Complex History of Codiversification and Host Switching of a Newfound Soricid-Borne Orthohantavirus in North America

Abstract
:1. Introduction
2. Materials and Methods
2.1. Specimens
2.2. RNA Extraction, cDNA Synthesis, and RT-PCR Amplification
2.3. Sequence Dataset
2.4. Phylogenetic Analysis
2.5. Tanglegrams, Diversity Analyses, Codiversification Tests, and Reconciliation
3. Results
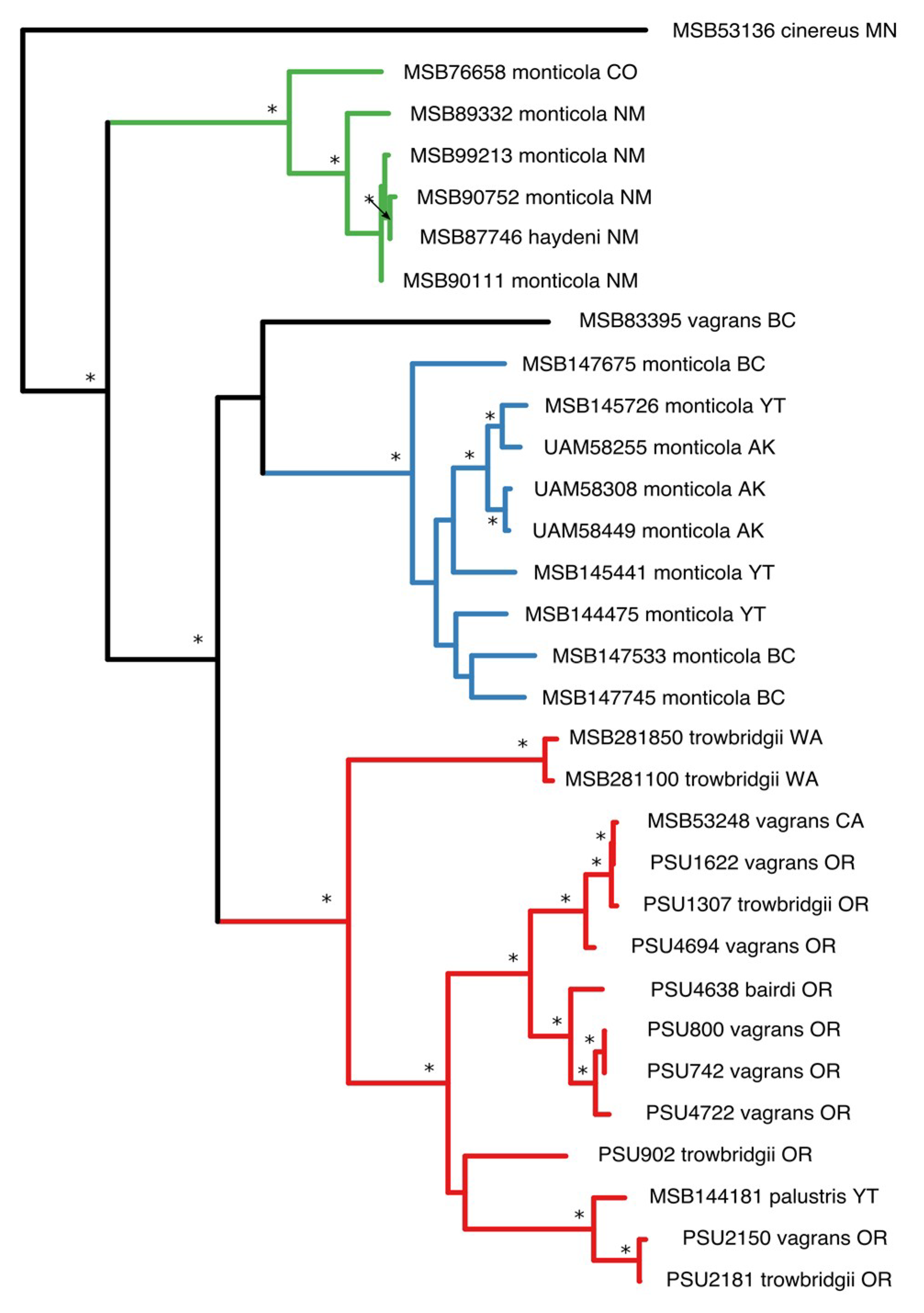
3.1. Phylogenetic Analysis
3.2. Population Demographics
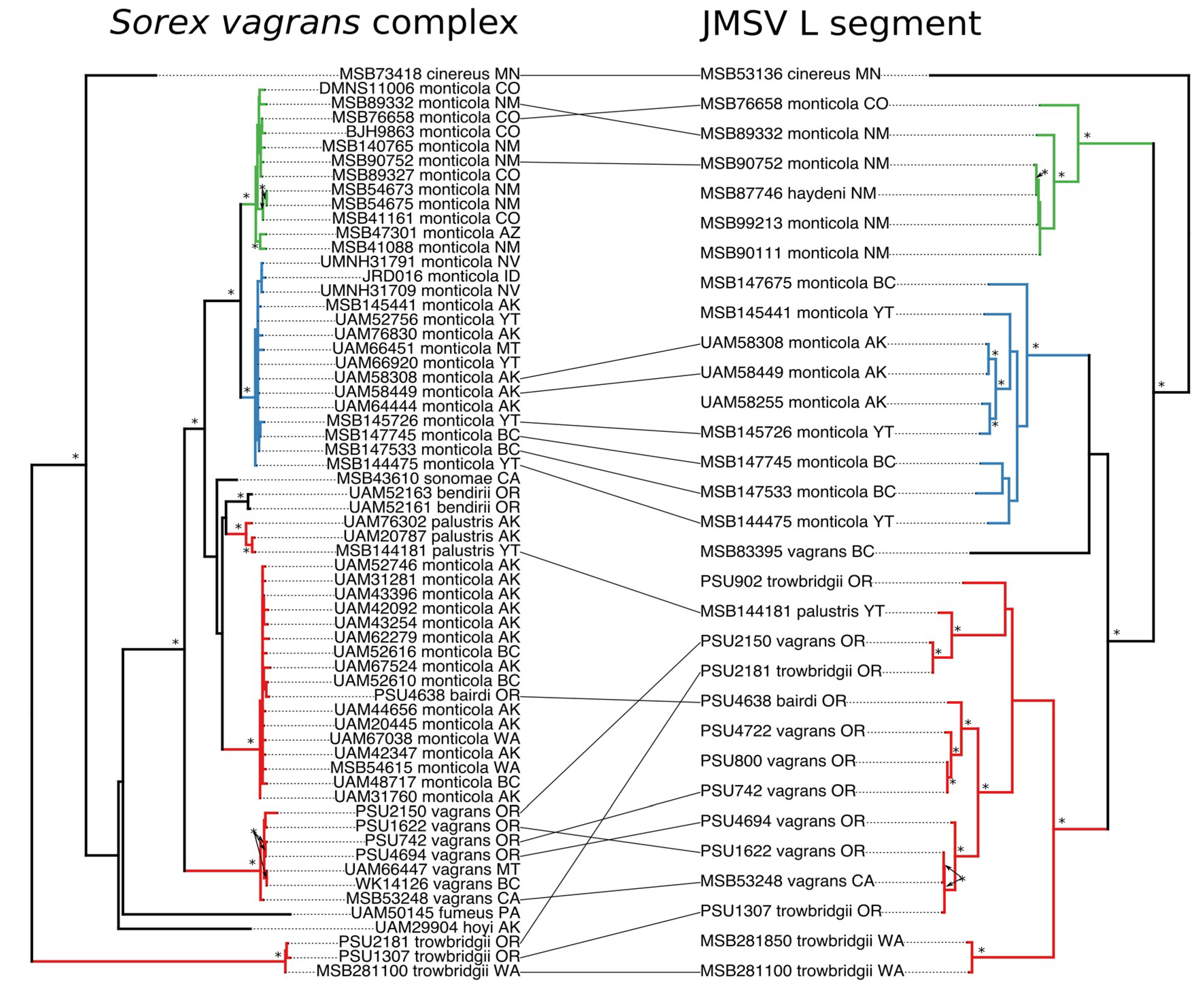
3.3. Cophylogeny Tanglegrams
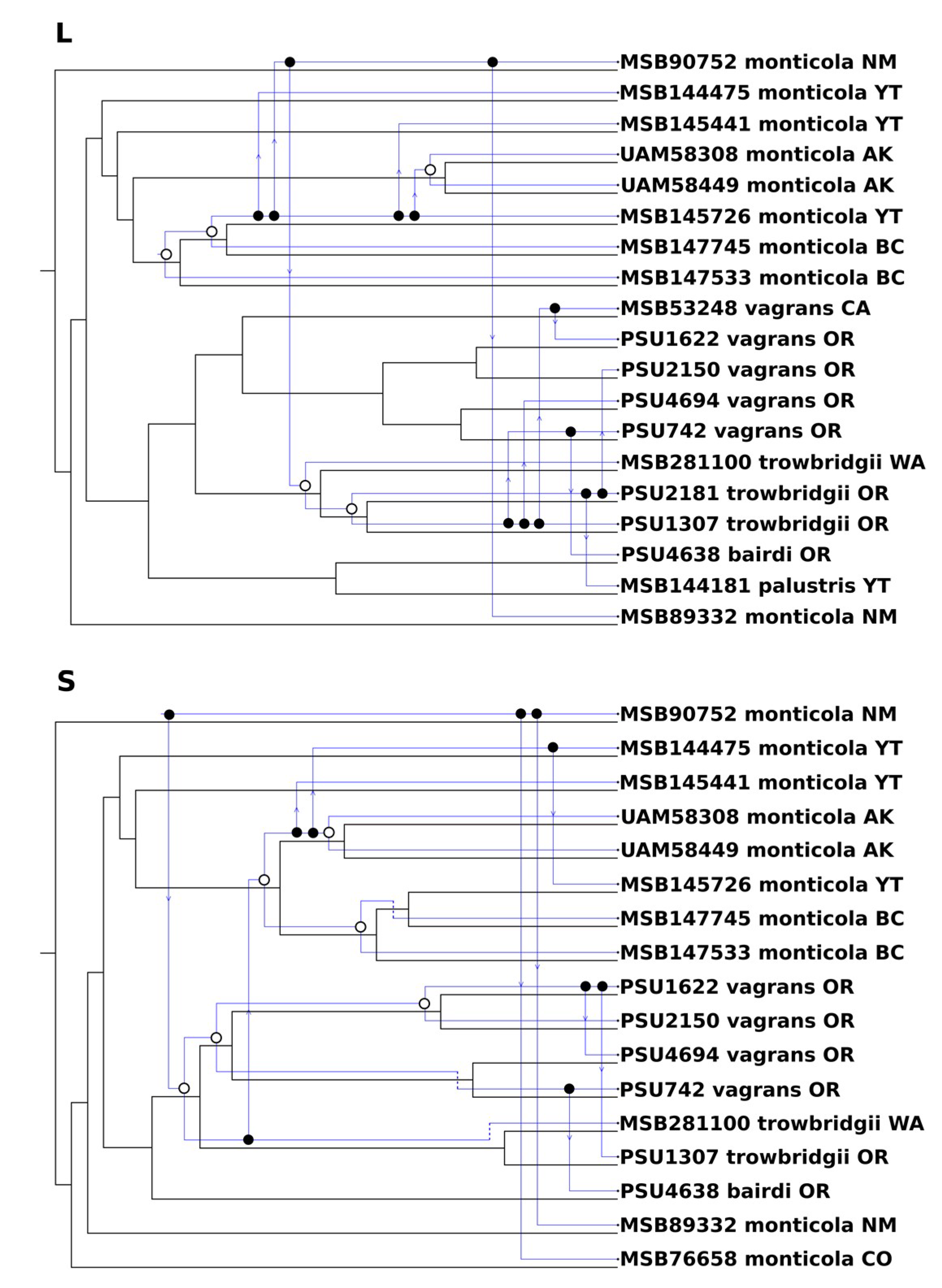
3.4. Codivergence and Phylogenetic Reconciliation
4. Discussion
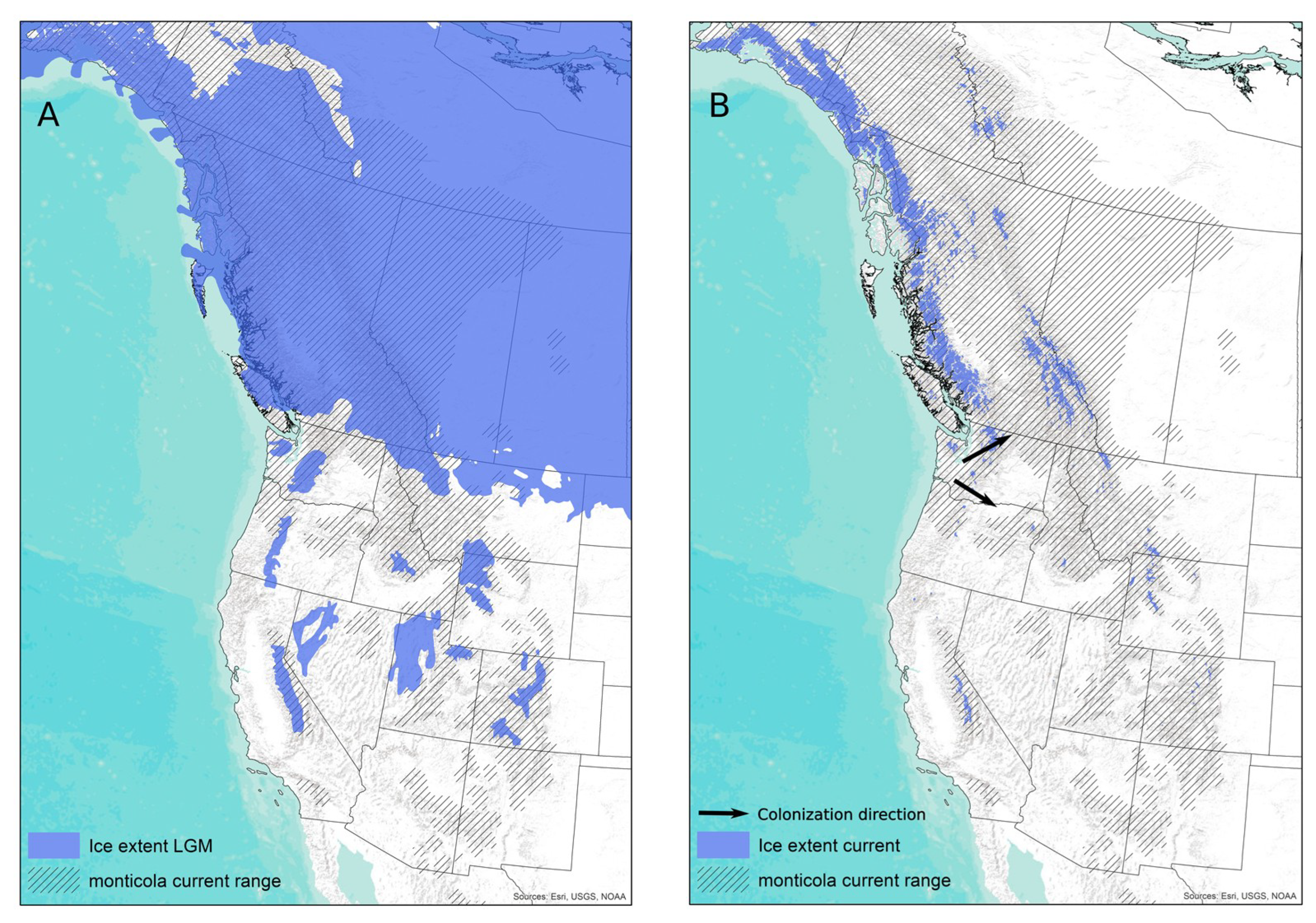
4.1. Codiversification Processes
4.2. Viral Reassortment
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brooks, D.R.; Ferrao, A.L. The historical biogeography of co-evolution: Emerging infectious diseases are evolutionary accidents waiting to happen. J. Biogeogr. 2005, 32, 1291–1299. [Google Scholar] [CrossRef]
- Geoghegan, J.L.; Holmes, E.C. Predicting virus emergence amid evolutionary noise. Open Biol. 2017, 7, 170189. [Google Scholar] [CrossRef] [PubMed]
- Nieberding, C.; Morand, S.; Libois, R.; Michaux, J.R. A parasite reveals cryptic phylogeographic history of its host. Proc. R. Soc. B Biol. Sci. 2004, 271, 2559–2568. [Google Scholar] [CrossRef] [PubMed]
- Araujo, S.B.L.; Braga, M.P.; Brooks, D.R.; Agosta, S.J.; Hoberg, E.P.; Von Hartenthal, F.W.; Boeger, W.A. Undestanding host-switching by ecological fitting. PLoS ONE 2015, 10, e0139225. [Google Scholar] [CrossRef] [PubMed]
- Geoghegan, J.L.; Duchêne, S.; Holmes, E.C. Comparative analysis estimates the relative frequencies of co-divergence and cross-species transmission within viral families. PLoS Pathog. 2017, 13, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Maes, P.; Adkins, S.; Alkhovsky, S.V.; Avšič-Županc, T.; Ballinger, M.J.; Bente, D.A.; Beer, M.; Bergeron, É.; Blair, C.D.; Briese, T.; et al. Taxonomy of the order Bunyavirales: Second update 2018. Arch. Virol. 2019, 164, 927–941. [Google Scholar] [CrossRef] [PubMed]
- Torres-Pérez, F.; Palma, R.E.; Hjelle, B.; Holmes, E.C.; Cook, J.A. Spatial but not temporal co-divergence of a virus and its mammalian host. Mol. Ecol. 2011, 20, 4109–4122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemirov, K.; Henttonen, H.; Vaheri, A.; Plyusnin, A. Phylogenetic evidence for host switching in the evolution of hantaviruses carried by Apodemus mice. Virus Res. 2002, 90, 207–215. [Google Scholar] [CrossRef]
- Kang, H.J.; Bennett, S.N.; Dizney, L.; Sumibcay, L.; Arai, S.; Ruedas, L.A.; Song, J.W.; Yanagihara, R. Host switch during evolution of a genetically distinct hantavirus in the American shrew mole (Neurotrichus gibbsii). Virology 2009, 388, 8–14. [Google Scholar] [CrossRef]
- Klempa, B. Reassortment events in the evolution of hantaviruses. Virus Genes 2018, 54, 638–646. [Google Scholar] [CrossRef] [Green Version]
- Arai, S.; Gu, S.H.; Baek, L.J.; Tabara, K.; Bennett, S.N.; Oh, H.S.; Takada, N.; Kang, H.J.; Tanaka-Taya, K.; Morikawa, S.; et al. Divergent ancestral lineages of newfound hantaviruses harbored by phylogenetically related crocidurine shrew species in Korea. Virology 2012, 424, 99–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briese, T.; Calisher, C.H.; Higgs, S. Viruses of the family Bunyaviridae: Are all available isolates reassortants? Virology 2013, 446, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Trifonov, V.; Khiabanian, H.; Rabadan, R. Geographic dependence, surveillance, and origins of the 2009 influenza A (H1N1) virus. N. Engl. J. Med. 2009, 361, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Arai, S.; Bennett, S.N.; Sumibcay, L.; Cook, J.A.; Song, J.W.; Hope, A.; Parmenter, C.; Nerurkar, V.R.; Yates, T.L.; Yanagihara, R. Short report: Phylogenetically distinct hantaviruses in the masked shrew (Sorex cinereus) and dusky shrew (Sorex monticolus) in the United States. Am. J. Trop. Med. Hyg. 2008, 78, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Hennings, D.; Hoffmann, R.S. A review of the taxonomy of the Sorex vagrans species complex from western North America. Occas. Pap. Museum Nat. Hist. Univ. Kansas. 1977, 68, 1–35. [Google Scholar] [CrossRef]
- Demboski, J.R.; Cook, J.A. Phylogeography of the dusky shrew, Sorex monticolus (Insectivora, Soricidae): Insight into deep and shallow history in northwestern North America. Mol. Ecol. 2001, 10, 1227–1240. [Google Scholar] [CrossRef] [PubMed]
- Hope, A.G.; Panter, N.; Cook, J.A.; Talbot, S.L.; Nagorsen, D.W. Multilocus phylogeography and systematic revision of North American water shrews (genus: Sorex). J. Mammal. 2014, 95, 722–738. [Google Scholar] [CrossRef]
- Ling, J.; Smura, T.; Tamarit, D.; Huitu, O.; Voutilainen, L.; Henttonen, H.; Vaheri, A.; Vapalahti, O.; Sironen, T. Evolution and postglacial colonization of Seewis hantavirus with Sorex araneus in Finland. Infect. Genet. Evol. 2018, 57, 88–97. [Google Scholar] [CrossRef]
- Yashina, L.N.; Abramov, S.A.; Gutorov, V.V.; Dupal, T.A.; Krivopalov, A.V.; Panov, V.V.; Danchinova, G.A.; Vinogradov, V.V.; Luchnikova, E.M.; Hay, J.; et al. Seewis virus: Phylogeography of a shrew-borne hantavirus in Siberia, Russia. Vector-Borne Zoonotic Dis. 2010, 10, 585–591. [Google Scholar] [CrossRef]
- Kang, H.J.; Arai, S.; Hope, A.G.; Song, J.W.; Cook, J.A.; Yanagihara, R. Genetic diversity and phylogeography of Seewis virus in the Eurasian common shrew in Finland and Hungary. Virol. J. 2009, 6, 208. [Google Scholar] [CrossRef]
- Gu, S.H.; Hejduk, J.; Markowski, J.; Kang, H.J.; Markowski, M.; Połatyńska, M.; Sikorska, B.; Liberski, P.P.; Yanagihara, R. Co-circulation of soricid- and talpid-borne hantaviruses in Poland. Infect. Genet. Evol. 2014, 28, 296–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ling, J.; Sironen, T.; Voutilainen, L.; Hepojoki, S.; Niemimaa, J.; Isoviita, V.M.; Vaheri, A.; Henttonen, H.; Vapalahti, O. Hantaviruses in Finnish soricomorphs: Evidence for two distinct hantaviruses carried by Sorex araneus suggesting ancient host-switch. Infect. Genet. Evol. 2014, 27, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Asikainen, K.; Hanninen, T.; Henttonen, H.; Niemimaa, J.; Laakkonen, J.; Andersen, H.K.; Bille, N.; Leirs, H.; Vaheri, A.; Plyusnin, A. Molecular evolution of Puumala hantavirus in Fennoscandia: Phylogenetic analysis of strains from two recolonization routes, Karelia and Denmark. J. Gen. Virol. 2000, 81, 2833–2841. [Google Scholar] [CrossRef] [PubMed]
- Nemirov, K.; Leirs, H.; Lundkvist, A.; Olsson, G.E. Puumala hantavirus and Myodes glareolus in northern Europe: No evidence of co-divergence between genetic lineages of virus and host. J. Gen. Virol. 2010, 91, 1262–1274. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.C. The phylogeography of human viruses. Mol. Ecol. 2004, 13, 745–756. [Google Scholar] [CrossRef]
- Dizney, L.J.; Ruedas, L.A. Increased host species diversity and decreased prevalence of Sin Nombre virus. Emerg. Infect. Dis. 2009, 15, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- GenBank. Available online: https://www.ncbi.nlm.nih.gov/genbank/ (accessed on 2 March 2019).
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Geneious. Available online: https://www.geneious.com/ (accessed on 26 September 2016).
- Bruen, T.C.; Philippe, H.; Bryant, D. A simple and robust statistical test for detecting the presence of recombination. Genetics 2006, 172, 2665–2681. [Google Scholar] [CrossRef]
- Jakobsen, I.B.; Easteal, S. A program for calculating and displaying compatibility matrices as an aid in determining reticulate evolution in molecular sequences. Bioinformatics 2007, 12, 291–295. [Google Scholar] [CrossRef]
- Maynard, J.S. Analyzing the mosaic structure of genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar]
- Bruen, T. PhiPack: PHI test and other tests of recombination. McGill Univ. Montr. Quebec 2005, 1–8. [Google Scholar]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- R, version 3.6.0; A Language and Envionrment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019.
- Paradis, E. Pegas: An R package for population genetics with an integrated–modular approach. Bioinformatics 2010, 26, 419–420. [Google Scholar] [CrossRef] [PubMed]
- Lessa, E.P.; Cook, J.A.; Patton, J.L. Genetic footprints of demographic expansion in North America, but not Amazonia, during the Late Quaternary. Proc. Natl. Acad. Sci. 2003, 100, 10331–10334. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Ho, S.Y.W.; Duchêne, S.; Duchêne, D. Simulating and detecting autocorrelation of molecular evolutionary rates among lineages. Mol. Ecol. Resour. 2015, 15, 688–696. [Google Scholar] [CrossRef]
- Critchlow, D.E.; Pearl, D.K.; Qian, C. The triples distance for rooted bifurcating phylogenetic trees. Syst. Biol. 1996, 45, 323–334. [Google Scholar] [CrossRef]
- Kuhner, M.K.; Yamato, J. Practical performance of tree comparison metrics. Syst. Biol. 2015, 64, 205–214. [Google Scholar] [CrossRef]
- Avino, M.; Ng, G.T.; He, Y.; Renaud, M.S.; Jones, B.R.; Poon, A.F.Y. Tree shape-based approaches for the comparative study of cophylogeny. Ecol. Evol. 2019, ece3.5185. [Google Scholar] [CrossRef]
- Conow, C.; Fielder, D.; Ovadia, Y.; Libeskind-Hadas, R. Jane: A new tool for the cophylogeny reconstruction problem. Algorithms Mol. Biol. 2010, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Revell, L.J. phytools: An R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 2012, 3, 217–223. [Google Scholar] [CrossRef]
- Bennett, S.N.; Gu, S.H.; Kang, H.J.; Arai, S.; Yanagihara, R. Reconstructing the evolutionary origins and phylogeography of hantaviruses. Trends Microbiol. 2014, 22, 473–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, D.R.; Hoberg, E.P.; Boeger, W.A.; Gardner, S.L.; Galbreath, K.E.; Herczeg, D.; Mejía-Madrid, H.H.; Rácz, S.E.; Dursahinhan, A.T. Finding them before they find us: informatics, parasites, and environments in accelerating climate change. Comp. Parasitol. 2014, 81, 155–164. [Google Scholar] [CrossRef]
- Sawyer, Y.E.; MacDonald, S.O.; Lessa, E.P.; Cook, J.A. Living on the edge: Exploring the role of coastal refugia in the Alexander Archipelago of Alaska. Ecol. Evol. 2019, 1777–1797. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Z.; Holmes, E.C. What is the time-scale of hantavirus evolution? Infect. Genet. Evol. 2014, 25, 144–145. [Google Scholar] [CrossRef]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef]
- Raup, B.; Racoviteanu, A.; Khalsa, S.J.S.; Helm, C.; Armstrong, R.; Arnaud, Y. The GLIMS geospatial glacier database: A new tool for studying glacier change. Glob. Planet. Change 2007, 56, 101–110. [Google Scholar] [CrossRef]
- Razzauti, M.; Plyusnina, A.; Henttonen, H.; Plyusnin, A. Accumulation of point mutations and reassortment of genomic RNA segments are involved in the microevolution of Puumala hantavirus in a bank vole (Myodes glareolus) population. J. Gen. Virol. 2008, 89, 1649–1660. [Google Scholar] [CrossRef]
- Razzauti, M.; Plyusnina, A.; Sironen, T.; Henttonen, H.; Pyusnin, A. Analysis of Puumala hantavirus in a bank vole population in northern Finland: Evidence for co-circulation of two genetic lineages and frequent reassortment between strains. J. Gen. Virol. 2009, 90, 1923–1931. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Hu, J.; Wang, Z.-X.X.; Wang, D.-M.M.; Yu, C.; Zhou, J.-Z.Z.; Fu, Z.F.; Zhang, Y.-Z.Z. Genetic characterization of hantaviruses isolated from Guizhou, China: Evidence for spillover and reassortment in nature. J. Med. Virol. 2008, 80, 1033–1041. [Google Scholar] [CrossRef] [PubMed]
- Black, W.C.; Doty, J.B.; Hughes, M.T.; Beaty, B.J.; Calisher, C.H. Temporal and geographic evidence for evolution of Sin Nombre virus using molecular analyses of viral RNA from Colorado, New Mexico and Montana. Virol. J. 2009, 6, 102. [Google Scholar] [CrossRef] [PubMed]
- Laenen, L.; Vergote, V.; Kafetzopoulou, L.E.; Wawina, T.B.; Vassou, D.; Cook, J.A.; Hugot, J.P.; Deboutte, W.; Kang, H.J.; Witkowski, P.T.; et al. A novel hantavirus of the European mole, Bruges virus, is involved in frequent Nova virus coinfections. Genome Biol. Evol. 2018, 10, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.D.; Chen, X.; Tian, J.H.; Chen, L.J.; Li, K.; Wang, W.; Eden, J.S.; Shen, J.J.; Liu, L.; et al. The evolutionary history of vertebrate RNA viruses. Nature 2018, 556, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Souza, W.M.; Bello, G.; Amarilla, A.A.; Alfonso, H.L.; Aquino, V.H.; Figueiredo, L.T.M. Phylogeography and evolutionary history of rodent-borne hantaviruses. Infect. Genet. Evol. 2014, 21, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.P.; Lin, X.D.; Wang, W.; Tian, J.H.; Cong, M.L.; Zhang, H.L.; Wang, M.R.; Zhou, R.H.; Wang, J.B.; Li, M.H.; et al. Phylogeny and origins of hantaviruses harbored by bats, insectivores, and rodents. PLoS Pathog. 2013, 9, e1003159. [Google Scholar] [CrossRef]
- Castel, G.; Tordo, N.; Plyusnin, A. Estimation of main diversification time-points of hantaviruses using phylogenetic analyses of complete genomes. Virus Res. 2017, 233, 60–69. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Country | State | Year | RT-PCR Positive/Tested |
|---|---|---|---|---|
| Sorex monticola | Canada | British Columbia | 2006 | 3/10 |
| Yukon Territory | 2004 | 0/1 | ||
| Yukon Territory | 2005 | 3/17 | ||
| Yukon Territory | 2006 | 0/1 | ||
| USA | Alaska | 1998 | 0/1 | |
| Alaska | 2001 | 3/9 | ||
| Alaska | 2002 | 0/7 | ||
| Alaska | 2005 | 0/6 | ||
| Colorado | 1994 | 1/1 | ||
| New Mexico | 1995 | 0/1 | ||
| New Mexico | 1996 | 2/5 | ||
| New Mexico | 1998 | 1/4 | ||
| New Mexico | 2000 | 1/4 | ||
| Utah | 1994 | 0/1 | ||
| Utah | 1997 | 0/1 | ||
| Sorex palustris | Canada | Yukon Territory | 2005 | 1/4 |
| USA | New Mexico | 1998 | 0/3 | |
| New Mexico | 2007 | 0/2 | ||
| Wyoming | 2007 | 0/1 | ||
| Sorex trowbridgii | USA | Washington | 1996 | 2/3 |
| Oregon | 2003 | 2/24 | ||
| Oregon | 2004 | 1/8 | ||
| Sorex vagrans | Canada | British Columbia | 1996 | 1/9 |
| USA | California | 1983 | 1/3 | |
| New Mexico | 1994 | 0/1 | ||
| New Mexico | 1996 | 0/1 | ||
| Oregon | 1994 | 0/3 | ||
| Oregon | 2003 | 3/17 | ||
| Oregon | 2004 | 1/5 | ||
| Oregon | 2005 | 2/6 | ||
| Sorex bairdi | USA | Oregon | 2005 | 1/1 |
| Sorex bendirii | USA | Oregon | 2003 | 0/1 |
| Oregon | 2005 | 0/2 | ||
| Sorex haydeni | USA | New Mexico | 1996 | 1/1 |
| Total | 30/164 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liphardt, S.W.; Kang, H.J.; Dizney, L.J.; Ruedas, L.A.; Cook, J.A.; Yanagihara, R. Complex History of Codiversification and Host Switching of a Newfound Soricid-Borne Orthohantavirus in North America. Viruses 2019, 11, 637. https://doi.org/10.3390/v11070637
Liphardt SW, Kang HJ, Dizney LJ, Ruedas LA, Cook JA, Yanagihara R. Complex History of Codiversification and Host Switching of a Newfound Soricid-Borne Orthohantavirus in North America. Viruses. 2019; 11(7):637. https://doi.org/10.3390/v11070637
Chicago/Turabian StyleLiphardt, Schuyler W., Hae Ji Kang, Laurie J. Dizney, Luis A. Ruedas, Joseph A. Cook, and Richard Yanagihara. 2019. "Complex History of Codiversification and Host Switching of a Newfound Soricid-Borne Orthohantavirus in North America" Viruses 11, no. 7: 637. https://doi.org/10.3390/v11070637