1. Introduction
Nanostructured lipid carriers (NLCs) have been introduced as a new generation of lipid-based drug delivery systems, including liposomes and solid lipid nanoparticles [
1]. NLCs are composed of solid lipids and liquid oils, which result in a high loading capacity for poorly water-soluble drugs [
2]. They possess numerous advantages, such as biocompatibility, controlled drug release, storage stability, and the possibility of large-scale production. However, their clinical applications for effective drug therapy are still very limited because of the lack of selectivity [
3]. Recently, to achieve multifunctionality for effective drug targeting, surface modifications of NLCs have been widely tried [
4].
In our previous work, we successfully synthesized a 16-mer RIPL peptide (IPLVVPLRRRRRRRRC) as a cell-penetrating and homing peptide [
5] and prepared RIPL peptide-conjugated NLCs (RIPL-NLCs) as a platform for hepsin-specific drug delivery [
6]. RIPL-NLCs enhanced the intracellular delivery of the anticancer drug docetaxel (DTX) or a fluorescent probe. However, it is well known that most nanoparticles are cleared from circulation by the mononuclear phagocyte system (MPS) because of opsonization and subsequent uptake by the reticuloendothelial system [
7,
8]. To overcome these limitations, the concept of “stealth NLCs”, designed to reduce the nonspecific protein adsorption and prevent the MPS uptake, has been suggested [
9]. Various polymers have been used to increase the stability of NLCs in blood circulation (described as “steric stabilization”) by building a polymeric barrier on the surface of NLCs [
10].
Currently, polyethylene glycol (PEG) has been widely used as a polymeric steric stabilizer. PEG’s neutrality, hydrophilicity, molecular flexibility, and non-immunogenicity make it a natural initial choice [
11]. For optimizing a PEGylated NLC system, several factors are crucial, such as the type, length, molecular weight, and density of PEG chains [
12]. In particular, PEG chains with molecular weights of 2 kDa or higher have been used to achieve a successful surface modification [
13,
14]. In an earlier study, we stabilized a liposomal nanocarrier by using PEG3000 at 5 mole % [
15] since the density of PEG chains has been recognized as an important factor. In particular, at a density between 5 and 9 mole %, polymer chains can effectively block opsonization while maintaining their flexibility [
16].
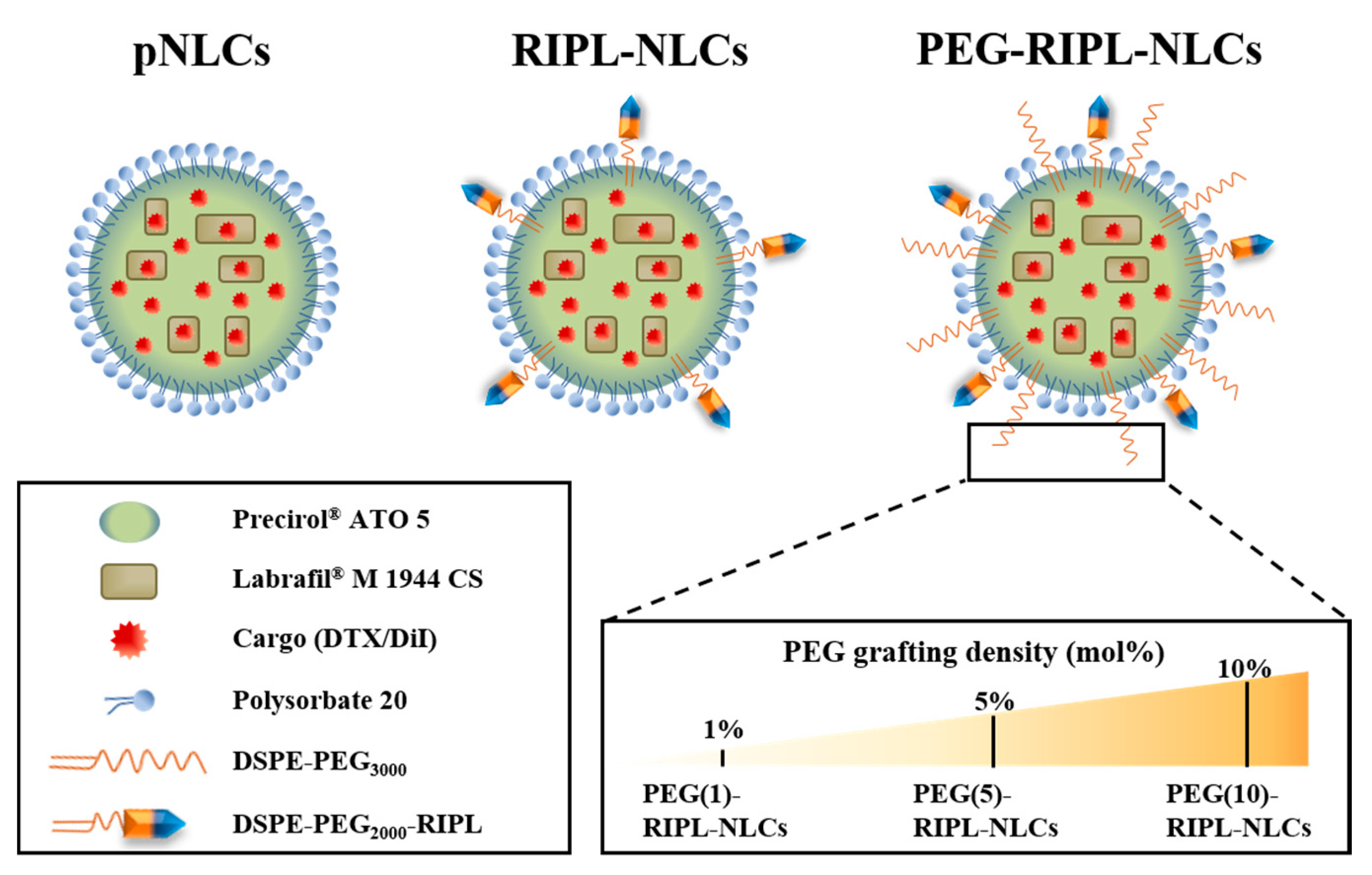
In this study, to reduce the nonspecific protein adsorption and to prevent the recognition by the MPS, PEG-modified RIPL-NLCs (PEG-RIPL-NLCs) were prepared using PEG3000 as a steric stabilizer at different grafting ratios (1, 5, and 10 mole %), with either DTX or a fluorescent probe encapsulated. Physicochemical properties of various NLCs were evaluated, including their particle size, zeta potential (ZP), polydispersity index (PDI), encapsulation efficiency (EE), and drug loading (DL). The conformational structure and drug release properties of NLCs were also characterized. The steric stabilization effect was evaluated by a protein adsorption assay and a macrophage phagocytosis inhibition study. The cellular uptake behavior of PEG-RIPL-NLCs was assessed using hepsin-expressing SKOV3, MCF7, and LNCaP cells, and the uptake mechanism was investigated in the presence of endocytosis inhibitors. Cytotoxicity and hemolytic activity were also assessed. Finally, an in vivo biodistribution study was performed after intravenous injection of the formulations to normal mice.
2. Materials and Methods
2.1. Materials
DTX (purity > 99%) was kindly provided by Chong Kun Dang Pharm. Co. (Yongin, Korea). Oleoyl macrogol-6 glycerides (Labrafil® M 1944 CS) and glyceryl distearate (Precirol® ATO 5) were received as a gift from Gattefossé (Saint-Priest, France). 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI), a micro bicinchoninic acid (BCA) protein assay kit, 5,5′-dithio-bis-(2-nitrobenzoic acid) (DTNB), cysteine hydrochloride anhydrous, phosphate-buffered saline (PBS) tablets, chlorpromazine hydrochloride, sodium azide, amiloride hydrochloride hydrate, and nystatin were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(PEG2000)] (DSPE-PEG2000-Mal) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(PEG3000)] (DSPE-PEG3000) were purchased from Avanti Polar Lipids (Alabaster, AL, USA). The RIPL peptide was synthesized by Peptron Co. (Daejeon, Korea). All other chemicals and reagents purchased from commercial sources were of analytical or cell culture grade.
Cell lines were purchased from the Korean Cell Line Bank (Seoul, Korea). PBS (10×, pH 7.4), and cell culture materials, including Roswell Park Memorial Institute (RPMI) 1640 medium, Dulbecco′s modified Eagle′s medium (DMEM), fetal bovine serum, penicillin-streptomycin, and trypsin-Ethylenediaminetetraacetic acid (EDTA) (0.25%), were obtained from Invitrogen (Carlsbad, CA, USA). Female Institute of Cancer Research (ICR) mice (5-week-old, 20 ± 2 g) were purchased from the Hanlim Experimental Animal Laboratory (Gyeonggi-do, Korea).
2.2. Preparations of Various NLC Formulations
The following DTX-loaded NLC formulations were prepared using a previously published solvent emulsification-evaporation method [
6]: plain NLCs (DTX-pNLCs), RIPL-NLCs (DTX-RIPL-NLCs), and PEGylated RIPL-NLCs (DTX-PEG-RIPL-NLCs) (
Figure 1). To observe the cellular uptake of NLCs, DiI (a lipophilic red fluorescent probe) was loaded instead of DTX. The unencapsulated DTX or DiI was removed by ultra-centrigufation at 13,000×
g for 20 min using Amicon
® ultra-centrifugal filters (MWCO 100 kDa, Millipore, Billerica, MA, USA). Empty NLCs were prepared without DTX or DiI. All prepared NLCs were stored at 4 °C. The NLC samples were used for the experiments within 2 weeks, and no changes were observed until use.
2.2.1. Preparation of DTX-pNLCs
Labrafil® M 1944 CS (liquid oil), Precirol® ATO 5 (solid lipid), and DTX (or DiI) were dissolved in dichloromethane as an organic phase and then mixed with an aqueous solution containing polysorbate 20 (Tween 20; 1%, w/v) and polyvinyl alcohol (PVA; 0.5%, w/v). The mixture was homogenized at 15,000 rpm for 2 min using an Ultra-Turrax® T25 basic homogenizer (IKA Labortechnik, Staufen, Germany), then sonicated using a probe-type sonicator (Sonoplus, HD 2070; Bandelin Electronics, Berlin, Germany) operating at a power of 45% for 3 min under cooling at 5 °C, and finally subjected to solvent evaporation with magnetic stirring at 300 rpm for 3 h.
2.2.2. Preparation of DTX-RIPL-NLCs
The RIPL peptide was conjugated to maleimide-derivatized DTX-NLCs via a thiol-maleimide reaction as reported [
6,
17]. Briefly, DTX, Precirol
® ATO 5, Labrafil
® M 1944 CS, and DSPE-PEG2000-Mal (1 mole %) were dissolved in dichloromethane, mixed with the Tween 20/PVA aqueous solution, and then homogenized, sonicated, and evaporated as described above. Subsequently, the RIPL peptide was added to the maleimide-derivatized DTX-NLC dispersion and allowed to react for 12 h at 25 °C. The unreacted RIPL peptide was removed by dialysis against distilled water using a cellulose ester dialysis membrane [50 kDa molecular weight cutoff (MWCO)] for 24 h at 4 °C.
2.2.3. Preparation of DTX-PEG-RIPL-NLCs
PEGylation of RIPL-NLCs was performed by a premodification technique as reported [
18]. Briefly, different amounts of DSPE-PEG3000 (1, 5, and 10 mole %) were dissolved in dichloromethane, containing predissolved DTX, Precirol
® ATO 5, Labrafil
® M 1944 CS, and DSPE-PEG2000-Mal (1 mole %), and homogenized with the Tween 20/PVA aqueous solution, followed by the same procedure as described in
Section 2.2.2 for RIPL-NLC preparation. Based on the amount of DSPE-PEG3000 added (1, 5, or 10 mole %), the prepared DTX-RIPL-NLCs were designated PEG(1)-, PEG(5)-, and PEG(10)-RIPL-NLCs, respectively (
Figure 1).
2.3. Preparation of a Reference DTX Solution
To reproduce a commercial DTX product, Taxotere®, a DTX solution (DTX-Sol) was prepared by dissolving DTX at a concentration of 20 mg/mL in distilled water containing polysorbate 80 (25%, w/v) and ethanol (9.75%, v/v). DTX-Sol was appropriately diluted with a cell culture medium or normal saline for in vitro cytotoxicity and in vivo biodistribution studies, respectively.
2.4. Determination of EE and DL
EE and
DL of NLCs were determined by ultrafiltration using Amicon
® ultracentrifugal devices. Briefly, 500 μL of DTX- or DiI-loaded NLC samples was centrifuged for 20 min at 14,000×
g. The concentration of the free drug in the filtrate was analyzed by high-performance liquid chromatography (HPLC). The following equations were used for the calculations:
where
WT,
WF, and
WL represent the total amount of the cargo, the amount of free cargo, and the total amount of the lipids, respectively.
2.5. HPLC
DTX and DiI were quantified using a Waters Corporation (Milford, MA, USA) HPLC system consisting of a separations module (e2695), an ultraviolet (UV) detector (e2489), and a data station (Empower 3). DTX was separated on a Kromasil® C18 column (5 μm, 4.6 × 250 mm; Akzo Nobel, Bohus, Sweden) using an isocratic mobile phase consisting of acetonitrile and water (55:45, v/v) at a flow rate of 1 mL/min at 25 °C. The eluate was monitored at a UV wavelength of 230 nm, and the injection volume was 50 μL. For DiI quantification, a fluorescence detector (W2475) was used. Chromatography was performed on a C18 column (5 μm, 4.6 × 150 mm; Shiseido, Tokyo, Japan) at a flow rate of 1.5 mL/min using a mobile phase consisting of 0.05 M dimethyl sulfate and methanol (2:98, v/v). The injection volume was 50 μL, and the excitation and emission wavelengths were 549 and 565 nm, respectively. The standard calibration curves of DTX and DiI were linear in the ranges of 0.5–100 μg/mL and 0.005–10 μg/mL, respectively, with coefficient of determination (r2) values of greater than 0.999. The analysis method offered a limit of detection of 0.1 μg/mL for DTX and 0.001 μg/mL for DiI at a signal-to-noise ratio of 3:1.
2.6. Particle Size and ZP Analysis
NLC samples were diluted 1:100 in distilled water, which provided an adequate scattering intensity. The particle size, PDI, and ZP of NLCs were measured in triplicate using a dynamic light scattering particle size analyzer (Zetasizer Nano-ZS; Malvern Instruments, Worcestershire, UK).
2.7. Transmission Electron Microscopy
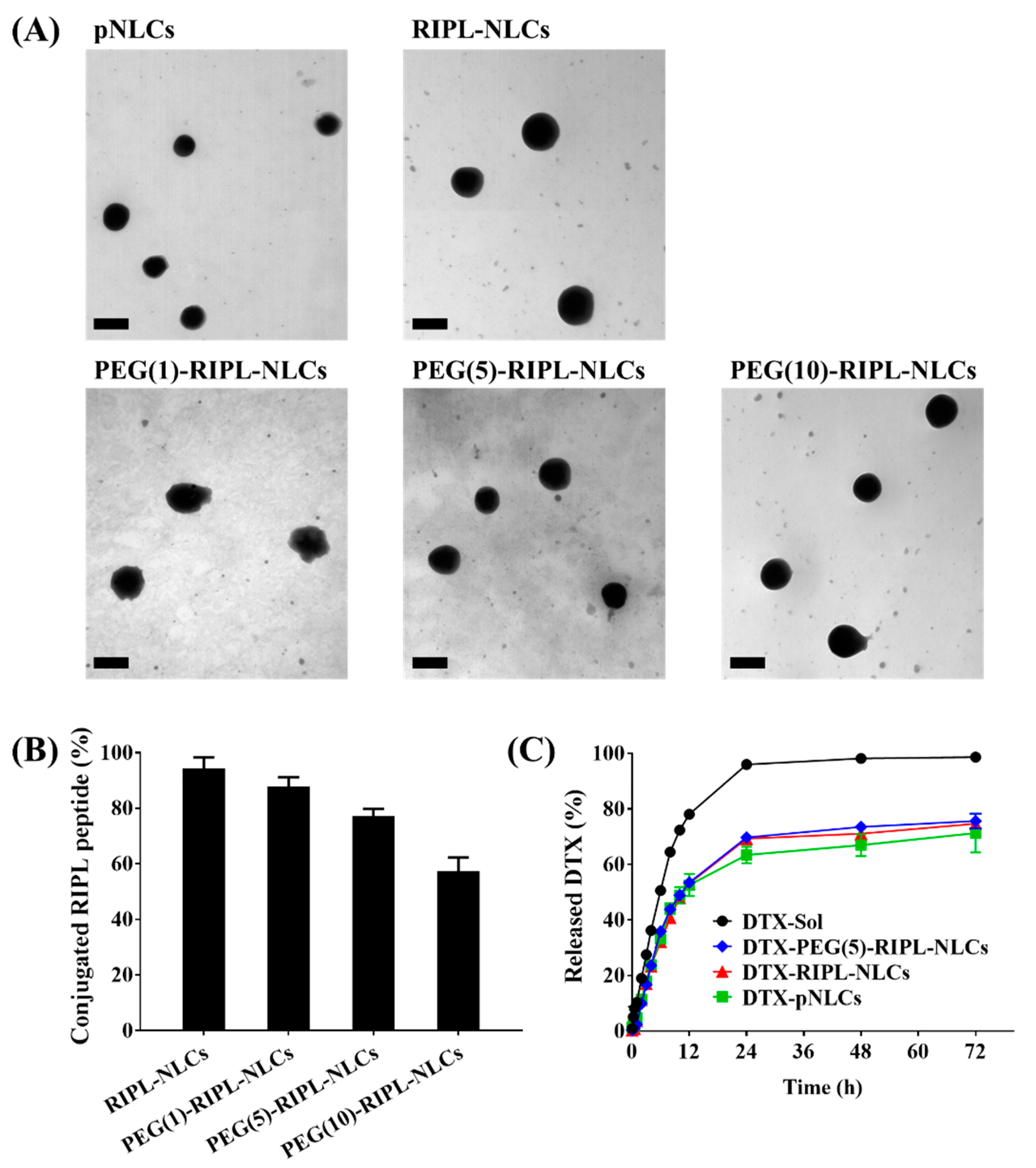
NLCs were imaged using a transmission electron microscope (JEM1010; JEOL, Tokyo, Japan) at an acceleration voltage of 80 kV. Briefly, NLC samples were diluted 10-fold with distilled water and placed onto a carbon film grid. The samples were stained with 2% phosphotungstic acid, washed with distilled water, and dried at room temperature before observation.
2.8. Conformational Characterization of Ligand Conjugation
The conjugation efficiencies of the RIPL peptide to RIPL-NLCs and PEG-RIPL-NLCs were calculated indirectly by determining the amount of unreacted cysteine in Ellman’s reaction as previously reported [
19,
20]. Briefly, RIPL-NLCs and PEG-RIPL-NLCs were incubated with a three-fold molar excess of cysteine to block the unreacted maleimide functional groups. Then, DTNB (1 mg/mL) was added to react with the unreacted cysteine, with the formation of a cysteine-5-thio-2-nitrobenzoic acid (TNB) adduct and a concomitant release of an equivalent amount of free TNB. The amount of liberated TNB was analyzed by HPLC at a flow rate of 1.0 mL/min, with UV detection at a wavelength of 420 nm. The mobile phase consisted of a mixture of methanol and a 10 mM ammonium formate solution (5:95,
v/
v).
2.9. In Vitro Drug Release
An in vitro drug release study was performed using a dialysis bag diffusion method [
21]. Briefly, 1 mL of DTX-Sol, DTX-pNLCs, DTX-RIPL-NLCs, or DTX-PEG(5)-RIPL-NLCs was placed in a dialysis bag (100 kDa MWCO; Spectrum Laboratories, Rancho Dominguez, CA, USA). Then, firmly clipped dialysis bags were completely soaked in 90 mL of a release medium [0.5% (
w/
v) sodium dodecyl sulfate in PBS, pH 7.4] and incubated at 37 °C with magnetic stirring at 100 rpm. To measure the amount of released DTX, 1 mL of the release medium was withdrawn at predetermined time points (0.25, 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 24, 48, and 72 h), and the dialysate volume was replenished with 1 mL of a fresh release medium. The concentration of DTX in the aliquots was analyzed by HPLC as described above.
2.10. Protein Adsorption Assay
Protein adsorption behaviors of the nanocarriers were investigated by measuring the amount of bovine serum albumin (BSA) bound to the NLC surface using a micro BCA protein assay kit as previously reported [
22]. Briefly, drug-free NLC formulations were incubated with a PBS solution (pH 7.4) containing BSA (250 μg/mL) at 37 °C for predetermined times (2, 8, 16, and 24 h). After the incubation, samples were centrifuged at 13,000×
g for 15 min to remove free BSA. The pellets were resuspended in 1 mL of PBS and vortexed, to ensure homogeneity, at 2000 rpm for 20 min. Afterward, 100 µL of the samples was transferred into 96-well plates, mixed with 100 µL of the BCA working reagent, and incubated at 37 °C for 30 min. The absorbance was measured spectrophotometrically at 562 nm using a microplate reader (FlexStation 3; Molecular Devices, Sunnyvale, CA, USA).
2.11. Cell Culture
Human ovarian carcinoma (SKOV3), human breast adenocarcinoma (MCF7), and human prostate carcinoma (LNCaP) cells were incubated in RPMI 1640 medium, and murine macrophage (RAW 264.7) cells were incubated in DMEM. Both media contained antibiotics (100 μg/mL streptomycin and 100 U/mL penicillin G) and 10% (v/v) fetal bovine serum. Cells were cultured every 2–4 days in a humidified incubator in an atmosphere of 5% CO2 at 37 °C and 95% relative humidity.
2.12. Macrophage Phagocytosis Inhibition Study
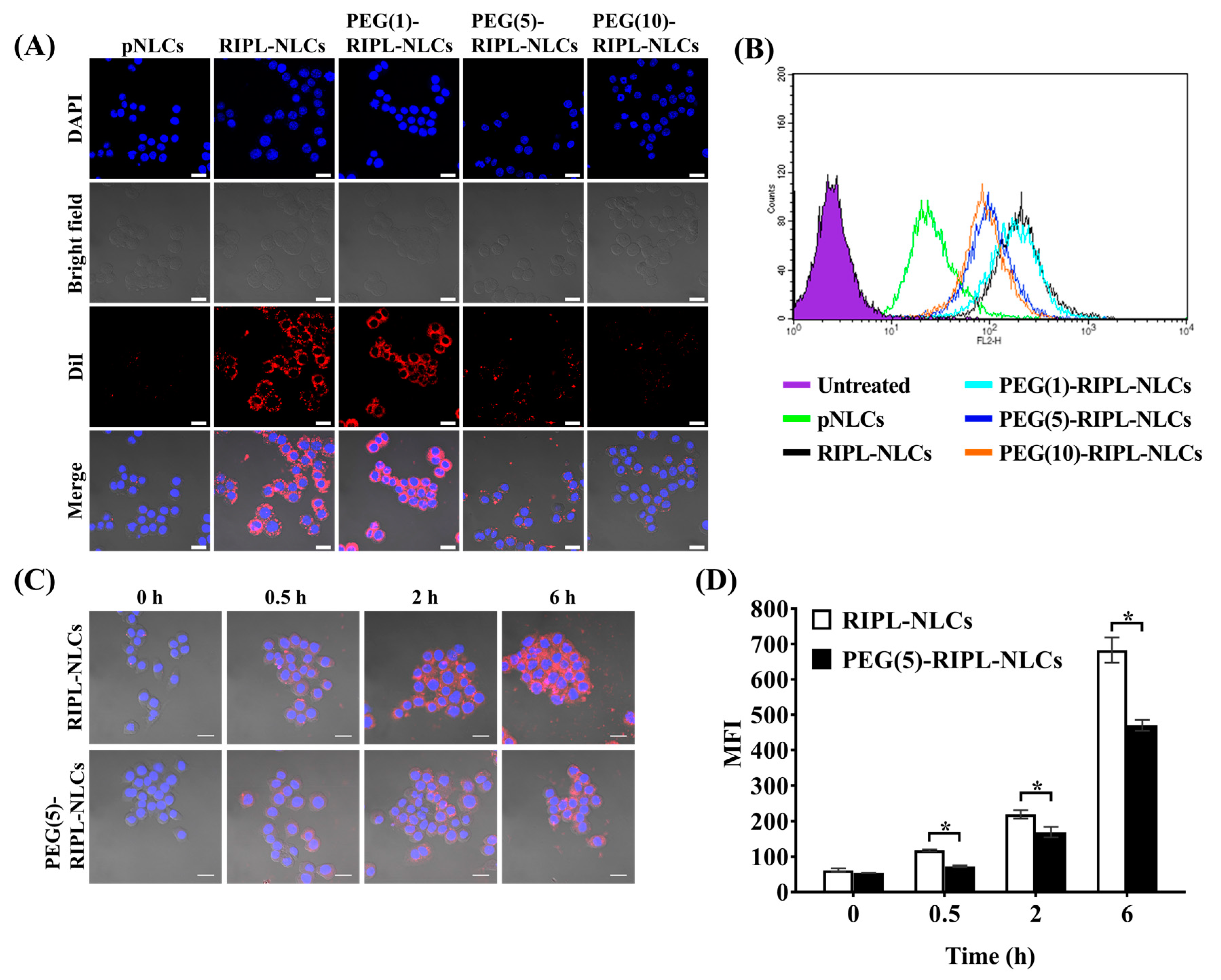
Macrophage phagocytosis was analyzed qualitatively and quantitatively using RAW 264.7 cells. Cells were seeded in a Lab-Tek II chamber slide with a cover (Thermo Scientific Nunc, Rochester, NY, USA) at a density of 5 × 104 cells/well for a qualitative assay and a in a 6-well plate at a density of 3 × 105 cells/well for a quantitative assay. After 24 h of incubation, the media were removed; the cells were washed twice with PBS and incubated with culture media containing various DiI-loaded samples (DiI concentration of 100 ng/mL) for 2 h. For qualitative observation, cells were fixed with 4% paraformaldehyde for 15 min, mounted using a mounting medium with 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, CA, USA) to prevent fading and to stain the nuclei, and visualized using a confocal microscope (LSM 700 Meta; Carl Zeiss, Jena, Germany) under a 400× magnification. For quantitative analysis, cells were detached from the wells using trypsin-EDTA, resuspended in 1 mL of PBS, and the mean fluorescence intensity (MFI) was measured using a flow cytometer (FACSCalibur; Becton Dickinson, Franklin Lakes, NJ, USA). Only viable cells were gated and analyzed by counting 10,000 events using the FL2 channel with the CellQuest Pro software (Becton Dickinson, Franklin Lakes, NJ, USA). All experiments were performed in triplicate. Additionally, time-dependent inhibition of phagocytosis was investigated for selected samples [DiI-loaded RIPL-NLCs and PEG(5)-RIPL-NLCs] at predetermined time points (0, 0.5, 2, and 6 h).
2.13. Cellular Uptake Study
The intracellular delivery of DiI-loaded NLC formulations was examined qualitatively and quantitatively using the hepsin-expressing SKOV3, MCF7, and LNCaP cancer cell lines incubated in a serum-free RPMI 1640 medium. In addition, the cellular uptake mechanism of the nanocarriers was investigated using endocytosis inhibitors and a low temperature as reported [
23]. Briefly, SKOV3 cells were seeded in 6-well plates at a density of 3 × 10
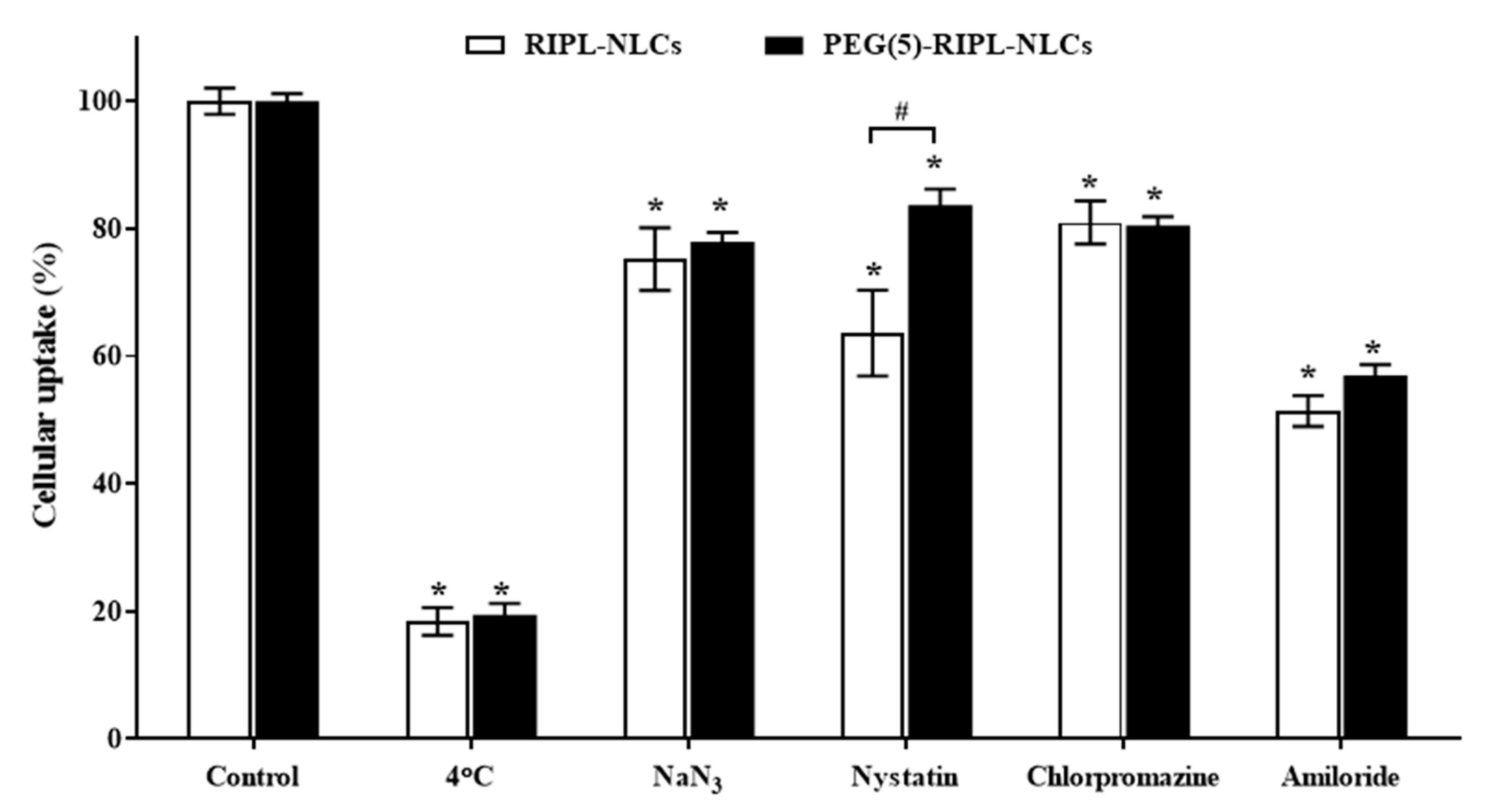
5 cells/well and preincubated at 37 °C for 1 h in the presence of endocytosis inhibitors such as sodium azide (1 mg/mL), amiloride (500 μg/mL), chlorpromazine (10 μg/mL), and nystatin (25 μg/mL), as well as at a lower temperature (4 °C). Untreated cells served as controls. Following preincubation, the cells were treated with DiI-loaded RIPL-NLCs and PEG(5)-RIPL-NLCs, and MFI was determined as described above.
2.14. Cytotoxicity Assessment
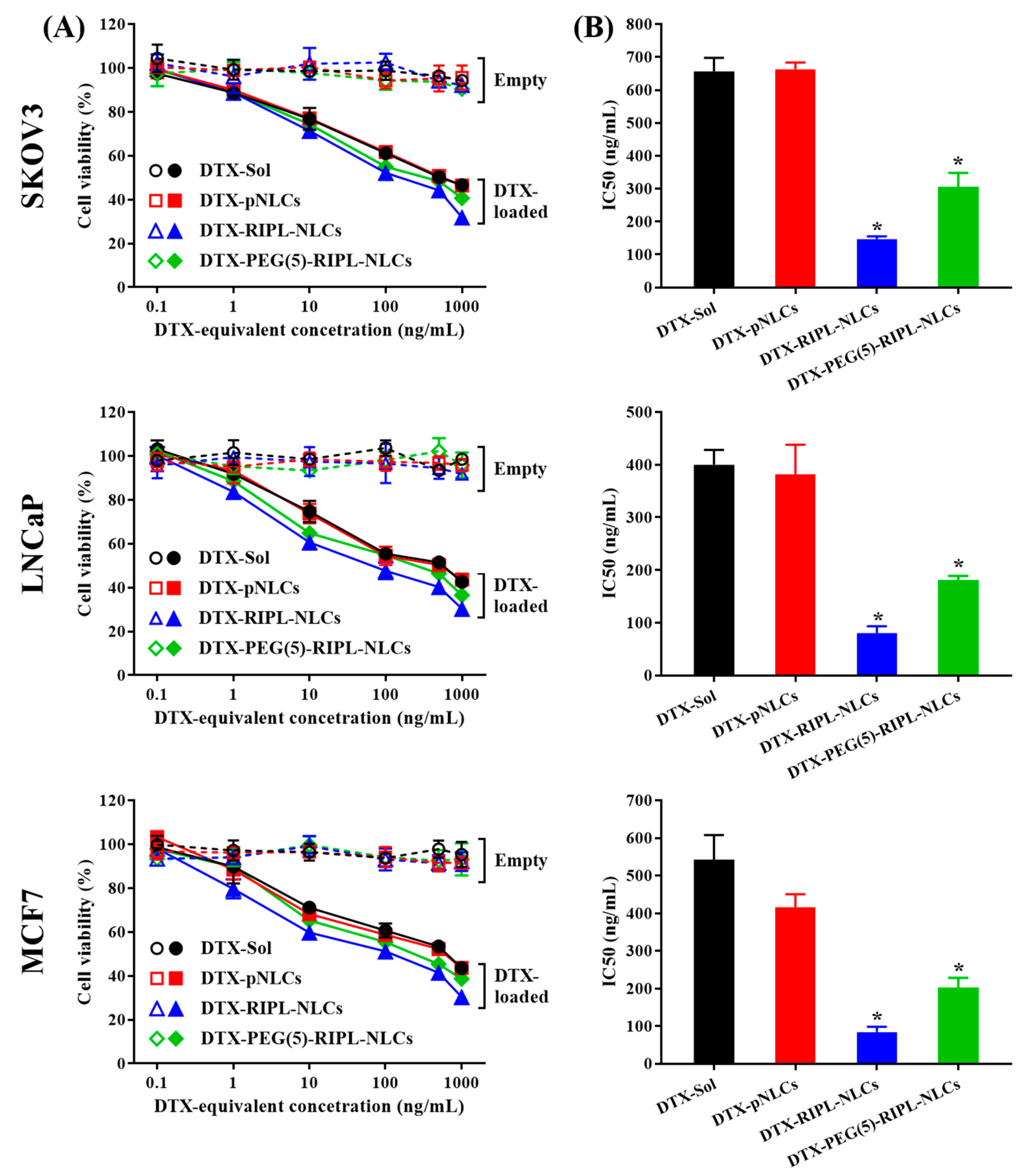
In vitro cytotoxicity of DTX-free and DTX-loaded formulations was determined by the (Water-soluble tetrazolium salt) WST-1 assay, as previously reported [
24]. Briefly, SKOV3, MCF7, and LNCaP cells were seeded in RPMI 1640 medium into 96-well plates at a density of 5000 cells per well. After reaching 70–80% confluence, the cells were incubated with the growth medium containing DTX-free or DTX-loaded NLC formulations (DTX-equivalent concentrations of 0.1, 1, 10, 100, 500, and 1000 ng/mL) at 37 °C for 24 h. Then, the cells were washed with PBS and incubated with a 10% WST-1 reagent (EZ-cytox; Daeil Lab Service, Seoul, Korea) for 30 min at 37 °C. The absorbance of the WST-1 formazan dye was measured at 450 nm using a microplate reader. Cell viability was measured as a percentage of viable cells relative to the untreated control. Half-maximal inhibitory concentration (IC
50) values were determined by plotting cell viability against the DTX-equivalent concentration on a log scale.
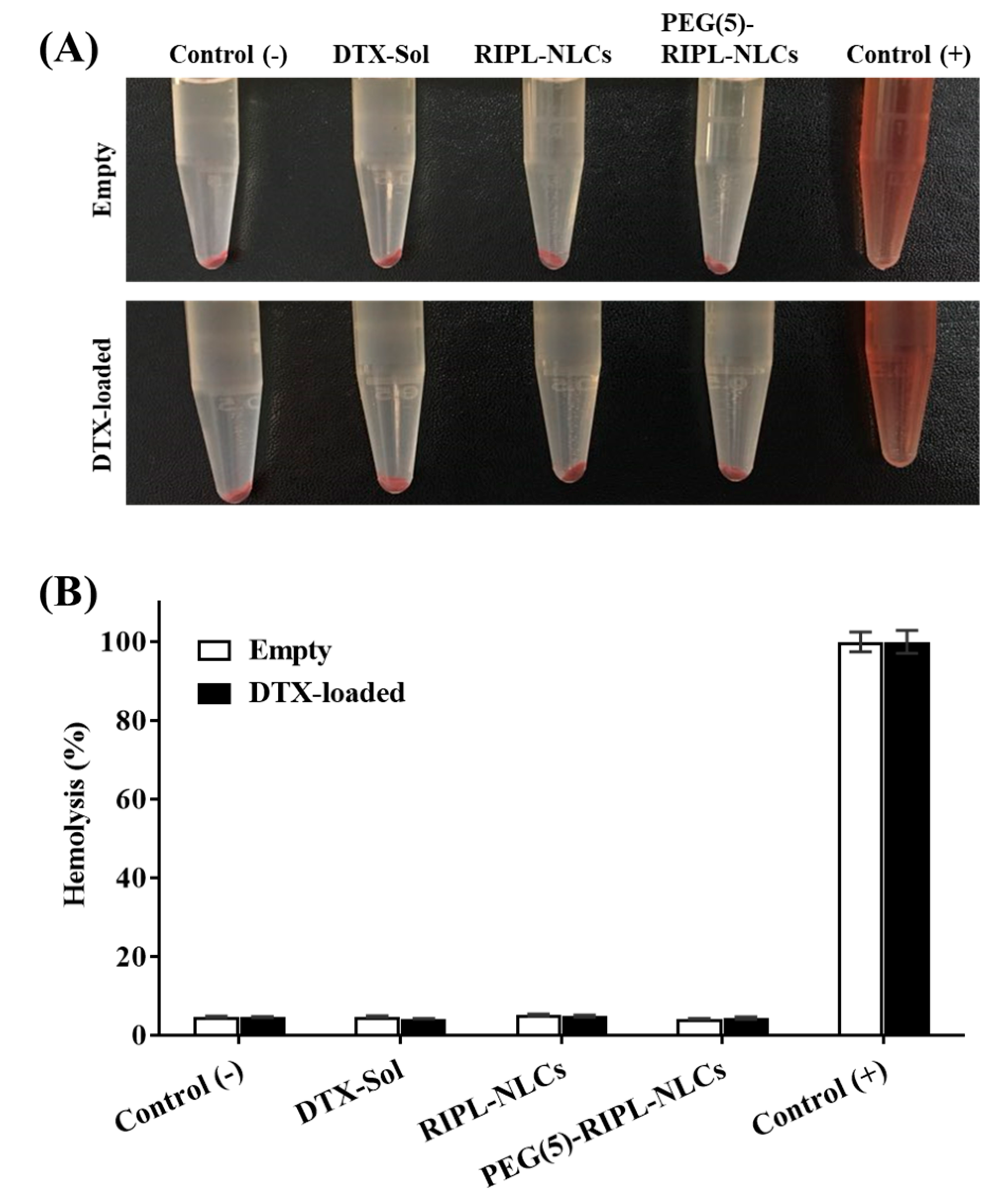
2.15. Hemolysis Test
The hemolytic activity of NLCs was determined as described previously to evaluate their biocompatibility for potential applications via intravenous injection [
25]. Fresh blood was collected from ICR mice into heparin-treated tubes and was immediately centrifuged at 1000×
g for 15 min at 4 °C to separate red blood cells (RBCs). RBCs were washed twice with an isotonic saline solution at a 1:4 (
v/
v) ratio by centrifugation at 1000×
g for 15 min and afterward resuspended, diluted with the saline solution, and stored at 4 °C. The obtained RBC suspension (2%) was used for the assay within 24 h.
DTX-free and DTX-loaded formulations were diluted with the saline solution to a DTX-equivalent concentration of 10 μg/mL and mixed with the RBC suspension at a 1:1 (v/v) ratio. The saline solution with/without Triton X-100 (1%, v/v) was used as a positive control (100% hemolysis) and a negative control (0% hemolysis), respectively. Samples were incubated in a shaking incubator (SI-900R; Jeio Tech, Gyeonggi-do, Korea) at 37 °C for 1 h at 100 rpm, followed by centrifugation at 1500× g for 10 min, and the tubes were then photographed. To quantify the amount of hemoglobin released, absorbance of the supernatant was measured spectrophotometrically at 540 nm using a microplate reader. The rate of hemolysis (%) was calculated as (A − A0)/(A100 − A0) × 100, where A is the absorbance of the sample, while A0 and A100 are the absorbance values of the negative and positive control, respectively.
2.16. Biodistribution
2.16.1. Administration of Formulations and Tissue Sampling
The animal experiment was approved by the Institutional Animal Care and Use Committee of Chung-Ang University (2018-00090, 3 August 2018, Seoul, Korea) and was carried out in accordance with the National Institute of Health Guidelines for the Care and Use of Laboratory Animals. Female ICR mice were randomly divided into three groups (n = 15–20) and injected intravenously via the tail vein with DTX-Sol, DTX-RIPL-NLCs, or DTX-PEG-RIPL-NLCs at a dose of 5 mg DTX-equivalent/kg. At 0.5, 2, and 6 h after the injection, the mice were anesthetized, and 0.5 mL of blood was collected from the retro-orbital plexus into a heparinized tube. The blood samples were centrifuged at 13,000× g for 20 min to separate the plasma, which was frozen at −80 °C until analysis. After blood collection, the mice were sacrificed, and the lung, heart, liver, spleen, and kidneys were harvested, washed with PBS, weighed after removing excess fluid, and preserved in a −80 °C freezer.
2.16.2. Sample Pretreatment
The frozen tissues were thawed at room temperature and sectioned. The sections were weighed and mixed with PBS at a ratio of 1:2 (w/v) for the liver, lung, and kidney and 1:4 (w/v) for the heart and spleen. The mixtures were placed into 2 mL tubes with four steel beads and homogenized at 3000× g using a Bead Blaster 24 homogenizer (Benchmark Scientific, Edison, NJ, USA) twice for 60 s each with a 15 s interval. The resulting homogenates were stored at −80 °C until analysis. For analysis of DTX, 100 μL of tissue homogenates or plasma samples were mixed with 20 μL of methanol and 900 μL of ether. The samples were vortexed for 5 min and centrifuged at 13,000 rpm for 5 min at 4 °C. The organic phase was transferred to another tube and evaporated to dryness at 40 °C under a stream of N2 gas. The residue was reconstituted in 100 μL of 30% acetonitrile, and the samples were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS).
2.16.3. DTX Determination by LC-MS/MS
An atmospheric pressure ionization (API) 2000 triple quadrupole mass spectrometer (ABI SCIEX, Foster City, CA, USA) and an LC-20 Prominence HPLC system (Shimadzu, Tokyo, Japan) were used for LC-MS/MS analysis of DTX. The systems were interfaced through electrospray ionization (ESI) in a positive ion mode. For LC, a Phenomenex Luna C18 column (2.0 × 150 mm, 5 μm) and an isocratic mobile phase consisting of 15% water and 85% acetonitrile (v/v) were used. The autosampler and the column oven were maintained at 4 and 40 °C, respectively. The ESI source parameters were set as follows: spray voltage, 5500 V; spray temperature, 350 °C; gas 1, 30 psi; gas 2, 34 psi; collision gas, 6 psi; curtain gas, 16 psi. For sensitive and selective identification of DTX, multiple reaction monitoring (MRM) was carried out. As the precursor ion for MRM, the Na adduct ion of DTX, observed at 829.9 m/z, was selected. The MRM precursor/product ion transitions used for DTX in the present study were m/z 829.9/548.9 (screening transition), 829.9/303.8 (the first confirmatory transition), and 829.9/248.0 (the second confirmatory transition). All data were acquired and analyzed using Analyst, version 1.5.2 (ABI SCIEX, Foster City, CA, USA).
2.17. Statistical Analysis
All values were expressed as the mean ± standard deviation (SD) (n = 3). Data were analyzed through statistical product and service solutions (SPSS) 19.0 software (USA). Statistical significance was determined using the analysis of variance (ANOVA) followed by a Tukey’s test for post hoc multiple comparisons, and differences were considered significant at p < 0.05.
4. Discussion
In the present study, RIPL-NLCs were sterically stabilized with PEG3000 (PEG-RIPL-NLCs) to prevent the recognition by MPS. The particle sizes of RIPL-NLCs (260 to 277 nm) were reduced by PEGylation (178 to 238 nm). As the modification ratio of PEG3000 increased from 1 to 10 mole %, the sizes of PEG-RIPL-NLCs decreased, which could be due to the amphiphilic nature of DSPE-PEG3000, leading to the reduction of the interfacial tension between the aqueous and organic phase during preparation [
29]. While nanoparticles with hydrodynamic radii of <10 nm can be rapidly cleared from the systemic circulation by renal filtration, NLC formulations of approximately 200 nm may effectively accumulate in tumors via the enhanced permeability and retention effect [
30]. In comparison to RIPL-NLCs, PEG-RIPL-NLCs showed a decreased tendency for surface charges, depending on the degree of PEGylation, probably due to reduced surface exposure of RIPL peptide molecules, which were overlaid with longer PEG segments [
31]. In general, accumulation of PEG3000 on the surface of NLCs may attract water molecules to the interior of the lipid-based carrier, resulting in the modulation of drug release [
32]. In this study, the surface modification of NLCs with the RIPL peptide and/or PEG3000 somewhat increased the DTX release. However, there was no significant difference between them, possibly due to the high solubilizing capacity of the release medium containing sodium dodecyl sulfate, which increased the wetting of nanoparticle, thereby facilitating the DTX dissolution.
We investigated steric stabilization of NLCs, depending on the PEG grafting density, by evaluating the protein adsorption by NLCs and the uptake of NLCs by macrophages. The matrix-type carrier, pNLCs, exhibited low protein adsorption and a negligible uptake by macrophages. This serum stability and phagocytosis avoidance of pNLCs are possibly due to the hydrophilic surfactant (polysorbate 20), which stabilizes the surface [
33]. However, in the case of RIPL-NLCs, large protein amounts were rapidly adsorbed, resulting in high phagocytosis. This high protein adsorption seems to be mainly due to the van der Waals interaction between the positively charged RIPL peptide, exposed on the NLC surface, and BSA [
15]. PEG-RIPL-NLCs showed increases in the inhibition of protein adsorption and phagocytosis as the PEG density increased. This stealth effect of PEGylation may be explained by steric repulsion and high mobility of longer PEG chains, which may prevent the RIPL peptide from interacting with serum proteins [
11]. In general, 1.5–5-kDa PEG chains, with a grafting density of 5–10 mole %, show a good stealth effect with increased hydrophilicity [
8,
34]. Furthermore, PEG chains can acquire different conformations, depending on the grafting density [
35]. At a low grafting density (up to 4 mole %), PEG chains show a “mushroom” conformation, whereas a “brush” conformation is acquired at a higher grafting density [
36]. The ideal coverage to provide the most effective opsonin repulsion has been described as intermediate between these configurations [
37]. Considering factors such as the molecular weight, grafting density, and the conformation of PEG, the modification with 5 mole % PEG3000 was selected to achieve an efficient surface coverage of RIPL-NLCs.
The in vitro cellular uptake study provided some evidence of the advantages of RIPL peptide-modified NLC formulations for effective internalization into cancer cells. After PEGylation, the degree of internalization of PEG(5)-RIPL-NLCs was reduced, but the uptake pathway was not greatly changed. The cellular uptake efficiencies of RIPL-NLCs and PEG(5)-RIPL-NLCs were remarkably higher than that of pNLCs, demonstrating that the RIPL peptide facilitated the uptake by hepsin-expressing target cells, which could be attributed to the cell-penetrating and homing function of the RIPL peptide, as previously reported [
5]. However, because of the shielding effect of PEG, which interferes with the ligand–receptor interaction [
38], the cellular uptake of PEG(5)-RIPL-NLCs was lower than that of RIPL-NLCs. On the other hand, to investigate the uptake mechanism of RIPL-modified NLC formulations, several inhibitors, such as nystatin, chlorpromazine, sodium azide, and amiloride, as well as a low temperature, were tested. It is well-known that the endocytic uptake is an energy-dependent process [
39]. The results of competition between low temperature and a metabolic inhibitor (sodium azide) displayed a strong impact on the cellular uptake, indicating that the uptake process of both NLC formulations was energy-dependent. Moreover, internalization was significantly restrained by pretreatment with amiloride, nystatin, and chlorpromazine. Among these inhibitors, amiloride caused the greatest inhibition, indicating that macropinocytosis may be the main mechanism of endocytosis of NLCs. Additionally, both caveolin- and clathrin-mediated endocytosis pathways are involved because pretreatments with nystatin and chlorpromazine showed inhibitory effects. In particular, nystatin pretreatment greatly inhibited the uptake of RIPL-NLCs, showing a significant difference from that of PEG(5)-RIPL-NLCs. Thus, we could conclude that PEGylation affected the caveolin-mediated uptake of RIPL-NLCs, even though the detailed mechanism remains unclear. Since the RIPL peptide is a cell-penetrating and homing peptide, containing an R8 domain [
5], non-specific intracellular uptake, including caveolin-mediated endocytosis, is expected. Consequently, we could suggest that the cellular uptake of both RIPL-NLCs and PEG(5)-RIPL-NLCs was energy-dependent and mainly governed by macropinocytosis, although complex mechanisms were involved.
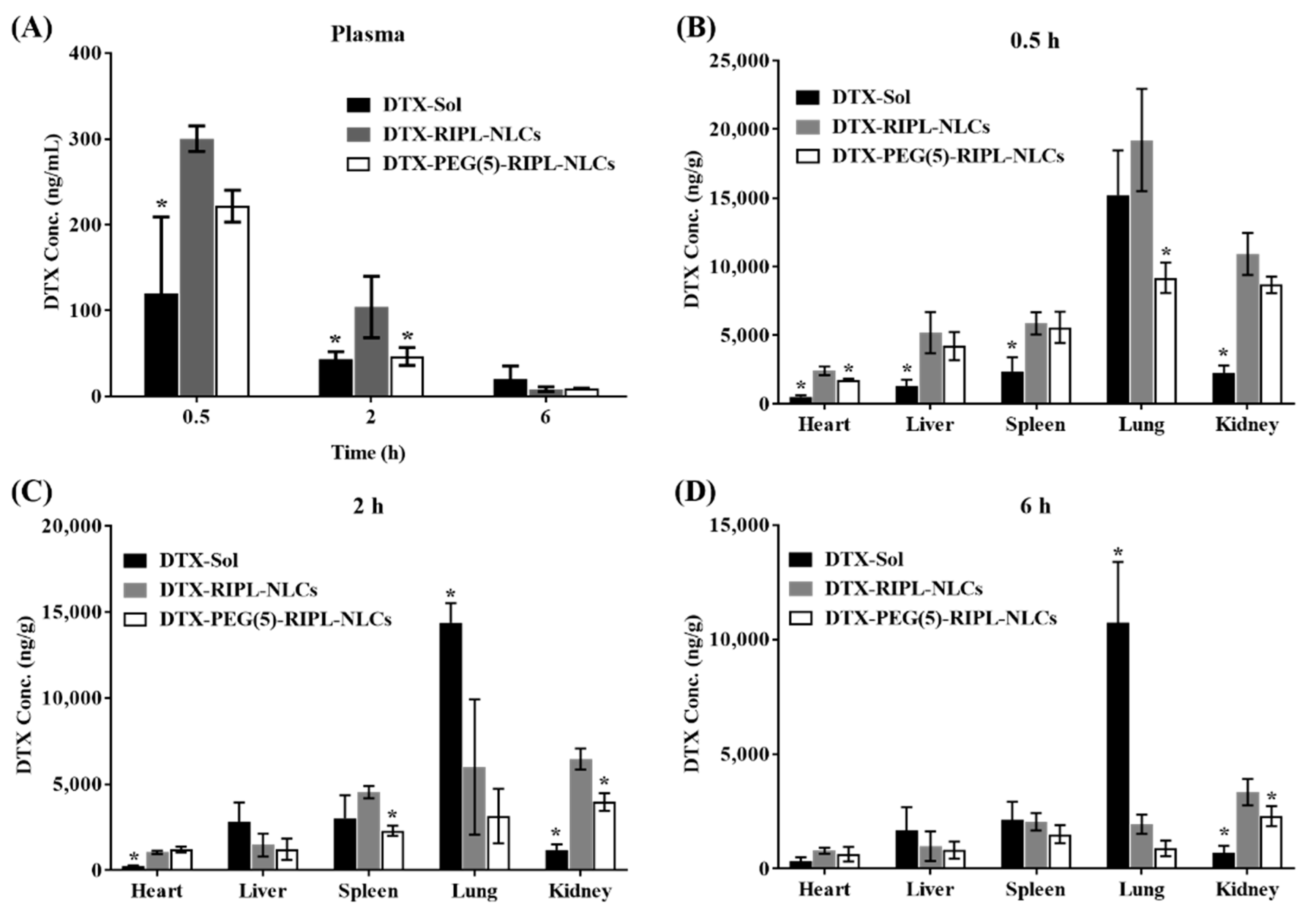
Throughout the in vivo biodistribution study in the plasma and representative organs of mice, treatment with DTX-loaded NLC formulations resulted in higher DTX levels than did that with DTX-Sol, even though the distribution patterns were inconsistent, likely due to DTX encapsulation in NLCs. Owing to the solid state of lipid matrix-type nanocarriers, NLCs undergo slow degradation in the body, which explains the slow, controlled DTX release [
40]. However, unexpectedly, the plasma DTX concentration in mice treated with RIPL-NLCs was higher than that in mice administered PEG(5)-RIPL-NLCs. It was considered that the high protein-binding capacity of RIPL-NLCs could affect their blood level. Especially for a highly protein-bound drug, as free drug is cleared by the kidney, the drug–protein complex dissociates, acting as a “depot” that helps maintain more stable plasma levels [
41]. In comparison, instead of the total drug measurement, the separated determination of protein-bound, free, and carrier-associated drugs is encouraged [
42,
43].
The organ distribution results demonstrated a close relationship with the structure of the blood capillary endothelium. DTX accumulation in the lung, which has a continuous endothelial barrier, was remarkably high compared with that in the other organs. DTX-Sol showed high accumulation up to 6 h, while DTX-RIPL-NLCs and DTX-PEG(5)-RIPL-NLCs exhibited decreased accumulation as the time passed, which may be attributed to the fact that the tissue/organ distribution should be accompanied by diffusion of drug molecules across the barrier [
44]. Thus, the nanoparticulates DTX-RIPL-NLCs and DTX-PEG(5)-RIPL-NLCs would not support continuous molecular diffusion since the drug molecules should be released from the nanoparticles prior to the diffusional absorption. On the contrary, DTX distribution to the kidney, which has a fenestrated endothelial barrier, was different. DTX-RIPL-NLCs and DTX-PEG(5)-RIPL-NLCs showed higher accumulation than did DTX-Sol, which may be explained by easy excretion of dissolved drug molecules in the urine, whereas the urinary excretion of both NLC formulations should be limited. Meanwhile, compared to DTX-Sol, both NLC formulations exhibited greater accumulation, especially at an earlier time point (0.5 h), in the liver and spleen, which possess a sinusoidal endothelial barrier. However, after 2 h, the accumulation pattern changed, resulting in a relatively high accumulation of DTX-Sol. More importantly, in virtue of PEGylation, DTX-PEG(5)-RIPL-NLCs could avoid the recognition by MPS and showed a reduced uptake by macrophages [
45]. Compared with that of DTX-RIPL-NLCs, the accumulation of DTX-PEG(5)-RIPL-NLCs in the liver and spleen was somewhat low and was further reduced after 2 h, particularly in the spleen. This behavior may be considered as proving the stealth effect of PEGylation. However, a pharmacodynamic investigation in a suitable animal cancer model and comprehensive pharmacokinetic studies of both NLC formulations are needed for their practical development in the future.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}