Promoting Light Hydrocarbons Yield by Catalytic Hydrodechlorination of Residual Chloromethanes Using Palladium Supported on Zeolite Catalysts

 , ,
, ,
Abstract
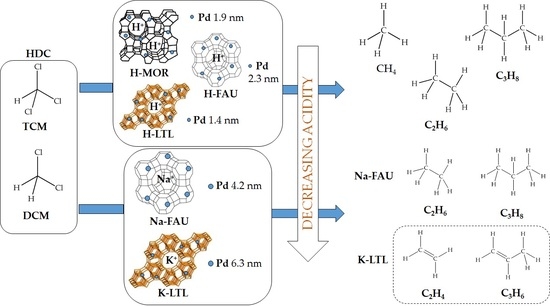
:
1. Introduction
2. Results and Discussion
2.1. Characterization of the Catalysts
2.2. HDC Tests
3. Materials and Methods
3.1. Materials and Chemicals
3.2. Synthesis of the Catalysts
3.3. Characterization
3.4. Gas-Phase HDC Tests
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Huang, B.; Lei, C.; Wei, C.; Zeng, G. Chlorinated volatile organic compounds (Cl-VOCs) in environment—Sources, potential human health impacts, and current remediation technologies. Environ. Int. 2014, 71, 118–138. [Google Scholar] [CrossRef] [PubMed]
- Kurylo, M.J.; Rodriguez, J.M.; Andreae, M.O.; Atlas, E.L.; Blake, D.R.; Butler, J.H.; Lal, S.; Lary, D.J.; Midgley, P.M.; Montzka, S.A.; et al. Scientific Assessment of Ozone Depletion: 2014; WMO: Geneva, Switzerland, 2014; ISBN 9789966076014. [Google Scholar]
- Zhao, J.; Chen, M. Leak Detection and Repair (LDAR) Standard Review for Self-Inspection and Management for VOC Emission in China’s Traditional Energy Chemical Industry. J. Environ. Prot. 2018, 9, 1155–1170. [Google Scholar] [CrossRef] [Green Version]
- Fraas, A.G.; Egorenkov, A. A Retrospective Study of EPA’s Rules Setting Best Available Technology Limits For Toxic Discharges to Water Under the Clean Water Act. SSRN Electron. J. 2015, 15–41. [Google Scholar] [CrossRef]
- Baran, R.; Kamińska, I.I.; Śrębowata, A.; Dzwigaj, S. Selective hydrodechlorination of 1,2-dichloroethane on NiSiBEA zeolite catalyst: Influence of the preparation procedure on a high dispersion of Ni centers. Microporous Mesoporous Mater. 2013, 169, 120–127. [Google Scholar] [CrossRef]
- Zichittella, G.; Aellen, N.; Paunović, V.; Amrute, A.P.; Pérez-Ramírez, J. Olefins from Natural Gas by Oxychlorination. Angew. Chem. Int. Ed. 2017, 56, 13670–13674. [Google Scholar] [CrossRef] [PubMed]
- Arevalo-Bastante, A.; Álvarez-Montero, M.A.; Bedia, J.; Gómez-Sainero, L.M.; Rodriguez, J.J. Gas-phase hydrodechlorination of mixtures of chloromethanes with activated carbon-supported platinum catalysts. Appl. Catal. B Environ. 2015, 179, 551–557. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Guo, T.; Li, K.; Sun, L. A facile approach to enhancing activity of Ni2P/SiO2 catalyst for hydrodechlorination of chlorobenzene: Promoting effect of water and oxygen. Catal. Sci. Technol. 2015, 5, 2670–2680. [Google Scholar] [CrossRef]
- López, E.; Ordóñez, S.; Sastre, H.; Díez, F.V. Kinetic study of the gas-phase hydrogenation of aromatic and aliphatic organochlorinated compounds using a Pd/Al2O3 catalyst. J. Hazard. Mater. 2003, 97, 281–294. [Google Scholar] [CrossRef]
- Martin-Martinez, M.; Gómez-Sainero, L.M.; Bedia, J.; Arevalo-Bastante, A.; Rodriguez, J.J. Enhanced activity of carbon-supported Pd–Pt catalysts in the hydrodechlorination of dichloromethane. Appl. Catal. B Environ. 2016, 184, 55–63. [Google Scholar] [CrossRef] [Green Version]
- Bedia, J.; Gómez-sainero, L.M.; Grau, J.M.; Busto, M.; Martin-martinez, M.; Rodriguez, J.J. Hydrodechlorination of dichloromethane with mono- and bimetallic Pd-Pt on sulfated and tungstated zirconia catalysts. J. Catal. 2012, 294, 207–215. [Google Scholar] [CrossRef]
- de Pedro, Z.M.; Gómez-Sainero, L.M.; González-Serrano, E.; Rodríguez, J.J. Gas-Phase Hydrodechlorination of Dichloromethane at Low Concentrations with Palladium/Carbon Catalysts. Ind. Eng. Chem. Res. 2006, 45, 7760–7766. [Google Scholar] [CrossRef]
- Chang, W.; Kim, H.; Oh, J.; Ahn, B.J. Hydrodechlorination of chlorophenols over Pd catalysts supported on zeolite Y, MCM-41 and graphene. Res. Chem. Intermed. 2018, 44, 3835–3847. [Google Scholar] [CrossRef]
- Díaz, E.; Mccall, A.; Faba, L.; Sastre, H.; Ordoñez, S. Trichloroethylene Hydrodechlorination in Water Using Formic Acid as Hydrogen Source: Selection of Catalyst and Operation Conditions. Environ. Prog. Sustain. Energy 2012, 32, 1217–1222. [Google Scholar] [CrossRef]
- Bonarowska, M.; Kaszkur, Z.; Kępiński, L.; Karpiński, Z. Hydrodechlorination of tetrachloromethane on alumina- and silica-supported platinum catalysts. Appl. Catal. B Environ. 2010, 99, 248–256. [Google Scholar] [CrossRef]
- Amorim, C.; Wang, X.; Keane, M.A. Application of Hydrodechlorination in Environmental Pollution Control: Comparison of the Performance of Supported and Unsupported Pd and Ni Catalysts. Chin. J. Catal. 2011, 32, 746–755. [Google Scholar] [CrossRef]
- Lan, L.; Liu, Y.; Liu, S.; Ma, X.; Li, X.; Dong, Z.; Xia, C. Effect of the supports on catalytic activity of Pd catalysts for liquid-phase hydrodechlorination/hydrogenation reaction. Environ. Technol. (UK) 2019, 40, 1615–1623. [Google Scholar] [CrossRef]
- Álvarez-Montero, M.A.; Gómez-Sainero, L.M.; Juan-Juan, J.; Linares-Solano, A.; Rodriguez, J.J. Gas-phase hydrodechlorination of dichloromethane with activated carbon-supported metallic catalysts. Chem. Eng. J. 2010, 162, 599–608. [Google Scholar] [CrossRef]
- Bueres, R.F.; Asedegbega-Nieto, E.; Díaz, E.; Ordóñez, S.; Díez, F.V. Performance of carbon nanofibres, high surface area graphites, and activated carbons as supports of Pd-based hydrodechlorination catalysts. Catal. Today 2010, 150, 16–21. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, M.; Zhang, C.; Ren, K.; Xin, Y.; Zhao, M.; Xing, E. Selectivity Control on Hydrogenation of Substituted Nitroarenes through End-On Adsorption of Reactants in Zeolite-Encapsulated Platinum Nanoparticles. Chem. Asian J. 2018, 13, 2077–2084. [Google Scholar] [CrossRef]
- Kamińska, I.I.; Lisovytskiy, D.; Casale, S.; Śrębowata, A.; Dzwigaj, S. Influence of preparation procedure on catalytic activity of PdBEA zeolites in aqueous phase hydrodechlorination of 1,1,2-trichloroethene. Microporous Mesoporous Mater. 2017, 237, 65–73. [Google Scholar] [CrossRef] [Green Version]
- Śrebowata, A.; Tarach, K.; Girman, V.; Góra-Marek, K. Catalytic removal of trichloroethylene from water over palladium loaded microporous and hierarchical zeolites. Appl. Catal. B Environ. 2016, 181, 550–560. [Google Scholar] [CrossRef]
- Śrębowata, A.; Kamińska, I.I.; Casale, S.; Brouri, D.; Calers, C.; Dzwigaj, S. The impact of the hydrodechlorination process on the physicochemical properties of bimetallic Ag-CuBeta zeolite catalysts. Microporous Mesoporous Mater. 2017, 243, 56–64. [Google Scholar] [CrossRef] [Green Version]
- Śrebowata, A.; Baran, R.; Łomot, D.; Lisovytskiy, D.; Onfroy, T.; Dzwigaj, S. Remarkable effect of postsynthesis preparation procedures on catalytic properties of Ni-loaded BEA zeolites in hydrodechlorination of 1,2-dichloroethane. Appl. Catal. B Environ. 2014, 147, 208–220. [Google Scholar] [CrossRef]
- Imre, B.; Hannus, I.; Kiricsi, I. Comparative IR spectroscopic study of Pt- and Pd-containing zeolites in the hydrodechlorination reaction of carbon tetrachloride. J. Mol. Struct. 2005, 744, 501–506. [Google Scholar] [CrossRef]
- Hannus, I.; Halász, J. Hydrodechlorination over Zeolite Supported Catalysts—Clarification of Reaction Mechanism. J. Jpn. Pet. Inst. 2006, 49, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Imre, B.; Kónya, Z.; Hannus, I.; Halász, J.; Nagy, J.B.; Kiricsi, I. Hydrodechlorination of chlorinated compounds on different zeolites. In Studies in Surface Science and Catalysis; Elsevier: Amsterdam, The Netherlands, 2002; Volume 142A, pp. 927–934. [Google Scholar]
- Elola, A.; Díaz, E.; Ordoñez, S. A new procedure for the treatment of organochlorinated off-gases combining adsorption and catalytic hydrodechlorination. Environ. Sci. Technol. 2009, 43, 1999–2004. [Google Scholar] [CrossRef]
- Fernandez-Ruiz, C.; Bedia, J.; Bonal, P.; Rodriguez, J.J.; Gómez-Sainero, L.M. Chloroform conversion into ethane and propane by catalytic hydrodechlorination with Pd supported on activated carbons from lignin. Catal. Sci. Technol. 2018, 8, 3926–3935. [Google Scholar] [CrossRef]
- Fernandez-Ruiz, C.; Bedia, J.; Andreoli, S.; Eser, S.; Rodriguez, J.J.; Gómez-Sainero, L.M. Selectivity to Olefins in the Hydrodechlorination of Chloroform with Activated Carbon-Supported Palladium Catalysts. Ind. Eng. Chem. Res. 2019, 58, 20592–20600. [Google Scholar] [CrossRef]
- Gómez-Sainero, L.M.; Palomar, J.; Omar, S.; Fernández, C.; Bedia, J.; Álvarez-Montero, A.; Rodriguez, J.J. Valorization of chloromethanes by hydrodechlorination with metallic catalysts. Catal. Today 2018, 310, 75–85. [Google Scholar] [CrossRef] [Green Version]
- Echeandia, S.; Pawelec, B.; Barrio, V.L.; Arias, P.L.; Cambra, J.F.; Loricera, C.V.; Fierro, J.L.G. Enhancement of phenol hydrodeoxygenation over Pd catalysts supported on mixed HY zeolite and Al2O3. An approach to O-removal from bio-oils. Fuel 2014, 117, 1061–1073. [Google Scholar] [CrossRef]
- Sato, K.; Nishimura, Y.; Matsubayashi, N.; Imamura, M.; Shimada, H. Structural changes of Y zeolites during ion exchange treatment: Effects of Si/Al ratio of the starting NaY. Microporous Mesoporous Mater. 2003, 59, 133–146. [Google Scholar] [CrossRef]
- Ma, Z.; Hu, H.; Sun, Z.; Fang, W.; Zhang, J.; Yang, L.; Zhang, Y.; Wang, L. Acidic Zeolite L as a Highly Efficient Catalyst for Dehydration of Fructose to 5-Hydroxymethylfurfural in Ionic Liquid. ChemSusChem 2017, 10, 1669–1674. [Google Scholar] [CrossRef]
- de Oliveira, A.M.; Baibich, I.M.; Machado, N.R.C.F.; Mignoni, M.L.; Pergher, S.B.C. Decomposition of nitric oxide on Pd-mordenite. Catal. Today 2008, 133–135, 560–564. [Google Scholar] [CrossRef]
- Sato, K.; Nishimura, Y.; Honna, K.; Matsubayashi, N.; Shimada, H. Role of HY zeolite mesopores in hydrocracking of heavy oils. J. Catal. 2001, 200, 288–297. [Google Scholar] [CrossRef]
- Sato, K.; Nishimura, Y.; Shimada, H. Preparation and activity evaluation of Y zeolites with or without mesoporosity. Catal. Lett. 1999, 60, 83–87. [Google Scholar] [CrossRef]
- Cano, M.; Guarín, F.; Aristizábal, B.; Villa, A.-L.; González, L.-M. Catalytic activity and stability of Pd/Co catalysts in simultaneous selective catalytic reduction of NOx with methane and oxidation of o -dichlorobenzene. Catal. Today 2017, 296, 105–117. [Google Scholar] [CrossRef]
- Lambrou, P.S.; Polychronopoulou, K.; Petallidou, K.C.; Efstathiou, A.M. Oxy-chlorination as an effective treatment of aged Pd/CeO2-Al2O3catalysts for Pd redispersion. Appl. Catal. B Environ. 2012, 111, 349–359. [Google Scholar] [CrossRef]
- Chandra Shekar, S.; Krishna Murthy, J.; Kanta Rao, P.; Rama Rao, K.S. Selective hydrogenolysis of dichlorodifluoromethane on carboncovered alumina supported palladium catalyst. Catal. Commun. 2003, 4, 39–44. [Google Scholar] [CrossRef]
- Bhogeswararao, S.; Srinivas, D. Catalytic conversion of furfural to industrial chemicals over supported Pt and Pd catalysts. J. Catal. 2015, 327, 65–77. [Google Scholar] [CrossRef]
- Feeley, J.S.; Sachtler, W.M.H. Palladium-enhanced reducibility of nickel in NaY. Zeolites 1990, 10, 738–745. [Google Scholar] [CrossRef]
- Seshu Babu, N.; Lingaiah, N.; Sai Prasad, P.S. Characterization and reactivity of Al2O3 supported Pd-Ni bimetallic catalysts for hydrodechlorination of chlorobenzene. Appl. Catal. B Environ. 2012, 111, 309–316. [Google Scholar] [CrossRef]
- McCusker, L.B.; Olson, D.H.; Baerlocher, C. Atlas of Zeolite Framework Types; Elsevier: Amsterdam, The Netherlands, 2007; ISBN 9780444530646. [Google Scholar]
- Velaga, B.; Parde, R.P.; Soni, J.; Peela, N.R. Synthesized hierarchical mordenite zeolites for the biomass conversion to levulinic acid and the mechanistic insights into humins formation. Microporous Mesoporous Mater. 2019, 287, 18–28. [Google Scholar] [CrossRef]
- Tangale, N.P.; Niphadkar, P.S.; Joshi, P.N.; Dhepe, P.L. Hierarchical K/LTL zeolite as solid base for aqueous phase hydrogenation of xylose to xylitol. Microporous Mesoporous Mater. 2019, 278, 70–80. [Google Scholar] [CrossRef]
- Treacy, M.M.J. Collection of Simulated XRD Powder Patterns for Zeolites; Elsevier Science: Amsterdam, The Netherlands, 2001. [Google Scholar]
- Pearce, H.A. Zeolite molecular sieves—Structure, chemistry and use. J. Chromatogr. A 1975, 106, 499. [Google Scholar] [CrossRef]
- Dantas Ramos, A.L.; da Silva Alves, P.; Aranda, D.A.G.; Schmal, M. Characterization of carbon supported palladium catalysts: Inference of electronic and particle size effects using reaction probes. Appl. Catal. A Gen. 2004, 277, 71–81. [Google Scholar] [CrossRef]
- Briggs, D.; Wanger, C.D.; Riggs, W.M.; Davis, L.E.; Moulder, J.F.; E. Muilenberg, G. Handbook of X-ray Photoelectron Spectroscopy; Chastain, J., Ed.; Perkin-Elmer Corporation, Physical Electronics Division: Eden Prairie, MN, USA, 1981. [Google Scholar]
- Gamero, M.; Aguayo, A.T.; Ateka, A.; Pérez-Uriarte, P.; Gayubo, A.G.; Bilbao, J. Role of Shape Selectivity and Catalyst Acidity in the Transformation of Chloromethane into Light Olefins. Ind. Eng. Chem. Res. 2015, 54, 7822–7832. [Google Scholar] [CrossRef]
- Bonarowska, M.; Kaszkur, Z.; Łomot, D.; Rawski, M.; Karpiński, Z. Effect of gold on catalytic behavior of palladium catalysts in hydrodechlorination of tetrachloromethane. Appl. Catal. B Environ. 2015, 162, 45–56. [Google Scholar] [CrossRef]
- Sánchez, C.A.G.; Patiño, C.O.M.; de Correa, C.M. Catalytic hydrodechlorination of dichloromethane in the presence of traces of chloroform and tetrachloroethylene. Catal. Today 2008, 133, 520–525. [Google Scholar] [CrossRef]
- Bedia, J.; Arevalo-Bastante, A.; Grau, J.M.; Dosso, L.A.; Rodriguez, J.J.; Mayoral, A.; Diaz, I.; Gómez-Sainero, L.M. Effect of the Pt-Pd molar ratio in bimetallic catalysts supported on sulfated zirconia on the gas-phase hydrodechlorination of chloromethanes. J. Catal. 2017, 352, 562–571. [Google Scholar] [CrossRef] [Green Version]
- Martin-Martinez, M.; Gómez-Sainero, L.M.; Alvarez-Montero, M.A.; Bedia, J.; Rodriguez, J.J. Comparison of different precious metals in activated carbon-supported catalysts for the gas-phase hydrodechlorination of chloromethanes. Appl. Catal. B Environ. 2013, 132, 256–265. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | ABET (m2g−1) | Vmicro (cm3g−1) | AEXT (m2g−1) | Vpore (cm3g−1) |
|---|---|---|---|---|
| KL | 145 | 0.075 | 28 | 0.109 |
| NaY | 342 | 0.177 | 67 | 0.287 |
| HMOR | 293 | 0.164 | 34 | 0.194 |
| HL | 98 | 0.043 | 33 | 0.094 |
| HY | 174 | 0.041 | 59 | 0.186 |
| Catalysts | TEM | NH3-TPD | ICP | |
|---|---|---|---|---|
| Mean Pd Particle Size (nm) | Dispersion (%) | Desorbed NH3 (mmolg−1) | Pd (%) | |
| KL | 6.3 | 18 | 0.37 | 1.02 |
| NaY | 4.2 | 26 | 1.49 | 0.97 |
| HMOR | 1.9 | 57 | 2.29 | 0.98 |
| HL | 1.4 | 79 | 1.75 | 0.99 |
| HY | 2.3 | 48 | 1.96 | 1.03 |
| Catalyst | Pd External Mass Content (%) | Pd0 (%) | Pdn+ (%) |
|---|---|---|---|
| KL | 0.38 | 72 | 28 |
| NaY | 0.67 | 95 | 5 |
| HMOR | 0.55 | 85 | 15 |
| HL | 0.27 | 87 | 13 |
| HY | 0.11 | 87 | 13 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandez-Ruiz, C.; Bedia, J.; Grau, J.M.; Romero, A.C.; Rodríguez, D.; Rodríguez, J.J.; Gómez-Sainero, L.M. Promoting Light Hydrocarbons Yield by Catalytic Hydrodechlorination of Residual Chloromethanes Using Palladium Supported on Zeolite Catalysts. Catalysts 2020, 10, 199. https://doi.org/10.3390/catal10020199
Fernandez-Ruiz C, Bedia J, Grau JM, Romero AC, Rodríguez D, Rodríguez JJ, Gómez-Sainero LM. Promoting Light Hydrocarbons Yield by Catalytic Hydrodechlorination of Residual Chloromethanes Using Palladium Supported on Zeolite Catalysts. Catalysts. 2020; 10(2):199. https://doi.org/10.3390/catal10020199
Chicago/Turabian StyleFernandez-Ruiz, Carlos, Jorge Bedia, Javier Mario Grau, Ana Clara Romero, Daniel Rodríguez, Juan José Rodríguez, and Luisa María Gómez-Sainero. 2020. "Promoting Light Hydrocarbons Yield by Catalytic Hydrodechlorination of Residual Chloromethanes Using Palladium Supported on Zeolite Catalysts" Catalysts 10, no. 2: 199. https://doi.org/10.3390/catal10020199