A Simple and Efficient Protocol for Proline-Catalysed Asymmetric Aldol Reaction
by
 ,
,
Marco Giuseppe Emma
, 
Alice Tamburrini
,
Ada Martinelli
,
Marco Lombardo
*,
Arianna Quintavalla
* and
Claudio Trombini
Department of Chemistry “G. Ciamician”, Alma Mater Studiorum, University of Bologna, Via Selmi 2, 40126 Bologna, Italy
*
Authors to whom correspondence should be addressed.
Catalysts 2020, 10(6), 649; https://doi.org/10.3390/catal10060649
Submission received: 1 May 2020
/
Revised: 26 May 2020
/
Accepted: 7 June 2020
/
Published: 10 June 2020
(This article belongs to the Special Issue New Trends in Asymmetric Catalysis)
Abstract
:The proline-catalysed asymmetric aldol reaction is usually carried out in highly dipolar aprotic solvents (dimethylsulfoxide, dimethylformamide, acetonitrile) where proline presents an acceptable solubility. Protic solvents are generally characterized by poor stereocontrol (e.g., methanol) or poor reactivity (e.g., water). Here, we report that water/methanol mixtures are exceptionally simple and effective reaction media for the intermolecular organocatalytic aldol reaction using the simple proline as the catalyst.
1. Introduction
The asymmetric intermolecular aldol addition catalysed by (S)-proline, proposed by List and co-workers in 2000 [1], is the prototype of enamine-based organocatalysis [2,3]. Proline is the smallest air- and water-stable bifunctional catalyst; it is inexpensive, non-toxic, and available in both enantiomeric forms. It has been proven to catalyse enantioselective α-functionalizations of carbonyl compounds (aldol and Mannich reactions, Michael additions, α-halogenations, oxygenations, aminations, and so on), adopting reaction protocols that do not require inert atmosphere and anhydrous conditions and are carried out at room temperature [4,5].
The proline-catalysed aldol reaction [4,6,7,8,9,10,11,12,13,14,15,16,17,18,19] has been object of in-depth analyses after the first mechanistic rationale proposed by List and Houck [20], in particular, fundamental contributions have been given by Seebach [21], Armstrong and Blackmond [22,23], Sharma and Sunoj [24], Benaglia [25], and Gschwind [26,27]. However, proline scarce solubility in most organic solvents has limited its use in dimethylsulfoxide (DMSO), acetonitrile, or dimethylformamide (DMF). Moreover, proline often displays poor activity, requiring the use of high catalyst loadings and high reaction times, sometimes with unsatisfactory stereocontrol [1,28,29,30,31,32,33,34,35,36]. Because, in several cases, proline-catalysed aldol reactions are still underdeveloped, the last 15 years have witnessed an intense effort aimed at modifying the proline scaffold, following two directions: (1) the carboxylic group is replaced by a new hydrogen-bonding donor, such as a tetrazole, or by a sterically demanding group, such as the Hayashi–Jørgensen diarylmethanol and related compounds, as exhaustively reviewed by Trost [37]; and (2) the carboxylic group is retained and a supplementary substituent is bound to the proline scaffold. The new substituent, generally installed on the 4-OH group of 4-hydroxyproline, may play different roles: (i) it modifies the solubility profile of the parent amino acid, expanding the solvent choice to further classes [38]; and/or (ii) it enhances the catalyst activity and stereoselectivity, allowing a reduction of catalyst loading and reaction time; and/or (iii) it allows the catalyst immobilization on a solid support [39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56], adopting a biphasic condition reaction protocol. Despite that high levels of reactivity and selectivity have been achieved with these modified prolines, most of the aforementioned proline derivatives require multi-step syntheses, dramatically increasing the catalyst cost, a severe limitation particularly when the catalyst cannot be efficiently recycled.
After having contributed to the synthesis of prolines, mostly modified with the incorporation of ionic tags on 4-position, and obtaining excellent results in terms of activity and stereoselectivity as well as catalyst recyclability [57,58,59,60,61,62,63,64,65,66], we decided to go back to the parent unmodified (S)-proline. We envisaged to improve the performance of this small, stable, inexpensive, non-toxic, and easily available organocatalyst exploring new experimental conditions.
The role of the solvent in determining the aldol reaction efficiency was also addressed by other authors. Invariably, the use of unmodified proline (without additives) forced to choose polar aprotic solvents to obtain acceptable yields and selectivities [1,11,13,14,15,28,29,30,31,32,33,34,35,36,44,45]. A peculiar case was represented by ionic liquids (ILs) [67,68,69,70,71,72], which allowed in a few cases to decrease the catalyst loading (up to 1 mol%) and, during the work-up, to confine proline in a separate phase, enabling a simple product isolation and the reuse of the catalytic system. In recent literature, attempts are reported where proline is used in acetone/CHCl3 mixtures [73], in DMF at 4 °C (a condition that often requires several days) [74], in tert-butyl methyl ether (MTBE) [75], in deep eutectic solvents [76,77], or under solvent-free conditions, with [78,79,80] or without [81] the ball milling approach. However, many issues associated with the use of proline remain unsolved and polar aprotic solvents are characterized by several undesirable features (toxicity, high production cost, high environmental impact, difficult product recovery) [82,83,84].
The use of unmodified proline can be also combined with additives [85,86], such as water [85,86,87], acids [85,86,88], diols [89,90], amines [85,86], or thioureas [91,92,93]. In these cases, the additive can tune the solubility, the reactivity, and/or the stereoselectivity of native proline, making the asymmetric aldol process more efficient [18]. In one case, the addition of achiral guanidinium salts as additives allows to switch the diastereoselectivity as a function of the counterion, for example, tetrafluoroborate versus tetraphenylborate [94]. Nevertheless, the achieved performance is not optimal yet (long reaction times, stereocontrol strongly depending on the substrate) and some drawbacks are still present, such as high proline loadings and the cost of the not recovered chiral additive. Significant advances were accomplished employing metal salts as additives [95,96,97,98,99,100,101,102,103,104,105,106]. In particular, Reiser and co-workers developed a strategy based on cobalt(II)-proline complexes, which ensured excellent results in direct aldol reactions involving aromatic aldehydes [106]. However, several disadvantages lead the avoidance of the use of metals, especially at an industrial level (costs, toxicity, environmental impact, limited sources) [107,108,109].
In the present work, we aim to avoid the use of both polar aprotic solvents and additives (being sometimes expensive, mostly non-recoverable, and contaminants, used in non-generalizable procedures), in order to develop an efficient and sustainable organocatalyzed aldol condensation protocol, which can be interesting from a scale-up and an industrial point of view. In particular, our goals are as follows: (1) a reduction of the process costs, related to employed solvents and reagents, but also purification and waste disposal; and (2) an improved reactivity, especially for poorly reactive substrates. We planned to achieve these objectives by using the following: (i) the native proline, a small, stable, inexpensive, and non-toxic organocatalyst; and (ii) the minimum amount of a low-cost, non-toxic reaction solvent, enabling a good process efficiency and a simple and inexpensive final reaction work-up.
A number of research groups noticed that protic media were not suitable for aldol condensations promoted exclusively by native proline [15,29,30,31,32,33,87]. However, despite a plethora of studies focused on the use and the role of water (as solvent, co-solvent, or additive) [31,32,34,87,100,101,102,103,104,105,110,111], very few authors extended their investigations to alcohols [15,29,31,42,102,106], discouraged by the generally observed poor diastereo- and enantioselectivity. Only when proline was used in combination with metal salts as additives, the use of methanol as solvent [106] or co-solvent [102] afforded acceptable results.
Intrigued by the few data available on the proline-mediated aldol condensation employing methanol, a prototypical green solvent [84,112] also in terms of LCA (life-cycle assessment) [113], we decided to explore in depth the impact of methanol on the asymmetric intermolecular aldol condensation promoted by unmodified (S)-proline. It should be stressed, however, that efficient organocatalyzed aldol condensations invariably require a large excess of a liquid donor ketone (5–10 equivalents) that must thus be considered as a part of the reaction solvent-system.
2. Results and Discussion
2.1. Optimization of the Reaction Protocol
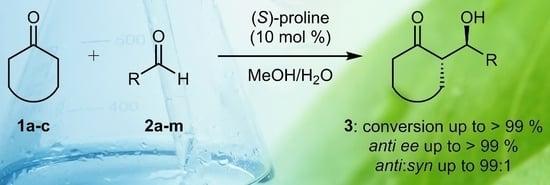
As model reaction, we selected the (S)-proline–catalysed aldol condensation between cyclohexanone 1a and aromatic aldehydes 2 (Scheme 1). At the outset, we confirmed the low performance of proline in terms of stereocontrol in pure methanol, but soon we realized that the simple use of a hydroalcoholic solution as the reaction medium was highly profitable. Here, we report a comparison among (S)-proline–catalysed reactions between cyclohexanone 1a and four different aromatic aldehydes 2a–d, carried out in methanol/water (2/1 V/V), pure water, and pure methanol, respectively, all other parameters being kept identical (Table 1). The 2/1 V/V methanol/water mixture composition ensures that the aldol reaction takes place under homogeneous conditions.
The data collected in Table 1 demonstrate the crucial role of water; if in pure water conversions are the lowest, enantioselectivity reaches the highest values. On the other hand, pure methanol displays the highest reactivity and the poorest stereocontrol. The 2/1 V/V methanol/water solution is able to combine the pros of the two pure solvents, providing the same conversions of pure methanol and almost the same ees and good drs observed in pure water.
In Table 2, the results are reported when the 2/1 V/V methanol/water solution was applied to aldol reactions between cyclohexanone 1a and other aromatic aldehydes 2e–i (Table 2).
With the most reactive electron-poor aldehydes (2a, 2b, 2e, and 2f), high conversion and high stereocontrol were achieved in only 19 h. Moreover, these results are excellent if compared with those reported in the literature for analogous transformations promoted by unmodified (S)-proline and exploiting more complex protocols [114,115,116,117]. Unfortunately, the limitations of the proline-catalysed aldol reactions were not completely overcome. In fact, electron-rich aromatic aldehydes were confirmed to be less reactive, requiring longer reaction times. More in detail, for substrates 2g and 2h, the conversions reached after 19 and 24 h, respectively, were modest; nevertheless, it is worth mentioning that the enantio- and the diastereoselectivities were both higher than those reported so far by proline-based protocols [118,119]. As far as the electron-rich p-methoxy benzaldehyde 2i is concerned, the only example with proline (20 mol%) in DMSO reported a low conversion (15%) and absence of diastereoselection [105]. The effect on product conversion was even poorer when a Lewis acid and water were added. Exploiting our MeOH/H2O-based protocol, the product conversion remained poor (18%), but the reaction proceeded with good enantio- (90% ee) and diastereoselectivity (anti/syn = 86:14).
Once the performance of native proline in 2/1 V/V methanol/water solution had been examined, we explored the effect of a more methanol-rich aqueous mixture. In Table 3, aldol reactions of cyclohexanone 1a and different aldehydes 2 in 2/1 V/V and 4/1 V/V solutions are compared.
Doubling the methanol volume (40 μL), the conversions significantly improved with all the tested aldehydes, while maintaining an excellent to remarkable level of enantiocontrol (Table 3). The most reactive substrates (2a, 2b, 2e, and 2f) provided excellent conversions in only 4 h, demonstrating an unprecedented reactivity of proline. Moreover, interesting amounts of product were obtained exploiting these reaction conditions for less electrophilic aldehydes as well (2c,2d, 2g,2i; Table 3). Concerning the diastereocontrol, a slight drop of the anti/syn ratio was observed with some aldehydes when the volume of methanol was increased. Conversely, for benzaldehyde 2d and 2-naphthyl aldehyde 2h, the diastereoselection lightly improved. In the case of benzaldehyde 2d, the better performance could be the result of the reduced amount of catalyst (10 mol%), exploitable thanks to the higher proline reactivity reached with larger amounts of methanol. In the case of the most reactive 4-nitrobenzaldehyde 2a, we solved the problem of diastereoselectivity drop by adding the methanol amount in two portions (one half after 2 h), completely restoring the diastereocontrol, while maintaining a high reaction rate. In general, a good diastereoselectivity level is retained with this protocol (4/1 V/V methanol/water) compared with the literature data [114,115,116,117,118,119]. At the same time, reaction rates are significantly enhanced. Therefore, these reaction conditions represent the best trade-off between reactivity and stereoselectivity. In Table 3, this optimized protocol was extended to some other aldehydes (2j–m), with good results compared with the literature data [120,121,122,123]. In particular, aliphatic aldehyde 2j, known as poorly responsive in this kind of organocatalysed reaction, reached an interesting conversion (63%) and remarkably high stereochemical results (>99% ee and anti/syn > 99:1), superior to those reported by other authors for unmodified proline [120].
The next step of our investigation was directed to the ketone partner 1 of the asymmetric aldol condensation. In proline-catalysed aldol reactions, a typical drawback is represented by the excess of ketone over the limiting aldehyde generally required to achieve good yields. To increase the sustainability of the process, we planned to lower the ketone excess (Table 4).
Some aldehydes characterized by high or medium reactivity were selected for this study, in which the ketone amount was reduced to 2 equivalents. With almost all the tested substrates, high conversions and excellent ee values were again obtained. Although longer reaction times were required, the reaction rates remained worthy of note, especially when compared with the performance of other protocols in similar conditions. The main drawback of this procedure was a slight decrease of diastereoselectivity, an effect that is not immediately obvious. Benaglia, using the reaction progress kinetic analysis (RPKA) approach [25], a technique that allowed Blackmond et al. to define the kinetic rate law of proline-catalysed aldol reactions [23], proved the reversibility of the aldol reaction. Lowering the ketone excess leads to the following: (i) longer reaction times to preserve the same level of product conversion; and (ii) a less efficient opposition to the retroaldol reaction, which is a slow process within the time scale of our reactions. Both factors promote equilibrium on a little extent, likely accounting for the slightly decreased diastereocontrol observed when reduced amounts of cyclohexanone 1a were used (Table 4). In conclusion, the high efficiency achieved by MeOH/H2O/(S)-proline-based protocol allows to reduce the ketone excess, involving (i) slight adverse effects on aldol reaction performance; and (ii) benefits, such as lower costs and easier work up and product purification.
2.2. Application of the Protocol to Other Ketones
Afterwards, we focused on the application of the developed catalytic protocol to two different donor ketones 1b,1c (Table 5). Considering the excellent performance (stereoselectivity and reaction rate) achieved employing the MeOH/H2O/(S)-proline protocol in the presence of 5 equivalents of 1a, we decided for convenience to apply these conditions to the ketones investigation.
At first, we tested cyclopentanone 1b with highly reactive 4-nitrobenzaldehyde 2a, observing a particularly high reaction rate, with the transformation being complete in only 4 hours. This result is unprecedented in the presence of unmodified proline or most of its derivatives [124,125,126,127], confirming once again the high reactivity achievable employing the MeOH/H2O protocol. The corresponding product 3ba was obtained with excellent ee, but low diastereoselectivity. This behaviour was expected because poorly diastereoselective aldol reactions catalyzed by proline or its derivatives were regularly reported for cyclopentanone 1b [87,124,125,126,127]. To improve the diastereoselctivity, we lowered the reaction temperature to 0 °C and we obtained a good 78:22 anti/syn ratio, maintaining a high conversion in a reasonable time.
Considering the excellent performance achievable with MeOH/H2O/(S)-proline-based protocol, we were particularly interested in the results obtainable with less reactive aldehydes. In fact, 4-Br benzaldehyde 2g and benzaldehyde 2d provided the corresponding products (3bg and 3bd, respectively) with good conversions and diastereoselectivities, and, noteworthy, with the best enantioselectivities ever achieved employing unmodified proline as catalyst [128].
As further confirmation, 2,2-dimethyl-1,3-dioxan-5-one 1c (Table 5) also displayed good reactivity and stereoselectivity when reacted with less reactive aldehydes 2g and 2d. In particular, our results represent the first examples of organocatalyzed aldol condensation between 2,2-dimethyl-1,3-dioxan-5-one 1c and 4-Br benzaldehyde 2g or benzaldehyde 2d, promoted by only 10 mol% of proline [129,130,131,132,133].
At last, we applied our protocol to acetone 1d as simple aliphatic ketone (Table S3, Section 2, Supplementary Materials). Although with 4-NO2 benzaldehyde 2a, we obtained an unprecedented high reaction rate if compared with the published corresponding transformations, the enantioselectivity was poor, as commonly reported for the proline-catalysed aldol additions involving these substrates.
2.3. Large-Scale Application of the Protocol
Our aim is the development of an efficient and sustainable organocatalyzed aldol condensation protocol, which can be interesting from a scale-up perspective. Therefore, as a first step, we confirmed the excellent performance of the MeOH/H2O/(S)-proline-based protocol by carrying out the aldol condensation between moderately reactive benzaldehyde 2d and cyclohexanone 1a on a 10 mmol scale (gram scale). The desired product 3ad was isolated in 78% yield and with high stereocontrol (90:10 dr, 95% ee), fully confirming the data obtained on small scale (Table 3).
The next step was the accomplishment of the same reaction on a 100 mmol scale of the limiting reagent benzaldehyde 2d (Scheme 2) in order to study some aspects in more detail.
At first, we investigated the impact of the aldehyde addition rate on the reaction outcome (Table 6). With benzaldehyde 2d not being very reactive, good conversions were recorded only after 23 h and we did not observe a significant difference depending on the addition rate of benzaldehyde (Table 6).
Then, we monitored product conversion and stereoselectivity for a longer reaction time (Table 6), to establish if a high conversion could be achieved without a significant loss in stereocontrol exploiting our reaction conditions. Indeed, as previously mentioned, aldol reaction is reversible and longer reaction times could make the retroaldol process competitive, providing a decreased diastereomeric ratio. Actually, we observed a slow increase of the product conversion, achieving 85% after 2 days, without a significant erosion of anti/syn ratio (in comparison with small scale-reactions, a slightly lower dr was recorded, which remained constant for the first 48 h). We confirmed that even the enantiomeric excess of the product remained at high levels (94% ee after 47 h).
The results reported in Table 6 clearly show that the best reaction outcome is obtained at a reaction time representing the best balance between product conversion and stereocontrol. To further explore this effect, we compared the data obtained with different moderately or poorly reactive aldehydes (Table 7).
Concerning the stereoselectivity, in this study, we focused our attention on diastereoselectivity variation, which is much more impaired by retroaldol reaction (see Supplementary Materials for a study on enantioselectivity variation). The data collected in Table 7 demonstrate that the aldol transformation reaches a position, after which the product conversion no longer grows, while the dr continues to drop. The time required to achieve this situation depends on the aldehyde reactivity. On the other hand, the rate of retroaldol process is less affected by the aldehyde nature; therefore, the retroaldol effects are less troublesome for reactive aldehydes (high conversion in short time with high dr) and more marked for poorly reactive aldehydes (long time required to reach acceptable conversion with low dr). This study proves that, in the asymmetric aldol process promoted by proline, the reaction time providing the best performance (balance between conversion and stereoselectivity) strongly depends on the substrate; therefore, a careful investigation should be done before tackling a large-scale application.
A further point that we evaluated to increase the sustainability of our large-scale protocol was the reduction of the ketone excess. For this purpose, we applied the MeOH/H2O/(S)-proline-based protocol to moderately reactive benzaldehyde 2d (50 mmol) in the presence of only 2 equivalents of cyclohexanone 1a, monitoring the results over the time. After 71 h, we achieved the highest product conversion (83%) with an excellent 89:11 dr. Prolonging the reaction time (98 h) only led to a drop in dr (84:16). These findings suggest that, exploiting our protocol, a large excess of ketone (5 equivalents) only enhances the initial reaction rate, but it is not necessary for the achievement of an excellent final performance.
2.4. Work-Up Investigations
As the last point, we investigated some different work-up approaches, in order to (i) compare the results (also in terms of sustainability), and (ii) establish if part of the organocatalyst could be easily recovered. The first 100 mmol-scale aldol condensation (Table 6, 45 min long aldehyde addition) was stopped after 49 h and the reaction mixture (total volume = 79 mL) was divided in six portions, treated as described in Table 8.
The first portion of reaction mixture (18 mL, 22.8 mmol) was filtered through a short pad of silica to remove water and proline (method A, Table 8). EtOAc was used as mobile phase to elute product 3ad (along with residual reagents 1a and 2d). Despite the significant polarity of EtOAc, a large amount of solvent was required to recover all the product and an undesirable high volume of organic solvent (242 mL) had to be evaporated under reduced pressure.
The second portion of reaction mixture (18 mL, 22.8 mmol) was subjected to a typical aqueous work-up to remove water and proline (method B, Table 8). NH4Cl (2 equivalents with respect to proline, solved in 20 mL of H2O) was employed to quench proline, the two phases were separated, and the aqueous phase was extracted two additional times with EtOAc, until complete recovery of the product (checked by thin-layer chromatography). The solution was dried with Na2SO4, which restrained a significant amount of aldol product 3ad, so that it was necessary to wash it three times with EtOAc. A considerable volume of organic solvent (90 mL) had to be evaporated.
At this point, we tried to develop a work-up method that allowed us in a simple way not only to remove the catalyst, but also to recover it, at least partially. Exploiting the very low amount of protic polar solvents used in our MeOH/H2O/(S)-proline-based protocol, we envisaged that the addition of a small portion of organic solvent could make the reaction environment sufficiently lipophilic to trigger the catalyst precipitation. Four different organic solvents were tested: EtOAc (method C, Table 8), Et2O (method D), dichloromethane (method E), and n-hexane (method F). The minimum amount of solvent able to provide an opalescent solution was added to each portion (10 mL, 12.6 mmol) and the mixtures were stored at −15 °C for 36 h. In the portions treated with EtOAc and Et2O (methods C and D, respectively), a white precipitate, corresponding to proline, was clearly observed; therefore, it was filtered under vacuum and washed with a small amount of cold solvent. The filtered solutions were stored at −15 °C for further 36 h and a second portion of catalyst was recovered in both cases. Afterwards, the mixtures were dried with Na2SO4, which was required to be washed three times with organic solvent. In these two cases (methods C and D), a considerable amount of organic solvent also had to be evaporated (46 and 52 mL, respectively). In the portions treated with DCM and n-hexane (methods E and F, respectively), two liquid phases were observed and we decided to directly use Na2SO4 to remove the small water-based phase. Na2SO4 was washed with solvent until complete recovery of the product (three times for DCM, four times for n-hexane). In these two cases (methods E and F), the lowest amounts of organic solvent were employed (36 and 43 mL, respectively).
By comparing the results obtained with the tested work-up methods, we can infer the following: (1) no work-up approach adversely affects reaction conversion and diastereoselectivity, in fact all the crude mixtures showed comparable good results (Table 8). (2) The least suitable and sustainable method to remove water and proline seems to be the silica-pad (method A), owing to the large amount of solvent required to recover all the desired product; (3) when two organic and aqueous phases are formed (methods B, E and F), the simplest and cheapest work-up appears the dilution with a very small amount of DCM, cooling, and directly drying with Na2SO4 (method E). This approach is practicable only thanks to the very low amount of protic polar solvents used in our MeOH/H2O/(S)-proline-based protocol. (4) Among the tested work-up approaches, the most convenient are those allowing an easy recovery of a large part of the organocatalyst (methods C and D). In particular, method D employing Et2O reached 86% of proline recovery using an acceptable volume of organic solvent. Moreover, this result is obtainable on the basis of very low amount of protic polar solvents used in our protocol.
Although the tested work-up methods are not optimized and, therefore, can be further improved, they give a clear indication of the advantages that our protocol can offer.
3. Materials and Methods
3.1. General Information
1H and 13C NMR spectra were recorded on Inova 400 NMR instrument (Agilent, Santa Clara, CA, United States) with a 5 mm probe. Chemical shifts (δ) are reported in ppm, relative to the residual peaks of deuterated solvent signals.
HPLC-MS analyses were performed on an Agilent Technologies HP1100 instrument (Agilent, Santa Clara, CA, United States) coupled with an Agilent Technologies MSD1100 single-quadrupole mass spectrometer (Agilent, Santa Clara, CA, United States). A Phenomenex Gemini C18, 3 μm (100 × 3 mm) column was employed for the chromatographic separation: mobile phase H2O/CH3CN, gradient from 30% to 80% of CH3CN in 8 min, 80% of CH3CN until 22 min, and then up to 90% of CH3CN in 2 min; flow rate 0.4 mL min−1 (Phenomenex, Torrance, CA, United States).
Chiral stationary phase (CSP)-HPLC analyses were performed on an Agilent Technologies Series 1200 instrument (Agilent, Santa Clara, CA, United States) using Daicel® chiral columns and n-hexane/2-propanol (n-Hex/IPA) mixtures (Daicel, Osaka, Japan).
Optical rotation measurements were performed on a polarimeter Schmidt+Haensch UniPol L1000 (Schmidt + Haensch GmbH & Co, Berlin, Germany).
Flash chromatography purifications were carried out using Merck silica gel 60 (230–400 mesh particle size). Thin layer chromatography was performed on Merck 60 F254 plates (Merck, Darmstadt, Germany).
Commercial reagents were used as received without additional purification, with exception of liquid aldehydes, which were distilled and stored under nitrogen atmosphere to avoid the formation of the corresponding acids. Dry methanol (Sure/Seal™ bottle) was used to ensure a reproducible water content.
The diastereomeric and enantiomeric compositions were checked on the crude products against the corresponding racemic products, obtained under the same reaction conditions using racemic proline.
3.2. Synthetic Procedures.
3.2.1. General Procedure for the Small-Scale Aldol Condensation Between Aldehydes 2 and Ketones 1
The aldol reaction was carried out in a 2 mL vial. In a typical reaction, the vial was charged at room temperature with the reactants in the following order: (S)-proline (0.03 mmol), methanol (40 µL), water (10 µL), the selected ketone 1 (1.5 mmol), and the selected aldehyde 2 (0.3 mmol). The flask was capped with a stopper and sealed. Then, the reaction mixture was stirred at room temperature for the desired time. The conversion was monitored by TLC (Merck, Darmstadt, Germany) and 1H-NMR (a small portion was taken, diluted, and immediately analyzed) (Agilent, Santa Clara, CA, United States). Then, the mixture was filtered on a short pad of silica with ethyl acetate and concentrated under reduced pressure.
The product conversion with respect to the limiting aldehyde and the diastereomeric ratio were determined by 1H-NMR in CDCl3 on the crude mixture. The enantiomeric excess was determined by chiral stationary phase (CSP)-HPLC (Agilent, Santa Clara, CA, United States) on the crude mixture.
3.2.2. Procedure for the Aldol Condensation Between Benzaldehyde 2d and Cyclohexanone 1a on 10 mmol Scale
The aldol reaction was conducted in a 25 mL flask. The flask was charged with (S)-proline (115 mg, 1 mmol), methanol (1.33 mL), water (330 µL), and cyclohexanone 1a (5.18 mL, 50 mmol) and the mixture was allowed to stir for 10 min at room temperature. Then, the mixture was cooled at 0 °C and benzaldehyde 2d (1.02 mL, 10 mmol) was slowly added by means of a syringe. The flask was capped with a stopper and sealed. The reaction mixture was stirred at room temperature for 30 h. Then, the mixture was filtered on a pad of silica with ethyl acetate and concentrated under reduced pressure. The conversion (85%) with respect to the limiting aldehyde and the diastereomeric ratio (90:10) were determined by 1H-NMR in CDCl3 on the crude mixture. The obtained residue was purified by column chromatography (ethyl acetate/cyclohexane = 2:8 as the eluent) to afford the product 3ad in 78% yield. The enantiomeric excess (95% ee) was determined by CSP-HPLC on the pure product.
3.2.3. Procedure for the Aldol Condensation between Benzaldehyde 2d and Cyclohexanone 1a on 100 mmol Scale (Table 6)
The aldol reaction was conducted in a 250 mL flask. The flask was charged with (S)-proline (1.15 g, 10 mmol), methanol (13.33 mL), water (3.33 mL), and cyclohexanone 1a (51.8 mL, 500 mmol) and the mixture was allowed to stir for 15 min at room temperature. Then, the mixture was cooled at 0 °C and benzaldehyde 2d (10.2 mL, 100 mmol) was slowly added by means of (i) addition funnel (addition rate = 45 min), or (ii) syringe for slow addition (addition rate = 6 h). Then, the flask was capped with a stopper and sealed. The reaction mixture was stirred at room temperature. The reaction performance was monitored over time (a small portion was taken, diluted, and immediately analyzed); the product conversion with respect to the limiting aldehyde and the diastereomeric ratio were determined by 1H-NMR in CDCl3 on the crude mixture, and the enantiomeric excess was determined by CSP-HPLC on the crude mixture. After 49 h, the first reaction (addition rate = 45 min) was stopped, the reaction mixture (total volume = 79 mL) was divided in six portions, and they were treated as described in Table 8 (see below for details).
3.2.4. General Procedure for the Study of Reaction Outcome as a Function of Reaction Time (Table 7)
The aldol reaction was conducted in a 100 mL flask. The flask was charged with (S)-proline (575 mg, 5 mmol), methanol (6.67 mL), water (1.67 mL), and cyclohexanone 1a (25.9 mL, 250 mmol) and the mixture was allowed to stir for 15 min at room temperature. Then, the mixture was cooled at 0 °C and the desired aldehyde 2 (50 mmol) was slowly added by means of an addition funnel. Then, the flask was capped with a stopper and sealed. The reaction mixture was stirred at room temperature. The reaction performance was monitored over time (a small portion was taken, diluted, and immediately analyzed); the product conversion with respect to the limiting aldehyde and the diastereomeric ratio were determined by 1H-NMR in CDCl3 on the crude mixture, and the enantiomeric excess was determined by CSP-HPLC on the crude mixture. At the reported time (Table 7), the mixture was filtered on a pad of silica with ethyl acetate and concentrated under reduced pressure. The obtained residue was purified by column chromatography (ethyl acetate/cyclohexane = 2:8 as the eluent) to afford the pure product (3ad: 76% yield, 3am: 67% yield, 3ai: 43% yield).
3.2.5. Procedure for the Aldol Condensation between Benzaldehyde 2d (50 mmol) and Cyclohexanone 1a (2 equivalents, 100 mmol)
The aldol reaction was conducted in a 100 mL flask. The flask was charged with (S)-proline (575 mg, 5 mmol), methanol (6.67 mL), water (1.67 mL), and cyclohexanone 1a (10.36 mL, 100 mmol) and the mixture was allowed to stir for 15 min at room temperature. Then, the mixture was cooled at 0 °C and benzaldehyde 2d (5.1 mL, 50 mmol) was slowly added by means of an addition funnel. Then, the flask was capped with a stopper and sealed. The reaction mixture was stirred at room temperature. The reaction performance was monitored over time (a small portion was taken, diluted, and immediately analyzed); the product conversion with respect to the limiting aldehyde and the diastereomeric ratio were determined by 1H-NMR in CDCl3 on the crude mixture, and the enantiomeric excess was determined by CSP-HPLC on the crude mixture. After 99 h, the reaction was stopped and it was filtered on a pad of silica with ethyl acetate and concentrated under reduced pressure. The obtained residue was purified by column chromatography (ethyl acetate/cyclohexane = 2:8 as the eluent) to afford the product 3ad in 76% yield. The enantiomeric excess (91% ee) was determined by CSP-HPLC on the pure product.
3.3. Work-Up Procedures
3.3.1. Procedure for Filtration on a Silica-Pad (Method A, Table 8)
A portion (18 mL corresponding to 22.8 mmol) of the first reaction (addition rate = 45 min) carried out on 100 mmol of limiting aldehyde 2d (see Section 3.2.3) was filtered on a silica-pad: 1.5 cm height, 9.6 cm diameter, gooch porosity = 4 (10–16 μm), mobile phase = EtOAc. In the last EtOAc portions (25 mL each), the presence of product was checked by TLC. The filtered reaction mixture (total volume = 242 mL) was concentrated under reduced pressure. The product conversion (87%) with respect to the limiting aldehyde and the diastereomeric ratio (84:16 = anti:/syn) were determined by 1H-NMR in CDCl3 on the obtained residue.
3.3.2. Procedure for Aqueous Work-Up Employing NH4Cl (Method B, Table 8)
A portion (18 mL corresponding to 22.8 mmol) of the first reaction (addition rate = 45 min) carried out on 100 mmol of limiting aldehyde 2d (see Section 3.2.3) was diluted with EtOAc (5 mL) and treated with an aqueous solution of NH4Cl (242 mg, 2 equivalents with respect to proline, in 20 mL of H2O). The two layers were separated and the aqueous phase was further extracted with EtOAc (2 x 20 mL, the complete product extraction was checked by TLC). The collected solution was dried with Na2SO4 (1.15 g), and then it was filtered and washed with EtOAc (3 x 9 mL, the complete product recovery was checked by TLC). The filtered reaction mixture (total volume = 90 mL) was concentrated under reduced pressure. The product conversion (86%) with respect to the limiting aldehyde and the diastereomeric ratio (83:17 = anti/syn) were determined by 1H-NMR in CDCl3 on the obtained residue.
3.3.3. Procedure for Dilution with EtOAc and Cooling (Method C, Table 8).
A portion (10 mL corresponding to 12.6 mmol) of the first reaction (addition rate = 45 min) carried out on 100 mmol of limiting aldehyde 2d (see Section 3.2.3) was diluted with EtOAc (4 mL) and placed at −15 °C for 36 h. A white precipitate was formed, was filtered under vacuum, and washed with cold EtOAc (2 × 4 mL). Here, 86.2 mg of white solid was recovered. The obtained solution was placed at −15 °C for further 36 h. A second portion of white solid was filtered under vacuum and washed with cold EtOAc (2 × 3 mL). Here, 27.1 mg of white solid was recovered. The collected solution was dried with Na2SO4 (650 mg), and then it was filtered and washed with EtOAc (3 × 6 mL, the complete product recovery was checked by TLC). The filtered reaction mixture (total volume = 46 mL) was concentrated under reduced pressure. The product conversion (89%) with respect to the limiting aldehyde and the diastereomeric ratio (84:16 = anti/syn) were determined by 1H-NMR in CDCl3 on the obtained residue. Total recovered proline = 113.3 mg (78%). The nature of the white solid was confirmed by 1H-NMR spectroscopy (see Supplementary Materials) and optical rotation measurement ([α]D 25 = −84; c = 0.135, water) in comparison with the commercial compound ([α]D 25 = −86; c = 0.133, water).
3.3.4. Procedure for Dilution with Et2O and Cooling (Method D, Table 8).
A portion (10 mL corresponding to 12.6 mmol) of the first reaction (addition rate = 45 min) carried out on 100 mmol of limiting aldehyde 2d (see Section 3.2.3) was diluted with Et2O (4 mL) and placed at −15 °C for 36 h. A white precipitate was formed, it was filtered under vacuum and washed with cold Et2O (2 × 4 mL). Here, 109.4 mg of white solid was recovered. The obtained solution was placed at −15 °C for further 36 h. A second portion of white solid was filtered under vacuum and washed with cold Et2O (2 × 3 mL). Here, 15.4 mg of white solid was recovered. The collected solution was dried with Na2SO4 (650 mg), and then it was filtered and washed with Et2O (3 × 8 mL, the complete product recovery was checked by TLC). The filtered reaction mixture (total volume = 52 mL) was concentrated under reduced pressure. The product conversion (90%) with respect to the limiting aldehyde and the diastereomeric ratio (85:15 = anti/syn) were determined by 1H-NMR in CDCl3 on the obtained residue. Total recovered proline = 124.8 mg (86%). The nature of the white solid was confirmed by 1H-NMR spectroscopy (see Supplementary Materials) and optical rotation measurement ([α]D 25 = −80; c = 0.131, water) in comparison with the commercial compound ([α]D 25 = −86; c = 0.133, water).
3.3.5. Procedure for Dilution with DCM and Cooling (Method E, Table 8)
A portion (10 mL corresponding to 12.6 mmol) of the first reaction (addition rate = 45 min) carried out on 100 mmol of limiting aldehyde 2d (see Section 3.2.3) was diluted with DCM (5 mL) and placed at −15 °C for 36 h. Two liquid phases were formed. The mixture was directly dried with Na2SO4 (650 mg), and then it was filtered and washed with DCM (3 × 7 mL, the complete product recovery was checked by TLC). The filtered reaction mixture (total volume = 36 mL) was concentrated under reduced pressure. The product conversion (88%) with respect to the limiting aldehyde and the diastereomeric ratio (85:15 = anti/syn) were determined by 1H-NMR in CDCl3 on the obtained residue.
3.3.6. Procedure for Dilution with n-Hexane and Cooling (Method F, Table 8)
A portion (10 mL corresponding to 12.6 mmol) of the first reaction (addition rate = 45 min) carried out on 100 mmol of limiting aldehyde 2d (see Section 3.2.3) was diluted with n-hexane (5 mL) and placed at −15 °C for 36 h. Two liquid phases were formed. The mixture was directly dried with Na2SO4 (650 mg), and then it was filtered and washed with n-hexane (4 × 7 mL, the complete product recovery was checked by TLC). The filtered reaction mixture (total volume = 43 mL) was concentrated under reduced pressure. The product conversion (89%) with respect to the limiting aldehyde and the diastereomeric ratio (86:14 = anti:syn) were determined by 1H-NMR in CDCl3 on the obtained residue.
3.4. Products Characterization.
All the synthesized products were known compounds and the obtained data were in agreement with the published ones:
[134] for products 3aa, 3ab, 3ac, 3ad, 3af, 3ag, and 3ah;
[135] for products 3ae, 3ai, 3ak, and 3ba;
[106] for product 3aj;
[136] for product 3bd;
[137] for product 3cd;
[138] for products 3al and 3am;
[139] for product 3bg;
[129] for product 3cg.
As an example, the complete characterization of the most studied aldol product 3ad (anti isomer) is reported in the Supplementary Materials. CSP-HPLC separation conditions and chromatograms of all the aldol products 3 are reported in the Supplementary Materials.
4. Conclusions
Since 2000, the time the first seminal publication by List, Lerner, and Barbas III on the intermolecular asymmetric aldol reaction catalyzed by proline appeared, a countless number of papers focused on enamine organocatalysis with the aim to solve a few critical issues inherent in the use of proline. Summarizing, high catalyst loading, long reaction times, solvent limitations owing to proline solubility, variable stereocontrol mainly dependent on the donor-acceptor aldol partners, difficult and/or expensive product isolation, and catalyst recovery characterize the proline-catalysed aldol protocol. On the other hand, advantages have been previously underlined such as low cost, no toxicity, no need for anhydrous solvents or controlled atmosphere, and process practicality.
Over these two decades, the greatest efforts have been dedicated to the design and synthesis of new catalysts, mostly sharing with proline the chiral pyrrolidine scaffold. These derivatives allow to enlarge the platform of solvent candidates, up to enabling the possibility of catalyst recycling. Reaction kinetics improve with shorter reaction times and lower catalyst loadings. If these improvements are beyond doubt, the costs coupled to their preparation are clearly a limiting factor. On the other hand, it is known that a number of common solvents have been questioned in recent years as their hazardous properties have come to light, for example, the environmental, safety, and health issues associated to the use of DCM, toluene, DMSO, and others.
The work presented here shows that very good results can be simply achieved using methanol/water mixtures as reaction medium. When only water is used, these reactions take place in a typical heterogeneous conditions (emulsions), where the interphase water has as many as about a quarter of the O–H bonds not being involved in hydrogen bonding. According to Jung and Marcus [140], the interactions of these unbound hydroxyl groups with organic reactants and, more importantly, with the transition states, lower the activation energies, enabling rate and yield enhancements. Faster reactions occur in pure methanol because of the homogeneous conditions, which allow all the amount of proline used to participate to catalysis, but this superior reactivity is characterized by a lower stereocontrol. Recent papers evidenced, by DFT calculations, the positive effects of co-additives such as water or methanol in stabilizing the transition states of the aldol reaction, with methanol displaying the larger effects [24,141,142,143]. These protic additives could directly participate in the reaction mechanism, acting as an active proton transfer relay between the proline carboxylic acid group and the incoming aldehyde. The amount and the nature of the protic additive could significantly change the reactivity and stereoselectivity of this transformation, as transition states with one, two, and even three molecules of the additive have been located and described.
If both methanol and water as pure solvents give largely unsatisfactory results that discouraged further investigations, we demonstrated that methanol/water mixtures provide the high reaction rates (good yields in short reaction times) typical of methanol and the high stereocontrol typical of water. The efficient, simple, and cost-effective reaction protocol proposed, easily scaled up here up to the 100 mmol scale, as well as the safe handling of the methanol/water mixture, positively impact the overall efficiency and sustainability of this proline-catalysed aldol protocol. However, we have to observe that, also following this procedure, the recurring dependence of relative reaction rates and stereochemical outcome on the nature of the donor-acceptor pair has not been overcome. Thus, cyclohexanone is the best donor in terms of reactivity and stereocontrol, while cyclopentanone works faster, but with a much lower stereocontrol. Electron-rich aromatic aldehydes are the slowest reaction acceptors, requiring long reaction times, while electron-poor aldehydes are the best. Nevertheless, given that the usual relative behavior of ketones and aldehydes is confirmed, the aldol protocol in methanol/water can be considered a useful contribution, enabling the achievement of performance never obtained before (also for less reactive compounds), employing the smallest and cheapest organocatalytic species, proline.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4344/10/6/649/s1, Table S1: Enantioselectivity variation as a function of reaction time; Figure S1: 1H-NMR spectrum of commercial proline; Figure S2: 1H-NMR spectrum of recovered proline employing work-up method C (Table 8); Figure S3: 1H-NMR spectrum of recovered proline employing work-up method D (Table 8), CSP-HPLC separation conditions and chromatograms of aldols 3 (racemic and enantio-enriched), full characterization of anti aldol product 3ad (CSP-HPLC chromatogram of enantio-enriched product, 1H-NMR spectrum, 13C-NMR spectrum, HPLC-MS chromatograms, ESI-MS spectrum).
Author Contributions
Conceptualization, M.L., A.Q., and C.T.; methodology, M.L., A.Q., and C.T.; validation, A.M. and A.Q.; investigation, M.G.E., A.T., and A.Q.; resources, M.L., A.Q., and C.T.; data curation, M.L., A.Q., and C.T.; writing—original draft preparation, A.Q.; writing—review and editing, A.M., M.L., A.Q., and C.T.; visualization, A.M. and A.Q.; supervision, C.T.; project administration, M.L. and A.Q.; funding acquisition, M.L., A.Q., and C.T.. All authors have read and agreed to the published version of the manuscript.
Funding
This research and the APC were funded by Università di Bologna (RFO) and MIUR (Rome).
Acknowledgments
We thank R. Miani for the execution of some experiments.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- List, B.; Lerner, R.A.; Barbas, C.F. Proline-Catalyzed Direct Asymmetric Aldol Reactions. J. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar] [CrossRef]
- Melchiorre, P.; Marigo, M.; Carlone, A.; Bartoli, G. Asymmetric Aminocatalysis-Gold Rush in Organic Chemistry. Angew. Chem. Int. Ed. 2008, 47, 6138–6171. [Google Scholar] [CrossRef] [PubMed]
- Macmillan, D.W.C. The advent and development of organocatalysis. Nature 2008, 455, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Pihko, P.M.; Majander, I.; Erkkilä, A. Enamine Catalysis. Top. Curr. Chem. 2009, 291, 145–200. [Google Scholar] [CrossRef]
- Schneider, J.F.; Ladd, C.L.; Bräse, S. Chapter 5: Proline as an Asymmetric Organocatalyst; RSC Publishing: Cambridge, UK, 2015; pp. 79–119. [Google Scholar] [CrossRef]
- Yamashita, Y.; Yasukawa, T.; Yoo, W.-J.; Kitanosono, T.; Kobayashi, S. Catalytic enantioselective aldol reactions. Chem. Soc. Rev. 2018, 47, 4388–4480. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, J. Recent Advances in Asymmetric Reactions Catalyzed by Proline and Its Derivatives. Synthesis 2016, 49, 960–972. [Google Scholar] [CrossRef]
- List, B. Proline-catalyzed asymmetric reactions. Tetrahedron 2002, 58, 5573–5590. [Google Scholar] [CrossRef]
- Barbas, C.F. Organocatalysis Lost: Modern Chemistry, Ancient Chemistry, and an Unseen Biosynthetic Apparatus. Angew. Chem. Int. Ed. 2008, 47, 42–47. [Google Scholar] [CrossRef]
- Palomo, C.; Oiarbide, M.; García, J.M. Current progress in the asymmetric aldol addition reaction. Chem. Soc. Rev. 2004, 33, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Wennemers, H. Peptides as Asymmetric Catalysts for Aldol Reactions. Chim. Int. J. Chem. 2007, 61, 276–278. [Google Scholar] [CrossRef]
- Notz, W.; Tanaka, F.; Barbas, C.F. Enamine-Based Organocatalysis with Proline and Diamines: The Development of Direct Catalytic Asymmetric Aldol, Mannich, Michael, and Diels−Alder Reactions. Accounts Chem. Res. 2004, 37, 580–591. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Yang, J.W.; Hoffmann, S.; List, B. Asymmetric Enamine Catalysis. Chem. Rev. 2007, 107, 5471–5569. [Google Scholar] [CrossRef] [PubMed]
- Alcaide, B.; Almendros, P. The Direct Catalytic Asymmetric Cross-Aldol Reaction of Aldehydes. Angew. Chem. Int. Ed. 2003, 42, 858–860. [Google Scholar] [CrossRef] [PubMed]
- Alcaide, B.; Almendros, P. The Direct Catalytic Asymmetric Aldol Reaction. Eur. J. Org. Chem. 2002, 2002, 1595–1601. [Google Scholar] [CrossRef]
- Guillena, G.; Najera, C.; Ramón, D.J. Enantioselective direct aldol reaction: The blossoming of modern organocatalysis. Tetrahedron Asymmetry 2007, 18, 2249–2293. [Google Scholar] [CrossRef]
- Geary, L.M.; Hultin, P.G. The state of the art in asymmetric induction: The aldol reaction as a case study. Tetrahedron Asymmetry 2009, 20, 131–173. [Google Scholar] [CrossRef]
- Schetter, B.; Mahrwald, R. Modern Aldol Methods for the Total Synthesis of Polyketides. Angew. Chem. Int. Ed. 2006, 45, 7506–7525. [Google Scholar] [CrossRef] [PubMed]
- Mestres, R. A green look at the aldol reaction. Green Chem. 2004, 6, 583. [Google Scholar] [CrossRef]
- Bahmanyar, S.; Houk, K.N.; Martin, H.J.; List, B. Quantum Mechanical Predictions of the Stereoselectivities of Proline-Catalyzed Asymmetric Intermolecular Aldol Reactions. J. Am. Chem. Soc. 2003, 125, 2475–2479. [Google Scholar] [CrossRef] [PubMed]
- Seebach, D.; Beck, A.K.; Badine, D.M.; Limbach, M.; Eschenmoser, A.; Treasurywala, A.M.; Hobi, R.; Prikoszovich, W.; Linder, B. Are Oxazolidinones Really Unproductive, Parasitic Species in Proline Catalysis?—Thoughts and Experiments Pointing to an Alternative View. Helvetica Chim. Acta 2007, 90, 425–471. [Google Scholar] [CrossRef]
- Zotova, N.; Franzke, A.; Armstrong, A.; Blackmond, D.G. Clarification of the Role of Water in Proline-Mediated Aldol Reactions. J. Am. Chem. Soc. 2007, 129, 15100–15101. [Google Scholar] [CrossRef] [PubMed]
- Zotova, N.; Broadbelt, L.J.; Armstrong, A.; Blackmond, D.G. Kinetic and mechanistic studies of proline-mediated direct intermolecular aldol reactions. Bioorganic Med. Chem. Lett. 2009, 19, 3934–3937. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.K.; Sunoj, R.B. Enamine versus Oxazolidinone: What Controls Stereoselectivity in Proline-Catalyzed Asymmetric Aldol Reactions? Angew. Chem. Int. Ed. 2010, 49, 6373–6377. [Google Scholar] [CrossRef] [PubMed]
- Orlandi, M.; Ceotto, M.; Benaglia, M. Kinetics versus thermodynamics in the proline catalyzed aldol reaction. Chem. Sci. 2016, 7, 5421–5427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haindl, M.H.; Hioe, J.; Gschwind, R.M. The Proline Enamine Formation Pathway Revisited in Dimethyl Sulfoxide: Rate Constants Determined via NMR. J. Am. Chem. Soc. 2015, 137, 12835–12842. [Google Scholar] [CrossRef] [PubMed]
- Renzi, P.; Hioe, J.; Gschwind, R.M. Enamine/Dienamine and Brønsted Acid Catalysis: Elusive Intermediates, Reaction Mechanisms, and Stereoinduction Modes Based on in Situ NMR Spectroscopy and Computational Studies. Accounts Chem. Res. 2017, 50, 2936–2948. [Google Scholar] [CrossRef] [PubMed]
- Notz, W.; List, B. Catalytic Asymmetric Synthesis ofanti-1,2-Diols. J. Am. Chem. Soc. 2000, 122, 7386–7387. [Google Scholar] [CrossRef]
- List, B.; Pojarliev, P.; Castello, C. Proline-Catalyzed Asymmetric Aldol Reactions between Ketones and α-Unsubstituted Aldehydes. Org. Lett. 2001, 3, 573–575. [Google Scholar] [CrossRef] [PubMed]
- Sakthivel, K.; Notz, W.; Bui, T.; Barbas, C.F. Amino acid catalyzed direct asymmetric aldol reactions: A bioorganic approach to catalytic asymmetric carbon-carbon bond-forming reactions. J. Am. Chem. Soc. 2001, 123, 5260–5267. [Google Scholar] [CrossRef] [PubMed]
- Córdova, A.; Notz, W.; Barbas, C.F. Direct organocatalytic aldol reactions in buffered aqueous media. Chem. Commun. 2002, 24, 3024–3025. [Google Scholar] [CrossRef] [PubMed]
- Pihko, P.M.; Nyberg, A.I.; Usano, A. Proline-Catalyzed Ketone-Aldehyde Aldol Reactions are Accelerated by Water. Synlett 2004, 2004, 1891–1896. [Google Scholar] [CrossRef]
- Peng, Y.-Y.; Ding, Q.-P.; Li, Z.; Wang, P.G.; Cheng, J.-P. Proline catalyzed aldol reactions in aqueous micelles: An environmentally friendly reaction system. Tetrahedron Lett. 2003, 44, 3871–3875. [Google Scholar] [CrossRef]
- Darbre, T.; Machuqueiro, M. Zn-Proline catalyzed direct aldol reaction in aqueous media. Chem. Commun. 2003, 1090–1091. [Google Scholar] [CrossRef]
- Hayashi, Y.; Tsuboi, W.; Shoji, M.; Suzuki, N. Application of high pressure, induced by water freezing, to the direct asymmetric aldol reaction. Tetrahedron Lett. 2004, 45, 4353–4356. [Google Scholar] [CrossRef]
- Wu, Y.-S.; Shao, W.-Y.; Zheng, C.-Q.; Huang, Z.-L.; Cai, J.; Deng, Q.-Y. Studies on Direct Stereoselective Aldol Reactions in Aqueous Media. Helvetica Chim. Acta 2004, 87, 1377–1384. [Google Scholar] [CrossRef]
- Trost, B.M.; Brindle, C.S. The direct catalytic asymmetric aldol reaction. Chem. Soc. Rev. 2010, 39, 1600–1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.-Y.; Chen, Y.; Zheng, C.-Q.; Yin, Y.; Wang, L.; Sun, X.-Q. Highly enantioselective aldol reactions catalyzed by reusable upper rim-functionalized calix[4]arene-based l -proline organocatalyst in aqueous conditions. Tetrahedron 2017, 73, 78–85. [Google Scholar] [CrossRef]
- Benaglia, M. Recoverable and Recyclable Catalysts, 1st ed.; Wiley & Sons: Chichester, UK, 2009. [Google Scholar]
- Liu, Y.-X.; Sun, Y.-N.; Tan, H.-H.; Tao, J.-C. Asymmetric Aldol Reaction Catalyzed by New Recyclable Polystyrene-supported l-proline in the Presence of Water. Catal. Lett. 2007, 120, 281–287. [Google Scholar] [CrossRef]
- Liu, Y.-X.; Sun, Y.-N.; Tan, H.-H.; Liu, W.; Tao, J.-C. Linear polystyrene anchored l-proline, new recyclable organocatalysts for the aldol reaction in the presence of water. Tetrahedron Asymmetry 2007, 18, 2649–2656. [Google Scholar] [CrossRef]
- Gruttadauria, M.; Giacalone, F.; Marculescu, A.M.; Meo, P.L.; Rielaa, S.; Noto, R. Hydrophobically Directed Aldol Reactions: Polystyrene-SupportedL-Proline as a Recyclable Catalyst for Direct Asymmetric Aldol Reactions in the Presence of Water. Eur. J. Org. Chem. 2007, 2007, 4688–4698. [Google Scholar] [CrossRef] [Green Version]
- Lu, A.; Smart, T.P.; Epps, T.H.; Longbottom, D.A.; O’Reilly, R.K. l-Proline Functionalized Polymers Prepared by RAFT Polymerization and Their Assemblies as Supported Organocatalysts. Macromolecules 2011, 44, 7233–7241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benaglia, M.; Cinquini, M.; Cozzi, F.; Puglisi, A.; Celentano, G. Poly(Ethylene Glycol)-Supported Proline: A Versatile Catalyst for the Enantioselective Aldol and Iminoaldol Reactions. Adv. Synth. Catal. 2002, 344, 533–542. [Google Scholar] [CrossRef]
- Benaglia, M.; Celentano, G.; Cozzi, F. Enantioselective Aldol Condensations Catalyzed by Poly(ethylene glycol)-Supported Proline. Adv. Synth. Catal. 2001, 343, 171–173. [Google Scholar] [CrossRef]
- Calderón, F.; Fernández, R.; Sánchez, F.; Fernández-Mayoralas, A. Asymmetric Aldol Reaction Using Immobilized Proline on Mesoporous Support. Adv. Synth. Catal. 2005, 347, 1395–1403. [Google Scholar] [CrossRef]
- Giacalone, F.; Gruttadauria, M.; Marculescu, A.M.; Noto, R. Polystyrene-supported proline and prolinamide. Versatile heterogeneous organocatalysts both for asymmetric aldol reaction in water and α-selenenylation of aldehydes. Tetrahedron Lett. 2007, 48, 255–259. [Google Scholar] [CrossRef]
- Font, D.; Jimeno, C.; Pericàs, M.A. Polystyrene-Supported Hydroxyproline: An Insoluble, Recyclable Organocatalyst for the Asymmetric Aldol Reaction in Water. Org. Lett. 2006, 8, 4653–4655. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhang, X.; Li, S.; Wang, X.; Ma, J. The high catalytic activity and reusability of the proline functionalized cage-like mesoporous material SBA-16 for the asymmetric aldol reaction proceeding in methanol–water mixed solvent. RSC Adv. 2014, 4, 9292–9299. [Google Scholar] [CrossRef]
- Angeloni, M.; Piermatti, O.; Pizzo, F.; Vaccaro, L. Synthesis of Zirconium Phosphonate SupportedL-Proline as an Effective Organocatalyst for Direct Asymmetric Aldol Addition. Eur. J. Org. Chem. 2014, 2014, 1716–1726. [Google Scholar] [CrossRef]
- Aghahosseini, H.; Ramazani, A.; Ślepokura, K.; Lis, T. The first protection-free synthesis of magnetic bifunctional l-proline as a highly active and versatile artificial enzyme: Synthesis of imidazole derivatives. J. Colloid Interface Sci. 2018, 511, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Doyagüez, E.G.; Calderón, F.; Sánchez, F.; Fernández-Mayoralas, A. Asymmetric Aldol Reaction Catalyzed by a Heterogenized Proline on a Mesoporous Support. The Role of the Nature of Solvents. J. Org. Chem. 2007, 72, 9353–9356. [Google Scholar] [CrossRef] [PubMed]
- Campisciano, V.; Giacalone, F.; Gruttadauria, M. Supported Ionic Liquids: A Versatile and Useful Class of Materials. Chem. Rec. 2017, 17, 918–938. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Tan, R.; Zhao, L.; Yin, D. l-Proline supported on ionic liquid-modified magnetic nanoparticles as a highly efficient and reusable organocatalyst for direct asymmetric aldol reaction in water. Green Chem. 2013, 15, 2422. [Google Scholar] [CrossRef]
- Ferré, M.; Pleixats, R.; Man, M.W.C.; Cattoën, X. Recyclable organocatalysts based on hybrid silicas. Green Chem. 2016, 18, 881–922. [Google Scholar] [CrossRef] [Green Version]
- Aprile, C.; Giacalone, F.; Gruttadauria, M.; Marculescu, A.M.; Noto, R.; Revell, J.D.; Wennemers, H. New ionic liquid-modified silica gels as recyclable materials for l-proline- or H–Pro–Pro–Asp–NH2-catalyzed aldol reaction. Green Chem. 2007, 9, 1328. [Google Scholar] [CrossRef]
- Lombardo, M.; Trombini, C. Ionic Tags in Catalyst Optimization: Beyond Catalyst Recycling. ChemCatChem 2010, 2, 135–145. [Google Scholar] [CrossRef]
- Lombardo, M.; Quintavalla, A.; Chiarucci, M.; Trombini, C. Multiphase Homogeneous Catalysis: Common Procedures and Recent Applications. Synlett 2010, 2010, 1746–1765. [Google Scholar] [CrossRef]
- Lombardo, M.; Pasi, F.; Easwar, S.; Trombini, C. An Improved Protocol for the Direct Asymmetric Aldol Reaction in Ionic Liquids, Catalysed by Onium Ion-Tagged Prolines. Adv. Synth. Catal. 2007, 349, 2061–2065. [Google Scholar] [CrossRef]
- Lombardo, M.; Pasi, F.; Easwar, S.; Trombini, C. Direct Asymmetric Aldol Reaction Catalyzed by an Imidazolium-Tagged trans-4-Hydroxy-l-proline under Aqueous Biphasic Conditions. Synlett 2008, 2471–2474. [Google Scholar] [CrossRef]
- Lombardo, M.; Easwar, S.; Pasi, F.; Trombini, C. The Ion Tag Strategy as a Route to Highly Efficient Organocatalysts for the Direct Asymmetric Aldol Reaction. Adv. Synth. Catal. 2009, 351, 276–282. [Google Scholar] [CrossRef]
- Bhati, M.; Upadhyay, S.; Easwar, S.; Srinivasan, E. Exploring “Through-Bond” Proximity between the Ion Tag and Reaction Site of an Imidazolium-Proline Catalyst for the Direct Asymmetric Aldol Reaction. Eur. J. Org. Chem. 2017, 1788–1793. [Google Scholar] [CrossRef]
- Rosso, C.; Emma, M.G.; Martinelli, A.; Lombardo, M.; Quintavalla, A.; Trombini, C.; Syrgiannis, Z.; Prato, M. A Recyclable Chiral 2-(Triphenylmethyl)pyrrolidine Organocatalyst Anchored to [60]Fullerene. Adv. Synth. Catal. 2019, 361, 2936–2944. [Google Scholar] [CrossRef]
- Bottoni, A.; Lombardo, M.; Miscione, G.P.; Montroni, E.; Quintavalla, A.; Trombini, C. Electrosteric Activation by using Ion-Tagged Prolines: A Combined Experimental and Computational Investigation. ChemCatChem 2013, 5, 2913–2924. [Google Scholar] [CrossRef]
- Montroni, E.; Lombardo, M.; Quintavalla, A.; Trombini, C.; Gruttadauria, M.; Giacalone, F. A Liquid-Liquid Biphasic Homogeneous Organocatalytic Aldol Protocol Based on the Use of a Silica Gel Bound Multilayered Ionic Liquid Phase. ChemCatChem 2012, 4, 1000–1006. [Google Scholar] [CrossRef]
- Montroni, E.; Sanap, S.P.; Trombini, C.; Dhavale, D.D.; Lombardo, M.; Quintavalla, A. A New Robust and Efficient Ion?Tagged Proline Catalyst Carrying an Amide Spacer for the Asymmetric Aldol Reaction. Adv. Synth. Catal. 2011, 353, 3234–3240. [Google Scholar] [CrossRef]
- Loh, T.-P.; Feng, L.-C.; Yang, H.-Y.; Yang, J.-Y. l-Proline in an ionic liquid as an efficient and reusable catalyst for direct asymmetric aldol reactions. Tetrahedron Lett. 2002, 43, 8741–8743. [Google Scholar] [CrossRef]
- Kotrusz, P.; Kmentová, I.; Gotov, B.; Toma, Š.; Solčániová, E. Proline-catalysed asymmetric aldol reaction in the room temperature ionic liquid [bmim]PF6. Chem. Commun. 2002, 2510–2511. [Google Scholar] [CrossRef]
- Gruttadauria, M.; Rielaa, S.; Aprile, C.; Meo, P.L.; D’Anna, F.; Noto, R. Supported Ionic Liquids. New Recyclable Materials for theL-Proline-Catalyzed Aldol Reaction. Adv. Synth. Catal. 2006, 348, 82–92. [Google Scholar] [CrossRef] [Green Version]
- Kitazume, T.; Jiang, Z.; Kasai, K.; Mihara, Y.; Suzuki, M. Synthesis of fluorinated materials catalyzed by proline or antibody 38C2 in ionic liquid. J. Fluor. Chem. 2003, 121, 205–212. [Google Scholar] [CrossRef]
- Córdova, A. Direct catalytic asymmetric cross-aldol reactions in ionic liquid media. Tetrahedron Lett. 2004, 45, 3949–3952. [Google Scholar] [CrossRef]
- Miao, W.; Chan, T.-H. Ionic-Liquid-Supported Organocatalyst: Efficient and Recyclable Ionic-Liquid-Anchored Proline for Asymmetric Aldol Reaction. Adv. Synth. Catal. 2006, 348, 1711–1718. [Google Scholar] [CrossRef]
- List, B.; Martínez, A.; Zumbansen, K.; Döhring, A.; Van Gemmeren, M. Improved Conditions for the Proline-Catalyzed Aldol Reaction of Acetone with Aliphatic Aldehydes. Synlett 2014, 25, 932–934. [Google Scholar] [CrossRef]
- Trajković, J.M.; Milanovic, V.D.; Ferjančić, Z.; Saicic, R.N. On the Asymmetric Induction in Proline-Catalyzed Aldol Reactions: Reagent-Controlled Addition Reactions of 2,2-Dimethyl-1,3-dioxane-5-one to Acyclic Chiral α-Branched Aldehydes. Eur. J. Org. Chem. 2017, 6146–6153. [Google Scholar] [CrossRef]
- Rougeot, C.; Situ, H.; Cao, B.H.; Vlachos, V.; Hein, J.E. Automated reaction progress monitoring of heterogeneous reactions: Crystallization-induced stereoselectivity in amine-catalyzed aldol reactions. React. Chem. Eng. 2017, 2, 226–231. [Google Scholar] [CrossRef]
- Brenna, D.; Massolo, E.; Puglisi, A.; Rossi, S.; Celentano, G.; Benaglia, M.; Capriati, V. Towards the development of continuous, organocatalytic, and stereoselective reactions in deep eutectic solvents. Beilstein J. Org. Chem. 2016, 12, 2620–2626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez, R.; Berbegal, L.; Guillena, G.; Ramón, D.J. Bio-renewable enantioselective aldol reaction in natural deep eutectic solvents. Green Chem. 2016, 18, 1724–1730. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, B.; Bruckmann, A.; Bolm, C. A Highly Efficient Asymmetric Organocatalytic Aldol Reaction in a Ball Mill. Chem. A Eur. J. 2007, 13, 4710–4722. [Google Scholar] [CrossRef] [PubMed]
- Bruckmann, A.; Krebs, A.; Bolm, C. Organocatalytic reactions: Effects of ball milling, microwave and ultrasound irradiation. Green Chem. 2008, 10, 1131. [Google Scholar] [CrossRef]
- Veverková, E.; Modrocká, V.; Sebesta, R. Organocatalyst Efficiency in the α-Aminoxylation and α-Hydrazination of Carbonyl Derivatives in Aqueous Media or in a Ball-Mill. Eur. J. Org. Chem. 2017, 1191–1195. [Google Scholar] [CrossRef]
- Kumar, A.; Gupta, M.K.; Kumar, M. l-Proline catalysed multicomponent synthesis of 3-amino alkylated indolesvia a Mannich-type reaction under solvent-free conditions. Green Chem. 2012, 14, 290–295. [Google Scholar] [CrossRef]
- Curzons, A.D.; Constable, D.J.C.; Cunningham, V.L. Expanding GSK’s Solvent Selection Guide?application of life cycle assessment to enhance solvent selections. Clean Technol. Environ. Policy 2004, 7, 42–50. [Google Scholar] [CrossRef]
- Byrne, F.; Jin, S.; Paggiola, G.; Petchey, T.H.M.; Clark, J.H.; Farmer, T.J.; Hunt, A.J.; McElroy, C.R.; Sherwood, J. Tools and techniques for solvent selection: Green solvent selection guides. Sustain. Chem. Process. 2016, 4, 1034. [Google Scholar] [CrossRef] [Green Version]
- Henderson, R.K.; Jiménez-González, C.; Constable, D.J.C.; Alston, S.R.; Inglis, G.G.A.; Fisher, G.; Sherwood, J.; Binks, S.P.; Curzons, A.D. Expanding GSK’s solvent selection guide – embedding sustainability into solvent selection starting at medicinal chemistry. Green Chem. 2011, 13, 854. [Google Scholar] [CrossRef]
- Pihko, P.M.; Laurikainen, K.M.; Usano, A.; Nyberg, A.I.; Kaavi, J.A. Effect of additives on the proline-catalyzed ketone–aldehyde aldol reactions. Tetrahedron 2006, 62, 317–328. [Google Scholar] [CrossRef]
- Kaur, N.; Kishore, D. Synthetic Strategies Applicable in the Synthesis of Privileged Scaffold: 1,4-Benzodiazepine. Synth. Commun. 2014, 44, 1375–1413. [Google Scholar] [CrossRef]
- Ward, D.; Jheengut, V. Proline-catalyzed asymmetric aldol reactions of tetrahydro-4H-thiopyran-4-one with aldehydes. Tetrahedron Lett. 2004, 45, 8347–8350. [Google Scholar] [CrossRef]
- Cai, J.; Wu, Y.-S.; Chen, Y.; Deng, D.-S. Proline-Catalyzed Asymmetric Direct Aldol Reaction Assisted by d-Camphorsulfonic Acid in Aqueous Media. Synlett 2005, 1627–1629. [Google Scholar] [CrossRef]
- Zhou, Y.; Shan, Z. (R)- or (S)-Bi-2-naphthol assisted, l-proline catalyzed direct aldol reaction. Tetrahedron Asymmetry 2006, 17, 1671–1677. [Google Scholar] [CrossRef]
- Zhou, Y.; Shan, Z. Chiral Diols: A New Class of Additives for Direct Aldol Reaction Catalyzed byl-Proline. J. Org. Chem. 2006, 71, 9510–9512. [Google Scholar] [CrossRef] [PubMed]
- Reis, O.; Eymur, S.; Reis, B.; Demir, A.S. Direct enantioselective aldol reactions catalyzed by a proline–thiourea host–guest complex. Chem. Commun. 2009, 1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Hamdouni, N.; Companyó, X.; Rios, R.; Moyano, A. Substrate-Dependent Nonlinear Effects in Proline-Thiourea-Catalyzed Aldol Reactions: Unraveling the Role of the Thiourea Co-Catalyst. Chem. A Eur. J. 2010, 16, 1142–1148. [Google Scholar] [CrossRef] [PubMed]
- Demir, A.S.; Basceken, S. Study of asymmetric aldol and Mannich reactions catalyzed by proline–thiourea host–guest complexes in nonpolar solvents. Tetrahedron Asymmetry 2013, 24, 515–525. [Google Scholar] [CrossRef]
- Martinez-Castaneda, A.; Rodriguez-Solla, H.; Concellón, C.; Del Amo, V. Switching Diastereoselectivity in Proline-Catalyzed Aldol Reactions. J. Org. Chem. 2012, 77, 10375–10381. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Daka, P.; Wang, H. Primary amine-metal Lewis acid bifunctional catalysts: The application to asymmetric direct aldol reactions. Chem. Commun. 2009, 6825. [Google Scholar] [CrossRef] [PubMed]
- Daka, P.; Xu, Z.; Alexa, A.; Wang, H. Primary amine-metal Lewis acid bifunctional catalysts based on a simple bidentate ligand: Direct asymmetric aldol reaction. Chem. Commun. 2011, 47, 224–226. [Google Scholar] [CrossRef] [PubMed]
- Paradowska, J.; Stodulski, M.; Mlynarski, J. Direct Catalytic Asymmetric Aldol Reactions Assisted by Zinc Complex in the Presence of Water. Adv. Synth. Catal. 2007, 349, 1041–1046. [Google Scholar] [CrossRef]
- Lu, Z.; Mei, H.; Han, J.; Pan, Y. The Mimic of Type II Aldolases Chemistry: Asymmetric Synthesis of β-Hydroxy Ketones by Direct Aldol Reaction. Chem. Boil. Drug Des. 2010, 76, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Itoh, S.; Kitamura, M.; Yamada, Y.; Aoki, S. Chiral Catalysts Dually Functionalized with Amino Acid and Zn2+Complex Components for Enantioselective Direct Aldol Reactions Inspired by Natural Aldolases: Design, Synthesis, Complexation Properties, Catalytic Activities, and Mechanistic Study. Chem. A Eur. J. 2009, 15, 10570–10584. [Google Scholar] [CrossRef]
- Kofoed, J.; Darbre, T.; Reymond, J. Dual mechanism of zinc-proline catalyzed aldol reactions in water. Chem. Commun. 2006, 1482. [Google Scholar] [CrossRef] [PubMed]
- Kofoed, J.; Machuqueiro, M.; Reymond, J.; Darbre, T. Zinc–proline catalyzed pathway for the formation of sugars. Chem. Commun. 2004, 1540. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Lopez, R.; Kofoed, J.; Machuqueiro, M.; Darbre, T. A Selective Direct Aldol Reaction in Aqueous Media Catalyzed byZinc-Proline. Eur. J. Org. Chem. 2005, 5268–5276. [Google Scholar] [CrossRef]
- Kofoed, J.; Reymond, J.; Darbre, T. Prebiotic carbohydrate synthesis: Zinc–proline catalyzes direct aqueous aldol reactions of α-hydroxy aldehydes and ketones. Org. Biomol. Chem. 2005, 3, 1850. [Google Scholar] [CrossRef] [PubMed]
- Akagawa, K.; Sakamoto, S.; Kudo, K. Direct asymmetric aldol reaction in aqueous media using polymer-supported peptide. Tetrahedron Lett. 2005, 46, 8185–8187. [Google Scholar] [CrossRef]
- Penhoat, M.; Barbry, D.; Rolando, C. Direct asymmetric aldol reaction co-catalyzed by l-proline and group 12 elements Lewis acids in the presence of water. Tetrahedron Lett. 2011, 52, 159–162. [Google Scholar] [CrossRef]
- Karmakar, A.; Maji, T.; Wittmann, S.; Reiser, O. L-Proline/CoCl2-Catalyzed Highly Diastereo- and Enantioselective Direct Aldol Reactions. Chem. A Eur. J. 2011, 17, 11024–11029. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Hou, Q.; Huang, G.; Fan, Q. Removal of heavy metals in aquatic environment by graphene oxide composites: A review. Environ. Sci. Pollut. Res. 2019, 27, 190–209. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Kumar, V.; Datta, S.; Dhanjal, D.S.; Sharma, K.; Samuel, J.; Singh, J. Current advancement and future prospect of biosorbents for bioremediation. Sci. Total. Environ. 2020, 709, 135895. [Google Scholar] [CrossRef] [PubMed]
- Vareda, J.P.; Valente, A.J.; Durães, L. Assessment of heavy metal pollution from anthropogenic activities and remediation strategies: A review. J. Environ. Manag. 2019, 246, 101–118. [Google Scholar] [CrossRef] [PubMed]
- Lindström, U.M. Stereoselective Organic Reactions in Water. Chem. Rev. 2002, 102, 2751–2772. [Google Scholar] [CrossRef] [PubMed]
- Sinou, D. Asymmetric Organometallic-Catalyzed Reactions in Aqueous Media. Adv. Synth. Catal. 2002, 344, 221–237. [Google Scholar] [CrossRef]
- Prat, D.; Pardigon, O.; Flemming, H.-W.; Letestu, S.; Ducandas, V.; Isnard, P.; Guntrum, E.; Senac, T.; Ruisseau, S.; Cruciani, P.; et al. Sanofi’s Solvent Selection Guide: A Step Toward More Sustainable Processes. Org. Process. Res. Dev. 2013, 17, 1517–1525. [Google Scholar] [CrossRef]
- Capello, C.; Fischer, U.; Hungerbühler, K. What is a green solvent? A comprehensive framework for the environmental assessment of solvents. Green Chem. 2007, 9, 927–934. [Google Scholar] [CrossRef]
- Clegg, W.; Harrington, R.W.; North, M.; Pizzato, F.; Villuendas, P. Cyclic carbonates as sustainable solvents for proline-catalysed aldol reactions. Tetrahedron Asymmetry 2010, 21, 1262–1271. [Google Scholar] [CrossRef]
- Tan, R.; Li, C.; Luo, J.; Kong, Y.; Zheng, W.; Yin, D. An effective heterogeneous l-proline catalyst for the direct asymmetric aldol reaction using graphene oxide as support. J. Catal. 2013, 298, 138–147. [Google Scholar] [CrossRef]
- Qian, Y.; Zheng, X.; Wang, X.; Xiao, S.; Wang, Y. An Efficient Ionic Liquid Additive for Proline-catalyzed Direct Asymmetric Aldol Reactions between Cyclic Ketones and Aromatic Aldehydes. Chem. Lett. 2009, 38, 576–577. [Google Scholar] [CrossRef]
- Rodriguez, B.; Rantanen, T.; Bolm, C. Solvent-Free Asymmetric Organocatalysis in a Ball Mill. Angew. Chem. Int. Ed. 2006, 45, 6924–6926. [Google Scholar] [CrossRef] [PubMed]
- Obregon, A.; Milán, M.; Juaristi, E. Improving the Catalytic Performance of (S)-Proline as Organocatalyst in Asymmetric Aldol Reactions in the Presence of Solvate Ionic Liquids: Involvement of a Supramolecular Aggregate. Org. Lett. 2017, 19, 1108–1111. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Castañeda, A.; Poladura, B.; Rodriguez-Solla, H.; Concellón, C.; Del Amo, V. Direct Aldol Reactions Catalyzed by a Heterogeneous Guanidinium Salt/Proline System under Solvent-Free Conditions‡. Org. Lett. 2011, 13, 3032–3035. [Google Scholar] [CrossRef] [PubMed]
- Szőllősi, G.; Fekete, M.; Gurka, A.A.; Bartók, M. Reversal of Enantioselectivity in Aldol Reaction: New Data on Proline/γ-Alumina Organic–Inorganic Hybrid Catalysts. Catal. Lett. 2013, 144, 478–486. [Google Scholar] [CrossRef]
- North, M.; Villuendas, P. A Chiral Solvent Effect in Asymmetric Organocatalysis. Org. Lett. 2010, 12, 2378–2381. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Aratake, S.; Itoh, T.; Okano, T.; Sumiya, T.; Shoji, M. Dry and wet prolines for asymmetric organic solvent-free aldehyde–aldehyde and aldehyde–ketone aldol reactions. Chem. Commun. 2007, 957–959. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Wu, Y.; Zhao, X.; Wang, J.; Zhang, L.; Cui, Y. Polymerization of l-proline functionalized styrene and its catalytic performance as a supported organocatalyst for direct enantioselective aldol reaction. Tetrahedron Asymmetry 2016, 27, 740–746. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, L.; Wang, Z.; Yan, J. Merrifield Resin Supported Ionic Liquids/l-Proline as Efficient and Recyclable Catalyst Systems for Asymmetric Aldol Reaction. Synthesis 2009, 3744–3750. [Google Scholar] [CrossRef]
- Yang, H.; Li, S.; Wang, X.; Zhang, F.; Zhong, X.; Dong, Z.; Ma, J. Core–shell silica magnetic microspheres supported proline as a recyclable organocatalyst for the asymmetric aldol reaction. J. Mol. Catal. A Chem. 2012, 363, 404–410. [Google Scholar] [CrossRef]
- Yacob, Z.; Nan, A.; Liebscher, J. Proline-Functionalized Magnetic Core-Shell Nanoparticles as Efficient and Recyclable Organocatalysts for Aldol Reactions. Adv. Synth. Catal. 2012, 354, 3259–3264. [Google Scholar] [CrossRef]
- Liebscher, J.; Shah, J.; Khan, S.; Blumenthal, H. 1,2,3-Triazolium-Tagged Prolines and Their Application in Asymmetric Aldol and Michael Reactions. Synthesis 2009, 3975–3982. [Google Scholar] [CrossRef]
- Kucherenko, A.S.; Struchkova, M.I.; Zlotin, S.G. The (S)-Proline/Polyelectrolyte System: An Efficient, Heterogeneous, Reusable Catalyst for Direct Asymmetric Aldol Reactions. Eur. J. Org. Chem. 2006, 2000–2004. [Google Scholar] [CrossRef]
- Ibrahem, I.; Zou, W.; Xu, Y.; Córdova, A. Amino Acid-Catalyzed Asymmetric Carbohydrate Formation: Organocatalytic One-StepDe Novo Synthesis of Keto and Amino Sugars. Adv. Synth. Catal. 2006, 348, 211–222. [Google Scholar] [CrossRef]
- Suri, J.T.; Mitsumori, S.; Albertshofer, K.; Tanaka, F.; Barbas, C.F. Dihydroxyacetone Variants in the Organocatalytic Construction of Carbohydrates: Mimicking Tagatose and Fuculose Aldolases. J. Org. Chem. 2006, 71, 3822–3828. [Google Scholar] [CrossRef] [PubMed]
- Grondal, C.; Enders, D. Direct asymmetric organocatalytic de novo synthesis of carbohydrates. Tetrahedron 2006, 62, 329–337. [Google Scholar] [CrossRef]
- Majewski, M.; Niewczas, I.; Palyam, N. Acids as Proline Co-catalysts in the Aldol Reaction of 1,3-Dioxan-5-ones. Synlett 2006, 2387–2390. [Google Scholar] [CrossRef]
- Ibrahem, I.; Córdova, A. Amino acid catalyzed direct enantioselective formation of carbohydrates: One-step de novo synthesis of ketoses. Tetrahedron Lett. 2005, 46, 3363–3367. [Google Scholar] [CrossRef]
- Gong, Z.; Wei, C.; Shi, Y.; Zheng, Q.; Song, Z.; Liu, Z. Novel chiral bifunctional l-thiazoline-amide derivatives: Design and application in the direct enantioselective aldol reactions. Tetrahedron 2014, 70, 1827–1835. [Google Scholar] [CrossRef]
- Miura, T.; Kasuga, H.; Imai, K.; Ina, M.; Tada, N.; Imai, N.; Itoh, A. Highly efficient asymmetric aldol reaction in brine using a fluorous sulfonamide organocatalyst. Org. Biomol. Chem. 2012, 10, 2209. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, Y.; Gai, X.; Zeng, X. Highly Enantio- and Diastereoselective l-Proline Derived Acetylglucose Amide Catalyzed Aldol Reaction of Ketones to Aldehydes under Solvent-Free Conditions. Synlett 2015, 26, 2858–2862. [Google Scholar] [CrossRef] [Green Version]
- Majewski, M.; Gleave, D.M.; Nowak, P. 1,3-Dioxan-5-ones: Synthesis, deprotonation, and reactions of their lithium enolates. Can. J. Chem. 1995, 73, 1616–1626. [Google Scholar] [CrossRef] [Green Version]
- Ying, A.; Liu, S.; Li, Z.; Chen, G.; Yang, J.; Yan, H.; Xu, S. Magnetic Nanoparticles-Supported Chiral Catalyst with an Imidazolium Ionic Moiety: An Efficient and Recyclable Catalyst for Asymmetric Michael and Aldol Reactions. Adv. Synth. Catal. 2016, 358, 2116–2125. [Google Scholar] [CrossRef]
- Sai, M.; Yamamoto, H. Chiral Brønsted Acid as a True Catalyst: Asymmetric Mukaiyama Aldol and Hosomi–Sakurai Allylation Reactions. J. Am. Chem. Soc. 2015, 137, 7091–7094. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Marcus, R.A. On the Theory of Organic Catalysis “on Water”. J. Am. Chem. Soc. 2007, 129, 5492–5502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, A.; Boto, R.A.; Dingwall, P.; Contreras-Garcia, J.; Harvey, M.J.; Mason, N.; Rzepa, H. The Houk–List transition states for organocatalytic mechanisms revisited. Chem. Sci. 2014, 5, 2057–2071. [Google Scholar] [CrossRef] [Green Version]
- Patil, M.P.; Sunoj, R.B. Insights on Co-Catalyst-Promoted Enamine Formation between Dimethylamine and Propanal through Ab Initio and Density Functional Theory Study. J. Org. Chem. 2007, 72, 8202–8215. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, S.E.; Seguin, T.J.; Guan, Y.; Doney, A. Noncovalent Interactions in Organocatalysis and the Prospect of Computational Catalyst Design. Acc. Chem. Res. 2016, 49, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
Scheme 1.
The benchmark aldol reaction.

Scheme 2.
Process scale-up on 100 mmol of limiting aldehyde 2d.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Comparison of different protic reaction media 1.
| R (2) | Solvent | t [h] | 3 Conversion [%] 2 | ee [%] 3 | anti/syn2 |
|---|---|---|---|---|---|
| 4-NO2Ph (2a) | MeOH/H2O | 19 | >99 | 98 | 93:7 |
| H2O | 19 | 25 | 99 | 95:5 | |
| MeOH | 19 | >99 | 76 | 59:41 | |
| 4-CNPh (2b) | MeOH/H2O | 19 | 97 | 98 | 95:5 |
| H2O | 19 | 80 | 99 | 95:5 | |
| MeOH | 19 | >99 | 87 | 82:18 | |
| 4-ClPh (2c) | MeOH/H2O | 19 | 43 | 99 | 97:3 |
| H2O | 19 | 5 | >99 | nd | |
| MeOH | 19 | 64 | 98 | 85:15 | |
| Ph (2d) 4 | MeOH/H2O | 30 | 58 | 97 | 88:12 |
| H2O | 30 | 20 | >99 | >99:1 | |
| MeOH | 30 | 64 | 83 | 72:28 |
1 Reaction conditions: 1a (5 equiv.), 2 (0.3 mmol), (S)-proline (10 mol%), rt, MeOH/H2O (20 µL/10 µL, 2/1 V/V) or H2O (10 µL), or MeOH (20 µL). 2 Determined by 1H NMR on the crude mixture. 3 Determined by chiral stationary phase (CSP)-HPLC on the crude mixture. 4 Here, 20 mol% of (S)-proline was used. rt = room temperature, h = hours, nd = not determined.
Table 2.
MeOH/H2O-based protocol applied to different aromatic aldehydes 2 1.
| R (2) | t [h] | 3 Conversion [%] 2 | ee [%] 3 | anti/syn2 |
|---|---|---|---|---|
| C6F5 (2e) | 19 | >99 | 97 | >99:1 |
| 2-NO2Ph (2f) | 19 | 93 | 95 | 95:5 |
| 4-BrPh (2g) | 19 | 41 | 99 | 98:2 |
| 2-naphthyl (2h) | 24 | 37 | 93 | 91:9 |
| 4-MeOPh (2i) 4 | 70 | 18 | 90 | 86:14 |
1 Reaction conditions: 1a (5 equiv.), 2 (0.3 mmol), (S)-proline (10 mol%), MeOH/H2O (20 µL/10 µL), rt. 2 Determined by 1H NMR on the crude mixture. 3 Determined by CSP-HPLC on the crude mixture. 4 Here, 20 mol% of (S)-proline was used.
Table 3.
Optimization of the MeOH/H2O-based protocol 1.
| R (2) | MeOH/H2O [μL/μL] | t [h] | 3 Conversion [%] 2 | ee [%] 3 | anti/syn2 |
|---|---|---|---|---|---|
| 4-NO2Ph (2a) | 20/10 | 4 | 3aa, 47 | 97 | 94:6 |
| 40/10 | 4 | 3aa, 82 | 98 | 92:8 | |
| 20 + 20 4/10 | 4 | 3aa, 84 | 97 | 94:6 | |
| 4-CNPh (2b) | 20/10 | 4 | 3ab, 65 | 97 | 95:5 |
| 40/10 | 4 | 3ab, 77 | 95 | 94:6 | |
| 4-ClPh (2c) | 20/10 | 19 | 3ac, 43 | 99 | 97:3 |
| 40/10 | 19 | 3ac, 64 | 98 | 95:5 | |
| Ph (2d) | 20/10 5 | 30 | 3ad, 58 | 97 | 88:12 |
| 40/10 | 30 | 3ad, 75 | 96 | 90:10 | |
| C6F5 (2e) | 20/10 | 4 | 3ae, 63 | 97 | >99:1 |
| 40/10 | 4 | 3ae, 67 | 96 | >99:1 | |
| 2-NO2Ph (2f) | 20/10 | 4 | 3af, 34 | 97 | 98:2 |
| 40/10 | 4 | 3af, 59 | 97 | 97:3 | |
| 4-BrPh (2g) | 20/10 | 19 | 3ag, 41 | 99 | 98:2 |
| 40/10 | 19 | 3ag, 88 | 96 | 94:6 | |
| 2-naphthyl (2h) | 20/10 | 24 | 3ah, 37 | 93 | 91:9 |
| 40/10 | 24 | 3ah, 72 | 93 | 92:8 | |
| 4-MeOPh (2i) 5 | 20/10 | 68 | 3ai, 18 | 90 | 86:14 |
| 40/10 | 68 | 3ai, 43 | 89 | 80:20 | |
| i-Pr (2j) 5 | 40/10 | 72 | 3aj, 63 | >99 | >99:1 |
| 4-CF3Ph (2k) | 40/10 | 4 | 3ak, 78 | 97 | 97:3 |
| 2-thiophenyl (2l) 5 | 40/10 | 48 | 3al, 65 | 88 | 84:16 |
| 4-CH3Ph (2m) | 40/10 | 40 | 3am, 64 | 94 | 87:13 |
1 Reaction conditions: 1a (5 equiv.), 2 (0.3 mmol), (S)-proline (10 mol%), rt. 2 Determined by 1H NMR on the crude mixture. 3 Determined by CSP-HPLC on the crude mixture. 4 Here, 20 μL of MeOH was added after 2 h. 5 Here, 20 mol% of (S)-proline was used.
Table 4.
Effects of cyclohexanone 1a amount in the MeOH/H2O/(S)-proline-based protocol 1.
| R (2) | 1a [eq.] | t [h] | 3 Conversion [%] 2 | ee [%] 3 | anti/syn2 |
|---|---|---|---|---|---|
| 4-NO2Ph (2a) | 5 | 4 | 82 | 98 | 92:8 |
| 5 | 4 | 84 4 | 97 | 94:6 | |
| 2 | 19 | 99 | 95 | 92:8 | |
| 1.05 | 19 | 92 | 97 | 90:10 | |
| 4-CNPh (2b) | 5 | 4 | 77 | 95 | 94:6 |
| 2 | 24 | 98 | 91 | 90:10 | |
| C6F5 (2e) | 5 | 4 | 67 | 97 | >99:1 |
| 2 | 24 | >99 | 92 | >99:1 | |
| 2-NO2Ph (2f) | 5 | 4 | 59 | 97 | 97:3 |
| 2 | 24 | 92 | 93 | 94:6 | |
| 4-BrPh (2g) | 5 | 19 | 88 | 96 | 94:6 |
| 2 | 24 | 97 | 91 | 90:10 | |
| 4-CF3Ph (2k) | 5 | 4 | 78 | 97 | 97:3 |
| 3 | 20 | 96 | 96 | 95:5 | |
| 2 | 24 | 93 | 94 | 93:7 |
1 Reaction conditions: 2 (0.3 mmol), (S)-proline (10 mol%), MeOH/H2O (40 µL/10 µL), rt. 2 Determined by 1H NMR on the crude mixture. 3 Determined by CSP-HPLC on the crude mixture. 4 Here, 20 μL of MeOH was added after 2 h.
Table 5.
MeOH/H2O/(S)-proline-based protocol applied to ketones 1b,c 1.
| (1) | R (2) | t [h] | 3, Conversion [%] 2 | ee [%] 3 | anti/syn2 |
|---|---|---|---|---|---|
 (1b) | 4-NO2Ph (2a) | 4 | 3ba, >99 | 94 | 61:39 |
| 4-NO2Ph (2a) | 19 4 | 3ba, 91 | 94 | 78:22 | |
| 4-BrPh (2g) | 19 | 3bg, 64 | 93 | 75:25 | |
| Ph (2d) | 30 | 3bd, 70 | 93 | 73:27 | |
 (1c) | 4-BrPh (2g) | 24 | 3cg, 90 | 86 | 78:22 |
| Ph (2d) | 48 | 3cd, 82 | 86 | 77:23 |
1 Reaction conditions: 2 (0.3 mmol), 1 (5 eq.), (S)-proline (10 mol%), MeOH/H2O (40 µL/10 µL), rt. 2 Determined by 1H NMR on the crude mixture. 3 Determined by CSP-HPLC on the crude mixture. 4 Reaction carried out at 0 °C.
Table 6.
Process scale-up study 1.
| Aldehyde Addition Rate | t [h] | Conversion [%] 2 | anti/syn2 | ee [%] 3 |
|---|---|---|---|---|
| 45 min | 23 | 72 | 86:14 | 96 |
| 28 | 78 | 87:13 | - | |
| 47 | 83 | 85:15 | 94 | |
| 6 h | 23 | 71 | 87:13 | - |
| 28 | 80 | 85:15 | - | |
| 47 | 85 | 84:16 | - |
1 Reaction conditions: 2d (100 mmol), 1a (500 mmol), (S)-proline (10 mol%), MeOH/H2O (13.33 mL/3.33 mL), rt. Total volume = 79 mL. 2 Determined by 1H NMR on the crude mixture. 3 Determined by CSP-HPLC on the crude mixture.
Table 7.
Study of the reaction outcome as a function of the reaction time 1.
| R (2) | t [h] | Conversion [%] 2 | anti/syn2 |
|---|---|---|---|
| Ph (2d) | 24 | 75 | 86:14 |
| 46 | 81 | 84:16 | |
| 71 | 85 | 79:21 | |
| 94 | 85 | 71:29 | |
| 4-CH3Ph (2m) | 23 | 53 | 90:10 |
| 45 | 65 | 87:13 | |
| 71 | 74 | 84:16 | |
| 138 | 75 | 79:21 | |
| 4-MeOPh (2i) 3 | 68 | 43 | 80:20 |
| 115 | 49 | 72:28 | |
| 164 | 52 | 66:34 |
1 Reaction conditions: 2 (50 mmol), 1a (5 eq.), (S)-proline (10 mol%), MeOH/H2O (6.67 mL/1.67 mL), rt. 2 Determined by 1H NMR on the crude mixture. 3 Here, 20 mol% of (S)-proline was used.
Table 8.
Process work-up study 1.
| Method | Conditions | Final Volume (mL) | Crude Analysis 2 |
|---|---|---|---|
| A | Reaction mixture = 18 mL (22.8 mmol) filtered on a silica-pad, mobile phase = EtOAc | 242 | Conv. = 87% dr = 84:16 |
| B | Reaction mixture = 18 mL (22.8 mmol) diluted with EtOAc, quenched with aqueous NH4Cl, extracted with EtOAc, dried with Na2SO4 (washed with EtOAc) | 90 | Conv. = 86% dr = 83:17 |
| C | Reaction mixture = 10 mL (12.6 mmol) diluted with EtOAc and placed at −15 °C for 36 h. 1° vacuum filtration. At −15 °C for 36 h. 2° vacuum filtration. Solution dried with Na2SO4 (washed with EtOAc) | 46 | Conv. = 89% dr = 84:16 Proline recovery: 113.3 mg (78%) |
| D | Reaction mixture = 10 mL (12.6 mmol) diluted with Et2O and placed at −15 °C for 36 h. 1° vacuum filtration. At −15 °C for 36 h. 2° vacuum filtration. Solution dried with Na2SO4 (washed with Et2O) | 52 | Conv. = 90% dr = 85:15 Proline recovery: 124.8 mg (86%) |
| E | Reaction mixture = 10 mL (12.6 mmol) diluted with DCM and placed at −15 °C for 36 h. Two liquid phases obtained, dried with Na2SO4 (washed with DCM) | 36 | Conv. = 88% dr = 85:15 |
| F | Reaction mixture = 10 mL (12.6 mmol) diluted with n-hexane and placed at −15 °C for 36 h. Two liquid phases obtained, dried with Na2SO4 (washed with n-hexane) | 43 | Conv. = 89% dr = 86:14 |
1 Aldol condensation carried out on 100 mmol of 2d, reaction conditions described in Scheme 2, reaction stopped after 49 h, reaction mixture (total volume = 79 mL) divided in six portions and treated with six different work-up methods. 2 Determined by 1H NMR on the crude mixture. EtOAc = ethyl acetate, Et2O = diethyl ether, DCM = dichloromethane.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Emma, M.G.; Tamburrini, A.; Martinelli, A.; Lombardo, M.; Quintavalla, A.; Trombini, C. A Simple and Efficient Protocol for Proline-Catalysed Asymmetric Aldol Reaction. Catalysts 2020, 10, 649. https://doi.org/10.3390/catal10060649
AMA Style
Emma MG, Tamburrini A, Martinelli A, Lombardo M, Quintavalla A, Trombini C. A Simple and Efficient Protocol for Proline-Catalysed Asymmetric Aldol Reaction. Catalysts. 2020; 10(6):649. https://doi.org/10.3390/catal10060649
Chicago/Turabian StyleEmma, Marco Giuseppe, Alice Tamburrini, Ada Martinelli, Marco Lombardo, Arianna Quintavalla, and Claudio Trombini. 2020. "A Simple and Efficient Protocol for Proline-Catalysed Asymmetric Aldol Reaction" Catalysts 10, no. 6: 649. https://doi.org/10.3390/catal10060649
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.