Modeling of Effective Antimicrobials to Reduce Staphylococcus aureus Virulence Gene Expression Using a Two-Compartment Hollow Fiber Infection Model


Abstract
:1. Introduction
2. Results
2.1. Growth Curve of the USA300 Strain in HFM in the Presence of Five Individual Antimicrobials
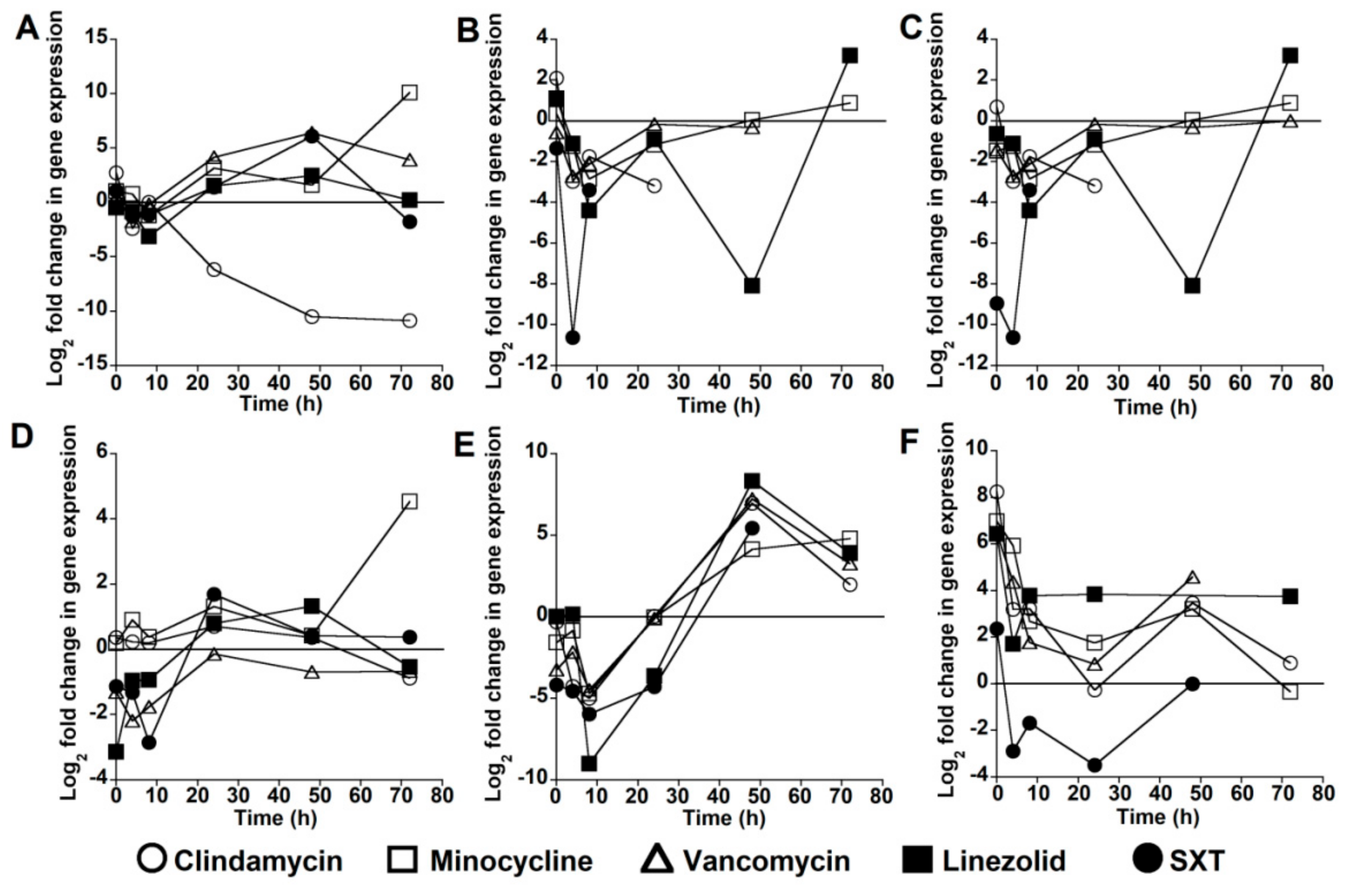
2.2. Virulence Gene Expression after Antibiotic Exposure
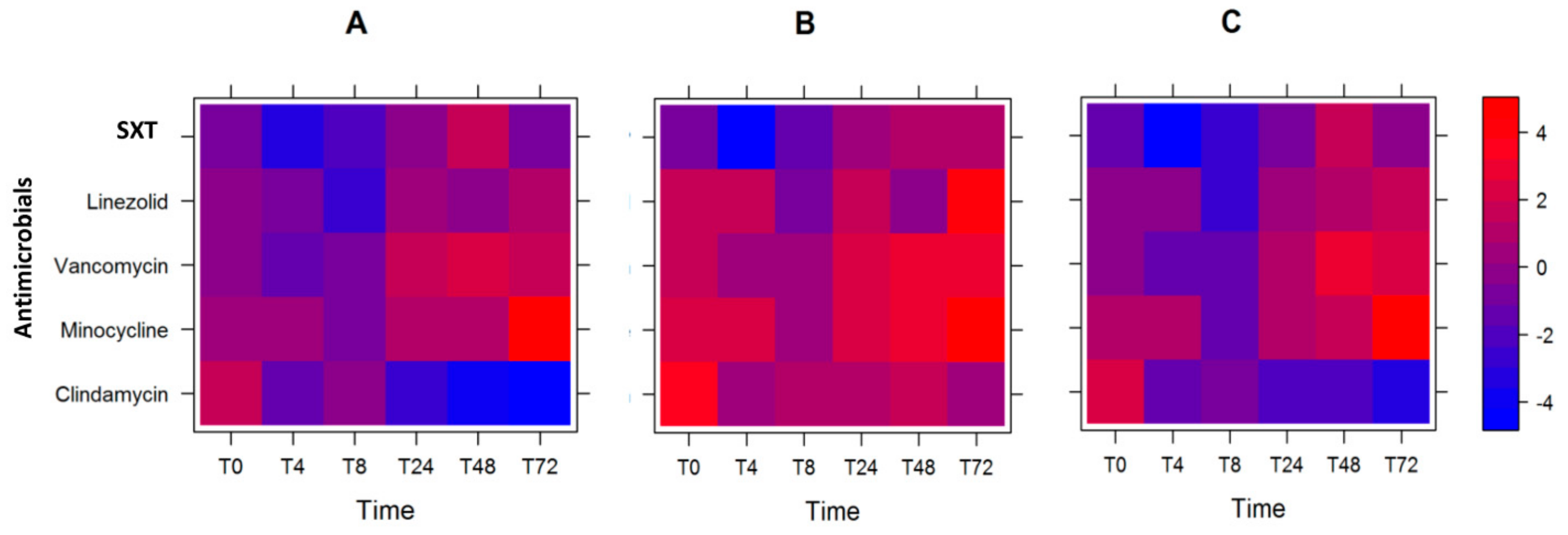
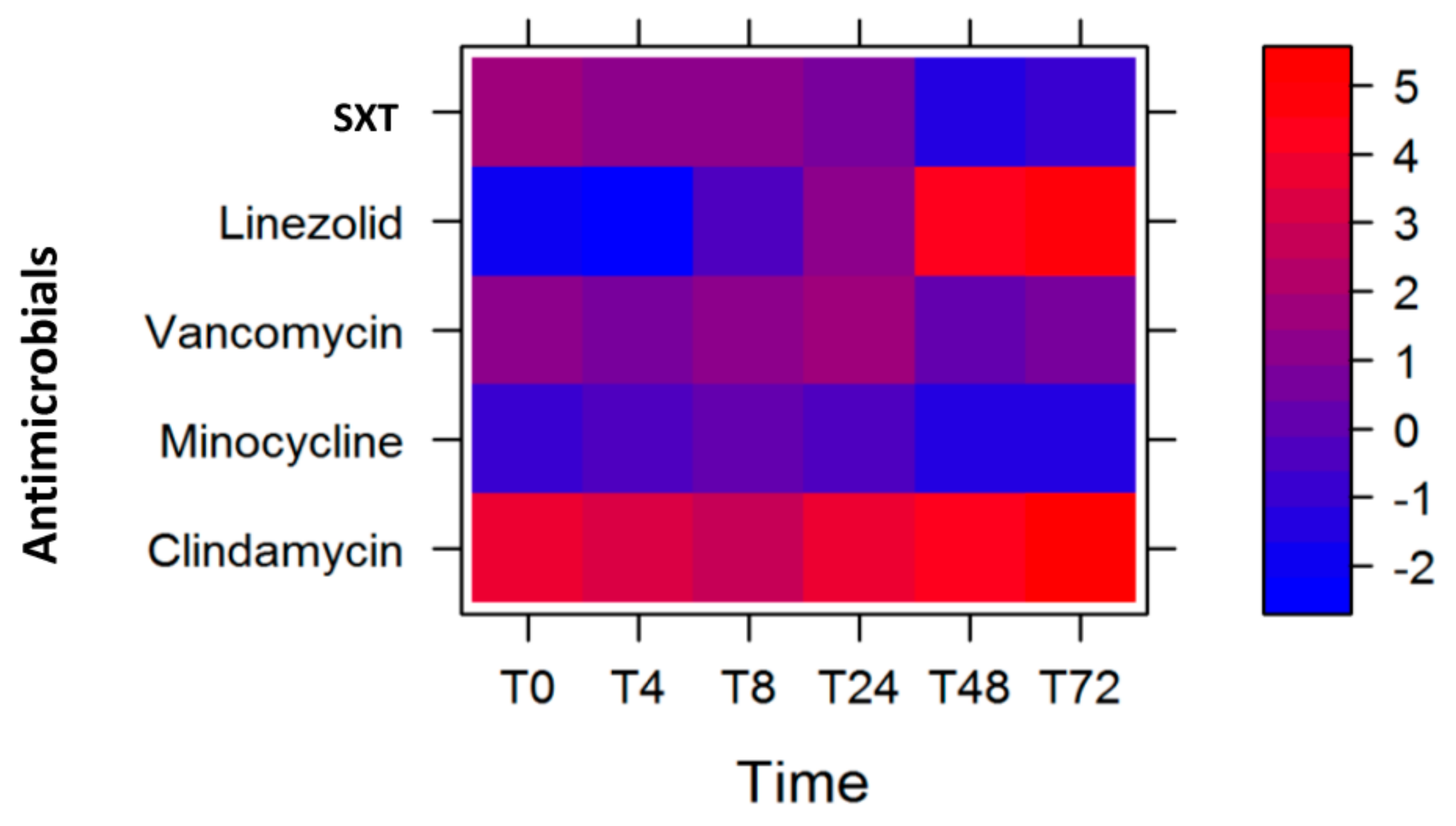
2.3. Optimal Antibiotic Course
2.4. Modeling Antibiotic Optimization to Reduce Expression of Selected MW2 Virulence Genes
3. Discussion
4. Materials and Methods
4.1. Strains
4.2. Media
4.3. Pharmacodynamic Model
4.4. Sample Collection for PK/PD and Gene Expression Studies
4.5. Antibiotic Concentration and Half-Life (t½) Estimation
4.6. Virulence Genes Expression Quantification
4.7. Statistical Model and Analysis
4.8. Evaluation of Empirical p-Values
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Baba, T.; Takeuchi, F.; Kuroda, M.; Yuzawa, H.; Aoki, K.-I.; Oguchi, A.; Nagai, Y.; Iwama, N.; Asano, K.; Naimi, T.; et al. Genome and virulence determinants of high virulence community-acquired MRSA. Lancet 2002, 359, 1819–1827. [Google Scholar] [CrossRef]
- Diep, B.A.; Chan, L.; Tattevin, P.; Kajikawa, O.; Martin, T.R.; Basuino, L.; Mai, T.T.; Marbach, H.; Braughton, K.R.; Whitney, A.R.; et al. Polymorphonuclear leukocytes mediate Staphylococcus aureus Panton-Valentine leukocidin-induced lung inflammation and injury. Proc. Natl. Acad. Sci. USA 2010, 107, 5587–5592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, S.K. Community-associated methicillin-resistant Staphylococcus aureus and its emerging virulence. Clin. Med. Res. 2005, 3, 57–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, S.K.; Stemper, M.E.; Ramaswamy, S.V.; Conradt, J.M.; Reich, R.; Graviss, E.A.; Reed, K.D. Molecular characteristics of nosocomial and Native American community-associated methicillin-resistant Staphylococcus aureus clones from rural Wisconsin. J. Clin. Microbiol. 2004, 42, 3752–3757. [Google Scholar] [CrossRef] [Green Version]
- Shukla, S.; Karow, M.E.; Brady, J.M.; Stemper, M.E.; Kislow, J.; Moore, N.; Wroblewski, K.; Chyou, P.H.; Warshauer, D.M.; Reed, K.D.; et al. Virulence genes and genotypic associations in nasal carriage, community-associated methicillin-susceptible and methicillin-resistant USA400 Staphylococcus aureus isolates. J. Clin. Microbiol. 2010, 48, 3582–3592. [Google Scholar] [CrossRef] [Green Version]
- Diep, B.A.; Carleton, H.A.; Chang, R.F.; Sensabaugh, G.F.; Pedreau-Remington, F. Roles of 34 virulence genes in the evolution of hospital- and community-associated strains of methicillin-resistant Staphylococcus aureus. J. Infect. Dis. 2006, 193, 1495–1503. [Google Scholar] [CrossRef] [Green Version]
- Tenover, F.C.; McDougal, L.K.; Goering, R.V.; Killgore, G.; Projan, S.J.; Patel, J.B.; Dunman, P.M. Characterization of a strain of community-associated methicillin-resistant Staphylococcus aureus widely disseminated in the United States. J. Clin. Microbiol. 2006, 44, 108–118. [Google Scholar] [CrossRef] [Green Version]
- Marrack, P.; Kappler, J. The staphylococcal enterotoxins and their relatives. Science 1990, 248, 1066. [Google Scholar] [CrossRef]
- McCormick, J.K.; Yarwood, J.M.; Schlievert, P.M. Toxic shock syndrome and bacterial superantigens: An update. Annu. Rev. Microbiol. 2001, 55, 77–104. [Google Scholar] [CrossRef]
- Miller, L.G.; Perdreau-Remington, F.; Rieg, G.; Mehdi, S.; Perlroth, J.; Bayer, A.S.; Tang, A.W.; Phung, T.O.; Spellberg, B. Necrotizing fasciitis caused by community-associated methicillin-resistant Staphylococcus aureus in Los Angeles. N. Engl. J. Med. 2005, 352, 1445–1453. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, B.E.; Martinez-Aguilar, G.; Hulten, K.G.; Hammerman, W.A.; Coss-Bu, J.; Avalos-Mishaan, A.; Mason, E.O.; Kaplan, S.L. Severe Staphylococcal sepsis in adolescents in the era of community-acquired methicillin-resistant Staphylococcus aureus. Pediatrics 2005, 115, 642–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillet, Y.; Issartel, B.; Vanhems, P.; Fournet, J.C.; Lina, G.; Bes, M.; Vandenesch, P.F.; Piemont, Y.P.; Brousse, N.; Floret, P.N.; et al. Association between Staphylococcus aureus strains carrying gene for Panton-Valentine leukocidin and highly lethal necrotizing pneumonia in young immunocompetent patients. Lancet 2002, 359, 753–759. [Google Scholar] [CrossRef]
- Crémieux, A.-C.; Dumitrescu, O.; Lina, G.; Vallée, C.; Côté, J.-F.; Muffat-Joly, M.; Lilin, T.; Etienne, J.; Vandenesch, F.; Saleh-Mghir, A. Panton-valentine leukocidin enhances the severity of community-associated methicillin-resistant Staphylococcus aureus rabbit osteomyelitis. PLoS ONE 2009, 4, e7204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuertz, K.; Schmidt, H.; Eiffert, H.; Schwartz, P.; Mäder, M.; Nau, R. Differential release of lipoteichoic and teichoic acids from Streptococcus pneumoniae as a result of exposure to beta-lactam antibiotics, rifamycins, trovafloxacin, and quinupristin-dalfopristin. Antimicrob. Agents Chemother. 1998, 42, 277–281. [Google Scholar]
- Otto, M. Community-associated MRSA: What makes them special? Int. J. Med. Microbiol. 2013, 303, 324–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pichereau, S.; Pantrangi, M.; Couet, W.; Badiou, C.; Lina, G.; Shukla, S.K.; Rode, W.E. Simulated antibiotic exposures in an in vitro hollow-fiber infection model influence toxin gene expression and production in community-associated methicillin-resistant Staphylococcus aureus strain MW2. Antimicrob. Agents Chemother. 2012, 56, 140–147. [Google Scholar] [CrossRef] [Green Version]
- Pantrangi, M.; Singh, V.K.; Wolz, C.; Shukla, S.K. Staphylococcal superantigen-like genes, ssl5 and ssl8, are positively regulated by Sae and negatively by Agr in the Newman strain. FEMS Microbiol. Lett. 2010, 308, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Aguilar, J.L.; Varshney, A.K.; Pechuan, X.; Dutta, K.; Nosanchuk, J.D.; Fries, B.C. Monoclonal antibodies protect from Staphylococcal Enterotoxin K (SEK) induced toxic shock and sepsis by USA300 Staphylococcus aureus. Virulence 2017, 8, 741–750. [Google Scholar] [CrossRef] [Green Version]
- Pantrangi, M.; Singh, V.K.; Shukla, S.K. Regulation of Staphylococcal Superantigen-Like Gene, ssl8, Expression in Staphylococcus aureus strain, RN6390. Clin. Med. Res. 2015, 13, 7–11. [Google Scholar] [CrossRef] [Green Version]
- Ono, H.K.; Hirose, S.; Naito, I.; Sato’o, Y.; Asano, K.; Hu, D.L.; Omoe, K.; Nakane, A. The emetic activity of staphylococcal enterotoxins, SEK, SEL, SEM, SEN and SEO in a small emetic animal model, the house musk shrew. Microbiol. Immunol. 2017, 61, 12–16. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.K.; Ring, R.P.; Aswani, V.; Stemper, M.E.; Kislow, J.; Ye, Z.; Shukla, S.K. Phylogenetic distribution and expression of a penicillin-binding protein homologue, Ear and its significance in virulence of Staphylococcus aureus. J. Med. Microbiol. 2017, 66, 1811–1821. [Google Scholar] [CrossRef] [PubMed]
- Novick, R.P.; Subedi, A. The SaPIs: Mobile pathogenicity islands of Staphylococcus. Chem. Immunol. Allergy 2007, 93, 42–57. [Google Scholar] [PubMed]
- Malachowa, N.; Whitney, A.R.; Kobayashi, S.D.; Sturdevant, D.E.; Kennedy, A.D.; Braughton, K.R.; Shabb, D.W.; Diep, B.A.; Chambers, H.F.; Otto, M.; et al. Global changes in Staphylococcus aureus gene expression in human blood. PLoS ONE 2011, 6, e18617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orwin, P.M.; Leung, D.Y.; Donahue, H.L.; Novick, R.P.; Schlievert, P.M. Biochemical and biological properties of Staphylococcal enterotoxin K. Infect. Immun. 2001, 69, 360–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orwin, P.M.; Leung, D.Y.; Tripp, T.J.; Bohach, G.A.; Earhart, C.A.; Ohlendorf, D.H.; Schlievert, P.M. Characterization of a novel staphylococcal enterotoxin-like superantigen, a member of the group V subfamily of pyrogenic toxins. Biochemistry 2002, 41, 14033–14040. [Google Scholar] [CrossRef]
- Itoh, S.; Yamaoka, N.; Kamoshida, G.; Takii, T.; Tsuji, T.; Hayashi, H.; Onozaki, K. Staphylococcal superantigen-like protein 8 (SSL8) binds to tenascin C and inhibits tenascin C-fibronectin interaction and cell motility of keratinocytes. Biochem. Biophys. Res. Commun. 2013, 433, 127–132. [Google Scholar] [CrossRef]
- van Langevelde, P.; van Dissel, J.T.; Ravensbergen, E.; Appelmelk, B.J.; Schrijver, I.A.; Groeneveld, P.H. Antibiotic-induced release of lipoteichoic acid and peptidoglycan from Staphylococcus aureus: Quantitative measurements and biological reactivities. Antimicrob. Agents Chemother. 1998, 42, 3073–3078. [Google Scholar] [CrossRef] [Green Version]
- Yanagihara, K.; Kihara, R.; Araki, N.; Morinaga, Y.; Seki, M.; Izumikawa, K.; Kakeya, H.; Yamamoto, Y.; Yamada, Y.; Kohno, S. Efficacy of linezolid against Panton-Valentine leukocidin (PVL)-positive methicillin-resistant Staphylococcus aureus (MRSA) in a mouse model of haematogenous pulmonary infection. Int. J. Antimicrob. Agents 2009, 34, 477–481. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.K.; Vaish, M.; Johansson, T.R.; Baum, K.R.; Ring, R.P.; Singh, S.; Shukla, S.K.; Moskovitz, J. Significance of four methionine sulfoxide reductases in Staphylococcus aureus. PLoS ONE 2015, 10, e0117594. [Google Scholar] [CrossRef]
- Stevens, D.L.; Ma, Y.; Salmi, D.B.; McIndoo, E.; Wallace, R.J.; Bryant, A.E. Impact of antibiotics on expression of virulence-associated exotoxin genes in methicillin-sensitive and methicillin-resistant Staphylococcus aureus. J. Infect. Dis. 2007, 195, 202–211. [Google Scholar] [CrossRef] [Green Version]
- Joo, H.S.; Chan, J.L.; Cheung, G.Y.; Otto, M. Subinhibitory concentrations of protein synthesis-inhibiting antibiotics promote increased expression of the agr virulence regulator and production of phenol-soluble modulin cytolysins in community-associated methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2010, 54, 4942–4944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, S.S.; Heilmann, K.P.; Dohrn, C.L.; Riahi, F.; Costello, A.J.; Kroeger, J.S.; Biek, D.; Critchley, I.A.; Diekema, D.J.; Doern, G.V. Activity of ceftaroline and epidemiologic trends in Staphylococcus aureus isolates collected from 43 medical centers in the United States in 2009. Antimicrob. Agents Chemother. 2011, 55, 4154–4160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erill, I.; Campoy, S.; Barbé, J. Aeons of distress: An evolutionary perspective on the bacterial SOS response. FEMS Microbiol. Rev. 2007, 31, 637–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirata, N.; Hiramatsu, K.; Kishi, K.; Yamasaki, T.; Ichimiya, T.; Nasu, M. Pretreatment of mice with clindamycin improves survival of endotoxic shock by modulating the release of inflammatory cytokines. Antimicrob. Agents Chemother. 2001, 45, 2638–2642. [Google Scholar] [CrossRef] [Green Version]
- Karlström, A.; Boyd, K.L.; English, B.K.; McCullers, J.A. Treatment with protein synthesis inhibitors improves outcomes of secondary bacterial pneumonia after influenza. J. Infect. Dis. 2009, 199, 311–319. [Google Scholar] [CrossRef]
- Schlievert, P.M.; Case, L.C. Molecular analysis of staphylococcal superantigens. Methods Mol. Biol. 2007, 391, 113–126. [Google Scholar]
- Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing; 19th informational supplement. CLSI document M100-S19; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2009. [Google Scholar]
- Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing; 24th informational supplement. CLSI document M100-S24; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2014. [Google Scholar]
- Drusano, G.L.; Liu, W.; Brown, D.L.; Rice, L.B.; Louie, A. Impact of short-course quinolone therapy on susceptible and resistant populations of Staphylococcus aureus. J. Infect. Dis. 2009, 199, 219–226. [Google Scholar] [CrossRef] [Green Version]
- Plaisance, K.I.; Drusano, G.L.; Forrest, A.; Townsend, R.J.; Standiford, H.C. Pharmacokinetic evaluation of two dosage regimens of clindamycin phosphate. Antimicrob. Agents Chemother. 1989, 33, 618–620. [Google Scholar] [CrossRef] [Green Version]
- Dehghanyar, P.; Bürger, C.; Zeitlinger, M.; Islinger, F.; Kovar, F.; Müller, M.; Kloft, C.; Joukhadar, C. Penetration of linezolid into soft tissues of healthy volunteers after single and multiple doses. Antimicrob. Agents Chemother. 2005, 49, 2367–2371. [Google Scholar] [CrossRef] [Green Version]
- Welling, P.G.; Shaw, W.R.; Uman, S.J.; Tse, F.L.; Craig, W.A. Pharmacokinetics of minocycline in renal failure. Antimicrob. Agents Chemother. 1975, 8, 532–537. [Google Scholar] [CrossRef] [Green Version]
- Varoquaux, O.; Lajoie, D.; Gobert, C.; Cordonnier, P.; Ducreuzet, C.; Pays, M.; Advenier, C. Pharmacokinetics of the trimethoprim-sulphamethoxazole combination in the elderly. Br. J. Clin. Pharmacol. 1985, 20, 575–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rybak, M.; Lomaestro, B.; Rotschafer, J.C.; Moellering, R.; Craig, W.; Billeter, M.; Dalovisio, J.R.; Levine, N.P. Therapeutic monitoring of vancomycin in adult patients: A consensus review of the American Society of Health-System Pharmacists, the Infectious Diseases Society of America, and the Society of Infectious Diseases Pharmacists. Am. J. Health. Syst. Pharm. 2009, 66, 82–98. [Google Scholar] [CrossRef] [PubMed]
- Rose, W.E.; Knier, R.M.; Hutson, P.R. Pharmacodynamic effect of clinical vancomycin exposures on cell wall thickness in heterogeneous vancomycin-intermediate Staphylococcus aureus. J. Antimicrob. Chemother. 2010, 65, 2149–2154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaPlante, K.L.; Leonard, S.N.; Andes, D.R.; Craig, W.A.; Rybak, M.J. Activities of clindamycin, daptomycin, doxycycline, linezolid, trimethoprim-sulfamethoxazole, and vancomycin against community associated methicillin-resistant Staphylococcus aureus with inducible clindamycin resistance in murine thigh infection and in vitro pharmacodynamic models. Antimicrob. Agents Chemother. 2008, 52, 2156–2162. [Google Scholar] [CrossRef] [Green Version]
- R Core Team. R: A language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013; Available online: http://www.R-project.org/ (accessed on 21 January 2020).




{kind=link}
{kind=link}
{kind=link}
| Antibiotic | Simulated Dosage Regimen | Half-Life (h) | Cmax (µg/mL) a | ||
|---|---|---|---|---|---|
| Predicted | Observed b | Predicted | Observed b | ||
| Clindamycin | 600 mg every 8 h | 2.4 | 2.6 ± 0.5 | 2.8 | 2.8 ± 0.3 |
| Minocycline | 100 mg every 12 h | 13.6 | 13.1 ± 2.1 | 0.6 | 0.7 ± 0.1 |
| Linezolid | 600 mg every 12 h | 7 | 8.5 ± 0.7 | 17.1 | 16.0 ± 0.6 |
| SXT | 160/800 mg every 12 h | 11/11 | 10.1 ± 0.3/ 10.9 ± 0.6 | 0.8/27 | 0.8 ± 0.1 28.5 ± 1.2 |
| Vancomycin | 1000 mg every 12 h | 6 | 6.2 ± 0.7 | 17 | 19.2 ± 1.3 |
| Weights of Gene | T0 | T4 | T8 | T24 | T48 | T72 |
|---|---|---|---|---|---|---|
| (0.4,0.3,0.2,0.1) | SXT | SXT | Linezolid | Clindamycin | Clindamycin | Clindamycin |
| (0.0,0.5,0.33,0.17) | SXT | SXT | SXT | SXT | Linezolid | Clindamycin |
| (0.25,0.25,0.25,0.25) | SXT | SXT | Linezolid | Clindamycin | Clindamycin | Clindamycin |
| Number of Permutations | WT = (0.4,0.3,0.2,0.1) | WT2 = (0,0.5,0.33,0.17) | WT3 = (0.25,0.25,0.25,0.25) | |||
|---|---|---|---|---|---|---|
| P1 | P2 | P1 | P2 | P1 | P2 | |
| 100 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 |
| 500 | <0.0001 | 0.002 | <0.0001 | 0.002 | <0.0001 | 0.004 |
| 1000 | <0.0001 | <0.0001 | <0.0001 | 0.003 | <0.0001 | <0.0001 |
| 10,000 | <0.0001 | 0.001 | <0.0001 | 0.0018 | <0.0001 | 0.0009 |
| Weights of MW2 Gene | T0 | T4 | T8 | T24 | T48 | T72 |
|---|---|---|---|---|---|---|
| (0.4,0.3,0.2,0.1) | Linezolid | Linezolid | Linezolid | Minocycline | Minocycline | Minocycline |
| (0.0,0.5,0.33,0.17) | Linezolid | Linezolid | Linezolid | Minocycline | Minocycline | Minocycline |
| (0.25,0.25, 0.25, 0.25) | Linezolid | Linezolid | Linezolid | Minocycline | Minocycline | Minocycline |
| Number of Permutations | WT = (0.4,0.3,0.2,0.1) | WT2 = (0.25,0.25,0.25,0.25) | WT3 = (0,0.5,0.33,0.17) | |||
|---|---|---|---|---|---|---|
| P1 | P2 | P1 | P2 | P1 | P2 | |
| 100 | 0 | 0 | 0 | 0 | 0 | 0 |
| 500 | 0 | 0.002 | 0 | 0 | 0 | 0.004 |
| 1000 | 0 | 0.002 | 0 | 0.001 | 0 | 0.001 |
| 10,000 | 0.0001 | 0.0014 | 0 | 0.0017 | 0.0001 | 0.0010 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shukla, S.K.; Carter, T.C.; Ye, Z.; Pantrangi, M.; Rose, W.E. Modeling of Effective Antimicrobials to Reduce Staphylococcus aureus Virulence Gene Expression Using a Two-Compartment Hollow Fiber Infection Model. Toxins 2020, 12, 69. https://doi.org/10.3390/toxins12020069
Shukla SK, Carter TC, Ye Z, Pantrangi M, Rose WE. Modeling of Effective Antimicrobials to Reduce Staphylococcus aureus Virulence Gene Expression Using a Two-Compartment Hollow Fiber Infection Model. Toxins. 2020; 12(2):69. https://doi.org/10.3390/toxins12020069
Chicago/Turabian StyleShukla, Sanjay K., Tonia C. Carter, Zhan Ye, Madhulatha Pantrangi, and Warren E. Rose. 2020. "Modeling of Effective Antimicrobials to Reduce Staphylococcus aureus Virulence Gene Expression Using a Two-Compartment Hollow Fiber Infection Model" Toxins 12, no. 2: 69. https://doi.org/10.3390/toxins12020069