
Investigating Effects of Proteasome Inhibitor on Multiple Myeloma Cells Using Confocal Raman Microscopy

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
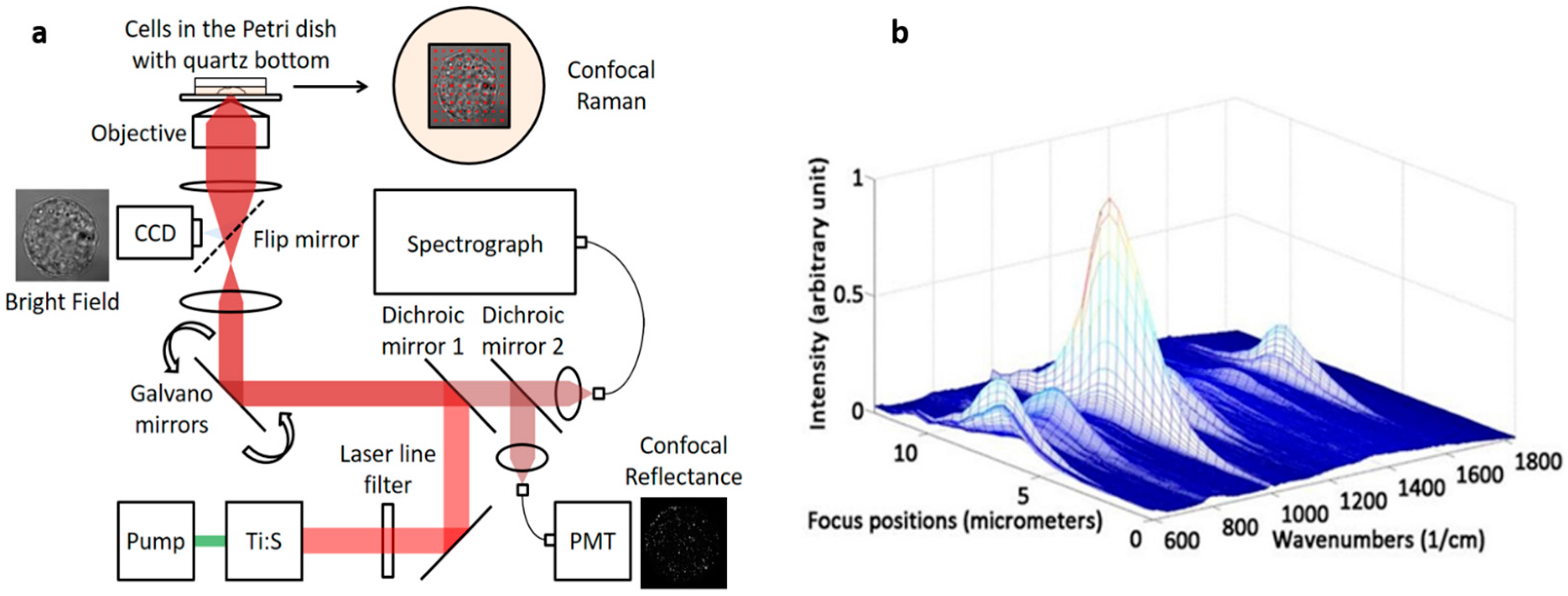
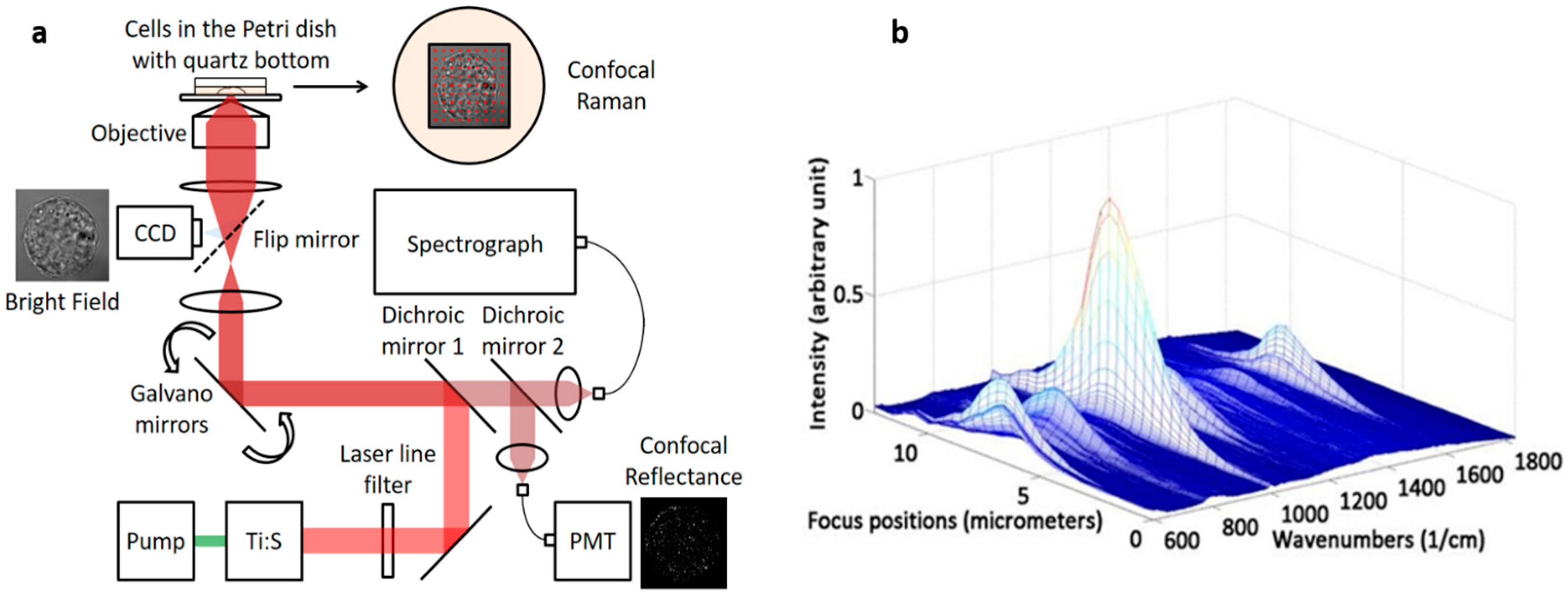
2.1. Instrumentation
2.2. Axial Resolution Test
2.3. Cell Culture
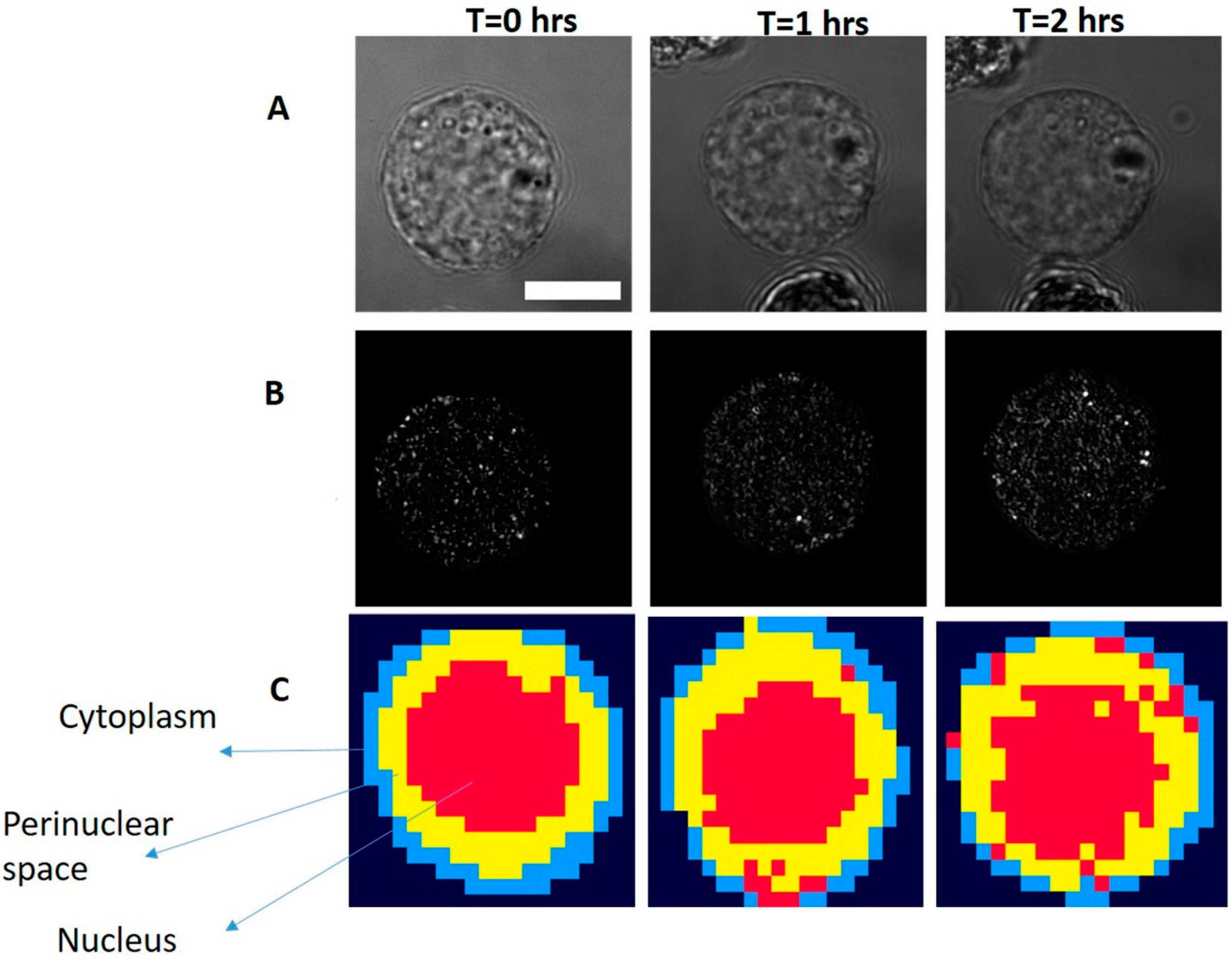
2.4. Spectral Acquisition from Single and Group of Cells
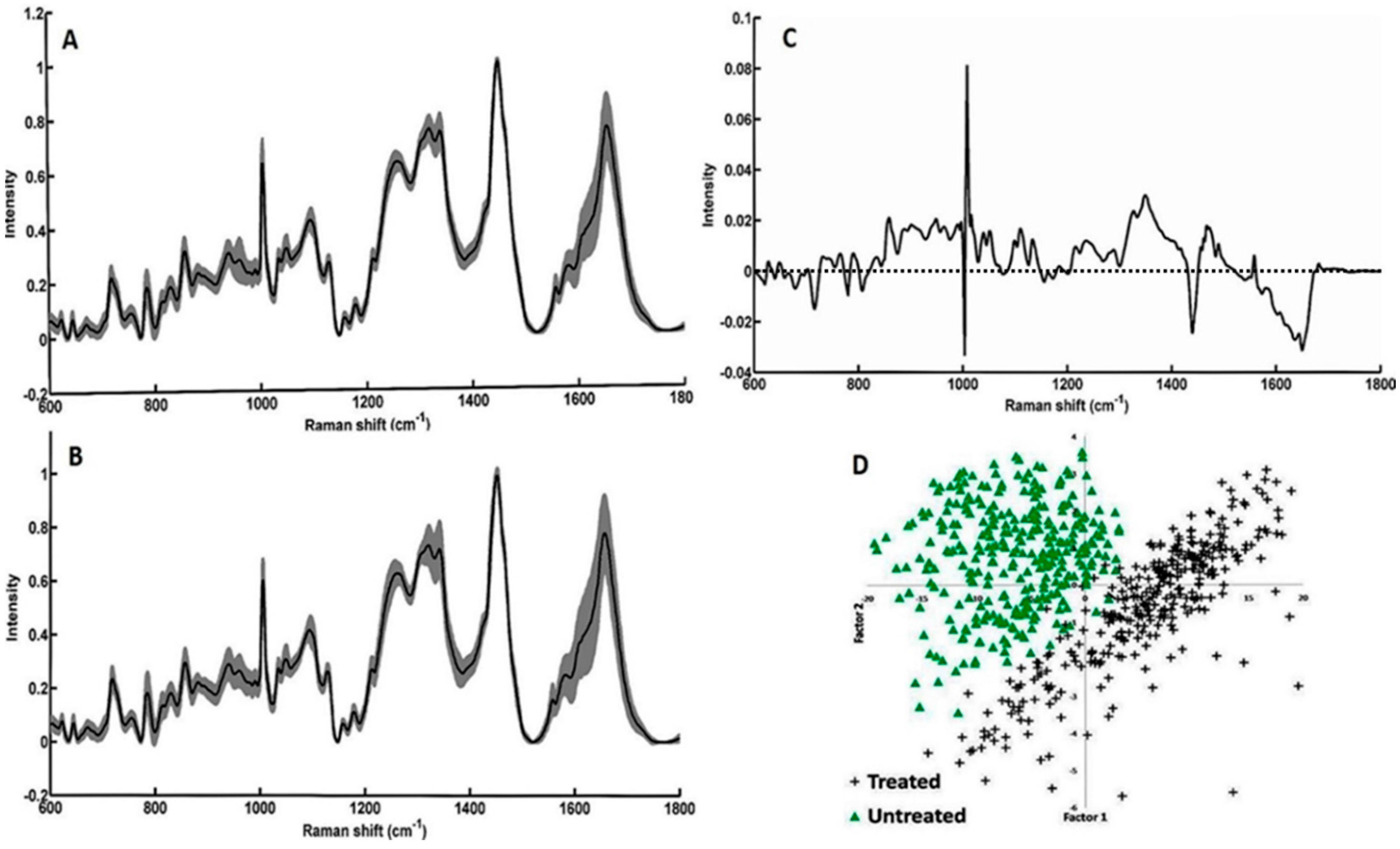
2.5. Multivariate Analysis
3. Results and Discussion
3.1. Testing Axial Resolution of the System
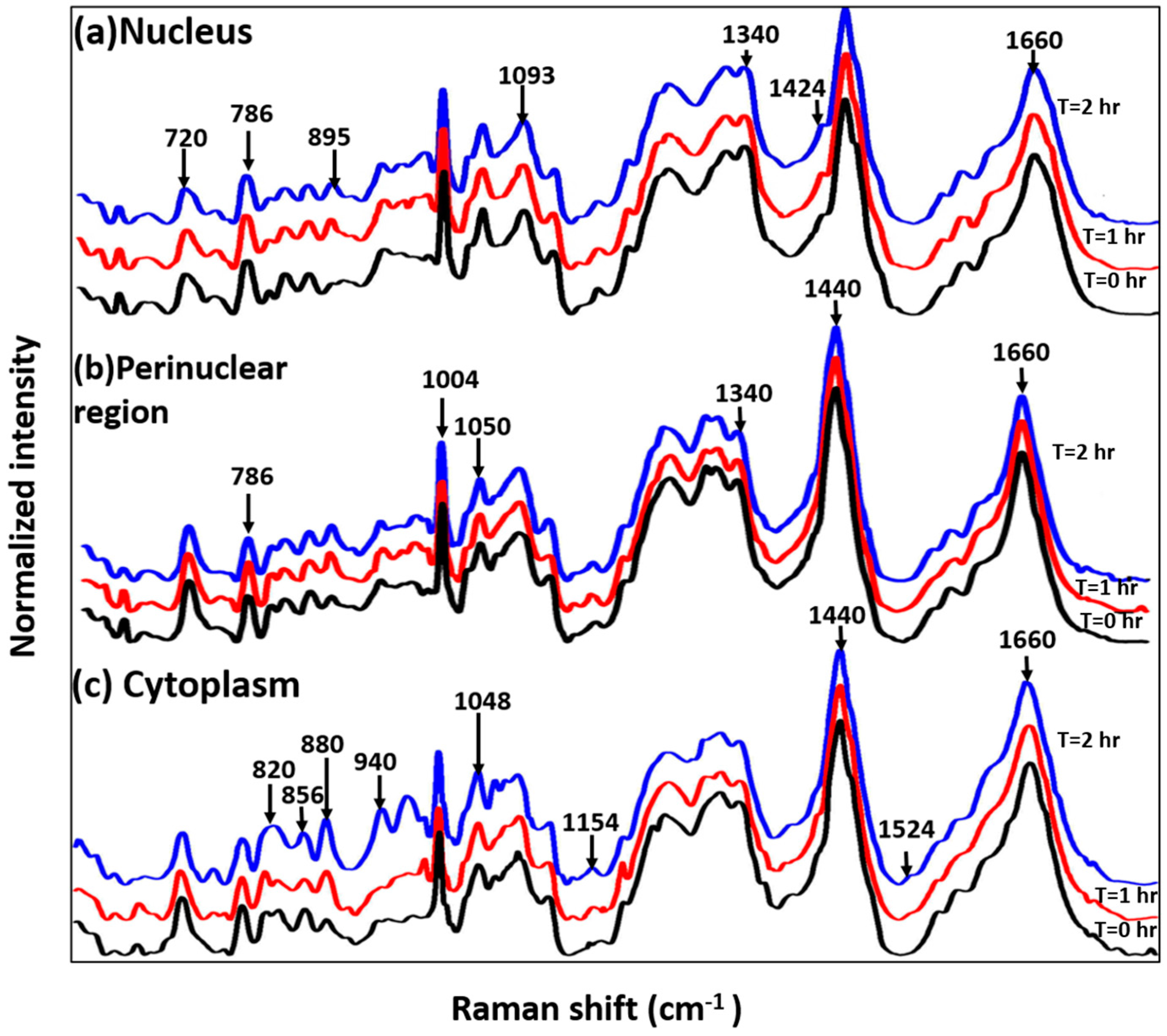
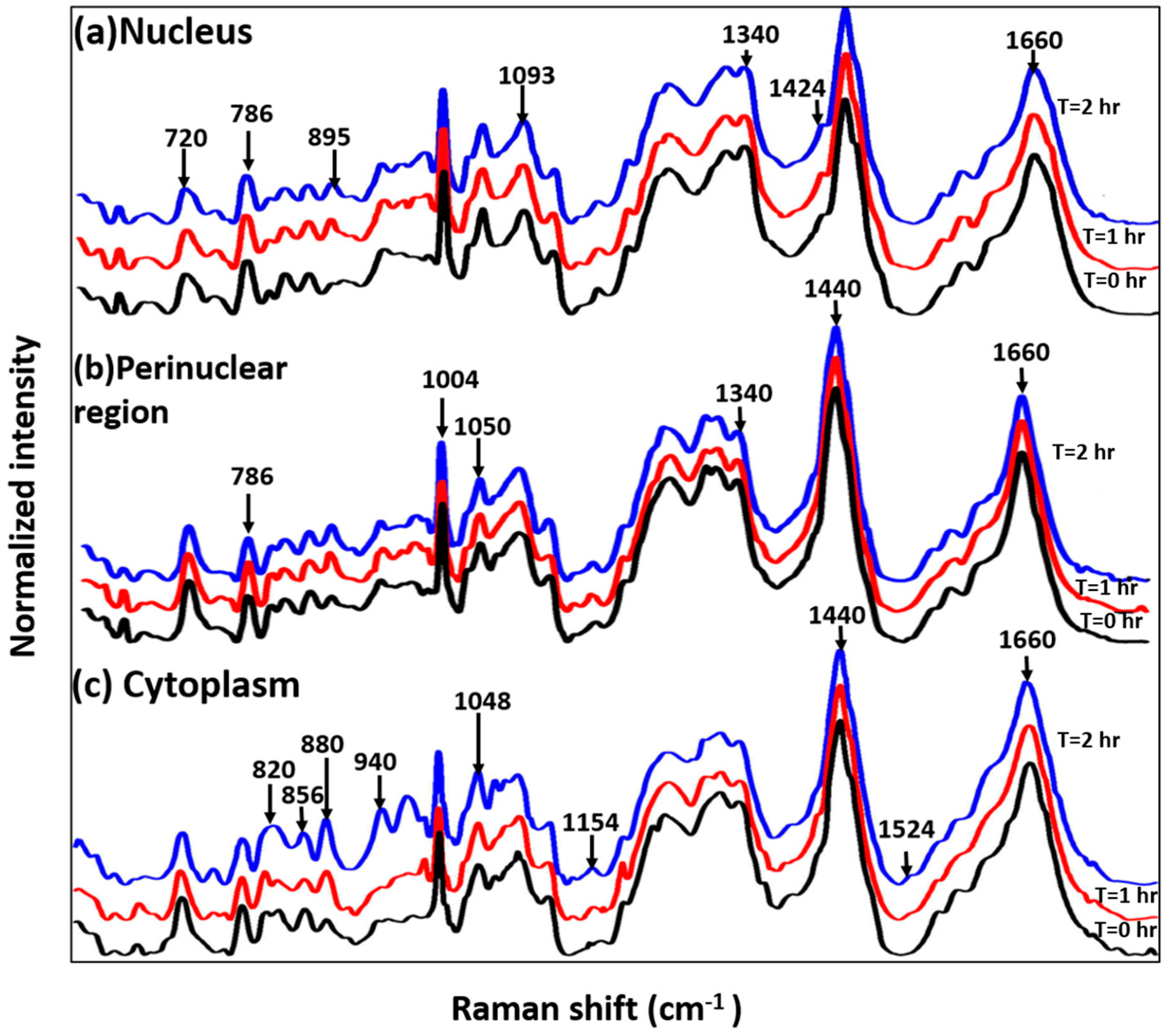
3.2. Study on Single Cells
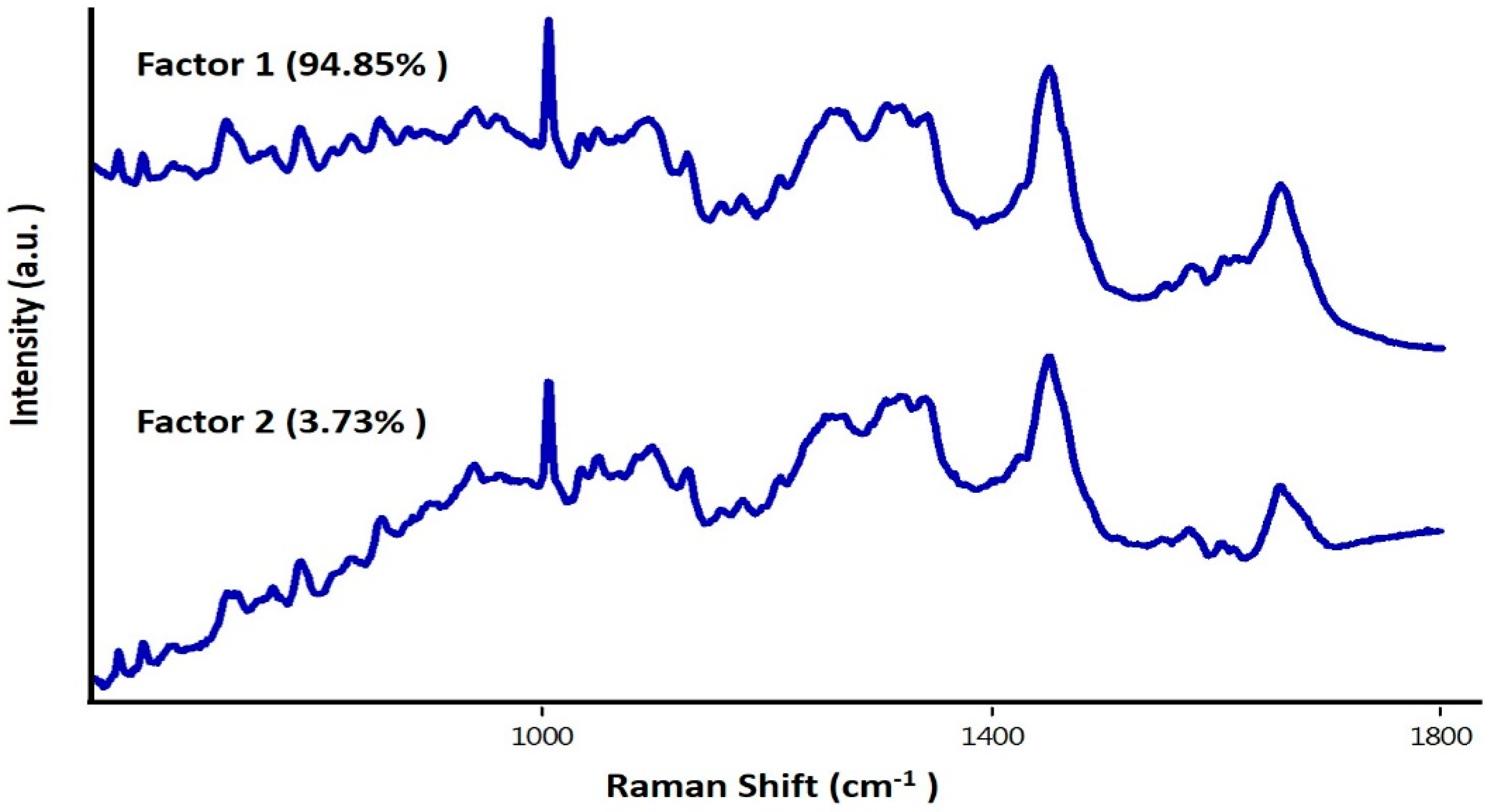
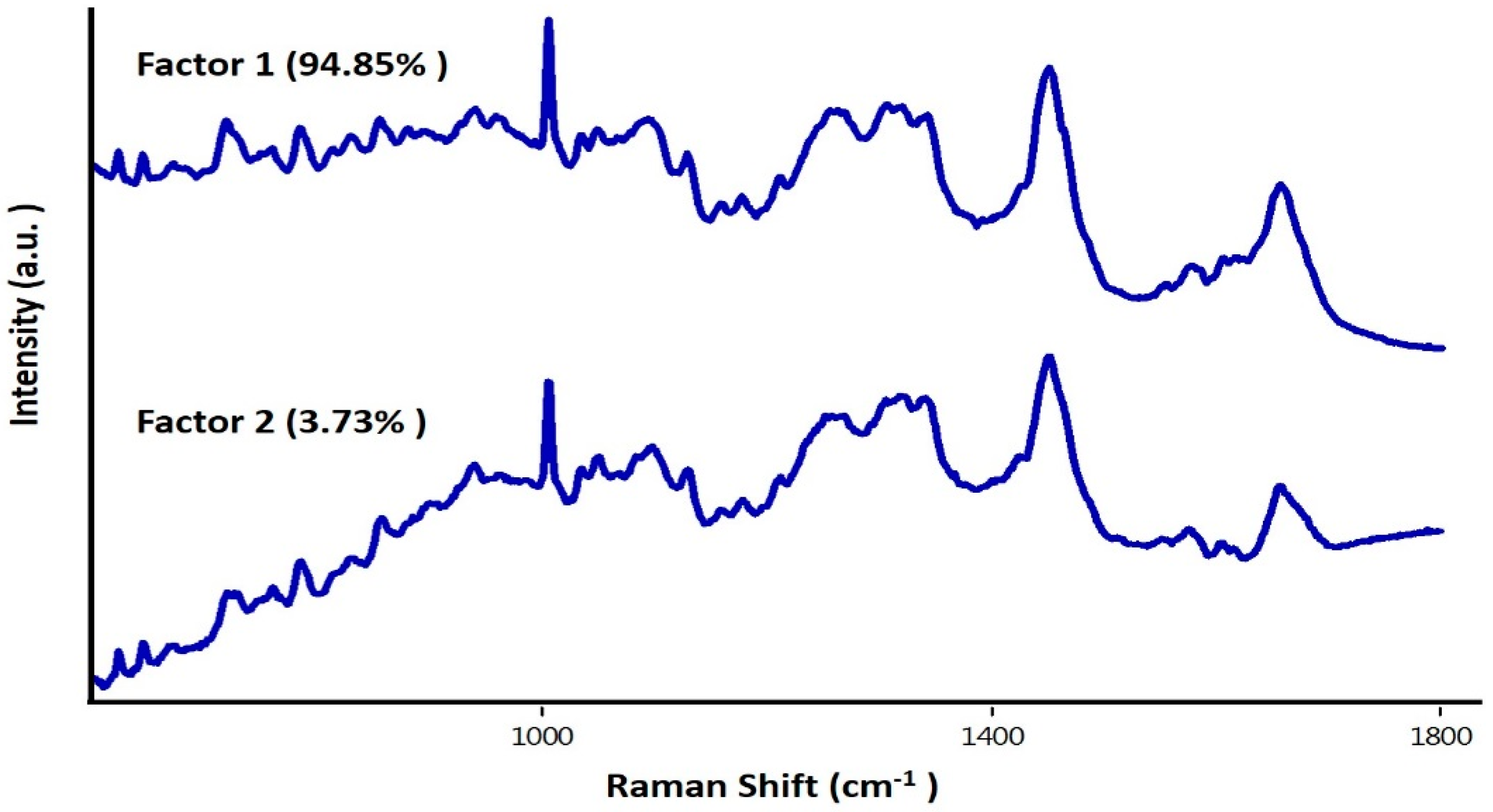
3.3. Study on Group of Cells
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ciechanover, A. The ubiquitin-proteasome proteolytic pathway. Cell 1994, 79, 13–21. [Google Scholar] [CrossRef]
- Ciechanover, A. The ubiquitin-proteasome pathway: On protein death and cell life. EMBO J. 1998, 17, 7151–7160. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Sonneveld, P.; Schuster, M.W.; Irwin, D.; Stadtmauer, E.A.; Facon, T.; Harousseau, J.L.; Ben-Yehuda, D.; Lonial, S.; Goldschmidt, H.; et al. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N. Engl. J. Med. 2005, 352, 2487–2498. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.; Oliva, L.; Cascio, P.; Pengo, N.; Fontana, F.; Cerruti, F.; Orsi, A.; Pasqualetto, E.; Mezghrani, A.; Calbi, V.; et al. The proteasome load versus capacity balance determines apoptotic sensitivity of multiple myeloma cells to proteasome inhibition. Blood 2009, 113, 3040–3049. [Google Scholar] [CrossRef] [PubMed]
- Bonvini, P.; Zorzi, E.; Basso, G.; Rosolen, A. Bortezomib-mediated 26S proteasome inhibition causes cell-cycle arrest and induces apoptosis in Cd-30+ anaplastic large cell lymphoma. Leukemia 2007, 21, 838–842. [Google Scholar] [CrossRef] [PubMed]
- Hideshima, T.; Richardson, P.; Chauhan, D.; Palombella, V.J.; Elliott, P.J.; Adams, J.; Anderson, K.C. The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001, 61, 3071–3076. [Google Scholar] [PubMed]
- Karin, M.; Cao, Y.; Greten, F.R.; Li, Z.W. NF-κB in cancer: From innocent bystander to major culprit. Nat. Rev. Cancer 2002, 2, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, S.V.; Richardson, P.G.; Hideshima, T.; Anderson, K.C. Proteasome inhibition as a novel therapeutic target in human cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 630–639. [Google Scholar] [CrossRef] [PubMed]
- Schie, I.W.; Huser, T. Methods and applications of Raman microspectroscopy to single-cell analysis. Appl. Spectrosc. 2013, 67, 813–828. [Google Scholar] [PubMed]
- Krafft, C.; Popp, J. The many facets of Raman spectroscopy for biomedical analysis. Anal. Bioanal. Chem. 2015, 407, 699–717. [Google Scholar] [CrossRef] [PubMed]
- Austin, L.A.; Osseiran, S.; Evans, C.L. Raman technologies in cancer diagnostics. Analyst 2016, 141, 476–503. [Google Scholar] [CrossRef] [PubMed]
- Raman, C.V.; Krishnan, K.S. A new type of secondary radiation. Nature 1928, 122, 12. [Google Scholar] [CrossRef]
- Puppels, G.J.; de Mul, F.F.; Otto, C.; Greve, J.; Robert-Nicoud, M.; Arndt-Jovin, D.J.; Jovin, T.M. Studying single living cells and chromosomes by confocal Raman microspectroscopy. Nature 1990, 347, 301–303. [Google Scholar] [CrossRef] [PubMed]
- Scarcelli, G.; Yun, S.H. Confocal Brillouin microscopy for three-dimensional mechanical imaging. Nat. Photonics 2008, 2, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Shafer-Peltier, K.E.; Haka, A.S.; Fitzmaurice, M.; Crowe, J.; Myles, J.; Dasari, R.R.; Feld, M.S. Raman microspectroscopic model of human breast tissue: Implications for breast cancer diagnosis in vivo. J. Raman Spectrosc. 2002, 33, 552–563. [Google Scholar] [CrossRef]
- Haka, A.S.; Shafer-Peltier, K.E.; Fitzmaurice, M.; Crowe, J.; Dasari, R.R.; Feld, M.S. Identifying microcalcifications in benign and malignant breast lesions by probing differences in their chemical composition using Raman spectroscopy. Cancer Res. 2002, 62, 5375–5380. [Google Scholar] [PubMed]
- Uzunbajakava, N.; Lenferink, A.; Kraan, Y.; Volokhina, E.; Vrensen, G.; Greve, J.; Otto, C. Nonresonant confocal Raman imaging of DNA and protein distribution in apoptotic cells. Biophys. J. 2003, 84, 3968–3981. [Google Scholar] [CrossRef]
- Kang, J.W.; Lue, N.; Kong, C.R.; Barman, I.; Dingari, N.C.; Goldfless, S.J.; Niles, J.C.; Dasari, R.R.; Feld, M.S. Combined confocal Raman and quantitative phase microscopy system for biomedical diagnosis. Biomed. Opt. Express 2011, 2, 2484–2492. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.W.; Nguyen, F.T.; Lue, N.; Dasari, R.R.; Heller, D.A. Measuring uptake dynamics of multiple identifiable carbon nanotube species via high-speed confocal Raman imaging of live cells. Nano Lett. 2012, 12, 6170–6174. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.W.; So, P.T.C.; Dasari, R.R.; Lim, D.-K. High resolution live cell Raman imaging using subcellular organelle-targeting SERS-sensitive gold nanoparticles with highly narrow intra-nanogap. Nano Lett. 2015, 15, 1766–1772. [Google Scholar] [CrossRef] [PubMed]
- Barman, I.; Dingari, N.C.; Kang, J.W.; Horowitz, G.L.; Dasari, R.R.; Feld, M.S. Raman spectroscopy-based sensitive and specific detection of glycated hemoglobin. Anal. Chem. 2012, 84, 2474–2482. [Google Scholar] [CrossRef] [PubMed]
- Dingari, N.C.; Horowitz, G.L.; Kang, J.W.; Dasari, R.R.; Barman, I. Raman spectroscopy provides a powerful diagnostic tool for accurate determination of albumin glycation. PLoS ONE 2012, 7, e32406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Candeloro, P.; Grande, E.; Raimondo, R.; Di Mascolo, D.; Gentile, F.; Coluccio, M.L.; Perozziello, G.; Malara, N.; Francardi, M.; Di Fabrizio, E. Raman database of amino acids solutions: A critical study of Extended Multiplicative Signal Correction. Analyst 2013, 138, 7331–7340. [Google Scholar] [CrossRef] [PubMed]
- Koljenovic, S.; Bakker Schut, T.C.; van Meerbeeck, J.P.; Maat, A.P.; Burgers, S.A.; Zondervan, P.E.; Kros, J.M.; Puppels, G.J. Raman microspectroscopic mapping studies of human bronchial tissue. J. Biomed. Opt. 2004, 9, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Koljenovic, S.; Schut, T.B.; Vincent, A.; Kros, J.M.; Puppels, G.J. Detection of meningioma in dura mater by Raman spectroscopy. Anal. Chem. 2005, 77, 7958–7965. [Google Scholar] [CrossRef] [PubMed]
- Cordella, C.B.Y.; Bertrand, D. SAISIR: A new general chemometric toolbox. TrAC Trends Analyt. Chem. 2014, 54, 75–82. [Google Scholar] [CrossRef]
- Charras, G.T. A short history of blebbing. J. Microsc. 2008, 231, 466–478. [Google Scholar] [CrossRef] [PubMed]
- Mitus, W.J.; Bergna, L.J.; Mednicoff, I.B.; Dameshek, W. Cytochemical in studies of glycogen content of lymphocytes in lymphocytic proliferations. Blood 1958, 13, 748–756. [Google Scholar] [PubMed]
- Pully, V.V.; Lenferink, A.T.M.; Otto, C. Time-lapse Raman imaging of single live lymphocytes. J. Raman Spectrosc. 2011, 42, 167–173. [Google Scholar] [CrossRef]
- Movasaghi, Z.; Rehman, S.; Rehman, I.U. Raman spectroscopy of biological tissues. Appl. Spectrosc. Rev. 2007, 42, 493–541. [Google Scholar] [CrossRef]
- Kann, B.; Offerhaus, H.L.; Windbergs, M.; Otto, C. Raman microscopy for cellular investigations—From single cell imaging to drug carrier uptake visualization. Adv. Drug Deliv. Rev. 2015, 89, 71–90. [Google Scholar] [CrossRef] [PubMed]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, J.W.; Singh, S.P.; Nguyen, F.T.; Lue, N.; Sung, Y.; So, P.T.C.; Dasari, R.R. Investigating Effects of Proteasome Inhibitor on Multiple Myeloma Cells Using Confocal Raman Microscopy. Sensors 2016, 16, 2133. https://doi.org/10.3390/s16122133
Kang JW, Singh SP, Nguyen FT, Lue N, Sung Y, So PTC, Dasari RR. Investigating Effects of Proteasome Inhibitor on Multiple Myeloma Cells Using Confocal Raman Microscopy. Sensors. 2016; 16(12):2133. https://doi.org/10.3390/s16122133
Chicago/Turabian StyleKang, Jeon Woong, Surya P. Singh, Freddy T. Nguyen, Niyom Lue, Yongjin Sung, Peter T. C. So, and Ramachandra R. Dasari. 2016. "Investigating Effects of Proteasome Inhibitor on Multiple Myeloma Cells Using Confocal Raman Microscopy" Sensors 16, no. 12: 2133. https://doi.org/10.3390/s16122133