Baeyer-Villiger-Including Domino Two-Step Oxidations of β-O-Substituted Primary Alcohols: Reflection of the Migratory Aptitudes of O-Substituted Alkyl Group in the Outcome of the Reaction


Abstract
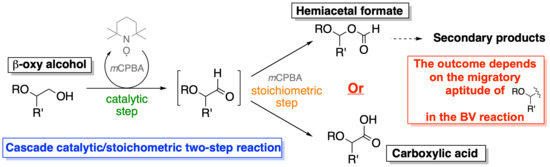
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Materials and Techniques
3.2. Synthesis of Substrates
3.2.1. Synthesis of [(2S,4S)-2-Phenyl-1,3-Dioxan-4-yl]Methanol (1b):
3.2.2. Synthesis of (5,5-Dimethyl-1,3-Dioxan-2-yl)Methanol (1e):
3.2.3. Synthesis of ((3ar,9as)-3a,4,9,9a-Tetrahydronaphtho[2,3-d][1,3]dioxol-2-yl)methanol (1f)
3.2.4. Synthesis of (4,4,5,5-Tetramethyl-1,3-dioxolan-2-yl)methanol (1g):
3.3. Oxidations and Characterization of Products
3.3.1. Typical Procedure for TEMPO/mCPBA Oxidation:
3.3.2. Oxidation of 2-Phenoxyethan-2-ol (1a):
3.3.3. Oxidation of [(2S,4S)-2-Phenyl-1,3-dioxan-4-yl]methanol (1b):
3.3.4. Oxidation of (2R,3R,4S,5R,6S)-2-(hydroxymethyl)-6-methoxytetrahydro-2H-pyran-3,4,5-triyl tribenzoate (1c):
3.3.5. Oxidation of (4R-cis)-6-Hydroxymethyl-2,2-dimethyl-1,3-dioxane-4-acetic acid 1,1-dimethylethyl ester (1d):
3.3.6. Oxidation of (5,5-Dimethyl-1,3-dioxan-2-yl)methanol (1e):
3.3.7. Oxidation of ((3aR,9aS)-3a,4,9,9a-Tetrahydronaphtho[2,3-d][1,3]dioxol-2-yl)methanol (1f):
3.3.8. Oxidation of (4,4,5,5-Tetramethyl-1,3-dioxolan-2-yl)methanol (1g):
3.3.9. Oxidation of Methyl 3,5-di-O-(2,4-dichlorobenzyl)- α-D-Ribofuranoside (1h):
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Notes
- Tojo, G.; Fernandez, M. Oxidation of Alcohols to Aldehydes and Ketones; Springer: New York, NY, USA, 2006. [Google Scholar]
- Krow, G.R. The Baeyer-Villiger oxidation of ketones and aldehydes. Org. React. 1993, 43, 251–798. [Google Scholar]
- ten Brink, G.J.; Arends, I.W.C.E.; Sheldon, R.A. The Baeyer-Villiger reaction: New developments toward greener procedures. Chem. Rev. 2004, 104, 4105–4123. [Google Scholar] [CrossRef]
- Strukul, G. Transition metal catalysis in the Baeyer-Villiger oxidation of ketones. Angew. Chem. Int. Ed. 1998, 37, 1199–1209. [Google Scholar] [CrossRef]
- Yaremenko, I.A.; Vil, V.A.; Demchuk, D.V.; Terent’ev, A.O. Rearrangements of organic peroxides and related processes. Beilstein. J. Org. Chem. 2016, 12, 1647–1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- For examples of oxidation of secondary alcohols to esters in two separate steps, see subsequent references 7-15, while for infrequent examples of one-pot oxidation of secondary alcohols to esters, see references 16-19.
- Tsang, R.; Fraser-Reid, B. Pyranose alpha-enones provide ready access to functionalized trans-decalins via bis-annulated pyranosides obtained by intramolecular Diels-Alder reactions—a key intermediate for Forskolin. J. Org. Chem. 1992, 57, 1065–1067. [Google Scholar] [CrossRef]
- Chida, N.; Tobe, T.; Ogawa, S. Regioselective Baeyer-Villiger reaction of polyhydroxycyclohexanone derivatives. Tetrahedron Lett. 1994, 35, 7249–7252. [Google Scholar] [CrossRef]
- Mecerreyes, D.; Atthoff, B.; Boduch, K.A.; Trollsas, M.; Hedrick, J.L. Unimolecular combination of an atom transfer radical polymerization initiator and a lactone monomer as a route to new graft copolymers. Macromolecules 1999, 32, 5175–5182. [Google Scholar] [CrossRef]
- Ishmuratov, G.Y.; Yakovleva, M.P.; Ganieva, V.A.; Gareeva, G.R.; Muslukhov, R.R.; Tolstikov, G.A. Synthesis of optically pure 3R-methylcyclopentan-1-one from L-(-)-menthol. Chem. Nat. Compd. 2005, 41, 549–551. [Google Scholar] [CrossRef]
- Rainbolt, E.A.; Washington, K.E.; Biewer, M.C.; Stefan, M.C. Towards smart polymeric drug carriers: Self-assembling γ-substituted polycaprolactones with highly tunable thermoresponsive behavior. J. Mater. Chem. B 2013, 1, 6532–6537. [Google Scholar] [CrossRef]
- Surnar, B.; Jayakannan, M. Stimuli-responsive poly(caprolactone) vesicles for dual drug delivery under the gastrointestinal tract. Biomacromolecules 2013, 14, 4377–4387. [Google Scholar] [CrossRef]
- Ercole, F.; Rodda, A.E.; Meagher, L.; Forsythe, J.S.; Dove, A.P. Surface grafted poly(ε-caprolactone) prepared using organocatalysed ring-opening polymerisation followed by SI-ATRP. Polym. Chem. 2014, 5, 2809–2815. [Google Scholar] [CrossRef]
- Yamauchi, S.; Nishimura, H.; Nishiwaki, H. Stereoselective syntheses of cryptocarya diacetate and all its stereoisomers in optically pure forms. Biosci. Biotechnol. Biochem. 2015, 79, 16–24. [Google Scholar] [CrossRef]
- Malhotra, M.; Surnar, B.; Jayakannan, M. Polymer topology driven enzymatic biodegradation in polycaprolactone block and random copolymer architectures for drug delivery to cancer cells. Macromolecules 2016, 49, 8098–8112. [Google Scholar] [CrossRef]
- Cella, J.A.; McGrath, J.P.; Kelley, J.A.; El Soukkary, O.; Hilpert, L. Applications of the peracid-mediated oxidation of alcohols. J. Org. Chem. 1977, 42, 2077–2080. [Google Scholar] [CrossRef]
- Chrobok, A. An efficient tandem oxidation of cyclohexanol to ε-caprolactone with peroxyacids and TEMPO catalyst in ionic liquids as solvents. Synlett 2011, 391–395. [Google Scholar] [CrossRef]
- Weisser, F.; Stevens, H.; Klein, J.; van der Meer, M.; Hohloch, S.; Sarkar, B. Tailoring Ru(II) pyridine/triazole oxygenation catalysts and using photoreactivity to probe their electronic properties. Chem. Eur. J. 2015, 21, 8926–8938. [Google Scholar] [CrossRef]
- Dijkmans, J.; Schutyser, W.; Dusselier, M.; Sels, B.F. Snβ-zeolite catalyzed oxido-reduction cascade chemistry with biomass-derived molecules. Chem. Commun. 2016, 52, 6712–6715. [Google Scholar] [CrossRef]
- Figadere, B.; Franck, X. Carboxylic Acids: Synthesis from alcohols. In Science of Synthesis; Panek, J.S., Ed.; Georg Thieme Verlag: Stuttgart, Germany, 2006; Volume 20a, pp. 173–204. [Google Scholar]
- For reactions of electron-rich benzaldehydes (Dakin reaction), see subsequent references 22–23, while for reactions of aldehydes with a nitrogen- or an oxygen-substituted α-carbon, see references 24–31.
- Hocking, M.B.; Bhandari, K.; Shell, B.; Smyth, T.A. Steric and pH effects on the rate of Dakin oxidation of acylphenols. J. Org. Chem. 1982, 47, 4208–4215. [Google Scholar] [CrossRef]
- Saikia, B.; Borah, P. A new avenue to the Dakin reaction in H2O2–WERSA. RSC Adv. 2015, 5, 105583–105586. [Google Scholar] [CrossRef]
- Alcaide, B.; Aly, M.F.; Sierra, M.A. Stereoselective synthesis of 3-substituted 4-(formyloxy)-2-azetidinones by the unusual Baeyer-Villiger reaction of beta-lactam aldehydes. Scope and synthetic applications. J. Org. Chem. 1996, 61, 8819–8825. [Google Scholar] [CrossRef]
- Deboer, A.; Ellwanger, R.E. Baeyer-Villiger oxidation of Δ1(9)-Octalone-2 and Δ1(8)-Indanone-2. J. Org. Chem. 1974, 39, 77–83. [Google Scholar] [CrossRef]
- Labadie, G.R.; Luna, L.E.; Gonzalez-Sierra, M.; Cravero, R.M. Synthesis of the tetracyclic bis(acetal) lactone portion of Saudin. Eur. J. Org. Chem. 2003, 3429–3434. [Google Scholar] [CrossRef]
- Jeso, V.; Iqbal, S.; Hernandez, P.; Cameron, M.D.; Park, H.; LoGrasso, P.V.; Micalizio, G.C. Synthesis of benzoquinone Ansamycin-inspired macrocyclic lactams from Shikimic acid. Angew. Chem. Int. Ed. 2013, 52, 4800–4804. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Frederick, M.O.; Burtoloso, A.C.B.; Denton, R.M.; Rivas, F.; Cole, K.P.; Aversa, R.J.; Gibe, R.; Umezawa, T.; Suzuki, T. Chemical synthesis of the GHIJKLMNO ring system of maitotoxin. J. Am. Chem. Soc. 2008, 130, 7466–7476. [Google Scholar] [CrossRef]
- Chaubet, G.; Bourgeois, D.; Périgaud, C. Synthetic studies towards new nucleoside analogues: Preparation of (±)-1′,4′-dimethyladenosine. Eur. J. Org. Chem. 2011, 319–326. [Google Scholar] [CrossRef]
- Himmelbauer, M.; Farcet, J.B.; Gagnepain, J.; Mulzer, J. Palladium-catalyzed carbo-oxygenation: The Bielschowskysin case. Org. Lett. 2013, 15, 3098–3101. [Google Scholar] [CrossRef]
- Urabe, F.; Nagashima, S.; Takahashi, K.; Ishihara, J.; Hatakeyama, S. Total synthesis of (-)-Cinatrin C1 based on an In(OTf)3-catalyzed Conia-ene reaction. J. Org. Chem. 2013, 78, 3847–3857. [Google Scholar] [CrossRef]
- Targel, T.; Ramesh, P.; Portnoy, M. Domino two-step oxidation of β-alkoxy alcohols to hemiacetal esters: Linking a stoichiometric step to an organocatalytic step with a common organic oxidant. Eur. J. Org. Chem. 2018, 23, 3017–3021. [Google Scholar] [CrossRef]
- For cases when such transformation was conducted in two successive, but separate, steps, see refs. 7,8.
- For similar comparison of some of these migratory aptitudes in ketones, see ref. 8.
- The endocyclic character of the alkoxyalkyl should not be a factor in this comparison, since the related 3-hydroxytetrahydrofurane undergoes a rapid cascade reaction with the insertion of oxygen near this group.
- Jimenez, F.; del Carmen Cruz, M.; Zuniga, C.; Martinez, M.A.; Chamorro, G.; Diza, F.; Tamariz, J. Aryloxyacetic esters structurally related to α-Asarone as potential antifungal agents. Med. Chem. Res. 2010, 19, 33–57. [Google Scholar] [CrossRef]
- 1b was prepared from a triol precursor by trans-acetalization with benzaldehyde dimethylacetal, see Materials and Methods.
- 38. See, for instance: Xie, Y.; Zhang, J.; Tian, G.; Xu, M.; Hu, T.; Jiang, X.; Shen, J. A neighboring group participation strategy: Facile synthesis of 3,5-di-O-benzoyl-2-C-methyl-d-arabino-γ-lactone. Tetrahedron Lett. 2015, 56, 4345–4348. [Google Scholar] [CrossRef]
- In order to drive the reaction to completion, excess of mCPBA was applied.
- Lehtinen, C.; Nevalainen, V.; Brunow, G. Experimental and computational studies on solvent effects in reactions of peracid-aldehyde adducts. Tetrahedron 2001, 57, 4741–4751. [Google Scholar] [CrossRef]
- Ogata, Y.; Sawaki, Y. Kinetics of the Baeyer-Villiger reaction of benzaldehydes with perbenzoic acid in aquoorganic solvents. J. Org. Chem. 1969, 34, 3985–3991. [Google Scholar] [CrossRef]
- Flogel, O.; Okala Amombo, M.G.; Reissig, H.U.; Zahn, G.; Brudgam, I.; Hartl, H. A stereoselective and short total synthesis of the polyhydroxylated γ-amino acid (-)-detoxinine, based on stereoselective preparation of dihydropyrrole derivatives from lithiated alkoxyallenes. Chem. Eur. J. 2003, 9, 1405–1415. [Google Scholar] [CrossRef]
- Miyazawa, S.; Shinoda, M.; Kawahara, T.; Watanabe, N.; Harada, H.; Iida, D.; Terauchi, H.; Nagakawa, J.; Fujisaki, H.; Kubota, A.; et al. Preparation of Benzimidazole Derivatives as Gastric Acid Secretion Inhibitors. U.S. Patent Appl. Publ. US 2007/0010542 A1, 11 January 2007. [Google Scholar]
- Huo, C.; Yang, H.; Cui, Q.C.; Dou, Q.P.; Chan, T.H. Proteasome inhibition in human breast cancer cells with high catechol-O-methyltransferase activity by green tea polyphenol EGCG analogs. Bioorg. Med. Chem. 2010, 18, 1252–1258. [Google Scholar] [CrossRef] [Green Version]
- Lei, P.-s.; Ogawa, Y.; Kovac, P. New N-acylating reagent derived from 3-deoxy-L-glycero-tetronic acid. J. Carbohydr. Chem. 1996, 15, 485–500. [Google Scholar] [CrossRef]
- Esmurziev, A.M.; Reimers, A.; Andreassen, T.; Simic, N.; Sundby, E.; Hoff, B.H. Benzoylated uronic acid building blocks and synthesis of N-uronate conjugates of Lamotrigine. Molecules 2012, 17, 820–835. [Google Scholar] [CrossRef] [Green Version]
- 47. For characterization of 3d, see: Edwards, J.T.; Merchant, R.R.; McClymont, K.S.; Knouse, K.W.; Qin, T.; Malins, L.R.; Vokits, B.; Shaw, S.A.; Bao, D.H.; Wei, F.-L.; et al. Decarboxylative alkenylation. Nature 2017, 545, 213–219. [Google Scholar]
- For characterization of 4e, see: Filliatre, C.; Brigand, G.; Lalande, R. Peroxidation of oxygenated heterocyclic compounds. Bull. Soc. Chim. Fr. 1971, 170–176. [Google Scholar]
- Goosen, A.; McCleland, C.W. Reaction of 1,3-dioxolans with iodine monochloride: The scope and mechanism of formation of 1,3-dioxolan-2-ylium dichloroiodates(I). J. Chem. Soc. Perkin Trans. I 1981, 977–983. [Google Scholar] [CrossRef]
- Suzuki, M.; Sugai, T. Mechanistic studies on nitrosation–deaminocyclization of mono-carbamoylated vicinal amino alcohols and diols: A new preparative in situ formation of ethanediazo hydroxide for the ethylation of carboxylates under mild conditions. Bull. Chem. Soc. Jpn. 2004, 77, 1217–1227. [Google Scholar] [CrossRef]
- Pihlaja, K.; Rossi, K. Conformational analysis. Part XLIII. 13C Chemical shifts and coupling constants as proof of the nonplanarity of the 2-oxo-1,3-dioxalane ring. Acta Chem. Scand. B, 1977; 31, 899–902. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Substrate | Time (h) | Consumption (%) | Products (Product Ratio) |
|---|---|---|---|---|
| 1 |  | 3 | 100 |  (1: 0.4) (1: 0.4) |
| 2 |  | 17 | 82 |  |
| 3 |  | 3 | 100 |  |
| 4 |  | 3 | 87 |  (1: 6.7) (1: 6.7) |
| 5 |  | 1 | 100 |  |
| 6 |  | 1 | 100 |  |
| 7 |  | 1 | 100 |  |
| 8 |  | 4 | 100 |  |
| Entry | Substrate | Solvent | Consumption (%) | Product Ratio 2 |
|---|---|---|---|---|
| 1 | 1i | benzene | 100 | 14:1 |
| 2 | 1i | DCM | 100 | 11:1 |
| 3 3 | 1i | EtOAc | 47 | 10:1 |
| 4 | 1i | ACN | 96 | 3.1:1 |
| 5 | 1d | benzene | 86 | 1:4.9 |
| 6 | 1d | DCM | 87 | 1:6.7 |
| 7 3 | 1d | EtOAc | 79 | 1:24 |
| 8 | 1d | ACN | 100 | Acid only |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Targel, T.; Portnoy, M. Baeyer-Villiger-Including Domino Two-Step Oxidations of β-O-Substituted Primary Alcohols: Reflection of the Migratory Aptitudes of O-Substituted Alkyl Group in the Outcome of the Reaction. Catalysts 2020, 10, 1275. https://doi.org/10.3390/catal10111275
Targel T, Portnoy M. Baeyer-Villiger-Including Domino Two-Step Oxidations of β-O-Substituted Primary Alcohols: Reflection of the Migratory Aptitudes of O-Substituted Alkyl Group in the Outcome of the Reaction. Catalysts. 2020; 10(11):1275. https://doi.org/10.3390/catal10111275
Chicago/Turabian StyleTargel, Tom, and Moshe Portnoy. 2020. "Baeyer-Villiger-Including Domino Two-Step Oxidations of β-O-Substituted Primary Alcohols: Reflection of the Migratory Aptitudes of O-Substituted Alkyl Group in the Outcome of the Reaction" Catalysts 10, no. 11: 1275. https://doi.org/10.3390/catal10111275