
Convergent Synthesis of Two Fluorescent Ebselen-Coumarin Heterodimers



Abstract
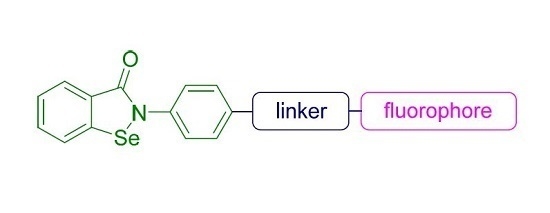
:
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General Methods and Materials
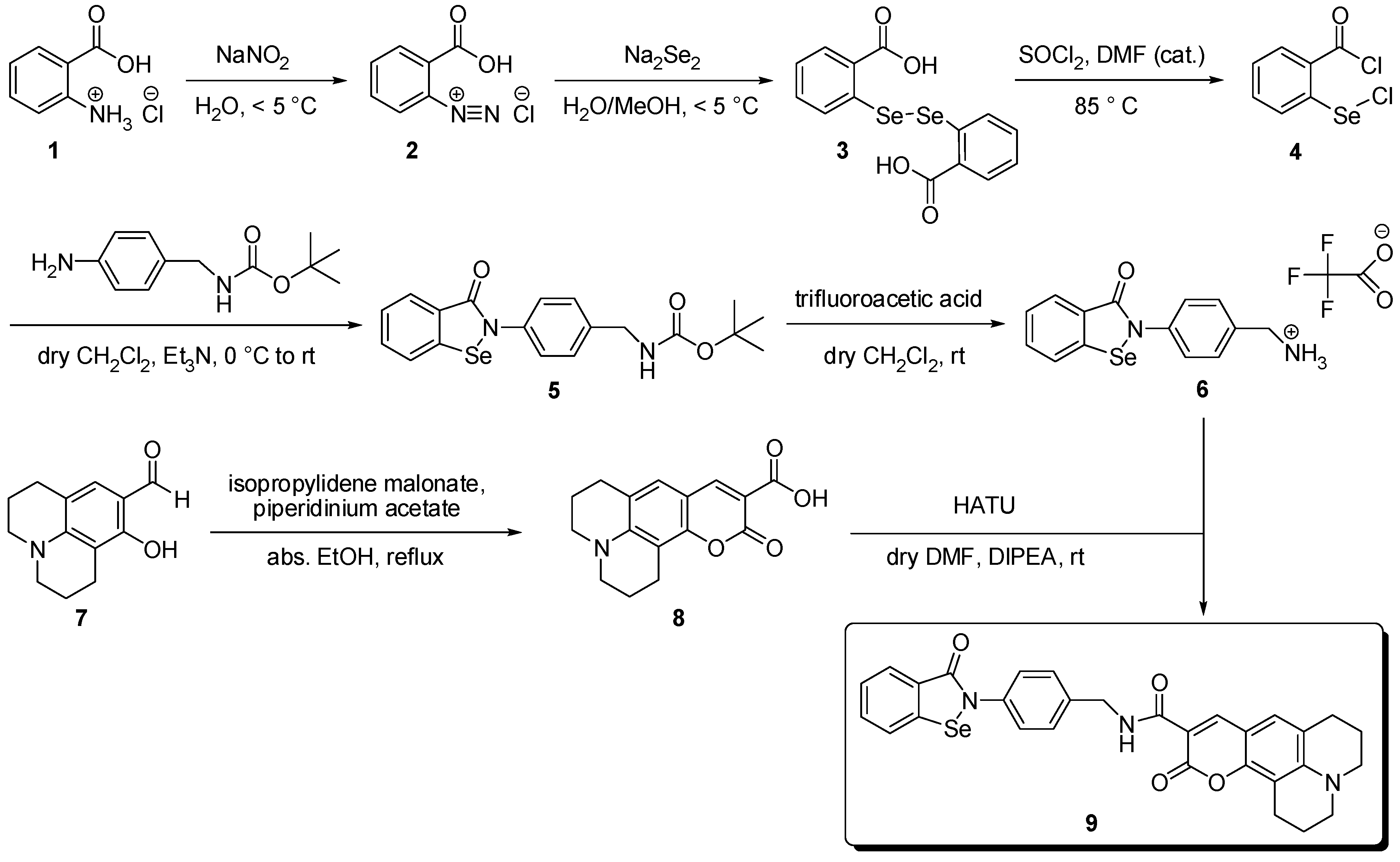
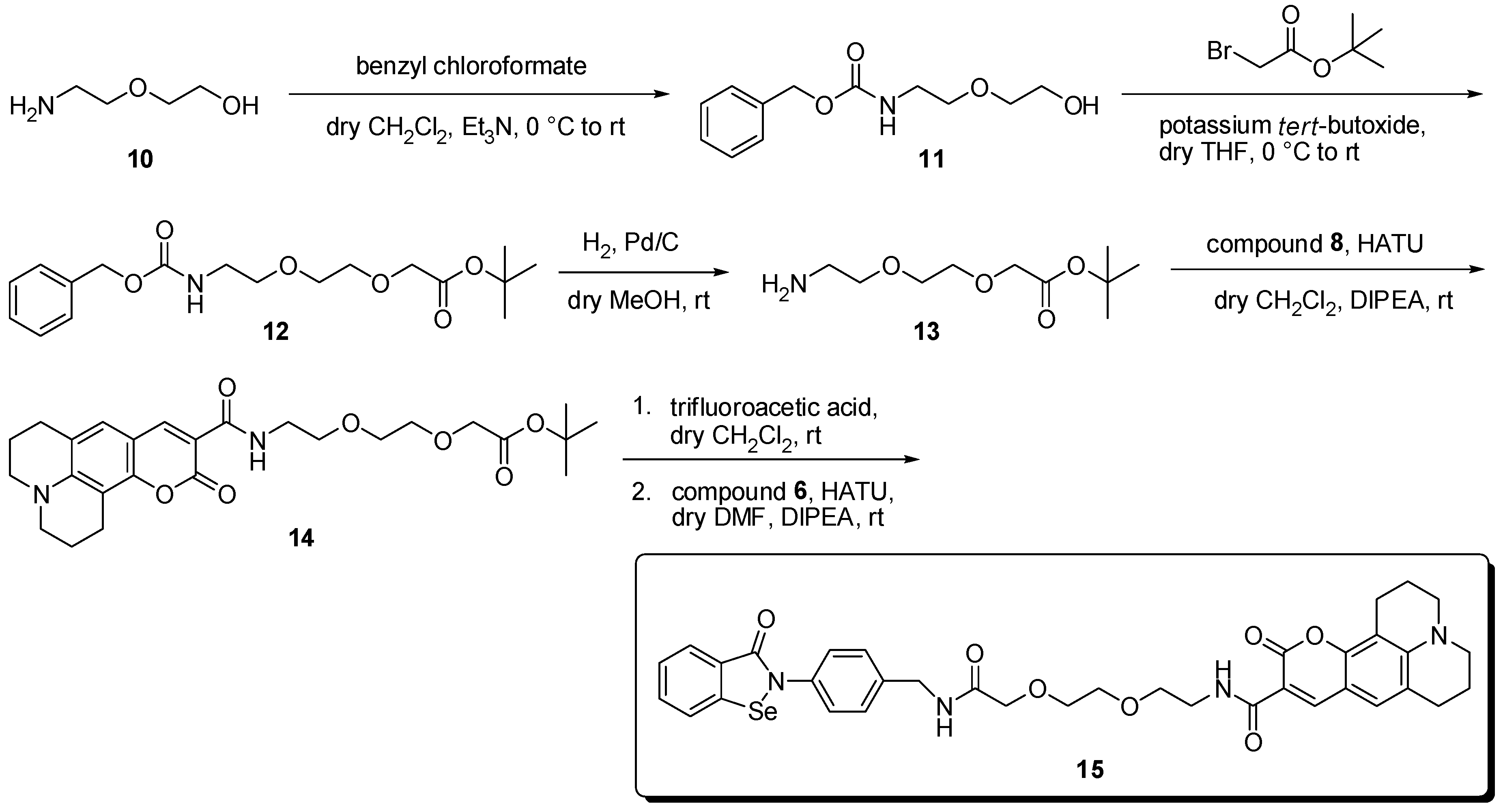
3.2. Syntheses
3.3. Modified Enzymatic Assays
Supplementary Material
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| DIPEA | ethyldiisopropylamine |
| DTT | dithiothreitol |
| GPx | glutathione peroxidase |
| HAUT | 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate |
| PMT | photomultiplier tube |
| TrxR | thioredoxin reductase |
References
- Azad, G.K.; Tomar, R.S. Ebselen, a promising antioxidant drug: Mechanisms of action and targets of biological pathways. Mol. Biol. Rep. 2014, 41, 4865–4879. [Google Scholar] [CrossRef] [PubMed]
- Kálai, T.; Mugesh, G.; Roy, G.; Sies, H.; Berente, Z.; Hideg, K. Combining benzo[d]isoselenazol-3-ones with sterically hindered alicyclic amines and nitroxides: Enhanced activity as glutathione peroxidase mimics. Org. Biomol. Chem. 2005, 3, 3564–3569. [Google Scholar]
- Parnham, M.; Sies, H. Ebselen: Prospective therapy for cerebral ischaemia. Exp. Opin. Investig. Drugs 2000, 9, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Stoyanovsky, D.A.; Jiang, J.; Murphy, M.P.; Epperly, M.; Zhang, X.; Li, S.; Greenberger, J.; Kagan, V.; Bayır, H. Design and synthesis of a mitochondria-targeted mimic of glutathione peroxidase, MitoEbselen-2, as a radiation mitigator. ACS Med. Chem. Lett. 2014, 5, 1304–1307. [Google Scholar] [CrossRef] [PubMed]
- Bhabak, K.P.; Mugesh, G. Synthesis, characterization, and antioxidant activity of some ebselen analogues. Chem. Eur. J. 2007, 13, 4594–4601. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Liu, S.; Miyake, M.; Liu, K.J. Ebselen induced C6 glioma cell death in oxygen and glucose deprivation. Chem. Res. Toxicol. 2006, 19, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Borges, V.C.; Rocha, J.B.; Nogueira, C.W. Effect of diphenyl diselenide, diphenyl ditelluride and ebselen on cerebral Na+, K+-ATPase activity in rats. Toxicology 2005, 215, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Terentis, A.C.; Freewan, M.; Sempértegui Plaza, T.S.; Raftery, M.J.; Stocker, R.; Thomas, S.R. The selenazal drug ebselen potently inhibits indoleamine 2,3-dioxygenase by targeting enzyme cysteine residues. Biochemistry 2010, 49, 591–600. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Masayasu, H.; Holmgren, A. Ebselen: A substrate for human thioredoxin reductase strongly stimulating its hydroperoxide reductase activity and a superfast thioredoxin oxidant. Proc. Natl. Acad. Sci. USA 2002, 99, 8579–8584. [Google Scholar] [CrossRef] [PubMed]
- Arnér, E.S.; Holmgren, A. The thioredoxin system in cancer. Semin. Cancer Biol. 2006, 16, 420–426. [Google Scholar]
- Kłossowski, S.; Muchowicz, A.; Firczuk, M.; Swiech, M.; Redzej, A.; Golab, J.; Ostaszewski, R. Studies toward novel peptidomimetic inhibitors of thioredoxin-thioredoxin reductase system. J. Med. Chem. 2012, 55, 55–67. [Google Scholar]
- Luo, Z.; Sheng, J.; Sun, Y.; Lu, C.; Yan, J.; Liu, A.; Luo, H.B.; Huang, L.; Li, X. Synthesis and evaluation of multi-target-directed ligands against Alzheimer’s disease based on the fusion of donepezil and ebselen. J. Med. Chem. 2013, 56, 9089–9099. [Google Scholar] [CrossRef] [PubMed]
- Terai, T.; Nagano, T. Fluorescent probes for bioimaging applications. Curr. Opin. Chem. Biol. 2008, 12, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Elsinghorst, P.W.; Härtig, W.; Goldhammer, S.; Grosche, J.; Gütschow, M. A gorge-spanning, high-affinity cholinesterase inhibitor to explore beta-amyloid plaques. Org. Biomol. Chem. 2009, 7, 3940–3946. [Google Scholar] [CrossRef] [PubMed]
- Nizamov, S.; Willig, K.I.; Sednev, M.V.; Belov, V.N.; Hell, S.W. Phosphorylated 3-heteroarylcoumarins and their use in fluorescence microscopy and nanoscopy. Chem. Eur. J. 2012, 18, 16339–16348. [Google Scholar] [CrossRef] [PubMed]
- Galdeano, C.; Viayna, E.; Sola, I.; Formosa, X.; Camps, P.; Badia, A.; Clos, M.V.; Relat, J.; Ratia, M.; Bartolini, M.; et al. Huprine-tacrine heterodimers as anti-amyloidogenic compounds of potential interest against Alzheimer’s and prion diseases. J. Med. Chem. 2012, 55, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Mertens, M.D.; Schmitz, J.; Horn, M.; Furtmann, N.; Bajorath, J.; Mareš, M.; Gütschow, M. A coumarin-labeled vinyl sulfone as tripeptidomimetic activity-based probe for cysteine cathepsins. ChemBioChem 2014, 15, 955–959. [Google Scholar] [CrossRef] [PubMed]
- Meimetis, L.G.; Carlson, J.C.; Giedt, R.J.; Kohler, R.H.; Weissleder, R. Ultrafluorogenic coumarin-tetrazine probes for real-time biological imaging. Angew. Chem. Int. Ed. 2014, 53, 7531–7534. [Google Scholar] [CrossRef] [PubMed]
- Ostrowska, K.; Hejchman, E.; Maciejewska, D.; Włodarczyk, A.; Wojnicki, K.; Matosiuk, D.; Czajkowska, A.; Młynarczuk-Biały, I.; Dobrzycki, Ł. Microwave-assisted preparation, structural characterization, lipophilicity and anti-cancer assay of some hydroxycoumarin derivatives. Monatshefte Chem. 2015, 146, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Kohl, F.; Schmitz, J.; Furtmann, N.; Schulz-Fincke, A.C.; Mertens, M.D.; Küppers, J.; Benkhoff, M.; Tobiasch, E.; Bartz, U.; Bajorath, J.; et al. Design, characterization and cellular uptake studies of fluorescence-labeled prototypic cathepsin inhibitors. Org. Biomol. Chem. 2015, 13, 10310–10323. [Google Scholar] [CrossRef] [PubMed]
- Kerkovius, J.K.; Menard, F. A practical synthesis of 6,8-difluoro-7-hydroxycoumarin derivatives for fluorescence applications. Synthesis 2016, 48, 1622–1629. [Google Scholar]
- Takechi, H.; Kamada, S.; Machida, M. 3-[4-(Bromomethyl)phenyl]-7-(diethylamino)-2H-1-benzopyran-2-one (MPAC-Br): A highly sensitive fluorescent derivatization reagent for carboxylic acids in high-performance liquid chromatography. Chem. Pharm. Bull. 1996, 44, 793–799. [Google Scholar] [CrossRef]
- Muller, C.; Even, P.; Viriot, M.L.; Carré, M.C. Protection and labelling of thymidine by a fluorescent photolabel group. Helv. Chim. Acta 2001, 84, 3735–3741. [Google Scholar] [CrossRef]
- Woodroofe, C.C.; Lippard, S.J. A novel two-fluorophore approach to ratiometric sensing of Zn2+. J. Am. Chem. Soc. 2003, 125, 11458–11459. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Zhou, X.; Huang, B.; Zhang, X.; Chen, C. On-gel fluorescent visualization and the site identification of S-nitrosylated proteins. Anal. Biochem. 2008, 377, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Kim, T.K.; Lee, J.H.; Kim, H.J.; Hong, J.I. Fluorescence turn-on probe for homocysteine and cysteine in water. Chem. Commun. 2008, 6173–6175. [Google Scholar] [CrossRef] [PubMed]
- Simmons, J.T.; Allen, J.R.; Moris, D.R.; Clark, R.J.; Levenson, C.W.; Davidson, M.W.; Zhu, L. Integrated and passive 1,2,3-triazolyl groups in fluorescent indicators for zink(II) ions: Thermodynamic and kinetic evaluations. Inorg. Chem. 2013, 52, 5838–5850. [Google Scholar] [CrossRef] [PubMed]
- Palus, J.; Młochowski, J.; Juchniewicz, L. 2,2’-Diselenobisbenzoates and 2,2’-Diselenobisbenzenesulfonates: New Chiral Aryl Diselenides. Polish J. Chem. 1998, 72, 1931–1936. [Google Scholar]
- Adamczyk, M.; Fishpaugh, J.R.; Thiruvazhi, M. Concise synthesis of N-protected carboxyalkyl ether amines. Org. Prep. Proced. Int. 2002, 34, 326–331. [Google Scholar] [CrossRef]
- Xu, K.; Qiang, M.; Gao, W.; Su, R.; Li, N.; Gao, Y.; Xie, Y.; Kong, F.; Tang, B. A near-infrared reversible fluorescent probe for real-time imaging of redox status changes in vivo. Chem. Sci. 2013, 4, 1079–1086. [Google Scholar] [CrossRef]
- Ghosh, H.N. Charge transfer emission in coumarin 343 sensitized TiO2 nanoparticle: A direct measurement of back electron transfer. J. Phys. Chem. B 1999, 103, 10382–10387. [Google Scholar] [CrossRef]
- Webb, M.R.; Corrie, J.E. Fluorescent coumarin-labeled nucleotides to measure ADP release from actomyosine. Biophys. J. 2001, 81, 1562–1569. [Google Scholar] [CrossRef]
- Murase, T.; Yoshihara, T.; Yamada, K.; Tobita, S. Fluorescent peptides labeled with environment-sensitive 7-aminocoumarins and their interactions with lipid bilayer membranes and living cells. Bull. Chem. Soc. Jpn. 2013, 86, 510–519. [Google Scholar] [CrossRef]
- He, J.; Li, D.; Xiong, K.; Ge, Y.; Jin, H.; Zhang, G.; Hong, M.; Tian, Y.; Yin, J.; Zeng, H. Inhibition of thioredoxin reductase by a novel series of bis-1,2-benzisoselenazol-3(2H)-ones: Organoselenium compounds for cancer therapy. Bioorg. Med. Chem. 2012, 20, 3816–3827. [Google Scholar] [CrossRef] [PubMed]
- Mao, F.; Chen, J.; Zhou, Q.; Luo, Z.; Huang, L.; Li, X. Novel tacrine-ebselen hybrids with improved cholinesterase inhibitory, hydrogen peroxide and peroxynitrite scavenging activity. Bioorg. Med. Chem. Lett. 2013, 23, 6737–6742. [Google Scholar] [CrossRef] [PubMed]
- Azad, G.K.; Singh, V.; Mandal, P.; Singh, P.; Golla, U.; Baranwal, S.; Chauhan, S.; Tomar, R.S. Ebselen induces reactive oxygen species (ROS)-mediated cytotoxicity in Saccharomyces cerevisiae with inhibition of glutamate dehydrogenase being a target. FEBS Open Bio 2014, 4, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Zhang, Y.; Tang, B.; Laskin, J.; Roach, P.J.; Chen, H. Study of highly selective and efficient thiol derivatization using selenium reagents by mass spectrometry. Anal. Chem. 2010, 82, 6926–6932. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Vlamis-Gardikas, A.; Kandasamy, K.; Zhao, R.; Gustafsson, T.N.; Engstrand, L.; Hoffner, S.; Engman, L.; Holmgren, A. Inhibition of bacterial thioredoxin reductase: An antibiotic mechanism targeting bacteria lacking glutathione. FASEB J. 2013, 27, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Frizler, M.; Lohr, F.; Lülsdorff, M.; Gütschow, M. Facing the gem-dialkyl effect in enzyme inhibitor design: Preparation of homocycloleucine-based azadipeptide nitriles. Chem. Eur. J. 2011, 17, 11419–11423. [Google Scholar] [CrossRef] [PubMed]
- Frizler, M.; Lohr, F.; Furtmann, N.; Kläs, J.; Gütschow, M. Structural optimization of azadipeptide nitriles strongly increases association rates and allows the development of selective cathepsin inhibitors. J. Med. Chem. 2011, 54, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Syper, L.; Młochowski, J. Lithium diselenide in aprotic medium—A convenient reagent for synthesis of organic diselenides. Tetrahedron 1988, 44, 6119–6130. [Google Scholar] [CrossRef]
- Van Gompel, J.; Schuster, G.B. Chemiluminescence of organic peroxides: Intramolecular electron-exchange luminescence from a secondary perester. J. Org. Chem. 1987, 52, 1465–1468. [Google Scholar] [CrossRef]


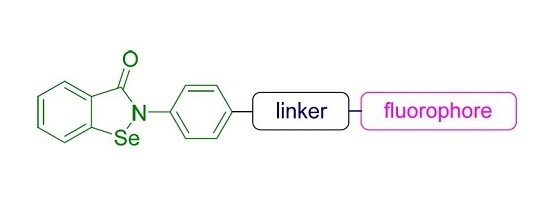{kind=link}
{kind=link}
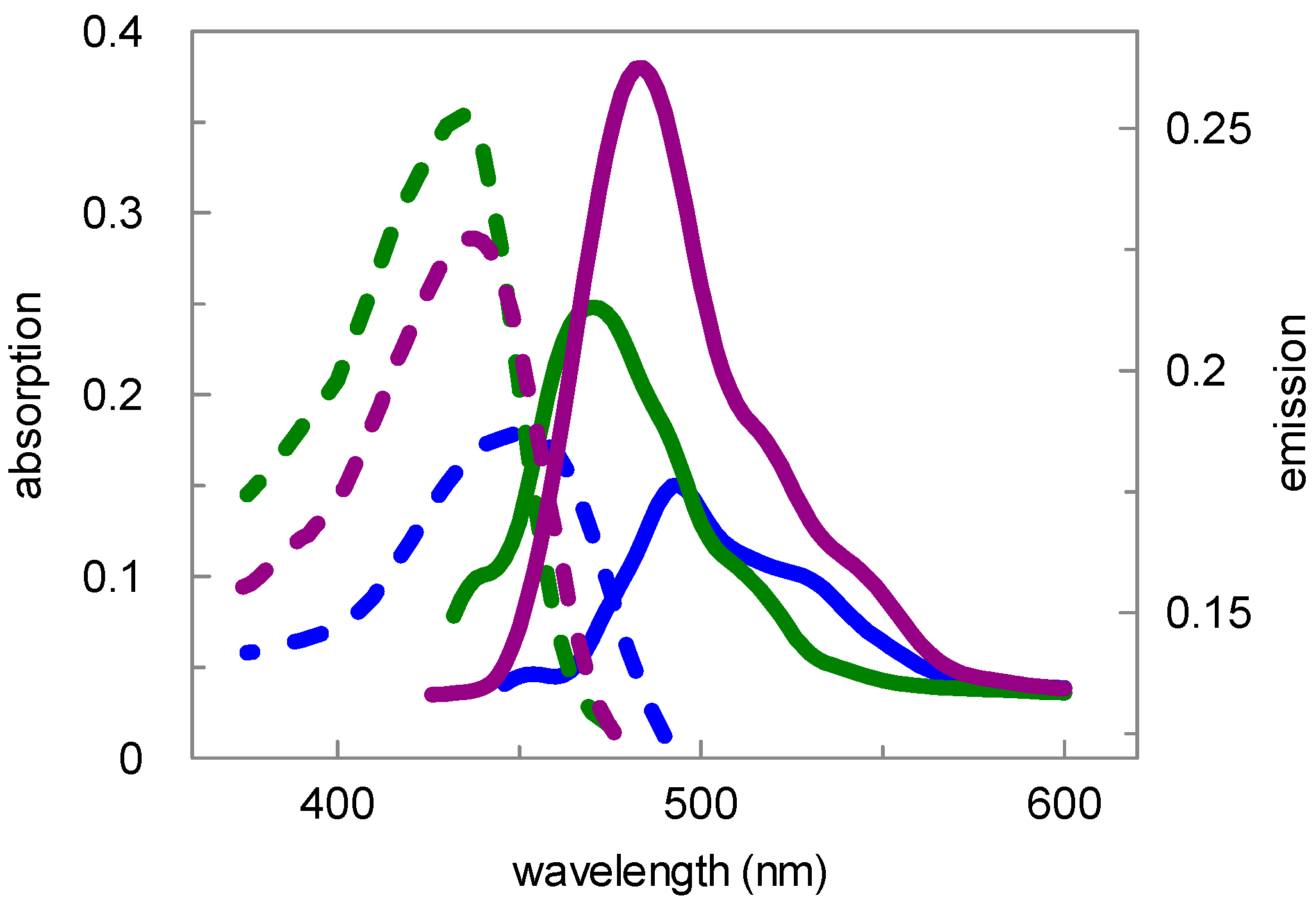{kind=link}
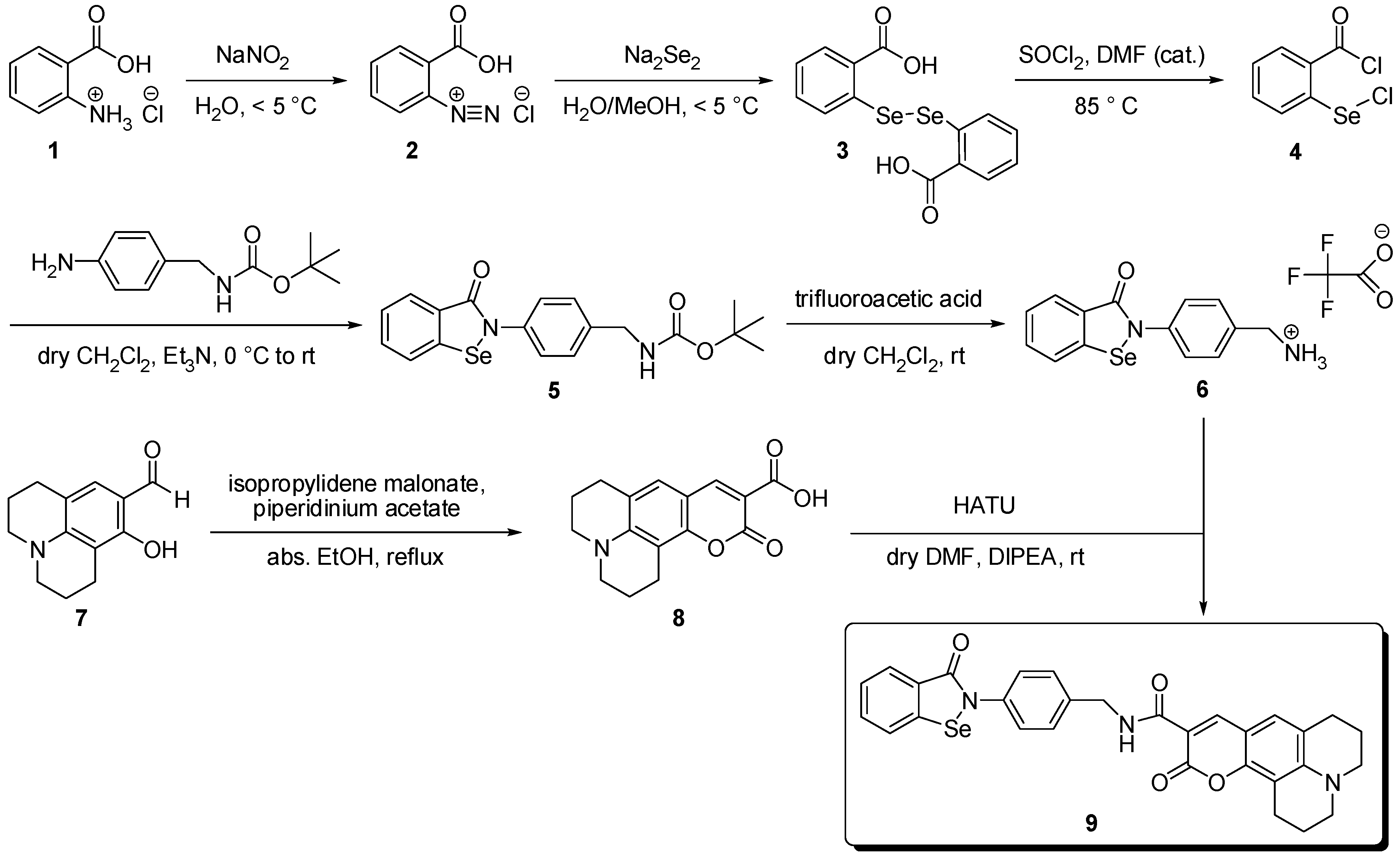{kind=link}
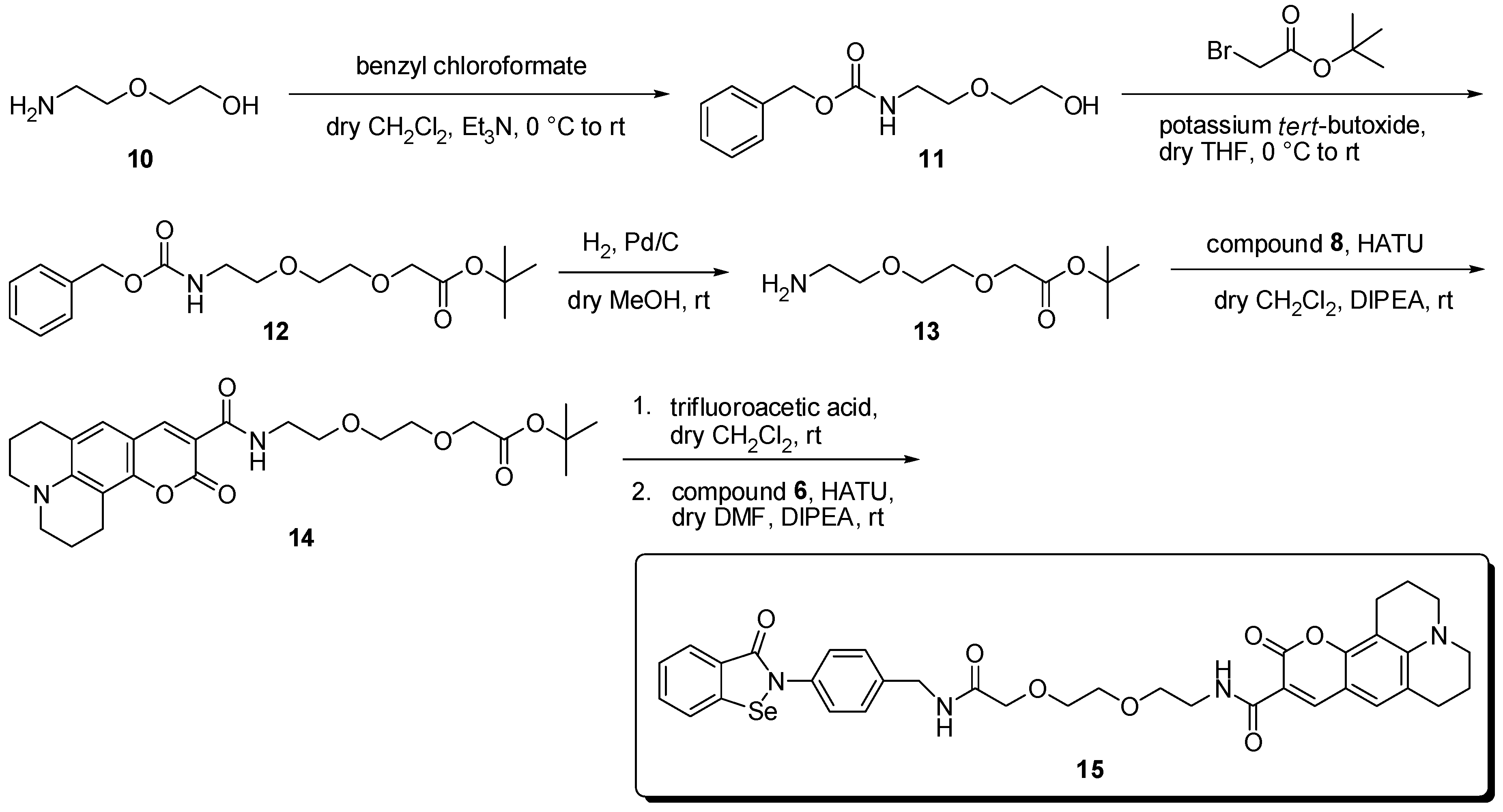{kind=link}
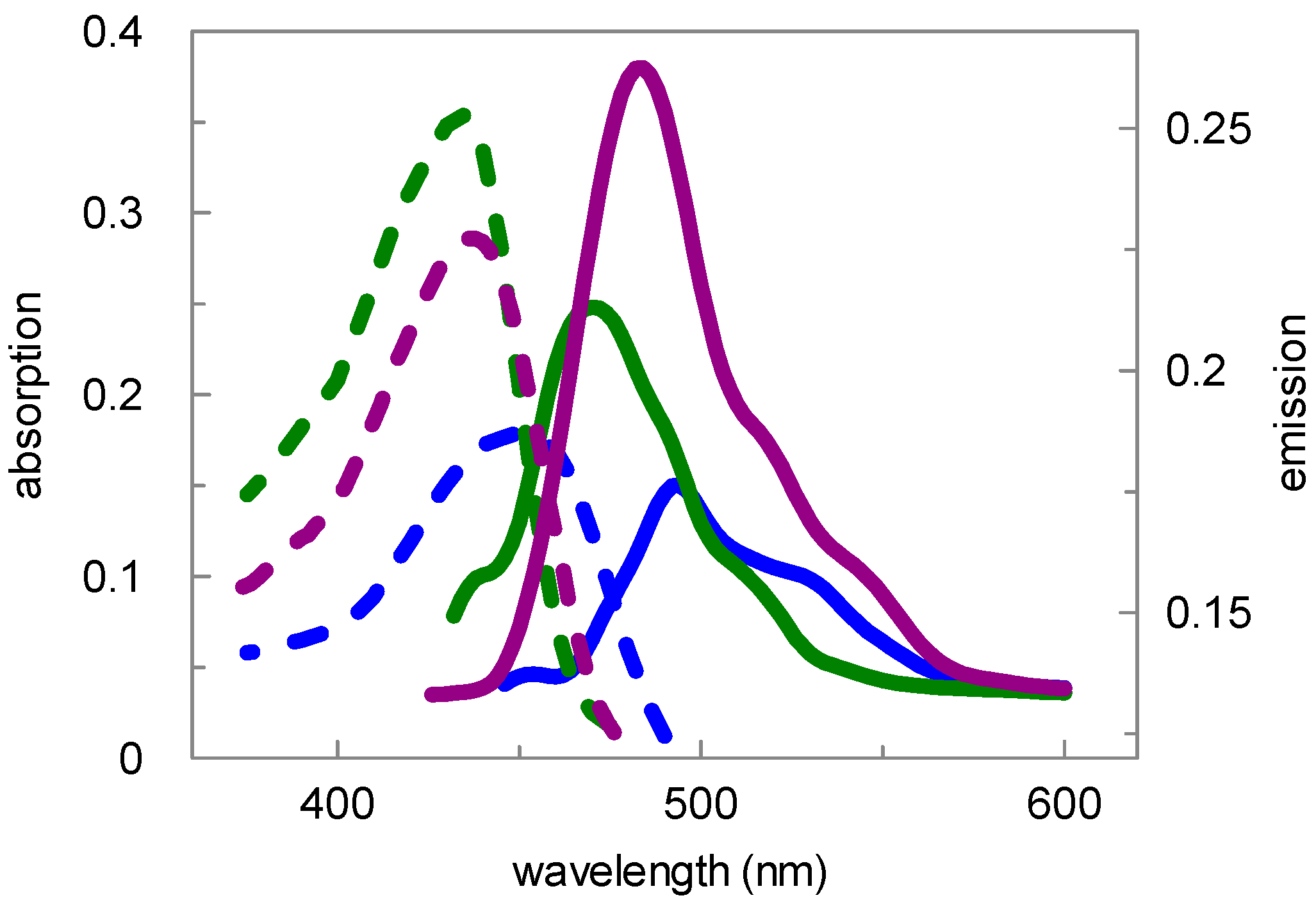
| Compd. | Absorption | Emission | ||||
|---|---|---|---|---|---|---|
| Buffer, pH 7.8 | MeOH | CH2Cl2 | Buffer, pH 7.8 | MeOH | CH2Cl2 | |
| 9 | 434 nm | 436 nm | 435 nm | 494 nm | 484 nm | 470 nm |
| 15 | 450 nm | 436 nm | 435 nm | 494 nm | 484 nm | 470 nm |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Küppers, J.; Schulz-Fincke, A.C.; Palus, J.; Giurg, M.; Skarżewski, J.; Gütschow, M. Convergent Synthesis of Two Fluorescent Ebselen-Coumarin Heterodimers. Pharmaceuticals 2016, 9, 43. https://doi.org/10.3390/ph9030043
Küppers J, Schulz-Fincke AC, Palus J, Giurg M, Skarżewski J, Gütschow M. Convergent Synthesis of Two Fluorescent Ebselen-Coumarin Heterodimers. Pharmaceuticals. 2016; 9(3):43. https://doi.org/10.3390/ph9030043
Chicago/Turabian StyleKüppers, Jim, Anna Christina Schulz-Fincke, Jerzy Palus, Mirosław Giurg, Jacek Skarżewski, and Michael Gütschow. 2016. "Convergent Synthesis of Two Fluorescent Ebselen-Coumarin Heterodimers" Pharmaceuticals 9, no. 3: 43. https://doi.org/10.3390/ph9030043