of weak and medium basic sites, produced smaller nanoparticles, and increased the CO2 adsorption ability of the catalyst. The increased uptake of the oxidant may have reduced, but not suppressed, the propensity of the catalyst towards coking. In a similar way, a catalyst of Ni/ 8%ZrPO4 + ZrO2 was prepared onto which varying amounts of cerium oxide were loaded [184]. All the catalysts promoted with ceria had higher conversions of CO2 and CH4 and reduced the amount of generated coke.
The influence of loading the zirconia-based catalyst with MgO [185] was recently investigated. Upon treatment, the Ni/MgO-ZrO2 catalysts developed a NiO–MgO-solid solution. The reaction with these catalysts achieved a high constant conversion of CH4 and CO2, and a syngas of ratio 0.96 could be generated. The authors attributed the highly constant performance of magnesium-oxide-modified catalysts to the ability of CO2 to oxidize the carbon deposits, thus maintaining the catalytic stability.
Magnesium oxide has also been applied successfully as a support of the DRM catalyst. A study about the performance of MgO supports [186] revealed some interesting features when the nanoparticulate catalyst was doped with molybdenum. A cubic single-crystalline magnesium oxide (MgO) support was first achieved by autothermal combustion of Mg in a flow of CO2 (Figure 14). Subsequently, the support nanopowder was suspended in a solution of nickel chloride and ammonium molybdate in ethyleneglycol. The Ni-Mo nanoparticles were obtained by reduction of the salts with hydrazine using polyvinylpyrrolidone as a size controller. Upon heating under DRM conditions, the Ni-Mo nanoparticles migrated on the oxide surface to step edges to form larger, highly stable nanoparticles of 17 nm. The production of synthesis gas was quantitative during 850 h of the experiment with no detectable coking. Without Mo, the conversion was much lower (~20%), and there was a NiO layer around the nanoparticles. The promising Ni-Mo catalyst could have a marked impact in many challenging scenarios, for instance, for the reforming of biogas containing sulfur impurities, thanks to its activity, poison resistance, and longevity.
The effect of promotion by the addition of Mg has also been investigated [187]. Starting from hydrotalcite-like precursors, a layered material was first achieved by co-precipitation of nitrate solutions that contained Ni, Mg, and Al, and the resulting solid was calcined at 600 °C and pre-reduced in H2 at 800–1000 °C. The catalysts contained Ni nanoparticles of 7–20 nm sizes with overgrowths of nickel aluminate (Figure 15). Catalysts of increasing particle sizes and Ni contents exhibited increasing tendencies to coking and smaller areas covered by overgrowths compared to catalysts with low Ni contents. The overgrowths favored the simultaneous presence of Ni2+ and Ni0, which is related to improved coking resistance and fast turnover for CH4 conversion. Complementarily, La-promoted Ni-containing catalysts were achieved from hydrotalcite-like precursors catalysts at low-moderate temperatures [188]. The authors found NiO segregation and enhanced Ni-reducibility in the La-promoted catalysts, with the generation of medium strength and weak basic sites for low La contents. Due to the improved reducibility of Ni, the activity of the La-promoted catalysts was found to be higher than that of the non-La-promoted analogs, especially for DRM at moderate temperatures (550 °C). The presence of La, however, enhanced undesirable side reactions, which were attributed to an increased rate of CH4 decomposition.
Rare Earth Oxide Supports
Rare earth supports such as mixed lanthanum or cerium oxides normally give moderate DRM performance. For instance, replacing part of the Zr with Ni in the pyrochlore La2ZrO7 led to higher carbon deposition than for a catalyst in which Rh was used as active metal instead of Ni [189]. These results suggested that Rh was a more active and selective center for the production of syngas compared to similar atomic loadings of Ni. When part of La was replaced by Ca, however, less coke was generated. The introduction of Ca2+ cations may have improved the oxygen mobility through the introduction of lattice defects which favor the oxidation of carbonaceous species deposited on the catalyst during the reaction. In each case, the syngas that was produced by the reaction had a very low ratio, close to 0.5. In line, when cerium oxide was used as a support for Ni-based catalyst [167], the produced H2/CO ratio also reached stabilization values significantly below unity. The authors investigated two different Ni/CeO2 catalysts prepared by either thermal or plasma treatment of pre-produced CeO2 nanoparticles onto which Ni was loaded by wet impregnation. The plasma-produced catalyst had an overall higher conversion (40%) and produced syngas with a ratio of 0.55, while the thermally-obtained catalyst only reached 15% and generated a syngas ratio of 0.42. Notably, large dendritic carbon deposits were observed on both spent catalysts, but the plasma-treated catalyst produced more of such carbon structures. We surmise that the promoting effect of the rare earth elements could also involve the occurrence of CO2 reduction with hydrogen and subsequent Boudouard carbon formation, because the formation of coke by the decomposition of methane seems to be slower at the temperatures of these tests (700–750 °C).
In another study [190], the authors utilized Fe modified natural clay supported Ni catalysts with La, Al, or Mn as promoters. The presence of the additives resulted in smaller Ni0 crystallite sizes and further promoted Ni dispersion compared to the unpromoted materials. Al-promoted catalysts improved the Ni reducibility with respect to La and Mn-promoted catalysts. Moreover, the Al-promoted Fe-clay-based catalysts presented the best catalytic performance in DRM from 850 to 600 °C. The reaction achieved thermodynamic conversions with syngas ratios of unity and negligible coking.
Carbide Supports
In this line, the effect of group V and group VI transition metal carbides as support materials for DRM catalysts has been investigated [191]. More specifically, the authors used molybdenum, tungsten, vanadium, niobium, and tantalum oxides which were converted to carbide prior to operating in DRM conditions. Vanadium carbide showed conversion in the magnitude of 70%, followed by a fast deactivation to 50% after 25 h. The H2/CO ratio was close to one for the first hours of the reaction, then slowly decreased to 0.7 after 25 h, which may be a sign that the catalyst is becoming oxidized by carbon dioxide leading to the production of carbon monoxide. The niobium carbide catalyst started in the range of 60% CH4 conversion and progressively deactivated below 50% conversion after 23 h. However, the H2/CO ratio remained stable after 10 h at 0.8. Tantalum carbide showed fast deactivation, although the H2/CO ratio was close to one once the catalyst was operating only at 25–30% conversion of the methane feed. Mo2C, however, showed one of the best patterns for this work, maintaining a methane conversion in the magnitude of 90% for 150 h. The authors also investigated the effect of temperature on their niobium carbide catalyst. The catalysts were tested at 1050 °C and 1100 °C and at 8 bar. At 1050 °C, the conversion of methane was slightly higher than the conversion of CO2, around 90%. After increasing the temperature to 1100 °C, the conversion was superior for CO2 than for methane. This change led to a slight increase in carbon monoxide production and a slight decrease in the H2/CO ratio. Since the conversion of methane was increasing as well, it could be a sign that Equation (7) occurred, which lowers the H2/CO ratio. Complementary molybdenum carbides have been studied on different supports (Al2O3, ZrO2, SiO2, and TiO2) [192]. The relative stability of the catalysts was found to be Mo2C/Al2O3 > Mo2C/ZrO2 > Mo2C/SiO2 > Mo2C/TiO2. Calcination of the oxide precursor for short periods was found to be beneficial to the catalyst stability. Although the support appears to play no beneficial role in the DRM reaction, the alumina-supported material was stable for long periods of time. The evidence suggested that the differences in the stabilities may be due to the interaction at the precursor stage between MoO3 and the support, while catalyst deactivation may occur by oxidation of the carbide to MoO2, which is inactive for DRM.
Perovskite Precursors
Perovskite oxides are crystalline materials that have a similar structure to the mineral CaTiO3. The general formula of these materials is ABO3, where “A” usually denotes a large alkaline, alkaline earth, rare earth cation located in dodecahedral sites of the structure, and “B” corresponds to a transition metal that occupies octahedral sites [15]. Another set of materials termed perovskite-like structures have the formula A2BO4 and feature alternating ABO3 and AO layers (Figure 16).
The popularity of perovskites as functional materials originates from the possibility of fine-tuning the material properties with a careful selection of A, B and substituting elements A’ and B’, which may be incorporated into the framework without altering the structure [193]. Perovskites are thermally stable, which enables their applications in catalytic reactors operating at high temperatures. The utilization of perovskites specifically for methane reforming became a topic of interest during the end of the 1990s [194]. Introspective reviews on the utilization of perovskites in DRM are recently available in the literature [15].
An overview of the literature shows that lanthanum-nickel perovskite is the model material utilized for DRM applications. In one study [195], the authors synthetized the LaNiO3 by an “explosion method” and then reduced the obtained material. The DRM was performed at 700 °C and 800 °C by a pulse strategy, in which the gas mixture of CH4 and CO2 in stoichiometric proportions was introduced over the catalyst. The results suggested that the first step of CO2 adsorption and CH4 cracking initiated the reaction. Subsequently, the DRM was transitorily accompanied by an RWGS reaction, finally leading to stabilization of the reaction products (about 25 pulses). However, a low carbon deposit occurred at the surface of the catalyst, although no catalyst deactivation was observed even after 90 pulses. After the catalytic behavior was stabilized at 800 °C, the detected phases were La2O3 and Ni0. Contrarily, when the reaction was performed at 700 °C, the spent catalyst was found to be composed of the spinel LaNiO4 and Ni0.
Another study utilized the same perovskite system [196], but this time the authors opted to investigate also the performance of the catalyst without pre-reduction. The study found that the perovskite phase was rapidly modified while the temperature was increased from room temperature to 700 °C under a stoichiometric mixture of CH4 and CO2. The reduction properties of methane led to the formation of metallic nickel and lanthanum oxide, while carbon dioxide adsorbed at the surface of the basic in situ formed La2O3 to give rise to oxycarbonate species. The CH4 and CO2 conversion reached levels of 90%, with a molar ratio for H2/CO close to one. For comparison, the authors of another study achieved a similar behavior on Ni/La2O3 catalyst [197] prepared by wet impregnation, with nevertheless much lower conversion and catalytic stability compared to the perovskite-based materials. These studies suggest that the perovskite is most likely not the active catalytic phase but a precursor which may produce in situ or during pre-reduction the relevant working structures, namely the metallic nickel, an intermediary spinel phase, and lanthanum oxides. The comparatively high activity and durability of the perovskite compared to catalysts achieved by wet impregnation could be the result of dynamic phenomena that increase the metal–support interaction and stabilize the working catalyst.
A very recent work explicitly addressed the local and global structural and chemical evolution of archetypical LaNiO3 under slightly reductive ultrahigh vacuum conditions [198]. The authors aimed at disentangling chemical and thermal effects, thus using vacuum conditions to slow the dynamics to measurable levels. The results achieved by in situ electron microscopy, in situ X-ray diffraction, and in situ X-ray photoelectron spectroscopy allowed the authors to visualize step-by-step the evolution of the perovskite into the mixed final state and to devise the role of reactive oxygen more closely. Starting at room temperature (Figure 17), the initial structure of polycrystalline LaNiO3 featured a homogeneous distribution of Ni and La. As the temperature was increased, local Ni enrichment was observed at 400 °C, which was more evident at 600 °C. The onset of detectable Ni exsolution, that is, the local enrichment of Ni from an initially homogenously distributed phase, seemed to be mediated by atomic diffusion of Ni toward the surface with the nucleation of Ni-rich particles occurring on the free surface upon heating to a higher temperature.
The nucleation/growth kinetics compiled in the study suggested that the Ni nucleation took place at low temperatures, while growth was favored at high temperatures. Furthermore, the grain size of the remaining La-rich perovskite structure was found to be smaller compared to the initial sample, which could indicate that the exsolution accompanying the decomposition of the perovskite into La2NiO4 and La2O3 compressed the crystal lattice [199]. The authors detected high amounts of NiO in the exsolved Ni. Hence, O loss occurred during the LaNiO3 decomposition, creating oxygen vacancies [200]. The authors argued that oxygen diffusion was driven by the pressure gradient from the bulk to the sample surface [201], which may be a general explanation of the high oxygen mobility in perovskite-like materials not only for DRM but also for oxygen evolution reaction. This is a well-known feature of perovskite catalysts and may be one of the effects that increase the overall resilience to coking. Subsequently, the migrated oxygen oxidized metallic Ni on the surface to NiO.
In addition to the exsolution effect, another feature of perovskite-like materials is the possibility of either partially or completely substituting La and Ni with other elements in both the A and B cation sublattices, giving rise to a complete set of functional materials. The chemical influences of partial substitutions are of the same nature as the promotion and bimetallic effects summarized in previous sections. For instance, the effect of introducing Co, Fe, Cu, Bi, and Cr in the Ni-B sublattice site was evaluated in a study [202]. The authors utilized materials of general composition La0.8Sr0.2Ni0.8M0.2O3, which were prepared by the sol–gel process in which citric acid was used as the complexing agent. The M group refers to various elements (M = Bi, Co, Cr, Cu, and Fe) utilized in the partial substitution of the B-site. The catalytic activity to DRM was negligible in the bismuth substitution, which the authors explained as a consequence of fast Bi2O3 formation encapsulating the catalytic particle. In this line, Co and Cr introduction led to catalysts initially very active, which deactivated after only 150 min on stream due to coking and Ni sintering. The Cu-doped catalyst also exhibited high initial activity, close to 90%, which started to decline after 12 h of treatment. The authors argued that the bimetallic Cu-Ni nanoparticle could be more easily reduced than the monometallic analog; however, it still has some tendency to form big metallic agglomerates, which may favor coking. On the other end, the Fe-doped catalyst had an initially low conversion of only ~50%, which was found to be maintained for 300 min on stream, and then slowly increased and stabilized after 500 min at values above 80%. Coke was not detected in this catalyst. The effect of Fe substitution on this study may be originated from a combination of stronger metal–support interactions, which hindered thermal agglomeration of the Ni particles and the presence of lattice oxygen species. These are less active for C–H bond activation but active to react with CO2 to form La2O2CO3, which enhances surface carbon oxidation.
In another study [203], the authors achieved mixed perovskite precursors with nominal compositions of La2NiO4, LaNiO3, La2Ni0.5Fe0.5O4, and LaNi0.5Fe0.5O3 by a wet impregnation method. The study found that the only-Ni catalysts had the highest activity for CH4 (90%) and CO2 (80%) conversions upon DRM at 750 °C. However, the catalyst further decomposed during the reaction and gave rise to the formation of large quantities of coke after only 4 h on stream. Notably, the Fe-substituted analogs exhibited much lower activities of both reactants, in the order of 50% to 70% for the initial values, which thoroughly increased over time and reached values similar to the monometallic Ni catalysts. They also produced comparatively much less carbon (eight-fold improvement) for similar times on stream. The authors attributed these observations to a limited rate of perovskite exsolution in the bimetallic materials, which imparted a higher durability and coke resistance due to strong metal–support interactions and tuned redox affinities.
A study exploring the systematic substitution of Ni by Co based on the LaNiO3 material and prepared by sol–gel resin synthesis [204] has shown that a bimetallic effect is achieved in these materials for substitution degrees below 60%. The bimetallic catalysts exhibited conversions and selectivities close to the thermodynamic equilibrium. However, monometallic catalysts were not so effective as single-Ni prompted coke formation, and excessive Co led to materials hard to be reduced. The authors argued that the in situ formation of Ni0-Co0 particles highly dispersed on a La2O2CO3 matrix might inhibit coke formation through interactions between the metallic phases. The closeness of Ni0-Co0 would provoke synergetic effects that stabilize active particles and suppress coke formation. In a similar study, the catalysts were prepared by co-precipitation [205]. Contrary to the previous results, the authors found that the catalytic activity and the stability of LaNi0.5Co0.5O3 were less than those of LaNiO3, while the coke resistance was higher. Controversially with these findings, another study in which a trimetallic (Ni-Co-Mn) catalyst was prepared by the microwave-assisted Pechini strategy [206] found that the addition of Co and Mn to give a LaNi0.34Co0.33Mn0.33O3 catalyst precursor had a different influence on DRM. Upon reaction, the trimetallic analog was found to have improved activity and stability compared to LaNiO3. The authors speculated that Mn migrated preferentially into the La2O3 matrix, where it could improve the stability of the catalyst through an increased rate of carbon oxidation, whereas Co could act as an additional active component that increased the intrinsic reaction rates. Strong metal–support interactions mediated by MnO were invoked to explain the synergistic effect of the tri-metals. We note that the apparent discrepancies in these studies may originate from the different synthetic strategies, which could strongly modulate the morphologies, reducibilities, and stabilities of the investigated materials. Nevertheless, a general observation that may be important in prospective catalyst preparations is that the Ni/Co ratio in the Ni-Co bimetallic perovskite derived catalysts is crucial, with low loadings of Co being insufficient to enhance the activity, whereas too high Co contents being able to suppress the reducibility and activity [15] because in essence Co may be less active alone for CH4 activation [207].
Another modification on perovskites may be achieved by partial or complete substitution of the A-site cation with alkaline, alkaline earth, and rare earth elements. As in other material types, the influence of such modifications can be understood as a promotion effect that increases the support basicity or enables the formation of mobile reactive surface O. Here, preferred substitution additives are K, Ca, Mg, Sr, Ba, Ce, Pr and Sm [15].
Apart from the high number of combinations and increasing reports on perovskite-like catalysts, we mention that other high-end catalytic architectures such as nanoporous silica and/or alumina may be superior in terms of catalytic performance. Carbon deposition and thermal stability are still areas of concern for perovskites in scenarios of long operation under industrial conditions. Moreover, perovskite usually exhibits a poor texture property, exemplified by small surface areas in the order of 15 m2 per gram and little porosity. As a solution, the perovskite material may be synthetized over supports of a large surface area. Another topic of concern relates to the utilization of harmful elements such as Ba and Sr, which are very common in perovskite preparations. These current limitations may hinder the implementation of these materials in large-scale DRM and suggest instead that further research efforts are needed to strengthen the capabilities of these promising materials.
5.1.4. Non-Traditional Catalytic DRM
Alternative approaches other than catalysts composed of metallic nanoparticles supported on oxidic materials may also be considered for present and future DRM developments. Moreover, there is no reason that the reaction should exclusively operate in fixed bed reactors, while other reactor designs such as membrane-based systems and cyclic operations involving separated DRM and regeneration steps may dramatically improve the current efficiency of the reaction with catalytic materials already at hand [108].
In the experiments undertaken in a Catformer [85,108] the authors have already shown that DRM could be operated sequentially in a reformer followed by a regeneration step. With a similar approach, the cyclic operation was achieved on supported Ni catalysts (Figure 18) [208]. After methane decomposition at 550 °C, coke was deposited. Subsequently, coke was oxidized in a pure CO2 atmosphere at 650 °C, which may have enlarged the longevity of the catalysts. With the same idea, other studies have successfully achieved the regeneration of Ni/ZrO2 catalyst after a stage of methane decomposition [209,210]. Overall, this would lead to a syngas ratio of 1. This represents an interesting approach in catalyst regeneration and may also be applied in other catalytic technologies where deactivation via coking is an issue, e.g., propane dehydrogenation [40].
In a recent approach [211], high conversion of CH4 and CO2 to syngas and solid carbon was achieved through simultaneous pyrolysis and DRM in a bubble column reactor using a molten metal alloy catalyst (65:35 mol% Ni:In). The H2/CO ratio could be controlled by the reactant’s ratio, reaction temperature, and residence time to produce stoichiometric solid carbon as a co-product that is separable from the molten metal. This facilitated the catalyst´s regeneration and improved the catalyst´s longevity. The reaction was discussed in terms of a Mars-van–Krevelen-like kinetics in which CO2 is reduced by a liquid metal species (for example, In) and methane is partially oxidized to syngas by the metal oxide intermediate (for example, In2O3), regenerating the native metal.
Another technology of interest is the utilization of so-called membrane reactors [212,213]. A feature of these reactors is the possibility of undertaking two unit operations, reaction and product purification, in a single system. The constant withdrawal of one of the products, in particular H2, may lead to apparent conversions above equilibrium, while in conventional fixed-bed reactors, conversion is limited by thermodynamic equilibrium. For endothermic reactions, this potentially allows achieving high conversion at comparably low temperatures. Most investigations have centered on the use of dense Pd or Pd/Ag membranes. Pd is chosen thanks to its high H2 solubility and permeability [214]. These membranes, while exhibiting excellent selectivity toward hydrogen, suffer from a number of drawbacks: they are expensive, exhibit relatively low H2 permeability, are susceptible to poisoning by sulfur and coke (both typically encountered in catalytic reforming), and are prone to pinhole/crack formation as a result of hydrogen embrittlement and fatigue [212]. To avoid these difficulties, other investigators [213] used porous ceramic membranes. These membranes exhibit, in general, better mechanical and thermal stability than dense film membranes but allow gases other than hydrogen to permeate through.
In a recent study [215], the authors highlighted the use of a porous Ni catalyst for the DRM reaction and a Pd-Cu membrane for H2 separation. According to the investigation, it is necessary to improve the DRM performance from the viewpoint of reaction kinetics and H2 separation. A Ni-based alloy catalyst is a promising catalyst in order to improve the performance of CH4 dry reforming since it may be that the catalytic reaction is still slow at moderate temperatures (400–600 °C) of the study. In another study [216], the authors achieved a high production of H2 (82%) with a purity of 100% in the permeate at 400 °C using SRM on a metallic membrane. The outcome showed that the methane conversion is above 57% in the membrane configuration and produced double the amount of hydrogen compared to a fixed-bed reactor. Initially, the membrane showed a near-infinite selectivity towards hydrogen permeation at pressures less than 1 bar. After 750 h on stream, the ideal selectivity of hydrogen dropped. Nevertheless, no CO was detected in the permeate side for all the experimental campaigns, which showed that as the membrane slowly degraded, it had the ability to still produce CO-free hydrogen for feeding a fuel cell or for other hydrogen-sourced industrial application. The authors utilized a porous stainless steel support clogged with Pd. Furthermore, in an interesting study that compared conventional and membrane reactors [217], the authors found that the membrane system (PdAgCu alloy) containing a catalyst of 5% Ni/Ce0.6Zr0.4O2 outperformed the fixed-bed reactor during DRM in terms of conversion and H2 yield. The authors found a 35% H2 yield compared to 11% achieved by a traditional packed catalyst bed under similar reaction conditions, including the ratio of the reactants (1:1) and temperature (550 °C). Furthermore, the efficiency of H2 recovery was maintained during the catalytic tests using the membrane. Until now, the membrane reactor has some drawbacks which may need careful attention in the future. One of these is the clogging of the membrane, which would slow the rate of hydrogen recovery. Furthermore, the membrane may be mechanically damaged or lose its properties during long-term operations. Apart from this, this technology is highly attractive because it could deliver a valuable pure H2 product without the need of further purification, thus helping in the decarbonation of the product stream which leads to a cleaner energy storage.
5.2. The Learning Curve of Catalytic DRM
From the above-mentioned overview of different catalytic systems, we can extract some important prerequisites for a high-performance DRM catalyst. The metallic nanoparticles have to be present in high quantities. In addition, they have to be as small and as dispersed as possible over the support. Alumina, magnesia, and ceria support usually provide high performance. However, functionalized porous silica or alumina supports with core-shell architectures may be preferable for high-performance catalysts. Incorporation of cerium or lanthanum compounds as well as potassium has been reported to increase the catalyst’s efficiency, while the promotion with manganese or tungsten compounds had the opposite effect. The utilization of a promoter or a co-catalyst was found efficient in such cases where its implementation into the catalyst system (for instance K, Ca, La, Ce, Co, Fe, etc.) allowed further spreading of the nickel particle or when it promoted the activation of carbon dioxide. The metal–support effect could be rationalized in terms of high dispersion and the formation of mobile oxygen species. In addition, the co-catalyst shall also function as an inhibitor for carbon formation. Furthermore, for industrial purposes, the catalytic system should be simple, economically friendly, and non-toxic. A schematic illustration of the structure of high-performance DRM catalysts derived from this literature overview is presented in Figure 19.
We also mention here that it is difficult to derive common features of reported high-performance DRM catalysts as most of the studies have been undertaken under different conditions, which limits direct comparisons. Due to the complexity of the chemical networks, variety of synthetic protocols, and combinations of operation parameters, the most efficient DRM catalyst has still to be optimized empirically for a particular application. Therefore, we suggest implementing a handbook for DRM testing in which important reaction parameters are defined. Apart from individual parameter selection, these handbook parameters have to be tested as supporting information. It is this comparison that will allow conclusions on how a study relates to the finding of others. A similar test protocol has recently been introduced for selective oxidation catalysis [218].
5.3. Non-Catalytic DRM: Plasma and Photochemistry
Apart from thermocatalysis, other approaches based on other energy sources such as light and electricity have been developed in recent years as a means to relieve the impact of atmospheric gas emissions [219] and to valorize the wasted gases in an energy-efficient approach. A very important part of the reforming reaction is energy, which has to be sufficient to overcome the thermokinetic barriers inherent to CH4 and CO2 activations. Energy input is also necessary to keep the endothermic reaction running. Hence, other ways of energy supply apart from heat should also be explored, which may be more efficient than current thermocatalytic approaches.
The future of DRM is linked to renewable energy sources. In a study about dry solar reforming of methane [220], the authors showed a significant accomplishment by avoiding coking with operation at low temperatures. The resilient system was energized by a Xe lamp that simulated (part of) the solar spectrum. The focused light heated the catalyst to temperatures in the range of 459–596 °C, depending on the part of the spectrum utilized. The approach exhibited impressive processing capacities of CH4 and CO2, with high production rates of H2 and CO (17.1 and 19.9 mmol min−1 g−1, respectively). The solar-to-fuel efficiency of the catalyst was as high as 12.5% [220]. This innovative system can potentially lead to technology with minimal energetic investment as it feeds on a free and environmentally friendly source. However, light-driven systems still seem to lack the ability to process high quantities of feed compared to other technologies. There are quite a few technical issues that have not been resolved yet, even after several decades of intensive research. As a result, widespread commercialization of solar reforming has not been achieved. Amongst others, drawbacks are high capital cost investments in solar installations, problems related to the thermal stability of the reformer materials, and, particularly, shortcomings related to solar thermal reforming catalysis [221].
Alternate strategies also include plasma-assisted chemical activation [17,222], which may help to operate the reaction at lower temperatures to avoid coking and with an overall lower energy investment [223]. A prominent feature of plasma technology is the simple scalability from small laboratory reactors to large industrial installations with megawatt input powers by a combination of smaller functional parts [224]. In a recent review [223], the authors discussed the benefits of plasma-based valorization strategies for CO2 and CH4. In general, plasma could help with the chemical activation of catalysis-based technologies and may help diminish the energy investment during (pre-)reaction stages.
There are comprehensive reviews in the literature [17,225,226,227] and further studies [228,229] which suggest that the plasma technology may be used to scale DRM up to commercial levels. Plasma is a state of matter characterized by the presence of charged (electrons, ions) and uncharged (atoms, radicals, molecules) species. Unlike conventional gases, plasmas are composed of a mixture of particles of different masses and charges which are expected to respond differentially to external stimuli. Therefore, the behavior of chemical species in plasmas is much more complex than in classical thermochemical reactors dealing with neutral gases.
The direct plasma reactor intends to generate plasma from the reactant mixture by external energy supply. Further recombination of the generated species may form syngas or may induce the formation of intermediaries necessary for catalytic DRM. There are a plethora of ways of generating plasma in a gas mixture, which diverge in the reactor geometry, thermal characteristics of the produced plasma, operating pressure, and electrical field applied. In general, plasma can be generated by the application of an electric field to the gap between two electrodes. For low-pressure applications and depending on the wave form of the electrical field, the plasma method may be termed as glow discharge (DC field or RF field of low frequency), capacitively coupled plasma (RF field of high frequency) [230], inductively coupled plasma (in which an electrode is coiled around the other) [231], and cascade arc devices [232]. Of major interest to achieve high processing efficiency is also the generation of plasma at atmospheric and higher pressures, including the methods of arc discharge (with either DC or AC voltage and high currents), corona discharge (generated on sharp tip electrodes with high voltages and low currents), dielectric barrier discharge (in which a dielectric material is placed on the electrode tip to avoid arc discharges), microwave plasma, spark discharge (which is the transition between corona discharge and glow discharge) [233] and atmospheric-pressure plasma jet [233,234].
When the electrons and heavy particles of the plasma have about the same thermal energy, the plasma is considered in thermodynamic equilibrium. [235] Hence, in equilibrium or “thermal” plasmas, the local gas temperature usually reaches thousands of Kelvin due to high-frequency collisions between electrons and the heavy chemical species. With increasing kinetic energy, activation energy is more easily achieved, and the rate of reforming can be significantly improved [17,226,236]. In addition, electron collisions on molecules may induce dissociation and ionization reactions, thus inducing additional reaction paths which may be conductive to the syngas product. Thermal plasmas have been applied for DRM and reforming of light hydrocarbons, with conversions over 80% and usually small carbon deposition [225,237]. In a study [237], the authors prepared a thermal plasma by the jet discharge of nitrogen gas and investigated the effect when the plasma was applied alone or in cooperation with a commercial catalyst to achieve the DRM. Both kinds of experiments were conducted in the same conditions except with or without catalyst in the reactor. The authors found that conversions of CH4 and CO2 amounted to 96.33% and 84.63%, and the selectivities of CO and H2 were 91.99% and 74.23%, respectively. Reaction observables were higher in the presence of a catalyst than those by thermal plasma alone. Although thermal plasmas showed excellent conversion and syngas selectivities, their operation required massive energy to maintain the high operating temperature [17].
A non-thermal plasma, on the other hand, is not in thermodynamic equilibrium because electrons may have a much higher temperature compared to the heavy species. Hence, the “gas” is usually at room temperature or may reach only a few hundred Kelvin above ambient conditions. Non-thermal plasmas, including corona discharge, gliding arc, dielectric barrier discharge, glow discharge, and microwave and spark discharge, have been investigated to generate syngas by DRM and other reforming reactions. Generally, CH4 and CO2 conversions achieved with non-thermal plasmas are lower than that of catalysis or thermal plasma. Moreover, side products including coke, ethane, acetylene, and higher hydrocarbons are usually formed during discharges and limit their application. In a study [235], a non-thermal plasma was generated by corona discharge in a mixture of CH4/CO2 with a variable feed ratio between 2.0 and 0.2 at ambient temperature and pressure. The authors achieved conversions in the order of 80% for CO2 and 91% for CH4. They also compared the AC corona plasma with previous reports involving DC corona plasma. The highest conversions were realized in a simple plasma reactor, and the H2/CO ratios in the products strongly depended on the CH4/CO2 ratios of the feeds. The conversions increased in the order +DC corona > AC corona > −DC corona, whereas H2/CO ratios in the products exhibited the opposite order. It was found that the corona plasma reactions often had higher conversions than the calculated values according to thermodynamics at room temperature. In another study [229], the authors evaluated the physical parameters of a dielectric barrier discharge reactor, including electric power, flow rate, gas pressure, and temperature. The results demonstrated that an arbitrary desired H2/CO ratio could be achieved from CH4/CO2 mixtures. The results suggested that the H2/CO molar ratio in the syngas was a simple function of the CH4/CO2 ratio in the feed. Specific input energy, gas pressure, and the temperature hardly influenced syngas composition. Only carbon and soot formation had a deviating effect on the syngas ratio. Syngas production strongly depended on the electric input energy, CO2-rich mixtures prevented carbon and wax formation. The highest CH4 conversion was 64%, and the CO2 conversion reached 54%. High operating temperatures led to coking in CH4-rich feeding mixtures, and a decreasing operating pressure favored the production of syngas.
In summary, the thermal plasma technology may be beneficial for a direct achievement of the syngas product by DRM. Some key factors have been identified in the past years in a cross-correlation study of the known plasma technologies [225]: electron density, plasma temperature, and reactor configuration. Corona discharge and dielectric barrier discharge are non-uniform plasmas with low electron density and limited reaction volume, which restrict their treatment capacity. Microwave discharge is a uniform discharge with high plasma temperature and large discharge space, but the equipment is more complicated and expensive, which may restrict its industrial application. Gliding arc discharge has high electron energy and electron density, as well as proper plasma temperature, but the reactor is difficult to enlarge, and the treatment capacity is still far from industrial requirements. Thermal plasma is efficient for DRM because of its high specific energy content, high temperature, high electron density, large treatment capacity, ease to enlarge, and relatively high energy conversion efficiency.
Furthermore, the energy conversion efficiency of plasma and light-driven processes may be further improved with the integration of thermocatalysis technology, which may be achieved by single-stage or two-stage hybrid systems, or by the utilization of plasma and light as strategies of catalyst pretreatment for tuning physicochemical properties to achieve higher activity and durability [17,225].
Combining plasma or photochemistry and catalysis can merge both advantages to overcome the shortcomings of these techniques. There are three ways to achieve this:
- (i)
- Operating non-thermal plasma or photochemical process as catalyst pre-treatment. Previous studies have shown that non-thermal plasma can change some of the properties of the catalyst. Since non-thermal plasma and photochemistry generate many species, e.g., electrons with high kinetic energy, oxygen atoms, and hydrogen atoms, the collision between particles and solid catalysts may induce various reactions to alter the structure of the catalyst, e.g., reducing or oxidizing the metals on the catalyst surface. Hence, the catalyst pretreated with these strategies can be improved to have higher activity, better durability, or higher selectivity [226].
- (ii)
- Sequential plasma-catalysis. Placing the catalyst after the discharge region of the plasma is a possible way to induce synergistic effects between plasma and catalyst, which is also called post plasma catalysis (PPC). Both thermal plasma and non-thermal plasma can be adopted before the catalyst bed to form a two-stage hybrid system. In a non-thermal plasma catalysis system, the long-lived reactive species produced by non-thermal plasma, e.g., radicals, vibration excited species, and ionized molecules can react with the catalyst to induce catalytic reactions by several surface mechanisms [226]. Thus, the conversion and selectivity of syngas generation can be enhanced by combining two reforming techniques.
- (iii)
- Hybrid single reactor systems that combine plasma or photochemistry and catalysis. The system is composed of a catalyst inserted in the discharge region of plasma or directly uses a photocatalyst [238]. The catalyst can be further modified in situ with the help of the plasma. Moreover, two kinds of interaction can be induced and may influence each other. The first is the effect of plasma/light on the catalyst, such as the modification of physicochemical characteristics and the work function of the catalyst. The second is the effect of the catalyst on the plasma, such as changes in the electric field distribution and the distribution of species in the plasma. It is noted that thermal plasma is not suitable in this system due to the deactivation of the catalyst at the high operating temperature (103–104 °C) that the gas may reach [226].
Until now, the mechanisms of plasma catalysis and photocatalysis for DRM are still vague. Even though many approaches have been adopted to enhance the DRM efficiency, a good hybrid system still needs further developments to commercialize this technology.
5.4. The Requirement of Operando Studies for DRM
The information about the thermodynamic state of the catalyst often involves complex interactions. For instance, catalysts adapt dynamically to the external conditions (chemical dynamics), favor the appearance of intermediaries, metastable phases, and transitions states which deviate largely from equilibrium (catalytic action) [239], and may fuel reaction paths that do not lead to equilibrium compositions of the products (selectivity control) [240]. The outcome from this combination of factors is difficult to predict or even theorize without knowledge of the catalyst state during the reaction. Complementarily and traditionally, information about the catalyst is collected ex situ by comparing the structure prior to and after the reaction, which has shed some light on the impact of reaction dynamics on the chemical reaction network. However, their informative depths may be limited as real-time information is missing, and the occurrence of external artifacts (e.g., cooling, removing the reaction atmosphere, transfer, etc.) cannot be disregarded. Hence, experimentation that aims at gathering information about the catalyst when it is performing its action is critical to shed light on the behavior of these reactions and bring our current knowledge closer to real processes.
Operando studies are widely absent for DRM. Recently, a study addressed Ni-Ru catalysts by operando DRIFTS, revealing in this case that Ru atoms block the most reactive Ni sites, thus leaving less reactive centers for methane activation. The effect also favored carbon gasification and prevented CO dissociation [80]. Another study aimed at investigations of the oxidation state of Ni and Co catalysts supported on zirconia has shown by operando XAS that the Co catalyst was progressively oxidized from the surface to the bulk during the reaction, whereas CoNi and Ni remained relatively reduced during the DRM. This demonstration highlights the successful achievement of real-time monitoring of the catalyst evolution under operando conditions, which is necessary for in-depth and accurate studies of structure-activity relationships [80]. A similar study [79] compared kinetic data and operando XAS and showed that the pure Co resulted in richer oxygen coverage, while pure Ni maintained a reduced state and clean surface at the steady state. The XAS results provided further evidence of the oxidation of pure Co during DRM, which progressively occurred from the surface to the bulk with time. The Ni catalyst experienced carbon deposition, while the bimetallic CoNi catalyst showed a negligible amount of deposited carbon, likely due to balancing the oxidative and reductive reactions on the catalyst surface. This information would have been inaccessible by other means, which in part explains the contradictory explanations of important aspects of the catalyst, such as the bimetallic and promotion effects.
An operando electron microscopy study (Figure 20) [78] has been presented, highlighting the importance of oxide/ metal phase transitions in the functioning of this reaction on a Ni catalyst. In this study, changes in the H2/CO ratio during activation and over time on stream under isothermal conditions have been found, pointing to different reaction mechanisms that were governed by the dynamic surface states. In addition, the apparent activation energies for hydrogen and CO production have been extracted during heating and cooling campaigns, showing that while the activation energies related to hydrogen remained constant during heating and cooling, the activation energies for CO decreased during cooling. This suggests that the formation of CO was favored after the stabilization of the performing structure, which led to a decreasing H2/CO ratio.
The latter operando experiment further suggests that DRM can be a valuable reaction to study syngas formation, which is the starting point of a complex C1 reaction network, simply by fine-tuning the reaction parameters. However, it requires that the structure and function are simultaneously detected.
6. On the Maturity of DRM in Industry
The overview compiled above suggests that critical aspects of catalytic DRM, such as its tendency to coking and sintering, may currently have been solved with the implementation of catalysts and operation procedures designed to cope with the hard conditions of an industrial process. Due to the fact that DRM enables the conversion of CO2 into valuable products, the hope has been raised that it could potentially serve as a “CO2 sink” which can help to improve the overall carbon footprints of industrial facilities [42] while valorizing a byproduct traditionally considered as waste. This scene has stimulated the application of DRM also at industrial scales, although with a somewhat less ample front compared to the other methane reforming technologies. We mention in this context some initiatives, projects, and process variants that have recently been put in operation and demonstrate the integration potential of DRM in large-scale production chains.
Linde AG, in collaboration with BASF SE, has paved the way for mature DRM applications. In their DRYREFTM process [241], the researchers solved the coking tendency of the reaction with two innovations. First, the process runs under combined SRM-DRM conditions. The co-feeding of steam reduces the amount of coke deposits because, as discussed above, water is a more potent oxidant than CO2 and may buffer out the concentration of gas-phase radicals that act as coke precursors. This modification also enables the production of syngas of any ratio between one and three by adjustment of the steam/carbon ratio. For comparison, conventional SRM needs steam/carbon ratios above two, which penalizes the overall energy efficiency as a large amount of steam has to be generated. Second is the utilization of the high-resilience, high-performance catalyst SYNSPIRETM G1–110 by BASF SE [242]. This catalyst features Ni species as active components and a state-of-the-art spinel-like metal oxide carrier, which helps to rapidly oxidize any carbon deposit. The catalyst has been tested for more than 1000 h on stream at the lab, pilot, and commercial reformers at typical industrial conditions of 20–40 bar at 800–950 °C with a projected lifetime of 5–8 years. Furthermore, the goal of achieving a dry reformer may now reach the stage of integration in a DME plant. The direct production of DME from the syngas generated by DRM may offer 30% CO2 emissions savings compared to a setup where a conventional reformer feeds a methanol plant intended to subsequently make DME. This may potentially lead to less carbon footprint than current technologies can do [243]. In line, Linde AG and BASF SE are currently investigating direct DME synthesis from the DRM syngas over catalysts based on zeolite supports modified for rapid carbon oxidation ability.
Investigators from the National Institute for Material Science, in collaboration with Kochi University of Technology and Tokyo Institute of Technology, recently developed a high-performance catalyst for low-temperature DRM [244,245]. The catalyst was prepared by melting pure Ni, Y, and Al in an Ar-protected arc melting furnace. The prepared ingots were ground into powder, and then the precursor powder was de-alloyed in a 30 wt.-% NaOH solution. The nanoporous nickel composite was stable against heat and coking due to its unique structure preventing pore coarsening. The inert and bundled Y2O3 support achieved by this method was found to be a suitable and robust substrate for metallic nanoporous Ni stabilization thanks to its high porosity [245]. Furthermore, in situ microscopic observations demonstrated that the carbon deposition occurring on traditionally supported catalysts was attributable to the sintering of metal nanoparticles, while the active center of the novel catalyst was found to be immobilized by its oxygen-deficient support. The catalyst outperformed conventional supported catalyst during 1000 h on stream at relevant pressures between 4–6 bar at 450 °C. This project is expected to be scaled up in the following years for pilot plant investigations.
Ironmaking is another sector of great potential for greener syngas technologies. Indeed, DRM may have a niche in this industry more prominent than in the preparation of chemicals. Normally, iron ores are reduced with coal during the blast furnace process, which generates a large portion of CO2 emissions. In the MIDREX processes, the ore is reduced directly by a share of strategies aiming at reduced carbon utilization. The company developed processes of ironmaking using natural gas, a mixture of natural gas and H2, and their greenest technology in which only H2 is used for the ore reduction [246]. In the most common process, the natural gas enters a reformer unit before meeting the ore at the shaft furnace. The hydrocarbons are reformed into syngas mixtures, which then react with the ore for reduction and with metallic iron for carburization. The exhaust gases containing CO2 and water are recycled to the reformer, in which further syngas is generated by combined SRM-DRM using a Ni-based catalyst [247,248]. Another initiative named Carbon2Value was recently put forward by an international consortium led by ArcelorMittal [249]. The objective of this project is to demonstrate the potential of reducing gas emissions in the steel sector by >30%, implementing a cost-efficient breakthrough solution for the separation of CO2 and CO, which are unavoidably emitted today. They plan to construct a pilot line that divides carbon-rich gases into two streams, one rich in CO and another one rich in CO2 that could be valorized into promising chemical building blocks in the future by syngas technologies.
A similar strategy was recently initiated by Saarstahl and Dillinger in cooperation with Paul Wurth of the SMS group and deals with the further development of the DRM technology utilizing blast furnace gases. A cooperation agreement to this effect was signed in 2020. The pilot plant is to be taken into operation at the site of the joint subsidiary of Dillinger and Saarstahl in Dillingen [250]. The first step of the project in the development of the DRM technology is the construction of a pilot plant for testing the process at a small scale with industrial gases. A further objective of the pilot plant is to test materials and components in regard to their suitability for the construction of an industrial-scale plant. The start-up of the pilot plant was scheduled for summer 2021 [250], but further information about the results of this initiative may still need some time.
In a very attractive strategy, the authors of an invention achieved the conversion of three equivalents of CO2 per mole of CH4 in a novel reaction called “super dry reforming” [251]. The reaction is based on the cyclic operation principles of combined “chemical looping” and “calcium looping” [252], which enables the undertaking of the catalytic reaction and regeneration in situ by external temperature modulations. The super-dry reforming instead operates isothermally, and the looping is achieved by changes in the composition of the feeding gas. The process features three catalytic components: a conventional reforming catalyst of NiO/MgAl2O4, a CO2 sorbent made of CaO/Al2O3, and a solid oxygen carrier made of Fe2O3/MgAl2O4. In the first step, the DRM reaction mixture enters the reactor, and methane reduces the iron oxide into metallic iron and/or oxides of a low oxidation state (e.g., FeO). The generated/fed CO2 is captured in situ by the CaO absorber, producing CaCO3 while water exits the reactor. The capture of CO2 shifts the equilibrium position to 99% conversion of methane by Le Chatelier’s principle. In a subsequent step, an inert gas stream is fed into the reactor, which stimulates the decomposition of the carbonate with the desorption of CO2. The released CO2 reacts in situ with the iron reduced in the previous step to regenerate the native iron oxide and produce CO as a byproduct. The combined cycles of “charge” and “regeneration” produce four equivalents of CO for a feed of three moles of CO2 and one mole of CH4 without the generation of H2. This technology would then efficiently “sink” the CO2 as an intensified stream of CO (H2/CO = 0) utilizing less energy-carrier CH4. The reaction has recently been targeted for large-scale applications in the frame of the Belgium project Moonshot SDR [253]. In addition to producing efficient materials, the running SDR project aims to build a pilot plant based on a multi-reactor configuration to demonstrate the transition from a linear to a circular economy of carbon in which the super dry reformer will be embedded.
Finally, we mention in this context the applications of alternative energy sources as process improvements which also aim at reducing the CO2 emission generated, for instance, during the heating of the catalytic reactor. Normally, the reformer is heated by burning natural gas. Today, companies aim at further reducing greenhouse gas emissions with the development of electrically-heated systems which are easily integrated with renewable sources of energy such as wind power and hydroelectricity. A proof of concept invention [254] described the integration of an electrically heated catalytic structure directly into an SRM reactor for hydrogen production. Intimate contact between the electric heat source and the reaction site set the reaction close to thermal equilibrium, increased catalyst utilization, and limited unwanted byproduct formation with minimal energy investment. The compact design potentially allows 100 times smaller reformers than current reformer platforms. The authors claim that the intimate contact between the electric heat source and the catalyst enables energy to be supplied directly to the active sites, removing thermal limitations and providing well-defined control of the reaction front. Electrification removes the fired section, substantially reducing reactor volume, CO2 emissions, and waste-heat streams [254]. In order to take this technology one step further, BASF SE, in collaboration with Linde AG and SABIC, has recently teamed in the development of “E-Furnace”, the first electrically heated furnace for large scale reforming applications [255]. The parties are evaluating the construction of a multi-megawatt demonstration plant at Ludwigshafen facilities which is intended for start-up in 2023.
In a preliminary study in 2011 [256], the authors investigated the DRM on an iron bed (iron wool acquired from a hardware store) activated with an electric current at temperatures ranging from 700 to 800 ° C and pressure close to 1 bar. The reaction, involving an excess of pure methane and carbon dioxide, produced syngas at production rates and compositions that depended strongly on the electron flow at the reactor. However, the power required for the conversion was found to be significantly higher than the amount of energy originally fed according to the calorific value of methane. However, the authors improved later the reactor design, achieving electricity investment in this case which corresponded to about 20% of the value of methane injected in the reactor [257], a value comparable to state-of-the-art SRM platforms. In 2018 the team aimed at the assembly of a pilot-scale reactor with a capacity of 40 L/min. The technology now relies on one patent and one patent-pending process and should be compatible with natural gas, biogas, and tail gas dry reforming [258].
7. Concluding Critical Remarks
7.1. General Considerations
From the academic perspective, it is clear that many interesting research questions remain unanswered without being explored by modern operando techniques. For instance, we can ask about the fundamental effect of the bimetallic catalyst or about the reasons that chemically different additives/supports such as La2O3 and MgO exhibit outstanding performance in the stabilization of the nanoparticles. Is it only the introduction of basic sites? Is there a modification of the functional interface between metal and oxide? Do properties of the pristine as-synthetized catalyst hold for the wide temperature range the reaction is meant to work on? How do surface hydroxides and carbonates appear at high DRM reaction temperatures? Is DRM improving energy storage?
From the industrial and practical perspective, the alleged immaturity of DRM was presented, which is mainly limited by its deactivation tendency due to coking. Nevertheless, some of the systems that have been presented in this review seem to imply that high-performance DRM with only marginal deactivation is already a reality, and the large-scale implementation of DRM has just started. In the following, we now critically explore further aspects of the reaction which may become relevant in future scenarios.
7.2. Energy Storage
Due to the intricate entanglement of the reactions associated with syngas technology, it is difficult to conclude the energy storage efficiency or final environmental impacts on a large-scale scenario of DRM. For instance, liquid fuels provide a larger energy density than gaseous analogs. However, synthetic liquid fuels achieved, for instance, by Fischer–Tropsch reaction feed on several upstream processes which have already required some significant energy investment. The assessment of actual energy investment has to be tracked across the whole production chain from the raw material.
To illustrate this, the scheme of Figure 2 summarizes the stream processes associated with syngas technology [3,68]. For example, most of the H2 intended for industrial methanol synthesis (Equation (10) is currently obtained by a combination of SRM (2)) consecutively followed by stages of WGS reaction (Equation (33) = inverse of Equation (7)) [14].
CO + H2O → CO2 + H2 ΔH298 = −41 kJ mol−1 WGS
The combination of Equation (2) and Equation (33) leads to Equation (34) which refers to the production of H2 from CH4.
2H2O + CH4 → CO2 + 4H2 cumulative ΔH298 = 165 kJ mol−1 H2 production
Subsequently, the synthesis of methanol may be achieved according to Equation (35) with an exceeding of one H2 molecule:
CO2 + 4H2 → CH3OH + H2O + H2 cumulative ΔH298 = 116 kJ mol−1 CO2 Hydrogenation: methanol synthesis
However, the carbon required for the synthesis of one equivalent of methanol as presented in Equation (35) is already in the CH4 feedstock, suggesting that steam reforming-based methanol synthesis is only marginally suitable for CO2 capturing and cycling apart from the H2 molecule produced during the successive reactions. In contrast to Equation (10), this route of methanol synthesis would make the whole process endothermic due to the high energy input for H2 production. Therefore, with current technologies, methane reforming methods for H2 production are inefficient from an economic and environmental perspective. It should be noted here that endothermic reactions cool the active sites, so additional energy, cost, and currently, fossil fuels are required to compensate for the energy loss of endothermic processes during catalysis. One approach to circumvent this is the direct energy delivery to the active sites using renewable energy sources such as light and green electricity [220]. However, this would also require the redesign of current reactors. Alternative and less energy-intensive processes for producing H2 are therefore more promising. Ideally, green H2 obtained “for free” from solar or hydroelectric energy would be used, which would make the exothermic methanol synthesis, shown in Equation (10), directly feasible.
In terms of energy and CO2 emissions, it should be mentioned here that the energy for a potential industrial process is traditionally provided by burning methane, and estimates of DRM under ideal operation conditions involve the use of 1.38 mol of CH4 to convert 0.62 mol of CO2 into valuable syngas [4]. During combustion, one mole of methane is converted into one mole of CO2 and two moles of water. In addition, DRM utilizes an equimolar educt ratio. Thus, the above numbers indicate that if 1 mole of CO2 is consumed in the catalytic reaction, 1.23 moles of CO2 are released into the atmosphere. A large amount of methane burning is required in order to efficiently heat the reaction and to overcome the high activation barriers of DRM, i.e., the activation of CO2. The entire process requires at least 340 kJmol−1 methane (without heat losses), more than is released by the burning of methane (−803 kJmol−1), which is 42.3 % of the energy value of methane [4]. From this perspective, it seems more energy-efficient to directly burn methane for energy applications instead of valorizing CO2 into synthetic fuels.
The focus now is to achieve the integration of CO2 and CH4 in the production chain either under the perspective of their utility as synthons, i.e., on the basis of their potentialities for the synthesis of chemicals or as energy carriers only in a situation that the reaction partner (H2 in particular) is achieved by renewable sources. A recent perspective article goes beyond DRM and concludes that all strategies of CO2 valorization, including electrochemistry, will only be a scientific curiosity unless efficient paths of green H2 generation are developed [259]. Because DRM cannot be decontextualized from the production chain, developments are necessary for several fields, including also upstream (energy and H2 generation) and downstream (methanol, Fischer–Tropsch, and CO2 capture) technologies to deal with the carbon stream. Therefore, DRM is not at the current stage a “CO2 sink” as it may have been publicized, but a single gear of a much larger machinery.
7.3. What to Do with the Carbon?
In addition to the energetic drawbacks, the current industrial carbon cycle (Figure 2) evinces an additional obstacle related to reaction stoichiometries. About 96% of H2 generation is achieved by SRM [13]. The subsequent WGS steps necessary to produce high-purity H2 transform the carbon in CO2, which can either be returned to the carbon cycle by DRM or reacted downstream with H2 to form methanol. Assuming that the subsequent WGS stages are arbitrarily efficient, the newly recovered syngas has a lower potential for H2 generation compared to the higher amount of CO2 that must be cycled.
It can be argued that CO2 can be used, for instance, for methanol synthesis, provided that H2 is obtained from a “green” source such as electrochemical water splitting. In addition to chemical energy storage, methanol is also used as a precursor for the synthesis of fine chemicals and plastics, as a solvent, fuel additive, or as a precursor for methyl tert-butyl ether, dimethyl ether, olefins, and formaldehyde. The syngas of ratio one itself would boost the industrial production of acetic acid, dimethyl ether, and hydroformylation processes (Equations (11)–(13)). In the scenario of DRM being scaled to process generated CO2 in situ coupled to a downstream DME plant in which both processes run with an arbitrary efficiency of 100%, the 49.4 gt a−1 of current CO2 emissions would be transformed alongside methane into 103 gt a−1 of DME. It is unlikely that the market for DME will grow such that these large volumes can be absorbed. Not to mention the CO2 already present in the environment. It could be further argued that DME itself can serve as a fuel, by which CO2 would then be injected back into the environment. In order to store carbon and effectively reduce the footprints, it has to be immobilized and taken out of the cycle. The production of plastics could be considered, as they have no further current use and act as another sink of manufactured carbon (Figure 2). However, plastic pollution is already recognized as the second-largest scourge of our planet [260], so new ways of CO2 storage have to be developed [4].
The CO2 released during the combustion of methanol and its derivatives ends up as a waste in the atmosphere. This would, at best, result in a net-zero carbon mass balance in an otherwise green hydrogen-based economy, in which CO2 is converted to methanol and fuels, burnt for energy production, and chemically recycled back at the expense of a huge energy source. Since, in DRM, CH4 is used as a reducing agent and energy carrier, DRM-based cycles would potentiate the carbon content in the atmosphere instead of reducing it, as the following equations can show:
Cycle 1:
CH4 + CO2 → 2 CO + 2 H2 DRM
2 CO + 2 H2O → 2 CO2 + 2 H2 WGS
2 CO2 + 6 H2 → 2 CH3OH + 2 H2O Methanol synthesis
2 CH3OH + 3 O2 → 2 CO2 + 4 H2O energy release
CH4 + CO2 → 2 CO + 2 H2 DRM
2 CO + 2 H2O → 2 CO2 + 2 H2 WGS
2 CO2 + 6 H2 → 2 CH3OH + 2 H2O Methanol synthesis
2 CH3OH + 3 O2 → 2 CO2 + 4 H2O energy release
In the first cycle, we start with one equivalent of CO2 and CH4 and end up with two equivalents of CO2. Further cycling of the generated CO2 would intensify the carbon content and also requires a higher energy investment:
Cycle 2:
2 CH4 + 2 CO2 → 4 CO + 4 H2 DRM
4 CO + 4 H2O → 4 CO2 + 4 H2 WGS
4 CO2 + 12 H2 → 4 CH3OH + 4 H2O Methanol synthesis
4 CH3OH + 6 O2 → 4 CO2 + 8 H2O energy release
2 CH4 + 2 CO2 → 4 CO + 4 H2 DRM
4 CO + 4 H2O → 4 CO2 + 4 H2 WGS
4 CO2 + 12 H2 → 4 CH3OH + 4 H2O Methanol synthesis
4 CH3OH + 6 O2 → 4 CO2 + 8 H2O energy release
Which now generates four equivalents of CO2, and then:
Cycle 3:
4 CH4 + 4 CO2 → 8 CO + 8 H2 DRM
8 CO + 8 H2O → 8 CO2 + 8 H2 WGS
8 CO2 + 24 H2 → 8 CH3OH + 8 H2O Methanol synthesis
8 CH3OH + 12 O2 → 8 CO2 + 16 H2O energy release
4 CH4 + 4 CO2 → 8 CO + 8 H2 DRM
8 CO + 8 H2O → 8 CO2 + 8 H2 WGS
8 CO2 + 24 H2 → 8 CH3OH + 8 H2O Methanol synthesis
8 CH3OH + 12 O2 → 8 CO2 + 16 H2O energy release
With further cycles, the total amount of created CO2 is increasing instead of being efficiently stored.
In the climate debate, the main application of methanol is therefore based mainly on the production of fuels and fuel additives to efficiently store chemical energy and ultimately recover the energy content through combustion. Thus, when DRM-based methanol is used as a synthetic fuel or fuel additive, we enter a spiral of steadily increasing CO2 levels in the atmosphere, which will not stop global warming. The scene is even more dramatic for the “super dry reforming” reaction as it converts more carbon equivalents per cycle while the useful H2 is wasted in the form of water.
Ironically, although at first glance, two greenhouse gases are converted into valuable products, tracking the carbon contents leads to conclude that DRM-based fuels have a negative efficiency in removing greenhouse gases from the environment. Efficient reactors running by solar, hydroelectric, or wind energy may have a more direct impact on climate change mitigation in other scenarios such as H2 generation. One should also consider the energy investment required to keep the processes shown in Figure 2 under operation. In other words: if the required energy is generated from conventional fossil sources, the impact of DRM on reducing the environmental footprint would be rather insignificant [4].
It can be argued that DRM itself, as we have shown, could be driven by solar light, hence making energy consumption significantly smaller, providing a promising and highly effective approach for converting renewable solar energy into H2 and CO fuels due to the highly endothermic properties [261,262]. However, the same could be achieved by direct H2 production from water splitting without the urge to deal with the carbonized byproducts. Alternative energy carriers which have no carbon in their structure, such as H2 and NH3, may become relevant in the future in the energy storage scene.
7.4. Quo Vadis DRM?
The above discussion of the negative economic and environmental impact of DRM begs the question, “Quo vadis DRM?” It is important to understand whether DRM has potential for industrial endeavors or whether its complexity remains for basic academic ventures to decipher principles and reaction mechanisms in heterogeneous catalysis.
First, from the academic point of view aiming at understanding the intimate interplay between structure and function, DRM is important as this reaction has the unique potential to provide a methane activation pathway in which carbon formation can be mitigated and, simultaneously, the hydrogen, which is produced, is not converted into water. It can, thus, be directly utilized in downstream reactions. Furthermore, if adequate operando techniques are applied, this reaction can be used to explore the C1 chemistry and to obtain unprecedented structure-function correlations that may be important to fine-tune respective catalysts for the individual reactions.
Second, the carbon-rich syngas, which are produced from DRM, may be beneficial in a prospective scenario to replace the currently applied iron production in blast furnaces. This alternative approach may allow the reduction of CO2 emissions during the iron formation processes. It is therefore essential to efficiently produce these syngas from CO2 and CH4 and to further develop this technology.
Third, an attractive scenario relates to the production of H2 from syngas, for instance, by membrane reactors, which may be beneficial for feeding “greener” energy devices such as fuel cells. The question holds on what to do with the high amounts of rich CO gas which is simultaneously generated and should be utilized.
Fourth is the construction of decentralized DRM units running on green energy for the transformation of biogas. In this aspect, biogas, which already amounts to 5% of global greenhouse gases emissions, could be effectively mitigated by small to mid-scale DRM reactors.
Fifth, methane pyrolysis (Equation (6)) could play an important role in answering this question. Methane decomposes into hydrogen and carbon over a catalyst. The resulting carbon residues must be disposed of, which is only possible to a limited extent due to the high proportions of transition metals in methane cracking catalysts. Here, DRM could step in. Inspired by the work of ref. [208] and by the general observations about DRM in cyclic operations such as in the Catformer [85], a consecutive process between reforming (Equations (1)–(3)) and the Boudouard reaction (Equation (5)) can be envisaged, as schematically illustrated in Figure 21.
In this process, syngas would be achieved by reforming methane. Subsequently, one can stimulate the decarbonation of the feed by the Boudouard reaction. The products would be solid carbon (coke), H2, and CO2. In this approach, we suggest that the high propensity of DRM feed to coking is paradoxically a big advantage since the carbon content of CH4 and CO2 would be safely immobilized in the solid byproduct. Moreover, the carbon byproduct could be further valorized by direct tailoring, for instance, as nanotubes [263]. The Boudouard reaction also produces CO2, which can be reintroduced into the reforming cycle. This combination of DRM and Boudouard reaction would be targeted for the generation of a “greener” H2, which can find application in conventional downstream technologies such as methanol synthesis and CO2 capture. H2 could subsequently be transformed into NH3 for energy storage and transportation. The disposal of coke can be alternatively optimized to release it back to the geological formations where the carbon was originally retrieved from or to be used in other refining industrial technologies. The Boudouard reaction could operate under mild conditions even without a metallic catalyst, which would facilitate the disposal of a relatively clean byproduct. The total reaction is formulated in (Equation (39))
CH4 + CO2 → CO2 + C + 2H2 ΔH298 = 75 kJ mol−1
We conclude from the discussion above that the question of which direction to go for DRM is a very live one. Considering the climate-related issues and application, DRM is particularly limited by its high energy input and negative CO2 balance unless one mol C per CO2 molecule used is removed from the system. Coupling with the Boudouard reaction could be interesting for this purpose. Industrially interesting systems already exist, which could be used to produce syngas mixtures with an H2/CO ratio in the range between one and three that may feed the production of a family of chemicals such as glacial acetic acid, dimethylether, and functionalized hydrocarbons directly from methane.
Author Contributions
Conceptualization, R.S., T.L. and L.E.S.-D.; formal analysis, R.S., T.L. and L.E.S.-D.; writing—original draft preparation, L.E.S.-D.; writing—review and editing, T.L. and L.E.S.-D.; supervision, R.S. and T.L.; funding acquisition, R.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Federal Ministry of Education and Research in the framework of the project Catlab (03EW0015A).
Acknowledgments
All authors thank the current members of the Department of Inorganic Chemistry at the Fritz-Haber-Institut der Max-Planck-Gesellschaft and of the Department of Heterogeneoues Reactions at the Max Planck Insitute of Chemical Energy Conversion for their support, work and discussions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rostrup-Nielsen, J. Sulfur-passivated nickel catalysts for carbon-free steam reforming of methane. J. Catal. 1984, 85, 31–43. [Google Scholar] [CrossRef]
- Olah, G.A. Beyond Oil and Gas: The Methanol Economy. Angew. Chem. Int. Ed. 2005, 44, 2636–2639. [Google Scholar] [CrossRef] [PubMed]
- Wittich, K.; Krämer, M.; Bottke, N.; Schunk, S.A. Catalytic Dry Reforming of Methane: Insights from Model Systems. ChemCatChem 2020, 12, 2130–2147. [Google Scholar] [CrossRef]
- Parsapur, R.K.; Chatterjee, S.; Huang, K.-W. The Insignificant Role of Dry Reforming of Methane in CO2 Emission Relief. ACS Energy Lett. 2020, 5, 2881–2885. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, T.; Blunt, M.J.; Anthony, E.J.; Park, A.-H.A.; Hughes, R.W.; Webley, P.A.; Yan, J. Advances in carbon capture, utilization and storage. Appl. Energy 2020, 278, 115627. [Google Scholar] [CrossRef]
- Fujikawa, S.; Selyanchyn, R.; Kunitake, T. A new strategy for membrane-based direct air capture. Polym. J. 2021, 53, 111–119. [Google Scholar] [CrossRef]
- Smit, B.; Park, A.-H.A.; Gadikota, G. The Grand Challenges in Carbon Capture, Utilization, and Storage. Front. Energy Res. 2014, 2, 2. [Google Scholar] [CrossRef] [Green Version]
- Eastman, E.D. Equilibria in the Systems Iron: Carbon Oxygen and Iron Hydrogen Oxygen, and the Free Energies of the Oxides of Iron1. J. Am. Chem. Soc. 1922, 44, 975–998. [Google Scholar] [CrossRef]
- Bazzanella, A.M.; Ausfelder, F. Low Carbon Energy and Feedstock for the European Chemical Industry. 2017, DECHEMA Gesellschaft für Chemische Technik und Biotechnologie e.V. Available online: https://dechema.de/dechema_media/Downloads/Positionspapiere/Technology_study_Low_carbon_energy_and_feedstock_for_the_European_chemical_industry.pdf (accessed on 7 April 2022).
- Hannah Ritchie, M.R. CO2 and Greenhouse Gas Emissions. 2017. Available online: https://ourworldindata.org/co2-and-other-greenhouse-gas-emissions (accessed on 9 February 2020).
- Liu, Z.; Guan, D.; Wei, W.; Davis, S.J.; Ciais, P.; Bai, J.; Peng, S.; Zhang, Q.; Hubacek, K.; Marland, G.; et al. Reduced carbon emission estimates from fossil fuel combustion and cement production in China. Nature 2015, 524, 335–338. [Google Scholar] [CrossRef] [Green Version]
- Kuramochi, T.; Elzen, M.D.; Peters, G.; Global Emissions Trends and G20 Status and Outlook in Emissions GAP Report 2020. United Nations. 2021, pp. 3–24. Available online: https://wedocs.unep.org/xmlui/bitstream/handle/20.500.11822/34428/EGR20ch2.pdf?sequence=3 (accessed on 6 April 2022).
- USEIA. Hydrogen Explained Production of Hydrogen. 2022. Available online: https://www.eia.gov/energyexplained/hydrogen/production-of-hydrogen.php. (accessed on 27 January 2020).
- Lavoie, J.-M. Review on dry reforming of methane, a potentially more environmentally-friendly approach to the increasing natural gas exploitation. Front. Chem. 2014, 2, 81. [Google Scholar] [CrossRef] [Green Version]
- Bhattar, S.; Abedin, A.; Kanitkar, S.; Spivey, J.J. A review on dry reforming of methane over perovskite derived catalysts. Catal. Today 2021, 365, 2–23. [Google Scholar] [CrossRef]
- Pakhare, D.; Spivey, J. A review of dry (CO2) reforming of methane over noble metal catalysts. Chem. Soc. Rev. 2014, 43, 7813–7837. [Google Scholar] [CrossRef]
- Chung, W.-C.; Chang, M.-B. Review of catalysis and plasma performance on dry reforming of CH4 and possible synergistic effects. Renew. Sustain. Energy Rev. 2016, 62, 13–31. [Google Scholar] [CrossRef]
- Edenhofer, O.; Kadner, S.; Pichs-Madruga, R.; Sokona, Y.; Farahani, E.; Kadner, S.; Seyboth, A.A.; Baum, I.; Brunner, S.; Eickemeier, P.; et al. IPCC, 2014: Climate Change 2014: Mitigation of Climate Change. Contribution of Working Group III to the Fifth Assessment. In Report of the Intergovernmental Panel on Climate Change; Savolainen, J., Schlömer, S., von Stechow, C., Zwickel, T.C.J., Eds.; Cambridge University Press: Cambridge, UK, 2014. [Google Scholar]
- United States Environmental Protection Agency. Global Greenhouse Gas Emissions Data. Available online: https://www.epa.gov/ghgemissions/global-greenhouse-gas-emissions-data (accessed on 20 January 2022).
- Ritchie, H.; Roser, M.; CO2 and Greenhouse Gas Emissions. OurWorldInData.org. Available online: https://ourworldindata.org/co2-emissions#citation (accessed on 7 April 2022).
- Lyu, L.; Zeng, X.; Yun, J.; Wei, F.; Jin, F. No Catalyst Addition and Highly Efficient Dissociation of H2O for the Reduction of CO2 to Formic Acid with Mn. Environ. Sci. Technol. 2014, 48, 6003–6009. [Google Scholar] [CrossRef]
- Ye, R.-P.; Ding, J.; Gong, W.; Argyle, M.; Zhong, Q.; Wang, Y.; Russell, C.K.; Xu, Z.; Russell, A.G.; Li, Q.; et al. CO2 hydrogenation to high-value products via heterogeneous catalysis. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Davis, S.J.; Caldeira, K.; Matthews, H.D. Future CO 2 Emissions and Climate Change from Existing Energy Infrastructure. Science 2010, 329, 1330–1333. [Google Scholar] [CrossRef] [Green Version]
- Jin, F.; Gao, Y.; Jin, Y.; Zhang, Y.; Cao, J.; Wei, Z.; Smith, R.L., Jr. High-yield reduction of carbon dioxide into formic acid by zero-valent metal/metal oxide redox cycles. Energy Environ. Sci. 2011, 4, 881–884. [Google Scholar] [CrossRef]
- Vansant, J.; Koziel, P.-W. Technical and Industrial Applications of CO2 in An Economy Based on Carbon Dioxide and Water: Potential of Large Scale Carbon Dioxide Utilization; Aresta, M., Karimi, I., Kaw, S.i., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 73–103. [Google Scholar]
- Romano, M.C.; Anantharaman, R.; Arasto, A.; Ozcan, D.C.; Ahn, H.; Dijkstra, J.W.; Carbo, M.; Boavida, D. Application of Advanced Technologies for CO2 Capture From Industrial Sources. Energy Procedia 2013, 37, 7176–7185. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Zhao, Z.-J.; Cheng, D.; Li, H.; Yu, J.; Wang, Q.; Gao, H.; Guo, J.; Wang, H.; Ozin, G.A.; et al. Efficient CO2 electroreduction on facet-selective copper films with high conversion rate. Nat. Commun. 2021, 12, 1–11. [Google Scholar] [CrossRef]
- Cauwenbergh, R.; Das, S. Photochemical reduction of carbon dioxide to formic acid. Green Chem. 2021, 23, 2553–2574. [Google Scholar] [CrossRef]
- Wareing, P.F.; Khalifa, M.M.; Treharne, K.J. Rate-limiting Processes in Photosynthesis at Saturating Light Intensities. Nature 1968, 220, 453–457. [Google Scholar] [CrossRef]
- Administration U.S.E.I. EIA Expects U.S. Fossil Fuel Production to Reach New Highs in 2023. Today in Energy 2022. Available online: https://www.eia.gov/todayinenergy/detail.php?id=50978 (accessed on 24 January 2022).
- Liu, Y.; Deng, D.; Bao, X. Catalysis for Selected C1 Chemistry. Chem 2020, 6, 2497–2514. [Google Scholar] [CrossRef]
- Fierro, J.L.G. Catalysis in C1 chemistry: Future and prospect. Catal. Lett. 1993, 22, 67–91. [Google Scholar] [CrossRef]
- Mesters, C. A Selection of Recent Advances in C1 Chemistry. Annu. Rev. Chem. Biomol. Eng. 2016, 7, 223–238. [Google Scholar] [CrossRef]
- Blanksby, S.J.; Ellison, G.B. Bond Dissociation Energies of Organic Molecules. Acc. Chem. Res. 2003, 36, 255–263. [Google Scholar] [CrossRef]
- Tomkins, P.; Ranocchiari, M.; van Bokhoven, J.A. Direct Conversion of Methane to Methanol under Mild Conditions over Cu-Zeolites and beyond. Acc. Chem. Res. 2017, 50, 418–425. [Google Scholar] [CrossRef]
- Horn, R.; Williams, K.; Degenstein, N.; Schmidt, L. Syngas by catalytic partial oxidation of methane on rhodium: Mechanistic conclusions from spatially resolved measurements and numerical simulations. J. Catal. 2006, 242, 92–102. [Google Scholar] [CrossRef]
- Zavyalova, U.; Holena, M.; Schlögl, R.; Baerns, M. Statistical Analysis of Past Catalytic Data on Oxidative Methane Coupling for New Insights into the Composition of High-Performance Catalysts. ChemCatChem 2011, 3, 1935–1947. [Google Scholar] [CrossRef]
- Guo, X.; Fang, G.; Li, G.; Ma, H.; Fan, H.; Yu, L.; Wu, X.; Deng, D.; Wei, M.; Tan, D.; et al. Direct, Nonoxidative Conversion of Methane to Ethylene, Aromatics, and Hydrogen. Science 2014, 344, 616–619. [Google Scholar] [CrossRef]
- Wang, L.; Tao, L.; Xie, M.; Xu, G.; Huang, J.; Xu, Y. Dehydrogenation and aromatization of methane under non-oxidizing conditions. Catal. Lett. 1993, 21, 35–41. [Google Scholar] [CrossRef]
- Pham, H.N.; Sattler, J.J.H.B.; Weckhuysen, B.M.; Datye, A. Role of Sn in the Regeneration of Pt/γ-Al2O3 Light Alkane Dehydrogenation Catalysts. ACS Catal. 2016, 6, 2257–2264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belgued, M.; Pareja, P.; Amariglio, A. Conversion of methane into higher hydrocarbons on platinum. Nature 1991, 352, 789–790. [Google Scholar] [CrossRef]
- Schwab, E.; Milanov, A.; Schunk, S.A.; Behrens, A.; Schödel, N. Dry Reforming and Reverse Water Gas Shift: Alternatives for Syngas Production? Chem. Ing. Tech. 2015, 87, 347–353. [Google Scholar] [CrossRef]
- Zhang, X.; Vajglova, Z.; Mäki-Arvela, P.; Peurla, M.; Palonen, H. Mono- and Bimetallic Ni−Co Catalysts in Dry Reforming of Methane. Chem. Sel. 2021, 6, 3424–3434. [Google Scholar] [CrossRef]
- Bown, R.M.; Joyce, M.; Zhang, Q.; Reina, T.R.; Duyar, M.S. Identifying Commercial Opportunities for the Reverse Water Gas Shift Reaction. Energy Technol. 2021, 9, 2100554. [Google Scholar] [CrossRef]
- Ramirez, A.; Lee, K.; Harale, A.; Gevers, L.; Telalovic, S.; Solami, B.A.; Gascon, J. Stable High-Pressure Methane Dry Reforming Under Excess of CO2. ChemCatChem 2020, 12, 5919–5925. [Google Scholar] [CrossRef]
- Rostrup-Nielsen, J.R. New aspects of syngas production and use. Catal. Today 2000, 63, 159–164. [Google Scholar] [CrossRef]
- Behrens, M. Promoting the Synthesis of Methanol: Understanding the Requirements for an Industrial Catalyst for the Conversion of CO2. Angew. Chem. Int. Ed. 2016, 55, 14906–14908. [Google Scholar] [CrossRef]
- Zhang, B.; Fuentes, D.P.; Börner, A. Hydroformylation. ChemTexts 2021, 8, 2. [Google Scholar]
- Rostrup-Nielsen, R.J. Production of synthesis gas. Catal. Today 1993, 18, 305–324. [Google Scholar] [CrossRef]
- Tan, Q.; Shi, Z.; Wu, D. CO2 Hydrogenation to Methanol over a Highly Active Cu–Ni/CeO2–Nanotube Catalyst. Ind. Eng. Chem. Res. 2018, 57, 10148–10158. [Google Scholar] [CrossRef]
- Laosiripojana, N.; Assabumrungrat, S. Catalytic dry reforming of methane over high surface area ceria. Appl. Catal. B Environ. 2005, 60, 107–116. [Google Scholar] [CrossRef]
- Nurunnabi, M.; Mukainakano, Y.; Kado, S.; Miyazawa, T.; Okumura, K.; Miyao, T.; Naito, S.; Suzuki, K.; Fujimoto, K.-I.; Kunimori, K.; et al. Oxidative steam reforming of methane under atmospheric and pressurized conditions over Pd/NiO–MgO solid solution catalysts. Appl. Catal. A Gen. 2006, 308, 1–12. [Google Scholar] [CrossRef]
- Armor, J. The multiple roles for catalysis in the production of H2. Appl. Catal. A Gen. 1999, 176, 159–176. [Google Scholar] [CrossRef]
- Alves, H.J.; Junior, C.B.; Niklevicz, R.R.; Frigo, E.P.; Frigo, M.; Coimbra-Araújo, C.H. Overview of hydrogen production technologies from biogas and the applications in fuel cells. Int. J. Hydrog. Energy 2013, 38, 5215–5225. [Google Scholar] [CrossRef]
- York, A.P.E.; Xiao, T.; Green, M.L.H.; Claridge, J. Methane Oxyforming for Synthesis Gas Production. Catal. Rev. 2007, 49, 511–560. [Google Scholar] [CrossRef]
- Kahle, L.C.S.; Roussière, T.; Maier, L.; Delgado, K.H.; Wasserschaff, G.; Schunk, S.A.; Deutschmann, O. Methane Dry Reforming at High Temperature and Elevated Pressure: Impact of Gas-Phase Reactions. Ind. Eng. Chem. Res. 2013, 52, 11920–11930. [Google Scholar] [CrossRef]
- Kikuchi, R.; Iwasa, Y.; Takeguchi, T.; Eguchi, K. Partial oxidation of CH4 and C3H8 over hexaaluminate-type oxides. Appl. Catal. A Gen. 2005, 281, 61–67. [Google Scholar] [CrossRef]
- Sheldon, D. Methanol Production—A Technical History. Johns. Matthey Technol. Rev. 2017, 61, 172–182. [Google Scholar] [CrossRef]
- Kim, J.; Park, J.; Qi, M.; Lee, I.; Moon, I. Process Integration of an Autothermal Reforming Hydrogen Production System with Cryogenic Air Separation and Carbon Dioxide Capture Using Liquefied Natural Gas Cold Energy. Ind. Eng. Chem. Res. 2021, 60, 7257–7274. [Google Scholar] [CrossRef]
- Zahedi nezhad, M.; Rowshanzamir, S.; Eikani, M.H. Autothermal reforming of methane to synthesis gas: Modeling and simulation. Int. J. Hydrog. Energy 2009, 34, 1292–1300. [Google Scholar] [CrossRef]
- Ghani, A.A.; Torabi, F.; Ibrahim, H. Autothermal reforming process for efficient hydrogen production from crude glycerol using nickel supported catalyst: Parametric and statistical analyses. Energy 2018, 144, 129–145. [Google Scholar] [CrossRef]
- Alyea, E.C.; He, D.; Wang, J. Alcohol synthesis from syngas: I. Performance of alkali-promoted Ni-Mo(MOVS) catalysts. Appl. Catal. A Gen. 1993, 104, 77–85. [Google Scholar] [CrossRef]
- Rostrup-Nielsen, J. Steam Reforming of Hydrocarbons. A Historical Perspective in Studies in Surface Science and Catalysis; Bao, X., Xu, Y., Eds.; Elsevier: Amsterdam, The Netherlands, 2004; pp. 121–126. [Google Scholar]
- Fischer, F.T.H. Conversion of methane into hydrogen and carbon monoxide. Brennst.Chem. 1928, 9, 23–27. [Google Scholar]
- Wei, J.; Iglesia, E. Mechanism and Site Requirements for Activation and Chemical Conversion of Methane on Supported Pt Clusters and Turnover Rate Comparisons among Noble Metals. J. Phys. Chem. B 2004, 108, 4094–4103. [Google Scholar] [CrossRef]
- Xiong, K.; Yin, Y.-L.; Cao, Y.; Liu, X.-T. Exergy Efficiency Promotion for the System of CO2 Hydrogenation to Methanol in Habitable Confined Space. Front. Energy Res. 2021, 9, 9. [Google Scholar] [CrossRef]
- Hank, C.; Gelpke, S.; Schnabl, A.; White, R.J.; Full, J.; Wiebe, N.; Smolinka, T.; Schaadt, A.; Henning, H.-M.; Hebling, C. Economics & carbon dioxide avoidance cost of methanol production based on renewable hydrogen and recycled carbon dioxide—Power-to-methanol. Sustain. Energy Fuels 2018, 2, 1244–1261. [Google Scholar]
- Wiesberg, I.L.; de Medeiros, J.L.; Alves, R.M.; Coutinho, P.L.; Araújo, O.Q. Carbon dioxide management by chemical conversion to methanol: Hydrogenation and Bi-Reforming. Energy Convers. Manag. 2016, 125, 320–335. [Google Scholar] [CrossRef]
- Borisut, P.; Nuchitprasittichai, A. Methanol Production via CO2 Hydrogenation: Sensitivity Analysis and Simulation—Based Optimization. Front. Energy Res. 2019, 7, 81. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Hyeon, S.; Lee, J.; Kim, W.D.; Lee, D.C.; Kim, J.; Lee, H. Energy-efficient CO2 hydrogenation with fast response using photoexcitation of CO2 adsorbed on metal catalysts. Nat. Commun. 2018, 9, 2425–2434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamu, S.; Bawah, A.; Muraza, O.; Malaibari, Z.; Hossain, M.M. Effects of metal support interaction on dry reforming of methane over Ni/ Ce-Al2O3 catalysts. Can. J. Chem. Eng. 2020, 98, 2425–2434. [Google Scholar] [CrossRef]
- le Saché, E.; Moreno, A.A.; Reina, T.R. Biogas Conversion to Syngas Using Advanced Ni-Promoted Pyrochlore Catalysts: Effect of the CH4/CO2 Ratio. Front. Chem. 2021, 9, 672419. [Google Scholar] [CrossRef]
- Chein, R.; Chen, Y.; Yu, C.; Chung, J. Thermodynamic analysis of dry reforming of CH4 with CO2 at high pressures. J. Nat. Gas Sci. Eng. 2015, 26, 617–629. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Y.; Zhang, X.; Mi, Z. Thermodynamic analysis of autothermal steam and CO2 reforming of methane. Int. J. Hydrog. Energy 2008, 33, 2507–2514. [Google Scholar] [CrossRef]
- Nikoo, M.K.; Amin, N. Thermodynamic analysis of carbon dioxide reforming of methane in view of solid carbon formation. Fuel Process. Technol. 2011, 92, 678–691. [Google Scholar] [CrossRef] [Green Version]
- Schulz, L.A.; Kahle, L.C.; Delgado, K.H.; Schunk, S.A.; Jentys, A.; Deutschmann, O.; Lercher, J.A. On the coke deposition in dry reforming of methane at elevated pressures. Appl. Catal. A Gen. 2015, 504, 599–607. [Google Scholar] [CrossRef]
- Roussière, T.; Schulz, L.; Schelkle, K.M.; Wasserschaff, G.; Milanov, A.; Schwab, E.; Deutschmann, O.; Jentys, A.; Lercher, J.; Schunk, S.A. Structure-Activity Relationships of Nickel-Hexaaluminates in Reforming Reactions Part II: Activity and Stability of Nanostructured Nickel-Hexaaluminate-Based Catalysts in the Dry Reforming of Methane. ChemCatChem 2014, 6, 1447–1452. [Google Scholar] [CrossRef]
- Sandoval-Diaz, L.; Plodinec, M.; Ivanov, D.; Poitel, S.; Hammud, A.; Nerl, H.C.; Schlögl, R.; Lunkenbein, T. Visualizing the importance of oxide-metal phase transitions in the production of synthesis gas over Ni catalysts. J. Energy Chem. 2020, 50, 178–186. [Google Scholar] [CrossRef]
- AlSabban, B.; Falivene, L.; Kozlov, S.M.; Aguilar-Tapia, A.; Ould-Chikh, S.; Hazemann, J.-L.; Cavallo, L.; Basset, J.-M.; Takanabe, M. In-operando elucidation of bimetallic CoNi nanoparticles during high-temperature CH4/CO2 reaction. Appl. Catal. B Environ. 2017, 213, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Tapia, A.A.; Ould-Chikh, S.; Lahera, E.; Prat, A.; Delnet, W.; Proux, O.; Kieffer, I.; Basset, J.-M.; Takanabe, K.; Hazemann, J.-L. A new high temperature reactor for operando XAS: Application for the dry reforming of methane over Ni/ZrO2 catalyst. Rev. Sci. Instrum. 2018, 89, 035109. [Google Scholar] [CrossRef] [Green Version]
- Álvarez, M.A.; Bobadilla, L.; Garcilaso, V.; Centeno, M.A.; Odriozola, J.A. CO2 reforming of methane over Ni-Ru supported catalysts: On the nature of active sites by operando DRIFTS study. J. CO2 Util. 2018, 24, 509–515. [Google Scholar] [CrossRef]
- Aramouni, N.A.K.; Zeaiter, J.; Kwapinski, W.; Ahmad, M.N. Thermodynamic analysis of methane dry reforming: Effect of the catalyst particle size on carbon formation. Energy Convers. Manag. 2017, 150, 614–622. [Google Scholar] [CrossRef]
- Xu, J.; Froment, G.F. Methane steam reforming, methanation and water-gas shift: I. Intrinsic kinetics. AIChE J. 1989, 35, 88–96. [Google Scholar] [CrossRef]
- Gokon, N.; Osawa, Y.; Nakazawa, D.; Kodama, T. Kinetics of CO2 reforming of methane by catalytically activated metallic foam absorber for solar receiver-reactors. Int. J. Hydrog. Energy 2009, 34, 1787–1800. [Google Scholar] [CrossRef]
- Ginsburg, J.M.; Piña, J.; El Solh, T.; de Lasa, H.I. Coke Formation over a Nickel Catalyst under Methane Dry Reforming Conditions: Thermodynamic and Kinetic Models. Ind. Eng. Chem. Res. 2005, 44, 4846–4854. [Google Scholar] [CrossRef]
- Zhang, Z.; Verykios, X. Carbon dioxide reforming of methane to synthesis gas over supported Ni catalysts. Catal. Today 1994, 21, 589–595. [Google Scholar] [CrossRef]
- Yuan, K.; Zhong, J.-Q.; Zhou, X.; Xu, L.; Bergman, S.L.; Wu, K.; Xu, G.Q.; Bernasek, S.L.; Li, H.X.; Chen, W. Dynamic Oxygen on Surface: Catalytic Intermediate and Coking Barrier in the Modeled CO2 Reforming of CH4 on Ni (111). ACS Catal. 2016, 6, 4330–4339. [Google Scholar] [CrossRef]
- Wei, J.; Iglesia, E. Isotopic and kinetic assessment of the mechanism of reactions of CH4 with CO2 or H2O to form synthesis gas and carbon on nickel catalysts. J. Catal. 2004, 224, 370–383. [Google Scholar] [CrossRef]
- Giehr, A.; Maier, L.; Angeli, S.; Schunk, S.A.; Deutschmann, O. Dry and Steam Reforming of CH4 on Co-Hexaaluminate: On the Formation of Metallic Co and Its Influence on Catalyst Activity. Ind. Eng. Chem. Res. 2020, 59, 18790–18797. [Google Scholar] [CrossRef]
- Keller, K.; Lott, P.; Stotz, H.; Maier, L.; Deutschmann, O. Microkinetic Modeling of the Oxidation of Methane Over PdO Catalysts—Towards a Better Understanding of the Water Inhibition Effect. Catalysts 2020, 10, 922. [Google Scholar] [CrossRef]
- Schmider, D.; Maier, L.; Deutschmann, O. Reaction Kinetics of CO and CO2 Methanation over Nickel. Ind. Eng. Chem. Res. 2021, 60, 5792–5805. [Google Scholar] [CrossRef]
- Akpan, E.; Sun, Y.; Kumar, P.; Ibrahim, H.; Aboudheir, A.; Idem, R. Kinetics, experimental and reactor modeling studies of the carbon dioxide reforming of methane (CDRM) over a new Ni/CeO2–ZrO2 catalyst in a packed bed tubular reactor. Chem. Eng. Sci. 2007, 62, 4012–4024. [Google Scholar] [CrossRef]
- Kathiraser, Y.; Oemar, U.; Saw, E.T.; Li, Z.; Kawi, S. Kinetic and mechanistic aspects for CO2 reforming of methane over Ni based catalysts. Chem. Eng. J. 2015, 278, 62–78. [Google Scholar] [CrossRef]
- Kroll, V.; Swaan, H.; Lacombe, S.; Mirodatos, C. Methane Reforming Reaction with Carbon Dioxide over Ni/SiO2 Catalyst: II. A Mechanistic Study. J. Catal. 1996, 164, 387–398. [Google Scholar] [CrossRef]
- Aparicio, L. Transient Isotopic Studies and Microkinetic Modeling of Methane Reforming over Nickel Catalysts. J. Catal. 1997, 165, 262–274. [Google Scholar] [CrossRef]
- Michaelides, A.; Hu, P. Methyl chemisorption on Ni(111) and CHM multicentre bonding: A density functional theory study. Surf. Sci. 1999, 437, 362–376. [Google Scholar] [CrossRef]
- Wang, S.-G.; Liao, X.-Y.; Hu, J.; Cao, D.-B.; Li, Y.-W.; Wang, J.; Jiao, H. Kinetic aspect of CO2 reforming of CH4 on Ni(111): A density functional theory calculation. Surf. Sci. 2007, 601, 1271–1284. [Google Scholar] [CrossRef]
- Muraza, O.; Galadima, A. A review on coke management during dry reforming of methane. Int. J. Energy Res. 2015, 39, 1196–1216. [Google Scholar] [CrossRef]
- Gao, X.; Wang, Z.; Ashok, J.; Kawi, S. A comprehensive review of anti-coking, anti-poisoning and anti-sintering catalysts for biomass tar reforming reaction. Chem. Eng. Sci. X 2020, 7, 100065. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, Z.; Das, S.; Kawi, S. Reforming of tar from biomass gasification in a hybrid catalysis-plasma system: A review. Appl. Catal. B Environ. 2019, 250, 250–272. [Google Scholar] [CrossRef]
- Ochoa, A.; Arregi, A.; Amutio, M.; Gayubo, A.G.; Olazar, M.; Bilbao, J.; Castaño, P. Coking and sintering progress of a Ni supported catalyst in the steam reforming of biomass pyrolysis volatiles. Appl. Catal. B Environ. 2018, 233, 289–300. [Google Scholar] [CrossRef]
- Yao, L.; Galvez, M.E.; Hu, C.; Da Costa, P. Synthesis Gas Production via Dry Reforming of Methane over Manganese Promoted Nickel/Cerium–Zirconium Oxide Catalyst. Ind. Eng. Chem. Res. 2018, 57, 16645–16656. [Google Scholar] [CrossRef]
- Bartholomew, C.H. Mechanisms of catalyst deactivation. Appl. Catal. A Gen. 2001, 212, 17–60. [Google Scholar] [CrossRef]
- Margossian, T.; Larmier, K.; Kim, S.M.; Krumeich, F.; Müller, C.; Copéret, C. Supported Bimetallic NiFe Nanoparticles through Colloid Synthesis for Improved Dry Reforming Performance. ACS Catal. 2017, 7, 6942–6948. [Google Scholar] [CrossRef]
- Goula, M.; Lemonidou, A.; Efstathiou, A. Characterization of Carbonaceous Species Formed during Reforming of CH4 with CO2 over Ni/CaO–Al2O3Catalysts Studied by Various Transient Techniques. J. Catal. 1996, 161, 626–640. [Google Scholar] [CrossRef]
- Wang, Z.; Cao, X.; Zhu, J.; Hu, P. Activity and coke formation of nickel and nickel carbide in dry reforming: A deactivation scheme from density functional theory. J. Catal. 2014, 311, 469–480. [Google Scholar] [CrossRef]
- Bermúdez, J.; Fidalgo, B.; Arenillas, A.; Menéndez, J. Dry reforming of coke oven gases over activated carbon to produce syngas for methanol synthesis. Fuel 2010, 89, 2897–2902. [Google Scholar] [CrossRef] [Green Version]
- Solh, T.E.; Jarosch, K.; de Lasa, H. Catalytic Dry Reforming of Methane in a CREC Riser Simulator Kinetic Modeling and Model Discrimination. Ind. Eng. Chem. Res. 2003, 42, 2507–2515. [Google Scholar] [CrossRef]
- Pekediz, A.; de Lasa, H.I. I. Methane oxidative coupling in a novel riser simulator reactor. Chem. Eng. Sci. 1994, 49, 4759–4770. [Google Scholar] [CrossRef]
- Becker, A.; Hüttinger, K. Chemistry and kinetics of chemical vapor deposition of pyrocarbon—IV pyrocarbon deposition from methane in the low temperature regime. Carbon 1998, 36, 213–224. [Google Scholar] [CrossRef]
- Li, A.; Deutschmann, O. Transient modeling of chemical vapor infiltration of methane using multi-step reaction and deposition models. Chem. Eng. Sci. 2007, 62, 4976–4982. [Google Scholar] [CrossRef]
- Li, A.; Norinaga, K.; Zhang, W.; Deutschmann, O. Modeling and simulation of materials synthesis: Chemical vapor deposition and infiltration of pyrolytic carbon. Compos. Sci. Technol. 2008, 68, 1097–1104. [Google Scholar] [CrossRef] [Green Version]
- Norinaga, K.; Deutschmann, O.; Saegusa, N.; Hayashi, J.-I. Analysis of pyrolysis products from light hydrocarbons and kinetic modeling for growth of polycyclic aromatic hydrocarbons with detailed chemistry. J. Anal. Appl. Pyrolysis 2009, 86, 148–160. [Google Scholar] [CrossRef]
- Dagaut, P.; Pengloan, G.; Ristori, A. Oxidation, ignition and combustion of toluene: Experimental and detailed chemical kinetic modelingElectronic supplementary information. Phys. Chem. Chem. Phys. 2002, 4, 1846–1854. [Google Scholar] [CrossRef]
- Becker, A.; Hu, Z.; Hüttinger, K. A hydrogen inhibition model of carbon deposition from light hydrocarbons. Fuel 2000, 79, 1573–1580. [Google Scholar] [CrossRef]
- Teuner, S.C.N.; Neumann, P.; von Linde, F. CO through CO2 Reforming. The Calcor Standard and Calcor Economy Processes. OIL GAS Eur. Mag. 2001, 3, 44–46. [Google Scholar]
- Tang, S.-B.; Qiu, F.-L.; Lu, S.-J. Effect of supports on the carbon deposition of nickel catalysts for methane reforming with CO2. Catal. Today 1995, 24, 253–255. [Google Scholar] [CrossRef]
- Kim, G.J.; Cho, D.-S.; Kim, K.-H.; Kim, J.-H. The reaction of CO2 with CH4 to synthesize H2 and CO over nickel-loaded Y-zeolites. Catal. Lett. 1994, 28, 41–52. [Google Scholar] [CrossRef]
- Mortensen, P.M.; Dybkjær, I. Industrial scale experience on steam reforming of CO2-rich gas. Appl. Catal. A Gen. 2015, 495, 141–151. [Google Scholar] [CrossRef]
- Zhu, Z.; Melaet, G.; Axnanda, S.; Alayoglu, S.; Liu, Z.; Salmeron, M.; A Somorjai, G. Structure and Chemical State of the Pt(557) Surface during Hydrogen Oxidation Reaction Studied by in Situ Scanning Tunneling Microscopy and X-ray Photoelectron Spectroscopy. J. Am. Chem. Soc. 2013, 135, 12560–12563. [Google Scholar] [CrossRef]
- Gabasch, H.; Hayek, K.; Klötzer, B.; Unterberger, W.; Kleimenov, E.; Teschner, D.; Zafeiratos, S.; Hävecker, M.; Knop-Gericke, A.; Schlögl, R.; et al. Methane Oxidation on Pd(111): In Situ XPS Identification of Active Phase. J. Phys. Chem. C 2007, 111, 7957–7962. [Google Scholar] [CrossRef]
- Teschner, D.; Pestryakov, A.; Kleimenov, E.; Havecker, M.; Bluhm, H.; Sauer, H.; Knopgericke, A.; Schlogl, R. High-pressure X-ray photoelectron spectroscopy of palladium model hydrogenation catalysts.: Part 1: Effect of gas ambient and temperature. J. Catal. 2005, 230, 186–194. [Google Scholar] [CrossRef]
- Mu, R.; Fu, Q.; Xu, H.; Zhang, H.; Huang, Y.; Jiang, Z.; Zhang, S.; Tan, D.; Bao, X. Synergetic Effect of Surface and Subsurface Ni Species at Pt−Ni Bimetallic Catalysts for CO Oxidation. J. Am. Chem. Soc. 2011, 133, 1978–1986. [Google Scholar] [CrossRef] [PubMed]
- Merte, L.R.; Knudsen, J.; Eichhorn, F.M.; Porsgaard, S.; Zeuthen, H.; Grabow, L.C.; Lægsgaard, E.; Bluhm, H.; Salmeron, M.; Mavrikakis, M.; et al. CO-Induced Embedding of Pt Adatoms in a Partially Reduced FeOx Film on Pt(111). J. Am. Chem. Soc. 2011, 133, 10692–10695. [Google Scholar] [CrossRef] [PubMed]
- Chin, Y.-H.; Buda, C.; Neurock, M.; Iglesia, E. Reactivity of Chemisorbed Oxygen Atoms and Their Catalytic Consequences during CH4–O2 Catalysis on Supported Pt Clusters. J. Am. Chem. Soc. 2011, 133, 15958–15978. [Google Scholar] [CrossRef]
- Chen, C.S.; Lin, J.H.; You, J.H.; Yang, K.H. Effects of Potassium on NiAl2O3−K/ Catalysts in the Synthesis of Carbon Nanofibers by Catalytic Hydrogenation of CO2. J. Phys. Chem. A 2009, 114, 3773–3781. [Google Scholar] [CrossRef]
- Oemar, U.; Ang, M.L.; Chin, Y.C.; Hidajat, K.; Kawi, S. Role of lattice oxygen in oxidative steam reforming of toluene as a tar model compound over Ni/La0.8Sr0.2AlO3 catalyst. Catal. Sci. Technol. 2015, 5, 3585–3597. [Google Scholar] [CrossRef]
- Li, C.; Hirabayashi, D.; Suzuki, K. A crucial role of O2− and O22− on mayenite structure for biomass tar steam reforming over Ni/Ca12Al14O33. Appl. Catal. B Environ. 2009, 88, 351–360. [Google Scholar] [CrossRef]
- Adnan, M.A.; Hidayat, A.; Ajumobi, O.O.; Adamu, S.; Muraza, O.; Hossain, M.M. Fluidizable Fe–Co/Ce–ZrO2 Catalysts for Steam Reforming of Toluene as a Tar Surrogate in Biomass Gasification. Energy Fuels 2018, 32, 12833–12842. [Google Scholar] [CrossRef]
- Alemán, J.V.; Chadwick, A.V.; He, J.; Hess, M.; Horie, K.; Jones, R.G.; Kratochvíl, P.; Meisel, I.; Mita, I.; Moad, G.; et al. Definitions of terms relating to the structure and processing of sols, gels, networks, and inorganic-organic hybrid materials (IUPAC Recommendations 2007). Pure Appl. Chem. 2007, 79, 1801–1829. [Google Scholar] [CrossRef]
- Jafarbegloo, M.; Tarlani, A.; Mesbah, A.W.; Muzart, J.; Sahebdelfar, S. NiO–MgO Solid Solution Prepared by Sol–Gel Method as Precursor for Ni/MgO Methane Dry Reforming Catalyst: Effect of Calcination Temperature on Catalytic Performance. Catal. Lett. 2016, 146, 238–248. [Google Scholar] [CrossRef]
- Mazumder, J.; de Lasa, H.I. Fluidizable La2O3 promoted Ni/γ-Al2O3 catalyst for steam gasification of biomass: Effect of catalyst preparation conditions. Appl. Catal. B Environ. 2015, 168-169, 250–265. [Google Scholar] [CrossRef]
- Ammendola, P.; Cammisa, E.; Chirone, R.; Lisi, L.; Ruoppolo, G. Effect of sulphur on the performance of Rh–LaCoO3 based catalyst for tar conversion to syngas. Appl. Catal. B Environ. 2012, 113-114, 11–18. [Google Scholar] [CrossRef]
- Yeo, T.Y.; Ashok, J.; Kawi, S. Recent developments in sulphur-resilient catalytic systems for syngas production. Renew. Sustain. Energy Rev. 2019, 100, 52–70. [Google Scholar] [CrossRef]
- Zuber, C.; Husmann, M.; Schroettner, H.; Hochenauer, C.; Kienberger, T. Investigation of sulfidation and regeneration of a ZnO-adsorbent used in a biomass tar removal process based on catalytic steam reforming. Fuel 2015, 153, 143–153. [Google Scholar] [CrossRef]
- Rangan, M.; Yung, M.M.; Medlin, J.W. Experimental and computational investigations of sulfur-resistant bimetallic catalysts for reforming of biomass gasification products. J. Catal. 2011, 282, 249–257. [Google Scholar] [CrossRef]
- Rangan, M.; Yung, M.M.; Medlin, J.W. NiW and NiRu Bimetallic Catalysts for Ethylene Steam Reforming: Alternative Mechanisms for Sulfur Resistance. Catal. Lett. 2012, 142, 718–727. [Google Scholar] [CrossRef]
- Sato, K.; Fujimoto, K. Development of new nickel based catalyst for tar reforming with superior resistance to sulfur poisoning and coking in biomass gasification. Catal. Commun. 2007, 8, 1697–1701. [Google Scholar] [CrossRef]
- Veksha, A.; Giannis, A.; Oh, W.-D.; Chang, V.W.-C.; Lisak, G.; Lim, T.-T. Catalytic activities and resistance to HCl poisoning of Ni-based catalysts during steam reforming of naphthalene. Appl. Catal. A Gen. 2018, 557, 25–38. [Google Scholar] [CrossRef]
- Han, J.W.; Park, J.S.; Choi, M.S.; Lee, H. Uncoupling the size and support effects of Ni catalysts for dry reforming of methane. Appl. Catal. B Environ. 2017, 203, 625–632. [Google Scholar] [CrossRef]
- Han, J.W.; Kim, C.; Park, J.S.; Lee, H. Highly Coke-Resistant Ni Nanoparticle Catalysts with Minimal Sintering in Dry Reforming of Methane. ChemSusChem 2014, 7, 451–456. [Google Scholar] [CrossRef]
- Akri, M.; Zhao, S.; Li, X.; Zang, K.; Lee, A.F.; Isaacs, M.A.; Xi, W.; Gangarajula, Y.; Luo, J.; Ren, Y.; et al. Atomically dispersed nickel as coke-resistant active sites for methane dry reforming. Nat. Commun. 2019, 10, 5181. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Das, S.; Hongmanorom, P.; Dewangan, N.; Wai, M.H.; Kawi, S. Silica-based micro- and mesoporous catalysts for dry reforming of methane. Catal. Sci. Technol. 2018, 8, 2763–2778. [Google Scholar] [CrossRef]
- Zuo, Z.; Liu, S.; Wang, Z.; Liu, C.; Huang, W.; Huang, J.; Liu, P. Dry Reforming of Methane on Single-Site Ni/MgO Catalysts: Importance of Site Confinement. ACS Catal. 2018, 8, 9821–9835. [Google Scholar] [CrossRef]
- Ryi, S.-K.; Lee, S.-W.; Park, J.-W.; Oh, D.-K.; Park, J.-S.; Kim, S.S. Combined steam and CO2 reforming of methane using catalytic nickel membrane for gas to liquid (GTL) process. Catal. Today 2014, 236, 49–56. [Google Scholar] [CrossRef]
- Gao, X.; Tan, Z.; Hidajat, K.; Kawi, S. Highly reactive Ni-Co/SiO2 bimetallic catalyst via complexation with oleylamine/oleic acid organic pair for dry reforming of methane. Catal. Today 2017, 281, 250–258. [Google Scholar] [CrossRef]
- San-José-Alonso, D.; Juan-Juan, J.; Illán-Gómez, M.; Román-Martínez, M. Ni, Co and bimetallic Ni–Co catalysts for the dry reforming of methane. Appl. Catal. A Gen. 2009, 371, 54–59. [Google Scholar] [CrossRef]
- Horlyck, J.; Lawrey, C.; Lovell, E.; Amal, R.; Scott, J. Elucidating the impact of Ni and Co loading on the selectivity of bimetallic NiCo catalysts for dry reforming of methane. Chem. Eng. J. 2018, 352, 572–580. [Google Scholar] [CrossRef]
- Bian, Z.; Kawi, S. Highly carbon-resistant Ni–Co/SiO2 catalysts derived from phyllosilicates for dry reforming of methane. J. CO2 Util. 2017, 18, 345–352. [Google Scholar] [CrossRef]
- Benrabaa, R.; Löfberg, A.; Caballero, J.G.; Bordes-Richard, E.; Rubbens, A.; Vannier, R.-N.; Boukhlouf, H.; Barama, A. Sol–gel synthesis and characterization of silica supported nickel ferrite catalysts for dry reforming of methane. Catal. Commun. 2015, 58, 127–131. [Google Scholar] [CrossRef]
- Kim, S.M.; Abdala, P.M.; Margossian, T.; Hosseini, D.; Foppa, L.; Armutlulu, A.; van Beek, W.; Comas-Vives, A.; Copéret, C.; Müller, C. Cooperativity and Dynamics Increase the Performance of NiFe Dry Reforming Catalysts. J. Am. Chem. Soc. 2017, 139, 1937–1949. [Google Scholar] [CrossRef]
- Chen, H.-W.; Wang, C.-Y.; Yu, C.-H.; Tseng, L.-T.; Liao, P.-H. Carbon dioxide reforming of methane reaction catalyzed by stable nickel copper catalysts. Catal. Today 2004, 97, 173–180. [Google Scholar] [CrossRef]
- Wu, T.; Zhang, Q.; Cai, W.; Zhang, P.; Song, X.; Sun, Z.; Gao, L. Phyllosilicate evolved hierarchical Ni- and Cu–Ni/SiO2 nanocomposites for methane dry reforming catalysis. Appl. Catal. A Gen. 2015, 503, 94–102. [Google Scholar] [CrossRef]
- García-Diéguez, M.; Pieta, I.; Herrera, C.; Larrubia, M.; Alemany, L.J. Improved Pt-Ni nanocatalysts for dry reforming of methane. Appl. Catal. A Gen. 2010, 377, 191–199. [Google Scholar] [CrossRef]
- Károlyi, J.; Németh, M.; Evangelisti, C.; Sáfrán, G.; Schay, Z.; Horváth, A.; Somodi, F. Carbon dioxide reforming of methane over Ni–In/SiO2 catalyst without coke formation. J. Ind. Eng. Chem. 2018, 58, 189–201. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Li, W.; Jin, M.; Jin, X. Strain-Induced Reverse Phase Transformation in Nanocrystalline Co-Ni Alloys. Mater. Res. Lett. 2014, 3, 107–113. [Google Scholar] [CrossRef]
- Shamsi, A. Partial Oxidation and Dry Reforming of Methane Over Ca/Ni/K(Na) Catalysts. Catal. Lett. 2006, 109, 189–193. [Google Scholar] [CrossRef]
- Luna, A.E.C.; Iriarte, M.E. Carbon dioxide reforming of methane over a metal modified Ni-Al2O3 catalyst. Appl. Catal. A Gen. 2008, 343, 10–15. [Google Scholar] [CrossRef]
- Nagaraja, B.M.; Bulushev, D.A.; Beloshapkin, S.; Ross, J.R. The effect of potassium on the activity and stability of Ni–MgO–ZrO2 catalysts for the dry reforming of methane to give synthesis gas. Catal. Today 2011, 178, 132–136. [Google Scholar] [CrossRef]
- Fan, M.-S.; Abdullah, A.Z.; Bhatia, S. Utilization of Greenhouse Gases through Dry Reforming: Screening of Nickel-Based Bimetallic Catalysts and Kinetic Studies. ChemSusChem 2011, 4, 1643–1653. [Google Scholar] [CrossRef]
- Vogt, E.T.C.; Weckhuysen, B.M. Fluid catalytic cracking: Recent developments on the grand old lady of zeolite catalysis. Chem. Soc. Rev. 2015, 44, 7342–7370. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Huang, K.; Wu, Q.; Dai, S. Solvent-Free Self-Assembly to the Synthesis of Nitrogen-Doped Ordered Mesoporous Polymers for Highly Selective Capture and Conversion of CO2. Adv. Mater. 2017, 29, 1700445. [Google Scholar] [CrossRef]
- Ferreira-Aparicio, P.; Rodríguez-Ramos, I.; Anderson, J.A.; Guerrero-Ruiz, A. Mechanistic aspects of the dry reforming of methane over ruthenium catalysts. Appl. Catal. A Gen. 2000, 202, 183–196. [Google Scholar] [CrossRef]
- Das, S.; Sengupta, M.; Patel, J.; Bordoloi, A. A study of the synergy between support surface properties and catalyst deactivation for CO 2 reforming over supported Ni nanoparticles. Appl. Catal. A Gen. 2017, 545, 113–126. [Google Scholar] [CrossRef]
- Múnera, J.; Faroldi, B.; Frutis, E.; Lombardo, E.; Cornaglia, L.; Carrazán, S.G. Supported Rh nanoparticles on CaO–SiO2 binary systems for the reforming of methane by carbon dioxide in membrane reactors. Appl. Catal. A Gen. 2014, 474, 114–124. [Google Scholar] [CrossRef]
- Steib, M.; Lou, Y.; Jentys, A.; Lercher, J.A. Enhanced Activity in Methane Dry Reforming by Carbon Dioxide Induced Metal-Oxide Interface Restructuring of Nickel/Zirconia. ChemCatChem 2017, 9, 3809–3813. [Google Scholar] [CrossRef]
- Odedairo, T.; Chen, J.; Zhu, Z. Metal–support interface of a novel Ni–CeO2 catalyst for dry reforming of methane. Catal. Commun. 2013, 31, 25–31. [Google Scholar] [CrossRef]
- Daoura, O.; Boutros, M.; Launay, F. 3 An overview of recent works on Ni silica-based catalysts for the dry reforming of methane. Hydrog. Prod. Energy Transit. 2021, 193–212. [Google Scholar] [CrossRef]
- Das, S.; Jangam, A.; Xi, S.; Borgna, A.; Hidajat, K.; Kawi, S. Highly Dispersed Ni/Silica by Carbonization–Calcination of a Chelated Precursor for Coke-Free Dry Reforming of Methane. ACS Appl. Energy Mater. 2020, 3, 7719–7735. [Google Scholar] [CrossRef]
- Zhang, J.; Li, F. Coke-resistant NiSiO2 catalyst for dry reforming of methane. Appl. Catal. B Environ. 2015, 176–177, 513–521. [Google Scholar] [CrossRef]
- Shen, D.; Huo, M.; Li, L.; Lyu, S.; Wang, J.; Wang, X.; Zhang, Y.; Li, J. Effects of alumina morphology on dry reforming of methane over Ni/Al2O3 catalysts. Catal. Sci. Technol. 2020, 10, 510–516. [Google Scholar] [CrossRef]
- Zhao, Y.; Kang, Y.; Li, H.; Li, H. CO2 conversion to synthesis gas via DRM on the durable Al2O3/Ni/Al2O3 sandwich catalyst with high activity and stability. Green Chem. 2018, 20, 2781–2787. [Google Scholar] [CrossRef]
- Jabbour, K.; El Hassan, N.; Davidson, A.; Casale, S.; Massiani, P. Factors affecting the long-term stability of mesoporous nickel-based catalysts in combined steam and dry reforming of methane. Catal. Sci. Technol. 2016, 6, 4616–4631. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.; Liao, X.; Zhao, Y. Comparative study of the dry reforming of methane on fluidised aerogel and xerogel Ni/Al2O3 catalysts. Appl. Petrochem. Res. 2013, 3, 91–99. [Google Scholar] [CrossRef] [Green Version]
- Djebarri, B.; Gonzalez-Delacruz, V.M.; Halliche, D.; Bachari, K.; Saadi, A.; Caballero, A.; Holgado, J.P.; Cherifi, O. Promoting effect of Ce and Mg cations in Ni/Al catalysts prepared from hydrotalcites for the dry reforming of methane. React. Kinet. Mech. Catal. 2014, 111, 259–275. [Google Scholar] [CrossRef]
- Zhou, L.; Li, L.; Wei, N.; Li, J.; Basset, J.-M. Corrigendum: Effect of NiAl2O4Formation on Ni/Al2O3Stability during Dry Reforming of Methane. ChemCatChem 2015, 7, 2406. [Google Scholar] [CrossRef]
- Alipour, Z.; Rezaei, M.; Meshkani, F. Effect of alkaline earth promoters (MgO, CaO, and BaO) on the activity and coke formation of Ni catalysts supported on nanocrystalline Al2O3 in dry reforming of methane. J. Ind. Eng. Chem. 2014, 20, 2858–2863. [Google Scholar] [CrossRef]
- Al-Fatesh, A.S.; Naeem, M.A.; Fakeeha, A.H.; Abasaeed, A.E. Role of La2O3 as Promoter and Support in Ni/γ-Al2O3 Catalysts for Dry Reforming of Methane. Chin. J. Chem. Eng. 2014, 22, 28–37. [Google Scholar] [CrossRef]
- Cao, Y.; Li, H.; Zhang, J.; Shi, L.; Zhang, D. Promotional effects of rare earth elements (Sc, Y, Ce, and Pr) on NiMgAl catalysts for dry reforming of methane. RSC Adv. 2016, 6, 112215–112225. [Google Scholar] [CrossRef]
- Kathiraser, Y.; Thitsartarn, W.; Sutthiumporn, K.; Kawi, S. Inverse NiAl2O4 on LaAlO3–Al2O3: Unique Catalytic Structure for Stable CO2 Reforming of Methane. J. Phys. Chem. C 2013, 117, 8120–8130. [Google Scholar] [CrossRef]
- Chen, W.; Zhao, G.; Xue, Q.; Chen, L.; Lu, Y. High carbon-resistance Ni/CeAlO3-Al2O3 catalyst for CH4/CO2 reforming. Appl. Catal. B Environ. 2013, 136–137, 260–268. [Google Scholar] [CrossRef]
- Wang, Y.; Yao, L.; Wang, Y.; Wang, S.; Zhao, Q.; Mao, D.; Hu, C. Low-Temperature Catalytic CO2 Dry Reforming of Methane on Ni-Si/ZrO2 Catalyst. ACS Catal. 2018, 8, 6495–6506. [Google Scholar] [CrossRef]
- Wang, Y.; Li, L.; Wang, Y.; Costa, P.D.; Hu, C. Highly Carbon-Resistant Y Doped NiO–ZrOm Catalysts for Dry Reforming of Methane. Catalysts 2019, 9, 1055. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, A.A.; Al-Fatesh, A.S.; Kumar, N.S.; Abasaeed, A.E.; Kasim, S.O.; Fakeeha, A.H. Dry Reforming of Methane Using Ce-modified Ni Supported on 8%PO4 + ZrO2 Catalysts. Catalysts 2020, 10, 242. [Google Scholar] [CrossRef] [Green Version]
- Titus, J.; Goepel, M.; Schunk, S.; Wilde, N. The role of acid/base properties in Ni/MgO-ZrO2–based catalysts for dry reforming of methane. Catal. Commun. 2017, 100, 76–80. [Google Scholar] [CrossRef]
- Song, Y.; Ozdemir, E.; Ramesh, S.; Adishev, A.; Subramanian, S.; Harale, A.; Albuali, M.; Fadhel, B.A.; Jamal, A.; Moon, D.; et al. Dry reforming of methane by stable Ni–Mo nanocatalysts on single-crystalline MgO. Science 2020, 367, 777–781. [Google Scholar] [CrossRef]
- Mette, K.; Kühl, S.; Tarasov, A.; Willinger, M.G.; Kröhnert, J.; Wrabetz, S.; Trunschke, A.; Scherzer, M.; Girgsdies, F.; Düdder, H.; et al. High-Temperature Stable Ni Nanoparticles for the Dry Reforming of Methane. ACS Catal. 2016, 6, 7238–7248. [Google Scholar] [CrossRef]
- Liu, H.; Wierzbicki, D.; Dębek, R.; Motak, M.; Grzybek, T.; Da Costa, P.; Galvez, M.E. La-promoted Ni-hydrotalcite-derived catalysts for dry reforming of methane at low temperatures. Fuel 2016, 182, 8–16. [Google Scholar] [CrossRef]
- Gaur, S.; Haynes, D.J.; Spivey, J.J. Rh, Ni, and Ca substituted pyrochlore catalysts for dry reforming of methane. Appl. Catal. A Gen. 2011, 403, 142–151. [Google Scholar] [CrossRef]
- Liu, H.; Hadjltaief, H.B.; Benzina, M.; Galvez, M.E.; Da Costa, P. Natural clay based nickel catalysts for dry reforming of methane: On the effect of support promotion (La, Al, Mn). Int. J. Hydrog. Energy 2019, 44, 246–255. [Google Scholar] [CrossRef]
- Brungs, A.J.; York, A.P.; Green, M.L. Comparison of the group V and VI transition metal carbides for methane dry reforming and thermodynamic prediction of their relative stabilities. Catal. Lett. 1999, 57, 65–69. [Google Scholar] [CrossRef]
- Brungs, A.J.; York, A.P.E.; Claridge, J.; Marquez-Alvarez, C.; Green, M.L.H. Dry reforming of methane to synthesis gas over supported molybdenum carbide catalysts. Catal. Lett. 2000, 70, 117–122. [Google Scholar] [CrossRef]
- Zhu, J.; Thomas, A. Perovskite-type mixed oxides as catalytic material for NO removal. Appl. Catal. B Environ. 2009, 92, 225–233. [Google Scholar] [CrossRef]
- Shiozaki, R.; Andersen, A.G.; Hayakawa, T.; Hamakawa, S.; Suzuki, K.; Shimizu, M.; Takehira, K. Sustainable Ni/BaTiO3 Catalysts for Partial Oxidation of Methane to Synthesis Gas in Studies in Surface Science and Catalysis; Grasselli, R.K., Oyama, S.T., Gaffney, A.M., Lyons, J.E., Eds.; Elsevier: Amsterdam, The Netherlands, 1997; pp. 701–710. [Google Scholar]
- Batiot-Dupeyrat, C.; Valderrama, G.; Meneses, A.; Martinez, F.; Barrault, J.; Tatibouët, J. Pulse study of CO2 reforming of methane over LaNiO3. Appl. Catal. A Gen. 2003, 248, 143–151. [Google Scholar] [CrossRef]
- Batiot-Dupeyrat, C.; Gallego, G.A.S.; Mondragon, F.; Barrault, J.; Tatibouët, J.-M. CO2 reforming of methane over LaNiO3 as precursor material. Catal. Today 2005, 107-108, 474–480. [Google Scholar] [CrossRef]
- Zhang, Z.; Verykios, X.E.; MacDonald, S.M.; Affrossman, S. Comparative Study of Carbon Dioxide Reforming of Methane to Synthesis Gas over Ni/La2O3 and Conventional Nickel-Based Catalysts. J. Phys. Chem. 1996, 100, 744–754. [Google Scholar] [CrossRef]
- Cao, P.; Tang, P.; Bekheet, M.F.; Du, H.; Yang, L.; Haug, L.; Gili, A.; Bischoff, B.; Gurlo, A.; Kunz, M.; et al. Atomic-Scale Insights into Nickel Exsolution on LaNiO3 Catalysts via In Situ Electron Microscopy. J. Phys. Chem. C 2021, 126, 786–796. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, D.; Saccoccio, M.; Lu, Z.; Ciucci, F. From material design to mechanism study: Nanoscale Ni exsolution on a highly active A-site deficient anode material for solid oxide fuel cells. Nano Energy 2016, 27, 499–508. [Google Scholar] [CrossRef]
- Malashevich, A.; Ismail-Beigi, S. First-principles study of oxygen-deficient LaNiO3 structures. Phys. Rev. B 2015, 92, 144102. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.S.; Liu, Y.; Rong, X.; Kolpak, A.M. Electronic Origin and Kinetic Feasibility of the Lattice Oxygen Participation During the Oxygen Evolution Reaction on Perovskites. J. Phys. Chem. Lett. 2018, 9, 1473–1479. [Google Scholar] [CrossRef]
- Sutthiumporn, K.; Maneerung, T.; Kathiraser, Y.; Kawi, S. CO2 dry-reforming of methane over La0.8Sr0.2Ni0.8M0.2O3 perovskite (M = Bi, Co, Cr, Cu, Fe): Roles of lattice oxygen on C–H activation and carbon suppression. Int. J. Hydrog. Energy 2012, 37, 11195–11207. [Google Scholar] [CrossRef]
- Song, X.; Dong, X.; Yin, S.; Wang, M.; Li, M.; Wang, H. Effects of Fe partial substitution of La2NiO4/LaNiO3 catalyst precursors prepared by wet impregnation method for the dry reforming of methane. Appl. Catal. A Gen. 2016, 526, 132–138. [Google Scholar] [CrossRef]
- Valderrama, G.; Kiennemann, A.; Goldwasser, M. Dry reforming of CH4 over solid solutions of LaNi1−xCoxO3. Catal. Today 2008, 133–135, 142–148. [Google Scholar] [CrossRef]
- Mousavi, M.; Pour, A.N. Performance and structural features of LaNi0.5Co0.5O3 perovskite oxides for the dry reforming of methane: Influence of the preparation method. New J. Chem. 2019, 43, 10763–10773. [Google Scholar] [CrossRef]
- Kim, W.Y.; Jang, J.S.; Ra, E.C.; Kim, K.Y.; Kim, E.H.; Lee, J.S. Reduced perovskite LaNiO3 catalysts modified with Co and Mn for low coke formation in dry reforming of methane. Appl. Catal. A Gen. 2019, 575, 198–203. [Google Scholar] [CrossRef]
- Gallego, G.S.; Batiot-Dupeyrat, C.; Barrault, J.; Florez, E.; Mondragóna, F. Dry reforming of methane over LaNi1−yByO3±δ (B = Mg, Co) perovskites used as catalyst precursor. Appl. Catal. A Gen. 2008, 334, 251–258. [Google Scholar] [CrossRef]
- Takenaka, S.; Kato, E.; Tomikubo, Y.; Otsuka, K. Structural change of Ni species during the methane decomposition and the subsequent gasification of deposited carbon with CO2 over supported Ni catalysts. J. Catal. 2003, 219, 176–185. [Google Scholar] [CrossRef]
- Steib, M.; Jentys, A.; A Lercher, J. Structural response of Ni/ZrO2 to feed modulations during CH4reforming reactions. J. Phys. Conf. Ser. 2016, 712, 12049. [Google Scholar] [CrossRef]
- Xu, B.-Q.; Wei, J.-M.; Yu, Y.-T.; Li, J.-L.; Zhu, Q.-M. Carbon Dioxide Reforming of Methane Over Nanocomposite Ni/ZrO2 Catalysts. Top. Catal. 2003, 22, 77–85. [Google Scholar] [CrossRef]
- Palmer, C.; Upham, D.C.; Smart, S.; Gordon, M.J.; Metiu, H.; McFarland, E.W. Dry reforming of methane catalysed by molten metal alloys. Nat. Catal. 2020, 3, 83–89. [Google Scholar] [CrossRef]
- Onstot, W.J.; Minet, R.G.; Tsotsis, T.T. Design Aspects of Membrane Reactors for Dry Reforming of Methane for the Production of Hydrogen. Ind. Eng. Chem. Res. 2000, 40, 242–251. [Google Scholar] [CrossRef]
- Chai, M.; Machida, M.; Eguchi, K.; Arai, H. Promotion of hydrogen permeation on metal-dispersed alumina membranes and its application to a membrane reactor for methane steam reforming. Appl. Catal. A Gen. 1994, 110, 239–250. [Google Scholar] [CrossRef]
- Hatlevik, Ø.; Gade, S.K.; Keeling, M.K.; Thoen, P.M.; Davidson, A.; Way, J.D. Palladium and palladium alloy membranes for hydrogen separation and production: History, fabrication strategies, and current performance. Sep. Purif. Technol. 2010, 73, 59–64. [Google Scholar] [CrossRef]
- Nishimura, A.; Takada, T.; Ohata, S.; Kolhe, M. Biogas Dry Reforming for Hydrogen through Membrane Reactor Utilizing Negative Pressure. Fuels 2021, 2, 194–209. [Google Scholar] [CrossRef]
- Anzelmo, B.; Wilcox, J.; Liguori, S. Natural gas steam reforming reaction at low temperature and pressure conditions for hydrogen production via Pd/PSS membrane reactor. J. Membr. Sci. 2017, 522, 343–350. [Google Scholar] [CrossRef]
- Sumrunronnasak, S.; Tantayanon, S.; Kiatgamolchai, S.; Sukonket, T. Improved hydrogen production from dry reforming reaction using a catalytic packed-bed membrane reactor with Ni-based catalyst and dense PdAgCu alloy membrane. Int. J. Hydrog. Energy 2016, 41, 2621–2630. [Google Scholar] [CrossRef]
- Trunschke, A.; Bellini, G.; Boniface, M.; Carey, S.J.; Dong, J.; Erdem, E.; Foppa, L.; Frandsen, W.; Geske, M.; Ghiringhelli, L.M.; et al. Towards Experimental Handbooks in Catalysis. Top. Catal. 2020, 63, 1683–1699. [Google Scholar] [CrossRef]
- Tavasoli, A.; Ozin, G. Green Syngas by Solar Dry Reforming. Joule 2018, 2, 571–575. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Mao, M.; Zhang, Q.; Li, Y.; Bai, J.; Yang, Y.; Zeng, M.; Zhao, X. Solar-Light-Driven CO2 Reduction by CH4 on Silica-Cluster-Modified Ni Nanocrystals with a High Solar-to-Fuel Efficiency and Excellent Durability. Adv. Energy Mater. 2018, 8, 1702472. [Google Scholar] [CrossRef]
- Simakov, D.S.A.; Wright, M.M.; Ahmed, S.; Mokheimer, E.M.A.; Román-Leshkov, Y. Solar thermal catalytic reforming of natural gas: A review on chemistry, catalysis and system design. Catal. Sci. Technol. 2015, 5, 1991–2016. [Google Scholar] [CrossRef]
- Vakili, R.; Gholami, R.; Stere, C.E.; Chansai, S.; Chen, H.; Holmes, S.M.; Jiao, Y.; Hardacre, C.; Fan, X. Plasma-assisted catalytic dry reforming of methane (DRM) over metal-organic frameworks (MOFs)-based catalysts. Appl. Catal. B Environ. 2020, 260, 118195. [Google Scholar] [CrossRef]
- Puliyalil, H.; Jurković, D.L.; Dasireddy, V.D.B.C.; Likozar, B. A review of plasma-assisted catalytic conversion of gaseous carbon dioxide and methane into value-added platform chemicals and fuels. RSC Adv. 2018, 8, 27481–27508. [Google Scholar] [CrossRef] [Green Version]
- Kogelschatz, U. Dielectric-barrier discharges: Their history, discharge physics, and industrial applications. Plasma Chem. Plasma Process. 2003, 23, 1–46. [Google Scholar] [CrossRef]
- Tao, X.; Bai, M.; Li, X.; Long, H.; Shang, S.; Yin, Y.; Dai, X. CH4–CO2 reforming by plasma—challenges and opportunities. Prog. Energy Combust. Sci. 2011, 37, 113–124. [Google Scholar] [CrossRef]
- Chen, H.L.; Lee, H.M.; Chen, S.H.; Chao, Y.; Chang, M.B. Review of plasma catalysis on hydrocarbon reforming for hydrogen production—Interaction, integration, and prospects. Appl. Catal. B Environ. 2008, 85, 1–9. [Google Scholar] [CrossRef]
- Chen, H.L.; Lee, H.M.; Chen, S.H.; Chang, M.B.; Yu, S.J.; Li, S.N. Removal of Volatile Organic Compounds by Single-Stage and Two-Stage Plasma Catalysis Systems: A Review of the Performance Enhancement Mechanisms, Current Status, and Suitable Applications. Environ. Sci. Technol. 2009, 43, 2216–2227. [Google Scholar] [CrossRef]
- Gesser, H.D.; Hunter, N.R.; Probawono, D. The CO2 Reforming of Natural Gas in a Silent Discharge Reactor. Plasma Chem. Plasma Process. 1998, 18, 241–245. [Google Scholar] [CrossRef]
- Zhou, L.M.; Xue, B.; Kogelschatz, U.; Eliasson, B. Nonequilibrium Plasma Reforming of Greenhouse Gases to Synthesis Gas. Energy Fuels 1998, 12, 1191–1199. [Google Scholar] [CrossRef]
- Sobolewski, M.A.; Langan, J.G.; Felke, B.S. Electrical optimization of plasma-enhanced chemical vapor deposition chamber cleaning plasmas. J. Vac. Sci. Technol. B Microelectron. Nanometer Struct. Process. Meas. Phenom. 1998, 16, 173–182. [Google Scholar] [CrossRef] [Green Version]
- Okumura, T. Inductively Coupled Plasma Sources and Applications. Phys. Res. Int. 2010, 2010, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Kroesen, G.M.W.; Schram, D.C.; de Haas, J.C.M. Description of a flowing cascade arc plasma. Plasma Chem. Plasma Process. 1990, 10, 531–551. [Google Scholar] [CrossRef]
- Fridman, A.; Gutsol, A.; Cho, Y.I. Non-Thermal Atmospheric Pressure Plasma, in Advances in Heat Transfer; Fridman, A., Ed.; Elsevier: Amsterdam, The Netherlands, 2007; pp. 1–142. [Google Scholar]
- Lu, X.; Naidis, G.; Laroussi, M.; Ostrikov, K. Guided ionization waves: Theory and experiments. Phys. Rep. 2014, 540, 123–166. [Google Scholar] [CrossRef]
- Li, M.-W.; Tian, Y.-L.; Xu, G.-H. Characteristics of Carbon Dioxide Reforming of Methane via Alternating Current (AC) Corona Plasma Reactions. Energy Fuels 2007, 21, 2335–2339. [Google Scholar] [CrossRef]
- Drost, H.; Rutkowsky, J.; Mach, R.; Klotz, H.-D.; Schulz, G. Plasma-chemical methane conversion under nonthermal and thermal conditions: An attempt toward uniform kinetic modeling. Plasma Chem. Plasma Process. 1985, 5, 283–291. [Google Scholar] [CrossRef]
- Tao, X.; Qi, F.; Yin, Y.; Dai, X. CO2 reforming of CH4 by combination of thermal plasma and catalyst. Int. J. Hydrog. Energy 2008, 33, 1262–1265. [Google Scholar] [CrossRef]
- Rao, Z.; Cao, Y.; Huang, Z.; Yin, Z.; Wan, W.; Ma, M.; Wu, Y.; Wang, J.; Yang, G.; Cui, Y.; et al. Insights into the Nonthermal Effects of Light in Dry Reforming of Methane to Enhance the H2/CO Ratio Near Unity over Ni/Ga2O3. ACS Catal. 2021, 11, 4730–4738. [Google Scholar] [CrossRef]
- Schlögl, R. Heterogeneous Catalysis. Angew. Chem. Int. Ed. 2015, 54, 3465–3520. [Google Scholar] [CrossRef] [Green Version]
- Greiner, M.T.; JonesOrcid, T.E.; Klyushin, A.; Knop-Gericke, A.; Schlögl, R. Ethylene Epoxidation at the Phase Transition of Copper Oxides. J. Am. Chem. Soc. 2017, 139, 11825–11832. [Google Scholar] [CrossRef]
- Linde-Engineering. Technologies That Do More with Less. Available online: https://www.linde-engineering.com/en/about-linde-engineering/success-stories/technologies-more-with-less.html (accessed on 5 April 2022).
- BASF-Catalysts. SynspireTM Catalysts for Dry Reforming. 2019. Available online: https://catalysts.basf.com/industries/chemical/syngas-catalysts/synspire-dry-reforming-catalysts (accessed on 4 April 2022).
- Tullo, A.H. Dry reforming puts CO2 to work. C&EN Glob. Enterp. 2016, 94, 30. [Google Scholar]
- Fujita, T.; Peng, X.; Yamaguchi, A.; Cho, Y.; Zhang, Y.; Higuchi, K.; Yamamoto, Y.; Tokunaga, T.; Arai, S.; Miyauchi, M.; et al. Nanoporous Nickel Composite Catalyst for the Dry Reforming of Methane. ACS Omega 2018, 3, 16651–16657. [Google Scholar] [CrossRef]
- Shoji, S.; Peng, X.; Imai, T.; Kumar, P.S.M.; Higuchi, K.; Yamamoto, Y.; Tokunaga, T.; Arai, S.; Ueda, S.; Hashimoto, A.; et al. Topologically immobilized catalysis centre for long-term stable carbon dioxide reforming of methane. Chem. Sci. 2019, 10, 3701–3705. [Google Scholar] [CrossRef] [Green Version]
- Kappes, H.B.; Energy Transition in the European Steel Industry—Reality Not Exception, in Direct from MIDREX. 2021, MIDREX. Available online: https://www.midrex.com/tech-article/energy-transition-in-the-european-steel-industry-reality-not-exception/ (accessed on 5 April 2022).
- Chevrier, V.; Lauren, L.; Michishita, H. MIDREX® Process: Bridge to Ultra-low CO2 Ironmaking. Kobelco Technol. Rev. 2021, 39, 33–40. [Google Scholar]
- Atsushi, M.; Hiroshi, U.; Sakaguchi, T. MIDREX Processes. Kobelco Technol. Rev. 2010, 29, 50–57. [Google Scholar]
- ArcelorMittal. Carbon2Value. Available online: https://www.carbon2value.be/en/ (accessed on 4 April 2022).
- Group, S. Paul Wurth and the Steel Partners Dillinger and Saarstahl Join Forces on Development of Dry Reforming Technology. 2021. Available online: https://www.sms-group.com/press-media/press-releases/press-detail/paul-wurth-and-the-steel-partners-dillinger-and-saarstahl-join-forces-on-development-of-dry-reforming-technology (accessed on 5 April 2022).
- Buelens Lukas, C.; Galvita, V.V.; Poelman, H.; Detavernier, C.; Marin, G.B. Super-dry reforming of methane intensifies CO2 utilization via Le Chatelier’s principle. Science 2016, 354, 449–452. [Google Scholar] [CrossRef]
- Galvita, V.V.; Poelman, H.; Marin, G.B. Combined chemical looping for energy storage and conversion. J. Power Sources 2015, 286, 362–370. [Google Scholar] [CrossRef]
- Catalisti. Flanders Industry Innovation Moonshot SDR. Available online: https://moonshotflanders.be/mot3-sdr/ (accessed on 4 April 2022).
- Wismann Sebastian, T. Electrified methane reforming: A compact approach to greener industrial hydrogen production. Science 2019, 364, 756–759. [Google Scholar] [CrossRef] [Green Version]
- News, B.-B.F. BASF, SABIC and Linde Join Forces to Realize the World’s First Electrically Heated Steam Cracker Furnace 2021. Available online: https://www.basf.com/global/en/who-we-are/sustainability/whats-new/sustainability-news/2021/basf-sabic-and-linde-join-forces-to-realize-wolds-first-electrically-heated-steam-cracker-furnace.html (accessed on 5 April 2022).
- Labrecque, R.; Lavoie, J.-M. Dry reforming of methane with CO2 on an electron-activated iron catalytic bed. Bioresour. Technol. 2011, 102, 11244–11248. [Google Scholar] [CrossRef]
- Banville, M.L.; Lavoie, J.-M.R. Dry Reforming of Methane Under An Electro-catalytic Bed: Effect of Electrical Current And Catalyst Composition. Energy Sustain. 2015, 186, 9. [Google Scholar]
- Vasconcelos, B.R.L. Is dry reforming the solution to reduce natural gas carbon footprint? Int. J. Energy Prod. Manag. 2018, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- Dokania, A.; Ramirez, A.; Bavykina, A.; Gascon, J. Heterogeneous Catalysis for the Valorization of CO2: Role of Bifunctional Processes in the Production of Chemicals. ACS Energy Lett. 2019, 4, 167–176. [Google Scholar] [CrossRef]
- Parker, L. The World’s Plastic Pollution Crisis Explained 2019. Available online: https://www.nationalgeographic.com/environment/article/plastic-pollution (accessed on 28 January 2022).
- Han, B.; Wei, W.; Chang, L.; Cheng, P.; Hu, Y.H. Efficient Visible Light Photocatalytic CO2 Reforming of CH4. ACS Catal. 2016, 6, 494–497. [Google Scholar] [CrossRef]
- Liu, H.; Meng, X.; Dao, T.D.; Zhang, H.; Li, P.; Chang, K.; Wang, T.; Li, M.; Nagao, T.; Ye, J. Conversion of Carbon Dioxide by Methane Reforming under Visible-Light Irradiation: Surface-Plasmon-Mediated Nonpolar Molecule Activation. Angew. Chem. Int. Ed. 2015, 54, 11545–11549. [Google Scholar] [CrossRef]
- Challiwala, M.S.; Choudhury, H.A.; Wang, D.; El-Halwagi, M.M.; Weitz, E.; Elbashir, N.O. A novel CO2 utilization technology for the synergistic co-production of multi-walled carbon nanotubes and syngas. Sci. Rep. 2021, 11, 1–8. [Google Scholar] [CrossRef]
Figure 1.
Free energies of formation (ΔGf) of C1 compounds and related chemicals associated with valorization strategies.
Figure 1.
Free energies of formation (ΔGf) of C1 compounds and related chemicals associated with valorization strategies.

Figure 2.
A representation of carbon stream in industrial syngas technology and C1 chemistry. The carbon “footprint” is not reduced whenever the molecules produced in downstream processes are burnt back into CO2 for energy applications. Note, plastics are the second major sink of current C1 industries.
Figure 2.
A representation of carbon stream in industrial syngas technology and C1 chemistry. The carbon “footprint” is not reduced whenever the molecules produced in downstream processes are burnt back into CO2 for energy applications. Note, plastics are the second major sink of current C1 industries.

Figure 3.
Thermodynamic equilibrium plots of DRM under different gas mixtures at 1 bar [72].
Figure 3.
Thermodynamic equilibrium plots of DRM under different gas mixtures at 1 bar [72].

Figure 4.
Thermodynamic equilibrium plots of DRM as a function of temperature at varying pressures. (a) Carbon production, (b) H2 production, (c) syngas ratio and (d) water production. Reprinted from ref. [73], Copyright 2015, with permission from Elsevier.
Figure 4.
Thermodynamic equilibrium plots of DRM as a function of temperature at varying pressures. (a) Carbon production, (b) H2 production, (c) syngas ratio and (d) water production. Reprinted from ref. [73], Copyright 2015, with permission from Elsevier.

Figure 5.
A scheme summarizing the proposed chemical paths of syngas generation during DRM.

Figure 6.
Post-mortem TEM and the elemental mapping analysis of a catalyst composed of Ni supported on ceria-zirconia (a–f) and of another catalyst composed of bimetallic Ni–Mn supported on ceria zirconia (g–m). Reprinted with permission from ref [102]. Copyright 2018 American Chemical Society.
Figure 6.
Post-mortem TEM and the elemental mapping analysis of a catalyst composed of Ni supported on ceria-zirconia (a–f) and of another catalyst composed of bimetallic Ni–Mn supported on ceria zirconia (g–m). Reprinted with permission from ref [102]. Copyright 2018 American Chemical Society.

Figure 7.
Electron microscopy images and size distribution of Ni-hydroxyapatite-Ce samples. (a) HAADF-STEM images of 0.5Ni-hydroxyapatite-Ce, (b) 2Ni-hydroxyapatite-Ce, and (c) 10Ni-hydroxyapatite-Ce samples after 500 °C H2 reduction. (d) Particle size distributions of (a–c); yellow circles indicate atomically dispersed Ni and red squares indicate Ni metals nanoparticles. (e) EDX element maps of 0.5Ni1/HAP-Ce [142].
Figure 7.
Electron microscopy images and size distribution of Ni-hydroxyapatite-Ce samples. (a) HAADF-STEM images of 0.5Ni-hydroxyapatite-Ce, (b) 2Ni-hydroxyapatite-Ce, and (c) 10Ni-hydroxyapatite-Ce samples after 500 °C H2 reduction. (d) Particle size distributions of (a–c); yellow circles indicate atomically dispersed Ni and red squares indicate Ni metals nanoparticles. (e) EDX element maps of 0.5Ni1/HAP-Ce [142].

Figure 8.
Gas flow passage and membrane effect in catalytic nickel membrane. Reprinted from ref. [145], Copyright 2014, with permission from Elsevier.
Figure 8.
Gas flow passage and membrane effect in catalytic nickel membrane. Reprinted from ref. [145], Copyright 2014, with permission from Elsevier.

Figure 9.
A representation of the bimetallic effect in DRM activity. Reprinted from ref. [148] Copyright 2018, with permission from Elsevier.
Figure 9.
A representation of the bimetallic effect in DRM activity. Reprinted from ref. [148] Copyright 2018, with permission from Elsevier.

Figure 10.
The versatility of silica to generate materials of tunable pore structure may outperform current catalytic materials applied in DRM. Reproduced from ref. [143], Copyright 2018, with permission from Royal Society of Chemistry.
Figure 10.
The versatility of silica to generate materials of tunable pore structure may outperform current catalytic materials applied in DRM. Reproduced from ref. [143], Copyright 2018, with permission from Royal Society of Chemistry.

Figure 11.
The presence of a porous silica overgrowth stabilizes the performing catalyst. (a) TEM and (b) HAADF-STEM images of the Ni/SiO2 catalyst; and (c) TEM, and (d) HAADF-STEM images of the silica-coated Ni/SiO2 catalyst after DRM reaction for 170 h at 800 °C. Coke formation is clear in the uncoated catalyst. Reproduced from ref. [141], Copyright 2014, with permission from Wiley.
Figure 11.
The presence of a porous silica overgrowth stabilizes the performing catalyst. (a) TEM and (b) HAADF-STEM images of the Ni/SiO2 catalyst; and (c) TEM, and (d) HAADF-STEM images of the silica-coated Ni/SiO2 catalyst after DRM reaction for 170 h at 800 °C. Coke formation is clear in the uncoated catalyst. Reproduced from ref. [141], Copyright 2014, with permission from Wiley.

Figure 12.
Representative TEM micrographs of the in situ reduced (800 °C/3 h) catalysts achieved by impregnation (a,b) and one-pot synthesis (c,d) of mesoporous alumina. Reproduced from ref. [173] Copyright 2016, with permission from Royal Society of Chemistry.
Figure 12.
Representative TEM micrographs of the in situ reduced (800 °C/3 h) catalysts achieved by impregnation (a,b) and one-pot synthesis (c,d) of mesoporous alumina. Reproduced from ref. [173] Copyright 2016, with permission from Royal Society of Chemistry.

Figure 13.
The paths of coke generation and catalytic deactivation are hindered by the immobilization of the active metallic particle. Reproduced from ref. [176], Copyright 2015, with permission from Wiley.
Figure 13.
The paths of coke generation and catalytic deactivation are hindered by the immobilization of the active metallic particle. Reproduced from ref. [176], Copyright 2015, with permission from Wiley.

Figure 14.
(a) A representation of the synthetic protocol of high-performance Ni-Mo catalyst on cubic magnesia support. (b) The cubic support is decorated with bimetallic nanoparticles (c). The spent catalyst does not develop carbon deposits nor sintering of the metallic phases (d). Reproduced from ref. [186], Copyright 2020, with permission of The American Association for the Advancement of Science.
Figure 14.
(a) A representation of the synthetic protocol of high-performance Ni-Mo catalyst on cubic magnesia support. (b) The cubic support is decorated with bimetallic nanoparticles (c). The spent catalyst does not develop carbon deposits nor sintering of the metallic phases (d). Reproduced from ref. [186], Copyright 2020, with permission of The American Association for the Advancement of Science.

Figure 15.
A representation of the metal–support interaction during different stages of catalyst synthesis of a DRM catalyst. The aluminate overgrowths (red) stabilize the Ni nanoparticle (green) and particle size (a–c) can be finely tuned with Ni loading. Reprinted with permission from ref. [187], Copyright 2018 American Chemical Society.
Figure 15.
A representation of the metal–support interaction during different stages of catalyst synthesis of a DRM catalyst. The aluminate overgrowths (red) stabilize the Ni nanoparticle (green) and particle size (a–c) can be finely tuned with Ni loading. Reprinted with permission from ref. [187], Copyright 2018 American Chemical Society.

Figure 16.
Models of perovskite-type oxides with ABO3 and A2BO4 structure. The red cycle means that an A-site cation was substituted by a foreign one; the blue square represents an oxygen vacancy. Reprinted from ref. [193], Copyright 2009, with permission from Elsevier.
Figure 16.
Models of perovskite-type oxides with ABO3 and A2BO4 structure. The red cycle means that an A-site cation was substituted by a foreign one; the blue square represents an oxygen vacancy. Reprinted from ref. [193], Copyright 2009, with permission from Elsevier.

Figure 17.
Exemplification of exsolution dynamics by EELS maps of Ni and La distribution during the vacuum heating process of LaNiO3 at selected temperatures (a–i). Green and red colors indicate La and Ni, respectively. Individual grains of LaNiO3 are marked by dashed lines in the inset HAADF image. In addition, an EDX map of the initial sample region is shown at the bottom of part a. Green and red colors indicate La and Ni, respectively [198].
Figure 17.
Exemplification of exsolution dynamics by EELS maps of Ni and La distribution during the vacuum heating process of LaNiO3 at selected temperatures (a–i). Green and red colors indicate La and Ni, respectively. Individual grains of LaNiO3 are marked by dashed lines in the inset HAADF image. In addition, an EDX map of the initial sample region is shown at the bottom of part a. Green and red colors indicate La and Ni, respectively [198].

Figure 18.
Time series of a cycling DRM reactor. Methane decomposition (100 min duration) stages are subsequently followed by carbon oxidation with CO2 (100 min duration). Reprinted from ref. [208], Copyright 2003, with permission from Elsevier.
Figure 18.
Time series of a cycling DRM reactor. Methane decomposition (100 min duration) stages are subsequently followed by carbon oxidation with CO2 (100 min duration). Reprinted from ref. [208], Copyright 2003, with permission from Elsevier.

Figure 19.
A schematic illustration of the high-performance DRM catalyst featuring small (5 nm) bimetallic (Ni-Cu, Ni-Mo or Ni-Co) nanoparticles anchored on modified (K addition) alumina support or by structured nanoporous overlayers.
Figure 19.
A schematic illustration of the high-performance DRM catalyst featuring small (5 nm) bimetallic (Ni-Cu, Ni-Mo or Ni-Co) nanoparticles anchored on modified (K addition) alumina support or by structured nanoporous overlayers.

Figure 20.
Operando SEM study of DRM on a metallic Ni catalyst in the temperature regime from 700 °C to 900 °C. (a) From bottom to top: time series of simultaneously recorded temperature of the catalyst, image intensities, and reaction traces of CH4, CO2, O2, H2, CO and H2O. Color-coded areas correspond to consecutive stages of the experiment. (b) Time series of the measured H2:CO ratio. (c) Arrhenius plots of the catalytic production of H2 and CO. The color region in (c) marks the temperature range at which oxides were observed. Reprinted from ref. [78], Copyright 2020, with permission from Elsevier.
Figure 20.
Operando SEM study of DRM on a metallic Ni catalyst in the temperature regime from 700 °C to 900 °C. (a) From bottom to top: time series of simultaneously recorded temperature of the catalyst, image intensities, and reaction traces of CH4, CO2, O2, H2, CO and H2O. Color-coded areas correspond to consecutive stages of the experiment. (b) Time series of the measured H2:CO ratio. (c) Arrhenius plots of the catalytic production of H2 and CO. The color region in (c) marks the temperature range at which oxides were observed. Reprinted from ref. [78], Copyright 2020, with permission from Elsevier.

Figure 21.
A representation of an alternative carbon cycle for industrial syngas technology. The proposed path links hydrogen generation by DRM or SRM and captures the generated organic molecules as carbon in the Boudouard stage subsequent to reforming.
Figure 21.
A representation of an alternative carbon cycle for industrial syngas technology. The proposed path links hydrogen generation by DRM or SRM and captures the generated organic molecules as carbon in the Boudouard stage subsequent to reforming.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
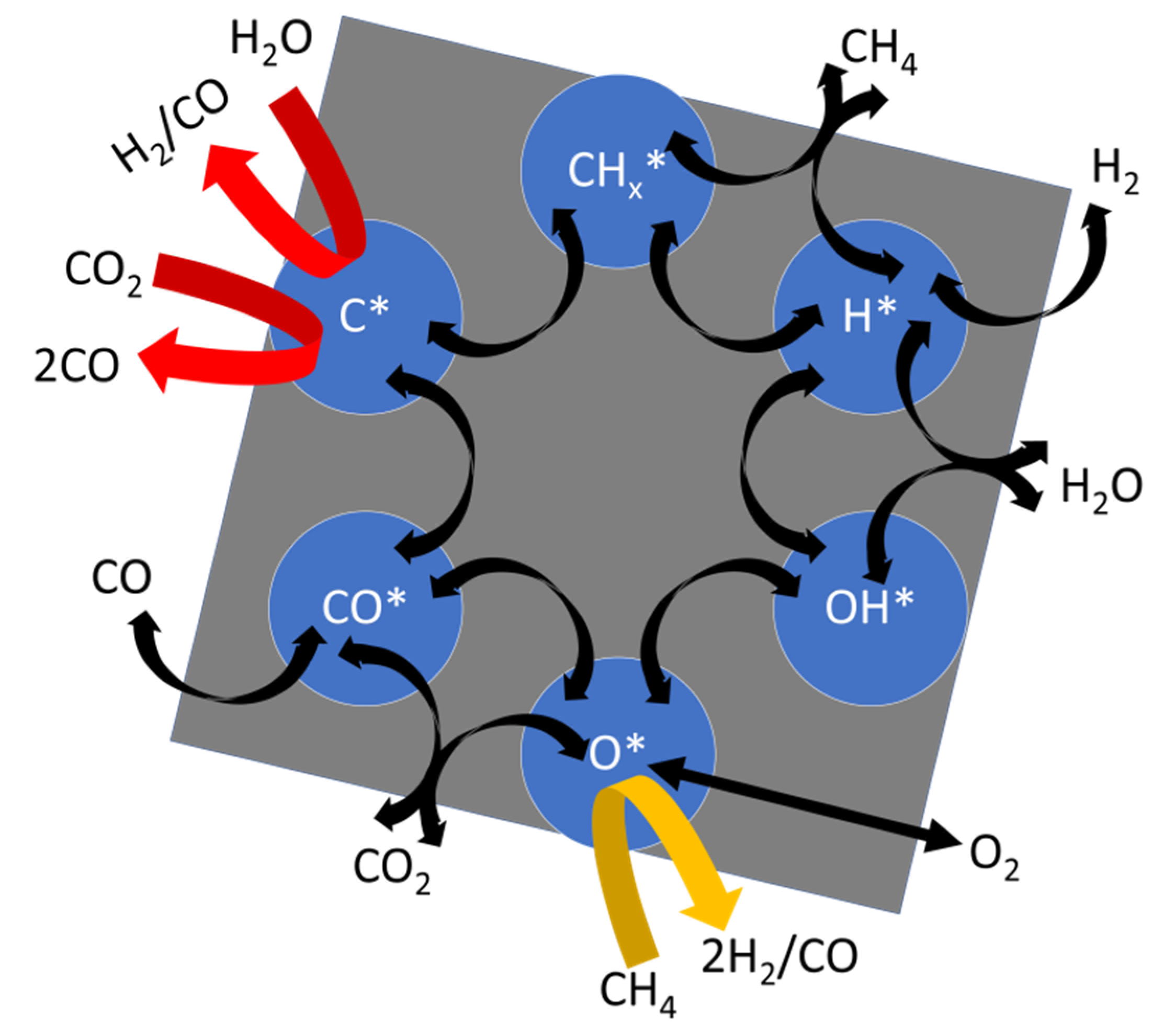{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Emission sources of greenhouse gases for the year 2016. Numbers are taken from reference [10].
Table 1.
Emission sources of greenhouse gases for the year 2016. Numbers are taken from reference [10].
| Source | Fraction (%) | Absolute (gt) | Sub-Source | Fraction (%) | Absolute (gt) |
|---|---|---|---|---|---|
| Energy | 73.2 | 36.2 | Industry * | 24.2 | 11.95 |
| Mobility ** | 16.2 | 8.00 | |||
| Heating | 17.5 | 8.54 | |||
| Other combustion | 7.8 | 3.85 | |||
| Leaks | 5.8 | 2.87 | |||
| Energy in agriculture | 1.7 | 0.84 | |||
| Industrial products | 5.2 | 2.57 | Cement | 3.0 | 1.48 |
| Chemicals | 2.2 | 1.09 | |||
| Waste | 3.2 | 1.58 | Sewage | 1.3 | 0.64 |
| Landfill | 1.9 | 0.94 | |||
| Agriculture | 18.4 | 9.09 | Animals | 5.8 | 2.86 |
| Cropland | 6.8 | 3.36 | |||
| Combustion of crop waste | 3.5 | 1.73 |
* steel 7.2%, process heat for chemicals 3.6%. ** air traffic 1.9%, shipping 1.7%.
Table 2.
Examples of strategies for chemical valorization of CO2 and CH4, the targeted products and generated waste.
Table 2.
Examples of strategies for chemical valorization of CO2 and CH4, the targeted products and generated waste.
| CO2 | CH4 | ||||
|---|---|---|---|---|---|
| Partner | Products | Waste | Partner | Products | Waste |
| H2 | Methanol Dimethylether CO Olefins | H2O H2O H2O H2O | O2 | Syngas Energy | -- CO2, H2O |
| CH4 | Syngas | -- | CO2 | Syngas | -- |
| NH3 | Urea | H2O | H2O | Syngas | H2O |
Table 3.
Bond dissociation energies of selected C1 molecules. Values are taken from reference [34].
Table 3.
Bond dissociation energies of selected C1 molecules. Values are taken from reference [34].
| Bond | Dissociation Energy [kJmol−1] | Molecule |
|---|---|---|
| H3C-H | 439 | Methane |
| OC=O | 532 | carbon dioxide |
| C≡O | 1077 | carbon monoxide |
| H3CO-H | 440 | Methanol |
| H-H | 436 | Hydrogen |
| O=O | 498 | Oxygen |
| N≡N | 945 | Nitrogen |
Table 4.
Summary of the effect on DRM of increasing the magnitude of an external parameter according to free energy minimization calculations.
Table 4.
Summary of the effect on DRM of increasing the magnitude of an external parameter according to free energy minimization calculations.
| External Parameter | Pressure | Temperature | CO2/CH4 |
|---|---|---|---|
| Effect of increasing the value of the external parameter while the other ones are constant | H2/CO increases | H2/CO decreases and stabilizes at 1.00 | H2/CO decreases |
| H2O increases | H2O decreases | H2O increases | |
| Coke increases | Coke decreases | Coke decreases | |
| Conversion decreases | Conversion increases | Conversion of CH4 increases |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sandoval-Diaz, L.E.; Schlögl, R.; Lunkenbein, T. Quo Vadis Dry Reforming of Methane?—A Review on Its Chemical, Environmental, and Industrial Prospects. Catalysts 2022, 12, 465. https://doi.org/10.3390/catal12050465
AMA Style
Sandoval-Diaz LE, Schlögl R, Lunkenbein T. Quo Vadis Dry Reforming of Methane?—A Review on Its Chemical, Environmental, and Industrial Prospects. Catalysts. 2022; 12(5):465. https://doi.org/10.3390/catal12050465
Chicago/Turabian StyleSandoval-Diaz, Luis E., Robert Schlögl, and Thomas Lunkenbein. 2022. "Quo Vadis Dry Reforming of Methane?—A Review on Its Chemical, Environmental, and Industrial Prospects" Catalysts 12, no. 5: 465. https://doi.org/10.3390/catal12050465
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.