Genome-Wide SNP Signal Intensity Scanning Revealed Genes Differentiating Cows with Ovarian Pathologies from Healthy Cows

 ,
,
Abstract
:1. Introduction
2. Material and Methods
2.1. Ethical Issues
2.2. Animal Samples and DNA Extraction
2.3. Post-Mortem Detection of Ovarian Pathologies and Genotyping
2.4. Signal Intensity Normalization
2.5. Gene Selection for the Study
2.6. Identification of Copy Number Variations
2.7. Principal Components Analysis
2.8. Haplotype Inference and Haplotype Blocks
2.9. Correction for Multiple Testing
3. Results
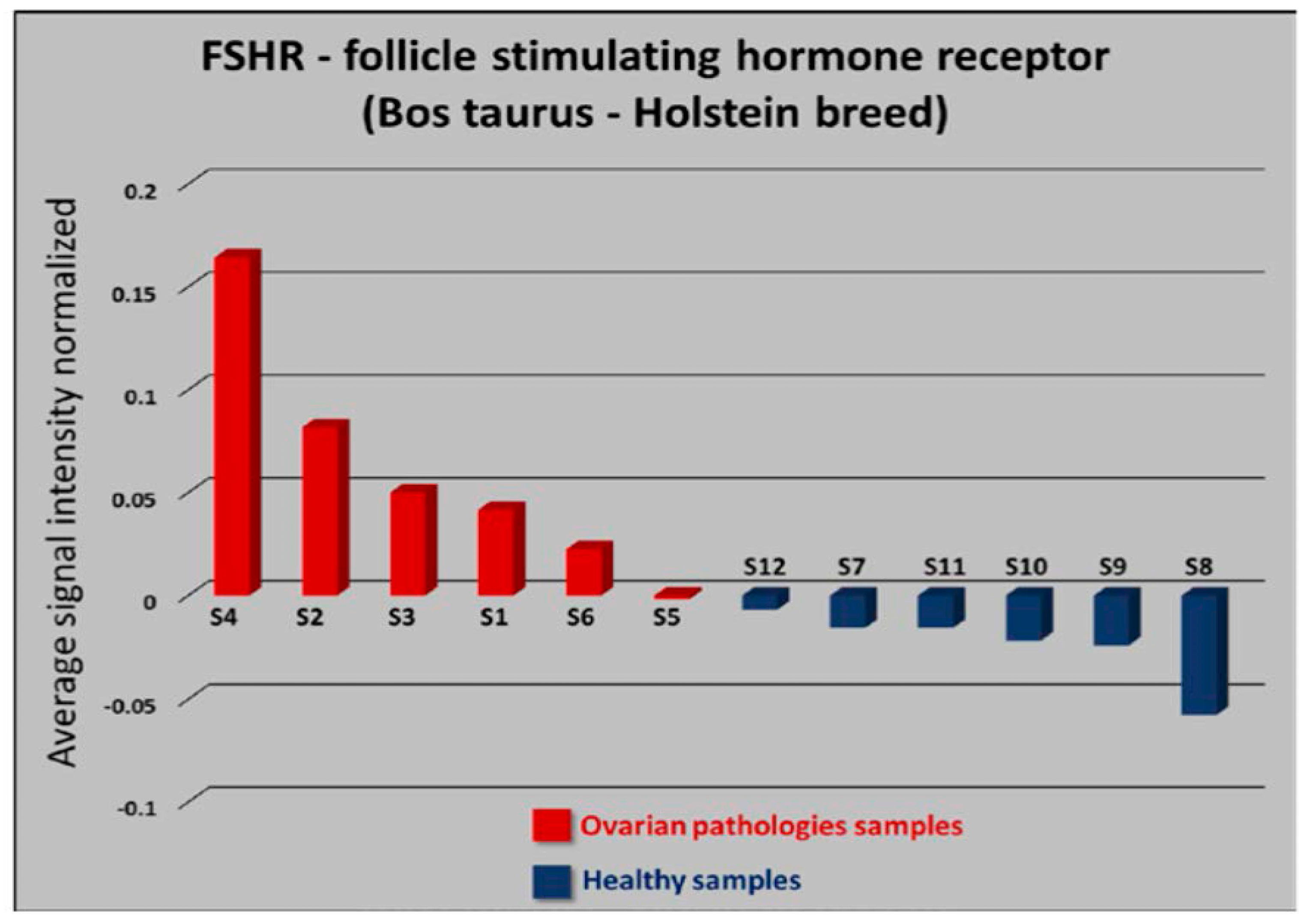
3.1. Differentiation between Disease and Healthy Samples
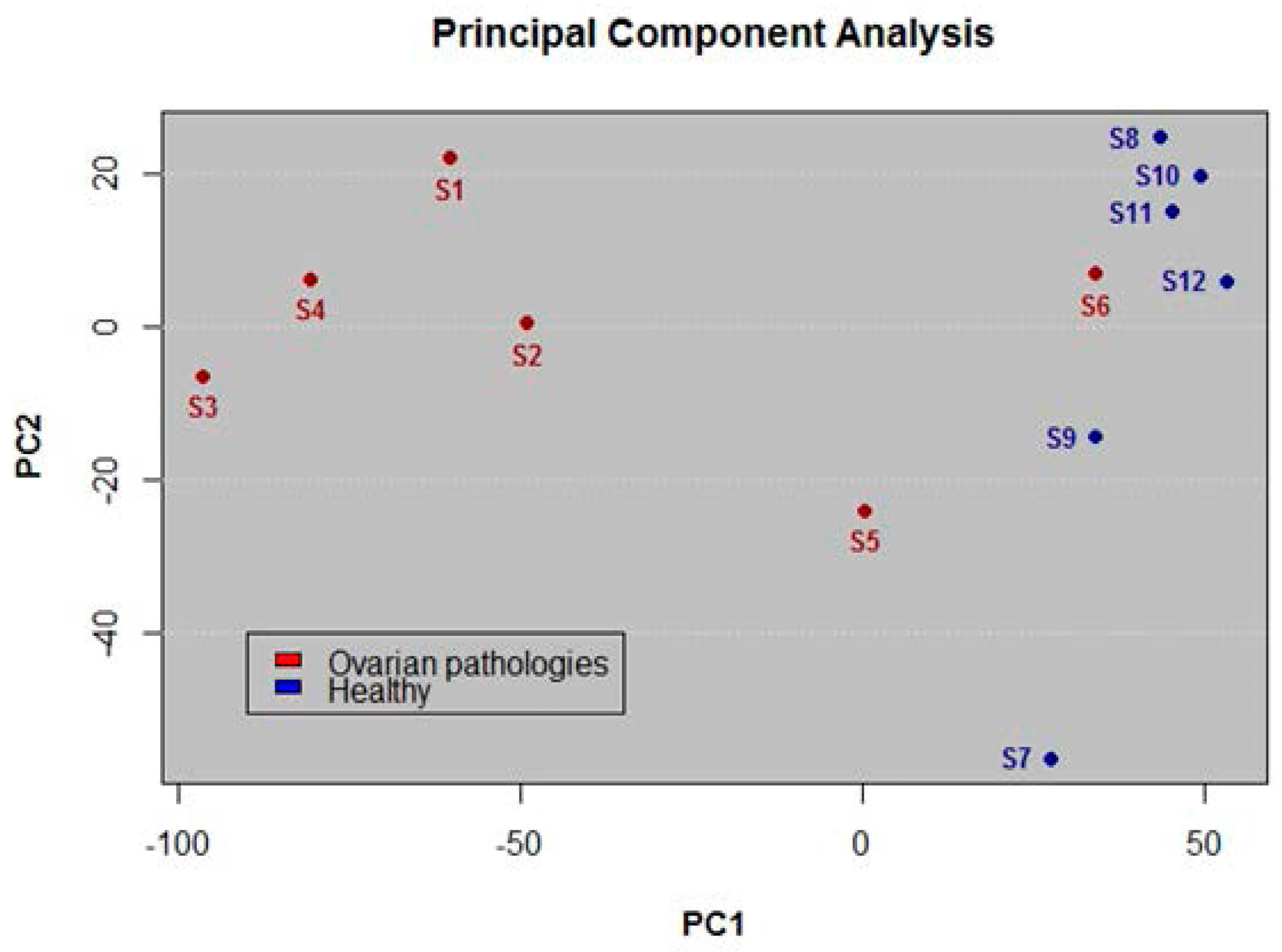
3.2. Application of PCA for Differentiation
3.3. Searching for Association between CNVs and COFs
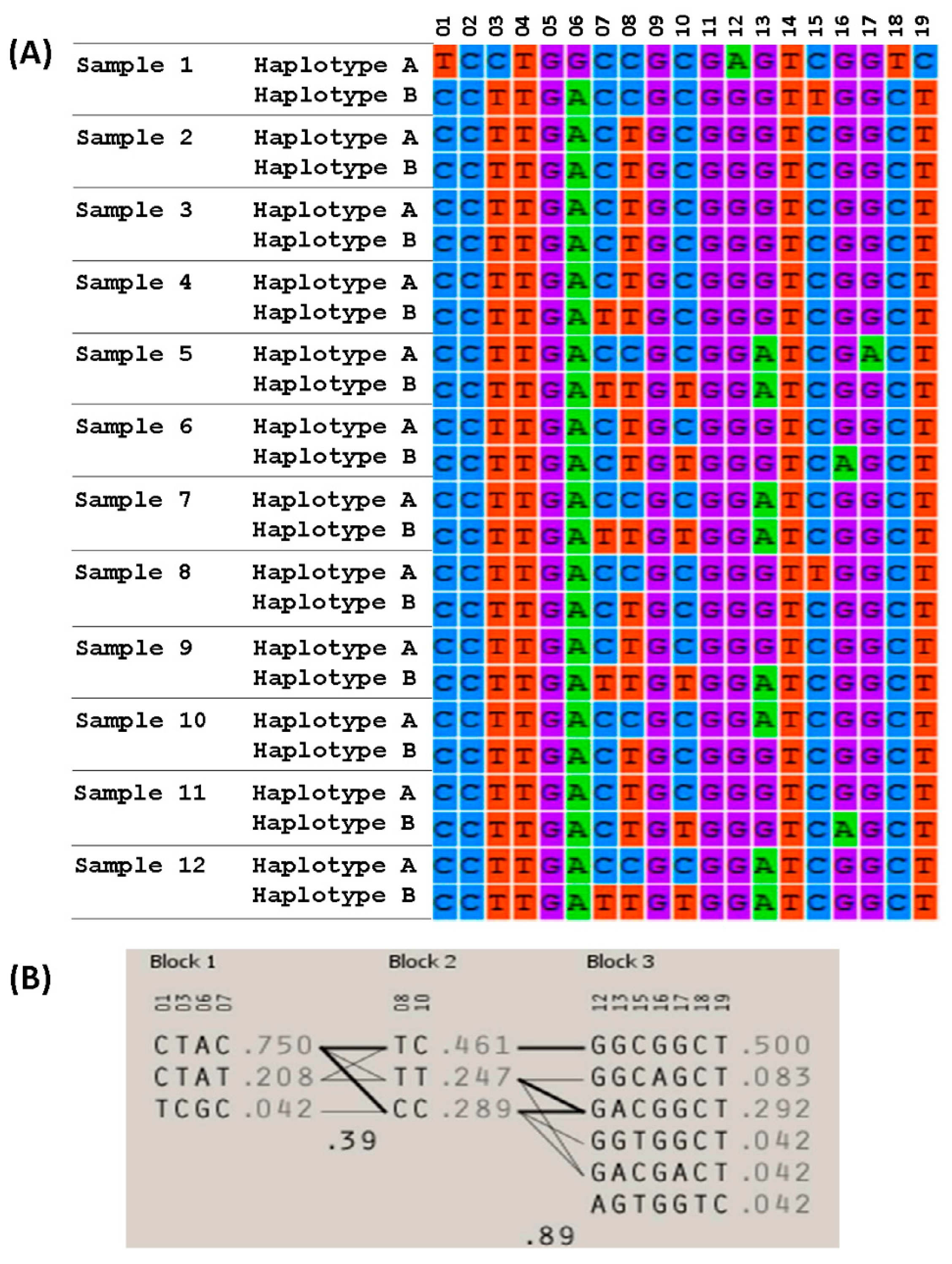
3.4. Searching for Association between Haplotypes and COFs
4. Discussion
5. Conclusions
Data Access
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- McEntee, K. Reproductive Pathology of Domestic Mammals, 1st ed.; Academic Press, Inc.: New York, NY, USA, 1990; p. 401. [Google Scholar]
- Bartolome, J.A.; Thatcher, W.W.; Melendez, P.; Risco, C.A.; Archbald, L.F. Strategies for the diagnosis and treatment of ovarian cysts in dairy cattle. J. Am. Vet. Med. Assoc. 2005, 9, 1409–1414. [Google Scholar] [CrossRef]
- Zulu, V.C.; Peeny, C. Risk factors of cystic ovarian disease in dairy cattle. J. Reprod. Physiol. 1998, 44, 191–195. [Google Scholar] [CrossRef]
- Dobson, H.; Ribadu, A.Y.; Noble, K.M.; Tebble, J.E.; Ward, W.R. Ultrasonography and hormone profiles of adrenocorticotrophic hormone (ACTH)-induced persistent ovarian follicles (cysts) in cattle. J. Reprod. Fertil. 2000, 2, 405–410. [Google Scholar]
- Peter, A.T. An update on cystic ovarian degeneration in cattle. Reprod. Domest. Anim. 2004, 1, 1–7. [Google Scholar] [CrossRef]
- Salvetti, N.R.; Rey, F.; Ortega, H.H. Enfermedad quistica ovarica bovina. Rev. FAVE Cienc. Vet. 2007, 6, 1–2. [Google Scholar] [CrossRef]
- Casida, L.E.; Chapman, A.B. Factors affecting the incidence of cystic ovaries in a herd of Holstein cows. J. Dairy 1951, 12, 1200–1205. [Google Scholar] [CrossRef]
- Kesler, D.J.; Garverick, H.A.; Caudle, A.B.; Elmore, R.G.; Youngquist, R.S.; Bierschwal, C.J. Reproductive hormone and ovarian changes in cows with ovarian cysts. J. Dairy 1980, 63, 166–170. [Google Scholar] [CrossRef]
- Laporte, H.M.; Hogeveen, H.; Schukken, Y.H.; Noordhuizen, J.P.T.M. Cystic ovarian disease in Dutch dairy cattle, I- Incidence, risk-factors and consequences. Livest. Prod. Sci. 1994, 38, 191–197. [Google Scholar] [CrossRef]
- Matukumalli, L.K.; Schroeder, S.; DeNise, S.K.; Sonstegard, T.; Lawley, C.T.; Georges, M. Analyzing LD Blocks and CNV Segments in Cattle: Novel Genomic Features Identified Using the BovineHD BeadChip; Pub No. 370-2011-002; Illumina Inc.: San Diego, CA, USA, 2011. [Google Scholar]
- Salomon-Torres, R.; Matukumalli, L.K.; Van Tassell, C.P.; Villa-Angulo, C.; Gonzalez-Vizcarra, V.M.; Villa-Angulo, R. High Density LD-Based Structural Variations Analysis in Cattle Genome. PLoS ONE 2014, 9, e103046. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Liu, G.E.; Bickhart, D.M.; Cardone, M.F.; Wang, K.; Kim, E.S.; Matukumalli, L.K.; Ventura, M.; Song, J.; VanRaden, P.M.; et al. Genomic characteristics of cattle copy number variations. BMC Genom. 2011, 12, 127. [Google Scholar] [CrossRef] [PubMed]
- Peiffer, D.A.; Le, J.M.; Steemers, F.J.; Chang, W.; Jenniges, T.; Garcia, F.; Haden, K.; Li, J.; Shaw, C.A.; Belmont, J.; et al. High-resolution genomic profiling of chromosomal aberrations using Infinium whole-genome genotyping. Genome Res. 2006, 9, 1136–1148. [Google Scholar] [CrossRef] [PubMed]
- Sisson, S.; Grossman, J.D.; Getty, R. The Anatomy of the Domestic Animals, 5th ed.; W B Saunders Co.: Philadelphia, PA, USA, 1975; 2130p. [Google Scholar]
- Wang, K.; Li, M.; Hadley, D.; Liu, R.; Glessner, J.; Grant, S.F.A.; Hakonarson, H.; Bucan, M. PennCNV: An integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007, 11, 1665–1674. [Google Scholar] [CrossRef] [PubMed]
- Rincon, G.; Weber, K.L.; Eenennaam, A.L.; Golden, B.L.; Medrano, J.F. Hot topic: Performance of bovine high-density genotyping platforms in Holsteins and Jerseys. J. Dairy Sci. 2011, 12, 6116–6121. [Google Scholar] [CrossRef] [PubMed]
- Colella, S.; Yau, C.; Taylor, J.M.; Mirza, G.; Butler, H.; Clouston, P.; Bassett, A.S.; Seller, A.; Holmes, C.C.; Ragoussis, J. QuantiSNP: An Objective Bayes Hidden-Markov Model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res. 2007, 6, 2013–2025. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.E.; Hou, Y.; Zhu, B.; Cardone, M.F.; Jiang, L.; Cellamare, A.; Mitra, A.; Alexander, L.J.; Coutinho, L.L.; Dell’Aquila, M.E.; et al. Analysis of copy number variations among diverse cattle breeds. Genome Res. 2010, 5, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Bickhart, D.M.; Hou, Y.; Schroeder, S.G.; Alkan, C.; Cardone, M.F.; Matukumalli, L.K.; Song, J.; Schnabel, R.D.; Ventura, M.; Taylor, J.F.; et al. Copy number variation of individual cattle genomes using next-generation sequencing. Genome Res. 2012, 4, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Bickhart, D.M.; Hvinden, M.L.; Li, C.; Song, J.; Boichard, D.A.; Fritz, S.; Eggen, A.; DeNise, S.; Wiggans, G.R.; et al. Fine mapping of copy number variations on two cattle genome assemblies using high density SNP array. BMC Genom. 2012, 13, 376. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Liu, G.E.; Bickhart, D.M.; Matukumalli, L.K.; Li, C.; Song, J.; Gasbarre, L.C.; Van Tassell, C.P.; Sonstegard, T.S. Genomic regions showing copy number variations associate with resistance or susceptibility to gastrointestinal nematodes in Angus cattle. Funct. Integr. Genom. 2012, 1, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Jiang, J.; Wang, J.; Ding, X.; Liu, J.; Zhang, Q. Genome-wide identification of copy number variations in Chinese Holstein. PLoS ONE 2012, 11, e48732. [Google Scholar] [CrossRef] [PubMed]
- Cicconardi, F.; Chillemi, G.; Tramontano, A.; Marchitelli, C.; Valentini, A.; Ajmone-Marsan, P.; Nardone, A. Massive screening of copy number population-scale variation in Bos taurus genome. BMC Genom. 2013, 14, 124. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Jiang, J.; Yang, J.; Liu, X.; Wang, J.; Wang, H.; Ding, X.; Liu, J.; Zhang, Q. Genome-wide detection of copy number variations using high-density SNP genotyping platforms in Holsteins. BMC Genom. 2013, 14, 131. [Google Scholar] [CrossRef] [PubMed]
- Jolliffe, I.T. Principal Component Analysis, 2nd ed.; Springer: New York, NY, USA, 2002; p. 488. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2015. [Google Scholar]
- Stephens, M.; Smith, N.J.; Donnelly, P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 2001, 4, 978–989. [Google Scholar] [CrossRef] [PubMed]
- Stephens, M.; Donnelly, P. A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am. J. Hum. Genet. 2003, 5, 1162–1169. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 2, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Drai, D.; Elmer, G.; Kafkafi, N.; Golani, I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 2001, 2, 279–284. [Google Scholar] [CrossRef]
- Webb, R.; Armstrong, D.G. Control of ovarian function; effect of local interactions and environmental influences on follicular turnover in cattle: A review. Livest. Prod. Sci. 1998, 53, 95–112. [Google Scholar] [CrossRef]
- Hunter, M.G.; Robinson, R.S.; Mann, G.E.; Webb, R. Endocrine and paracrine control of follicular development and ovulation rate in farm species. Anim. Reprod. Sci. 2004, 83, 461–477. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, T.; Opsomer, G.; de Kruif, A. Aetiology and pathogenesis of cystic ovarian follicles in dairy cattle: A review. Reprod. Nutr. Dev. 2006, 2, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Furumichi, M.; Tanabe, M.; Hirakawa, M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010, 38, D355–D360. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Entry | Name | KEGG Genes | Total Genes | Coverage Genes | Non-Significant Genes | Significant Genes |
|---|---|---|---|---|---|---|
| bta04020 | Calcium signaling pathway | 189 | 62 | 32.8% | 39 | 23 |
| bta04024 | cAMP signaling pathway | 199 | 59 | 29.64% | 39 | 20 |
| bta04080 | Neuroactive ligand-receptor interaction | 303 | 61 | 20.13% | 46 | 15 |
| bta04913 | Ovarian steroidogenesis | 52 | 19 | 36.53% | 9 | 10 |
| bta04917 | Prolactin signaling pathway | 76 | 19 | 25% | 12 | 7 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salomón-Torres, R.; Montaño-Gómez, M.F.; Villa-Angulo, R.; González-Vizcarra, V.M.; Villa-Angulo, C.; Medina-Basulto, G.E.; Ortiz-Uribe, N.; Mahadevan, P.; Yaurima-Basaldúa, V.H. Genome-Wide SNP Signal Intensity Scanning Revealed Genes Differentiating Cows with Ovarian Pathologies from Healthy Cows. Sensors 2017, 17, 1920. https://doi.org/10.3390/s17081920
Salomón-Torres R, Montaño-Gómez MF, Villa-Angulo R, González-Vizcarra VM, Villa-Angulo C, Medina-Basulto GE, Ortiz-Uribe N, Mahadevan P, Yaurima-Basaldúa VH. Genome-Wide SNP Signal Intensity Scanning Revealed Genes Differentiating Cows with Ovarian Pathologies from Healthy Cows. Sensors. 2017; 17(8):1920. https://doi.org/10.3390/s17081920
Chicago/Turabian StyleSalomón-Torres, Ricardo, Martin F. Montaño-Gómez, Rafael Villa-Angulo, Víctor M. González-Vizcarra, Carlos Villa-Angulo, Gerardo E. Medina-Basulto, Noé Ortiz-Uribe, Padmanabhan Mahadevan, and Víctor H. Yaurima-Basaldúa. 2017. "Genome-Wide SNP Signal Intensity Scanning Revealed Genes Differentiating Cows with Ovarian Pathologies from Healthy Cows" Sensors 17, no. 8: 1920. https://doi.org/10.3390/s17081920