
Ecology of Anti-Biofilm Agents II: Bacteriophage Exploitation and Biocontrol of Biofilm Bacteria

Abstract
:1. Introduction
2. Theory of Phage-Biofilm Ecological Interaction
2.1. Underlying Assumptions
2.1.1. There are Phages to Which Biofilm Bacteria are Sensitive
2.1.2. Bacteria Availability can be Insubstantial across Macroscopic Environments
2.1.3. Bacteria Availability can be Substantial across Microscopic Environments
2.1.4. Higher Bacteria Availability Should Support more Robust Phage Population Growth
2.2. General Ecological Scenario for Phage-Biofilm Interactions in Nature
2.2.1. Phage-Bacterial Steady States
2.2.2. Infection Foci Steady States
2.3. Parallels between Infection Focus and Biofilm Formation
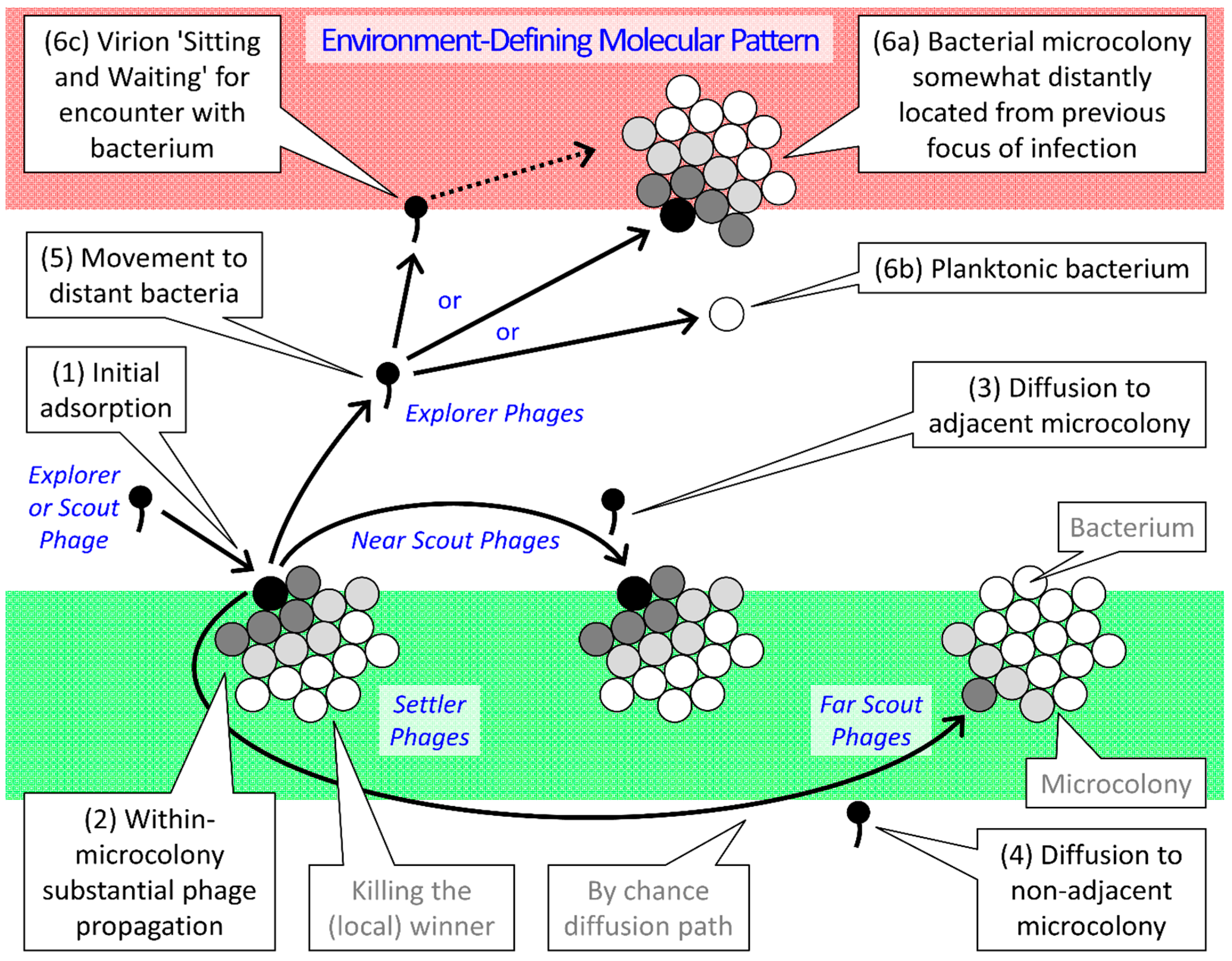

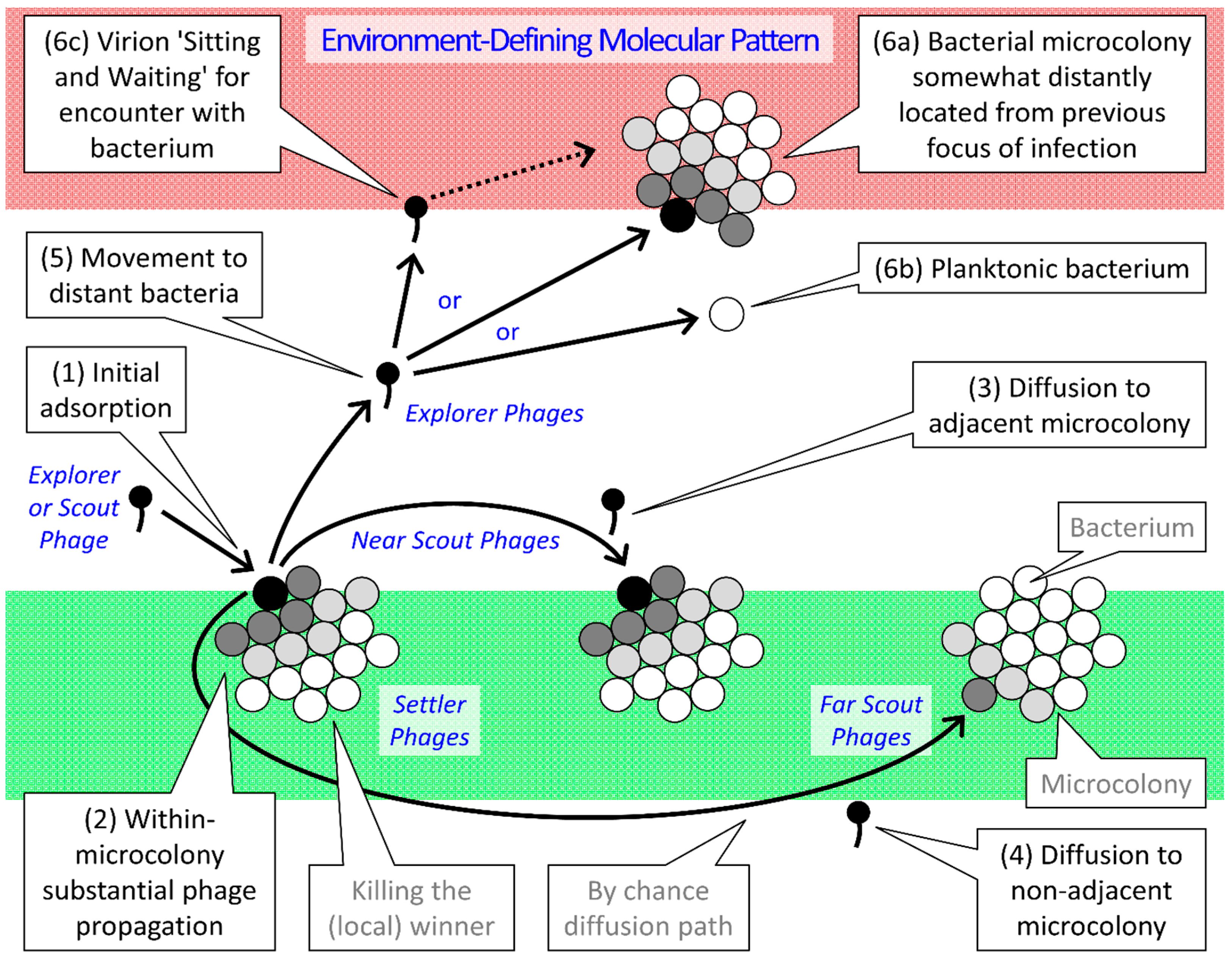{kind=link}
| Type | Gap Length 1 | Ecological as well as Infection-Focus Developmental Roles |
|---|---|---|
| Settler | No gap | Attachment; Virions exploit the same microcolony as that of their parental infection |
| Near Scout | Smaller or none | Maturation; Virions initiatiate the exploitation of individual bacterial microcolonies 2 |
| Far Scout | Larger or multiple | Maturation; Virions form the leading edge of individual infection foci |
| Explorer | Very large | Dispersion; Virons diffuse out of infection focus to found new infection foci |
2.4. Phage-Biofilm Coexistence
3. Phage-Mediated Biocontrol of Bacterial Biofilms
| Target Species | Context | Timing | Reference | |
|---|---|---|---|---|
| Acinetobacter baumannii | Microtiter plate | During | Thawal et al. (2012) | [69] |
| Acinetobacter baumannii | Microtiter plate | During | Mendes et al. (2014) | [70] |
| Acinetobacter johnsonii | Ultrafiltration membrane model | Before | Goldman et al. (2009) | [71] |
| Aggregatibacter actinomycetemcomitans | Polystyrene microtiter plate | During | Castillo-Ruiz et al. (2011) | [72] |
| Arthrobacter soli | Microtiter plate | Before? | Belgini et al. (2014) | [73] |
| Bacillus subtilis | Ultrafiltration membrane model | Before | Goldman et al. (2009) | [71] |
| Brevundimonas sp. | Microtiter plate | Before? | Belgini et al. (2014) | [73] |
| Campylobacter jejuni | Glass | During | Siringan et al. (2011) | [74] |
| Citrobacter freundii | “Environmental surface, stainless steel, high-density polyethylene plastic, and rubber” | During | Gong and Jiang (2015) | [75] |
| Delftia tsuruhatensis | Glass; Membrane bioreactor | During | Bhattacharjee et al. (2015) | [76] |
| Enterobacter agglomerans | Modified Robbins’ device | During | Hughes et al. (1998) | [77] |
| Enterobacter cloace | Glass | During | Tait et al. (2002) | [78] |
| Enterococcus faecalis | Ex vivo tooth root canal | Before? | Khalifa et al. (2015) | [79] |
| Enterococcus faecalis | Microtiter plate | During | Khalifa et al. (2015) | [79] |
| Escherichia coli | Polyvinylchloride coupons | During | Doolittle et al. (1995) | [80] |
| Escherichia coli | Flow cells | During | Doolittle et al. (1996) | [81] |
| Escherichia coli | Modified Robbins’ device | During | Corbin et al. (2001) | [82] |
| Escherichia coli | Stainless steel | During | Sharma et al. (2005) | [83] |
| Escherichia coli | 3-channel flow chamber | During | Moons et al. (2006) | [84] |
| Escherichia coli | Pegs in microtiter plates | During | Lu and Collins (2007) | [85] |
| Escherichia coli | Pegs in microtiter plates | During | Lu and Collins (2009) | [86] |
| Escherichia coli | Hydrogel-coated catheters | During | Carson et al. (2010) | [87] |
| Escherichia coli | Silicone rubber disks | During | Kay et al. (2011) | [88] |
| Escherichia coli | Microtiter plate | During | Chibeu et al. (2012) | [89] |
| Escherichia coli | Calgary biofilm device | During | Ryan et al. (2012) | [90] |
| Escherichia coli | Microtiter plate | During | Hosseinidoust et al. (2013) | [91] |
| Escherichia coli | 3-channel flow chamber | During | Moons et al. (2013) | [55] |
| Escherichia coli | Silicone Rubber Disks | During | Coulter et al. (2014) | [92] |
| Escherichia coli | Microtiter plate | Before | Pei and Lamas-Samanamud (2014) | [93] |
| Escherichia coli | Tissue culture plate | During | Schmerer et al. (2014) | [94] |
| Hafnia alvei | “Environmental surface, stainless steel, high-density polyethylene plastic, and rubber” | During | Gong and Jiang (2015) | [75] |
| Klebsiella pneumoniae | Microtiter plate | During | Bedi et al. (2009) | [95] |
| Klebsiella pneumoniae | Microtiter plate | During | Verma et al. (2009) | [96] |
| Klebsiella pneumoniae | Microtiter plate; Glass | During | Verma et al. (2010) | [97] |
| Klebsiella pneumoniae | Microtiter plate; Glass | During | Chhibber et al. (2013) | [98] |
| Klebsiella pneumoniae | Polycarbonate discs | During | Chhibber et al. (2015) | [99] |
| Klebsiella pneumoniae | Microtiter plate | During | Jamal et al. (2015) | [100] |
| Listeria monocytogenes | Stainless steel | During | Roy et al. (1993) | [101] |
| Listeria monocytogenes | Stainless steel | During | Hibma et al. (1997) | [102] |
| Listeria monocytogenes | Stainless steel | During | Soni and Nannapaneni (2010) | [103] |
| Listeria monocytogenes | Stainless steel | During | Montañez-Izquierdo et al. (2012) | [104] |
| Listeria monocytogenes | Stainless steel | During | Ganegama Arachchi et al. (2013) | [105] |
| Listeria monocytogenes | Stainless steel | During | Chaitiemwong et al. (2014) | [106] |
| Proteus mirabilis | Hydrogel-coated catheters | During | Carson et al. (2010) | [87] |
| Proteus mirabilis | Microtiter plate; Hydrogel-coated catheters | Before | Lehman and Donlan (2015) | [107] |
| Pseudomonas aeruginosa | Flow cells | During | Doolittle et al. (1996) | [81] |
| Pseudomonas aeruginosa | Poly(methyl)methacrylate discs | During | Hanlon et al. (2001) | [108] |
| Pseudomonas aeruginosa | Microtiter plate | During | Knezevic and Petrovic (2008) | [109] |
| Pseudomonas aeruginosa | Ultrafiltration membrane model | Before | Goldman et al. (2009) | [71] |
| Pseudomonas aeruginosa | Hydrogel-coated catheters | Before | Fu et al. (2010) | [110] |
| Pseudomonas aeruginosa | Microtiter plate | During | Ahiwale et al. (2011) | [111] |
| Pseudomonas aeruginosa | Silicone rubber disks | During | Kay et al. (2011) | [88] |
| Pseudomonas aeruginosa | Microtiter plate | Before | Knezevic et al. (2011) | [112] |
| Pseudomonas aeruginosa | Microtiter plate | During | Pires et al. (2011) | [113] |
| Pseudomonas aeruginosa | Epithelial-cell monolayer | During | Alemayehu et al. (2012) | [114] |
| Pseudomonas aeruginosa | Silicone catheter segment | Before | Liao et al. (2012) | [115] |
| Pseudomonas aeruginosa | Microtiter plate | During | Hosseinidoust et al. (2013) | [91] |
| Pseudomonas aeruginosa | Microtiter plate; Ex vivo tooth root canal | During | Phee et al. (2013) | [116] |
| Pseudomonas aeruginosa | Rat implant model | During | Yilmaz et al. (2013) | [117] |
| Pseudomonas aeruginosa | Microtiter plate; Glass | During | Zhang and Hu (2013) | [118] |
| Pseudomonas aeruginosa | Water biofiltration systems (anthracite or granular activated carbon) | During | Zhang et al. (2013) | [119] |
| Pseudomonas aeruginosa | Silicone Rubber Disks | During | Coulter et al. (2014) | [92] |
| Pseudomonas aeruginosa | Microtiter plate | During | Mendes et al. (2014) | [70] |
| Pseudomonas aeruginosa | Microtiter plate | Before | Pei and Lamas-Samanamud (2014) | [93] |
| Pseudomonas aeruginosa | Polycarbonate discs | During | Chhibber et al. (2015) | [99] |
| Pseudomonas aeruginosa | Pegs in microtiter plates | During | Danis-Wlodarczyk ett al. (2015) | [120] |
| Pseudomonas aeruginosa | Microtiter plate; Hydrogel-coated catheters | Before | Lehman and Donlan (2015) | [107] |
| Pseudomonas fluorescens | Inox plate placed in microtiter tray | During | Sillankorva et al. (2004) | [121] |
| Pseudomonas fluorescens | Glass | During | Sillankorva et al. (2008a) | [122] |
| Pseudomonas fluorescens | Stainless steel | During | Sillankorva et al. (2008b) | [123] |
| Pseudomonas fluorescens | Stainless steel | During | Sillankorva et al. (2010) | [124] |
| Pseudomonas putida | Polystyrene peg in 96-well microtiter plate | During | Cornelissen et al. (2011) | [125] |
| Pseudomonas sp. | Microtiter plate | Before? | Belgini et al. (2014) | [73] |
| Salmonella typhimurium | Microtiter plate | During | Hosseinidoust et al. (2013) | [91] |
| Serratia marcescens | Modified Robbins’ device | During | Hughes et al. (1998) | [77] |
| Serratia marcescens | Polystyrene flasks | Before | Zhang et al. (2014) | [36] |
| Sphaerotilus natans | Stainless steel coupons and wire screen | During | Gino et al. (2010) | [126] |
| Staphylococcus aureus | Polystyrene microtiter plate | During | Del Pozo et al. (2007) | [127] |
| Staphylococcus aureus | Microtiter plate | During | Son et al. (2010) | [128] |
| Staphylococcus aureus | Microtiter plate | During | Rahman et al. (2011) | [129] |
| Staphylococcus aureus | Microtiter plate | During | Kelly et al. (2012) | [130] |
| Staphylococcus aureus | Rabbit wound model | During | Seth et al. (2013) | [131] |
| Staphylococcus aureus | Rat implant model | During | Yilmaz et al. (2013) | [117] |
| Staphylococcus aureus | Silicone discs | During | Lungren et al. (2013) | [132] |
| Staphylococcus aureus | Microtiter plate | During | Alves et al. (2014) | [133] |
| Staphylococcus aureus | Sheep model of sinusitis | During | Drilling et al. (2014a) | [134] |
| Staphylococcus aureus | Plastic pegs | During | Drilling et al. (2014b) | [135] |
| Staphylococcus aureus | Cuffed central venous catheters | During | Lungren et al. (2014) | [136] |
| Staphylococcus aureus | Microtiter plate | During | Mendes et al. (2014) | [70] |
| Staphylococcus aureus | Microtiter plate | During | Gutierrez et al. (2015) | [137] |
| Staphylococcus epidermidis | Catheter | During | Wood et al. (2001) | [138] |
| Staphylococcus epidermidis | Hydrogel-coated catheters | Before | Curtin and Donlan (2006) | [139] |
| Staphylococcus epidermidis | Microtiter plate | During | Cerca et al. (2007) | [140] |
| Staphylococcus epidermidis | Microtiter plate | During | Gutierrez et al. (2015) | [137] |
| Staphylococcus lentus | Stainless steel | During | Sillankorva et al. (2010) | [124] |
| Vibrio anguillarum | polypropylene plastic tubes | During | Tan et al. (2015) | [141] |
3.1. Combatting Phage-Bacterial Coexistence
3.1.1. Apply Phages for Longer
3.1.2. Apply Reasonable Quantities of Phages
3.1.3. Biofilm Disruption can Aid Biofilm Clearance
3.1.4. If at First you don’t Fully Succeed, try Applying Greater Quantities of Phages
4. General “Pros and Cons” of Phage Therapy
5. Conclusions
Conflict of Interest
References
- Diaz-Munoz, S.L.; Koskella, B. Bacteria-phage interactions in natural environments. Adv. Appl. Microbiol. 2014, 89, 135–183. [Google Scholar]
- Whitman, W.B.; Coleman, D.C.; Wiebe, W.J. Prokaryotes: The unseen majority. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 6578–6583. [Google Scholar] [CrossRef]
- Karner, M.B.; DeLong, E.F.; Karl, D.M. Archaeal dominance in the mesopelagic zone of the Pacific Ocean. Nature 2001, 409, 507–510. [Google Scholar] [CrossRef]
- Lipp, J.S.; Morono, Y.; Inagaki, F.; Hinrichs, K.U. Significant contribution of Archaea to extant biomass in marine subsurface sediments. Nature 2008, 454, 991–994. [Google Scholar] [CrossRef]
- Lloyd, K.G.; May, M.K.; Kevorkian, R.T.; Steen, A.D. Meta-analysis of quantification methods shows that archaea and bacteria have similar abundances in the subseafloor. Appl. Environ. Microbiol. 2013, 79, 7790–7799. [Google Scholar] [CrossRef]
- Hall, M.R.; McGillicuddy, E.; Kaplan, L.J. Biofilm: Basic principles, pathophysiology, and implications for clinicians. Surg. Infect. 2014, 15, 1–7. [Google Scholar] [CrossRef]
- Flemming, H.C.; Wingender, J. The biofilm matrix. Nat. Rev. Microbiol. 2010, 8, 623–633. [Google Scholar] [CrossRef]
- Bjarnsholt, T. The role of bacterial biofilms in chronic infections. APMIS, 2013, 121, 1–58. [Google Scholar] [CrossRef]
- Scali, C.; Kunimoto, B. An update on chronic wounds and the role of biofilms. J. Cutan. Med. Surg. 2013, 17, 371–376. [Google Scholar]
- Cooper, R.A.; Bjarnsholt, T.; Alhede, M. Biofilms in wounds: A review of present knowledge. J. Wound Care 2014, 23, 570–580. [Google Scholar] [CrossRef]
- Macia, M.D.; Rojo-Molinero, E.; Oliver, A. Antimicrobial susceptibility testing in biofilm-growing bacteria. Clin. Microbiol. Infect. 2014, 20, 981–990. [Google Scholar] [CrossRef]
- Solheim, H.T.; Sekse, C.; Urdahl, A.M.; Wasteson, Y.; Nesse, L.L. Biofilm as an environment for dissemination of stx genes by transduction. Appl. Environ. Microbiol. 2013, 79, 896–900. [Google Scholar] [CrossRef]
- Costerton, J.; Stewart, P.; Greenberg, E. Bacterial biofilms: A common cause of persistent infections. Science 1999, 284, 1318–1322. [Google Scholar] [CrossRef]
- Matz, C. Biofilms as refuge against predation. In The Biofilm Mode of Life: Mechanisms and Adaptations; Kjelleberg, S., Givskov, M., Eds.; Horizon Bioscience: Norfolk, UK, 2007; pp. 195–213. [Google Scholar]
- Abedon, S.T. Bacteriophages and Biofilms: Ecology, Phage Therapy, Plaques; Nova Science Publishers: New York, NY, USA, 2011. [Google Scholar]
- Abedon, S.T. Spatial vulnerability: Bacterial arrangements, microcolonies, and biofilms as responses to low rather than high phage densities. Viruses 2012, 4, 663–687. [Google Scholar] [CrossRef]
- Abedon, S.T. Thinking about microcolonies as phage targets. Bacteriophage 2012, 2, 200–204. [Google Scholar] [CrossRef]
- Chan, B.K.; Abedon, S.T. Bacteriophages and their enzymes in biofilm control. Curr. Pharm. Des. 2015, 21, 85–99. [Google Scholar] [CrossRef]
- Abedon, S.T. Ecology of anti-biofilm agents I. Antibiotics versus bacteriophages. Pharmaceuticals 2015, 8, 525–588. [Google Scholar]
- Harper, D.R. Biological control by microorganisms. In The Encyclopedia of Life Sciences; John Wiley & Sons: Chichester, UK, 2006; pp. 1–10. [Google Scholar]
- Abedon, S.T. Kinetics of phage-mediated biocontrol of bacteria. Foodborne Pathog. Dis. 2009, 6, 807–815. [Google Scholar] [CrossRef]
- Hyman, P.; Abedon, S.T. Bacteriophages in Health and Disease; CABI Press: Wallingford, UK, 2012. [Google Scholar]
- Borysowski, J.; Miedzybrodzki, R.; Górski, A. Phage Therapy: Current Research and Applications; Caister Academic Press: Norfolk, UK, 2014. [Google Scholar]
- Brüssow, H. Bacteriophage-host interaction: From splendid isolation into a messy reality. Curr. Opin. Microbiol. 2013, 16, 500–506. [Google Scholar] [CrossRef]
- Fan, X.; Li, W.; Zheng, F.; Xie, J. Bacteriophage inspired antibiotics discovery against infection involved biofilm. Crit. Rev. Eukaryot. Gene Expr. 2013, 23, 317–326. [Google Scholar] [CrossRef]
- Harper, D.R.; Parracho, H.M.R.; Walker, J.; Sharp, R.; Hughes, G.; Werthrén, M.; Lehman, S.; Morales, S. Bacteriophages and biofilms. Antibiotics 2014, 3, 270–284. [Google Scholar] [CrossRef]
- Parasion, S.; Kwiatek, M.; Gryko, R.; Mizak, L.; Malm, A. Bacteriophages as an alternative strategy for fighting biofilm development. Pol. J. Microbiol. 2014, 63, 137–145. [Google Scholar]
- Sillankorva, S.; Azeredo, J. Bacteriophage attack as an anti-biofilm strategy. Methods Mol. Biol. 2014, 1147, 277–285. [Google Scholar]
- Sillankorva, S.; Azeredo, J. The use of bacteriophages and bacteriophage-derived enzymes for clinically relevant biofilm control. In Phage Therapy: Current Research and Applications; Borysowski, J., Miedzybrodzki, R., Górski, A., Eds.; Caister Academic Press: Norfolk, UK, 2014. [Google Scholar]
- Abedon, S.T.; Thomas-Abedon, C. Phage therapy pharmacology. Curr. Pharm. Biotechnol. 2010, 11, 28–47. [Google Scholar] [CrossRef]
- Abedon, S. Phage therapy pharmacology: Calculating phage dosing. Adv. Appl. Microbiol. 2011, 77, 1–40. [Google Scholar]
- Hyman, P.; Abedon, S.T. Bacteriophage host range and bacterial resistance. Adv. Appl. Microbiol. 2010, 70, 217–248. [Google Scholar]
- Labrie, S.J.; Samson, J.E.; Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010, 8, 317–327. [Google Scholar] [CrossRef]
- Friman, V.P.; Buckling, A. Phages can constrain protist predation-driven attenuation of Pseudomonas aeruginosa virulence in multienemy communities. ISME. J. 2014, 8, 1820–1830. [Google Scholar] [CrossRef]
- Ly, M.; Abeles, S.R.; Boehm, T.K.; Robles-Sikisaka, R.; Naidu, M.; Santiago-Rodriguez, T.; Pride, D.T. Altered oral viral ecology in association with periodontal disease. MBio 2014, 5. [Google Scholar] [CrossRef]
- Zhang, J.; Ormala-Odegrip, A.M.; Mappes, J.; Laakso, J. Top-down effects of a lytic bacteriophage and protozoa on bacteria in aqueous and biofilm phases. Ecol. Evol. 2014, 4, 4444–4453. [Google Scholar] [CrossRef]
- Rodriguez-Brito, B.; Li, L.; Wegley, L.; Furlan, M.; Angly, F.; Breitbart, M.; Buchanan, J.; Desnues, C.; Dinsdale, E.; Edwards, R.; et al. Viral and microbial community dynamics in four aquatic environments. ISME J. 2010, 4, 739–751. [Google Scholar] [CrossRef]
- Winter, C.; Bouvier, T.; Weinbauer, M.G.; Thingstad, T.F. Trade-offs between competition and defense specialists among unicellular planktonic organisms: The "killing the winner" hypothesis revisited. Microbiol. Mol. Biol. Rev. 2010, 74, 42–57. [Google Scholar] [CrossRef]
- Costerton, J.W.; Cheng, J.-J.; Geesey, G.G.; Ladd, T.I.; Nickel, J.C.; Dasgupta, M.; Marrie, T.J. Bacterial biofilms in nature and disease. Ann. Rev. Microbiol. 1987, 41, 435–464. [Google Scholar] [CrossRef]
- Costerton, J.W.; Lewandowski, Z.; Caldwell, D.E.; Korber, D.R.; Lappin-Scott, H.M. Microbial biofilms. Ann. Rev. Microbiol. 1995, 49, 711–745. [Google Scholar] [CrossRef]
- Tait, K.; Sutherland, I.W. Antagonistic interactions amongst bacteriocin-producing enteric bacteria in dual species biofilms. J. Appl. Microbiol. 2002, 93, 345–352. [Google Scholar] [CrossRef]
- Rao, D.; Webb, J.S.; Kjelleberg, S. Competitive interactions in mixed-species biofilms containing the marine bacterium Pseudoalteromonas tunicata. Appl. Environ. Microbiol. 2005, 71, 1729–1736. [Google Scholar] [CrossRef]
- Chan, B.K.; Abedon, S.T. Bacteriophage adaptation, with particular attention to issues of phage host range. In Bacteriophages in Dairy Processing; Quiberoni, A., Reinheimer, J., Eds.; Nova Science Publishers: New York, NY, USA, 2012; pp. 25–52. [Google Scholar]
- Bohannan, B.J.M.; Lenski, R.E. Effect of resource enrichment on a chemostat community of bacteria and bacteriophage. Ecology 1997, 78, 2303–2315. [Google Scholar] [CrossRef]
- Pace, M.L.; Cole, J.J. Comparative and experimental approaches to top-down and bottom-up regulation of bacteria. Microb. Ecol. 1994, 28, 181–193. [Google Scholar] [CrossRef]
- Abedon, S.T.; Culler, R.R. Optimizing bacteriophage plaque fecundity. J. Theor. Biol. 2007, 249, 582–592. [Google Scholar] [CrossRef]
- Anderson, T.F. Bacteriophages and their action on host cells. In Research in Medical Science; Green, D.E., Knox, W.E., Eds.; Macmillan: New York, 1950; pp. 1–16. [Google Scholar]
- Bjarnsholt, T.; Alhede, M.; Alhede, M.; Eickhardt-Sorensen, S.R.; Moser, C.; Kuhl, M.; Jensen, P.O.; Hoiby, N. The in vivo biofilm. Trends Microbiol. 2013, 21, 466–474. [Google Scholar] [CrossRef]
- Barr, J.J.; Auro, R.; Furlan, M.; Whiteson, K.L.; Erb, M.L.; Pogliano, J.; Stotland, A.; Wolkowicz, R.; Cutting, A.S.; Doran, K.S.; et al. Bacteriophage adhering to mucus provide a non-host-derived immunity. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 10771–10776. [Google Scholar] [CrossRef]
- Barr, J.J.; Youle, M.; Rohwer, F. Innate and acquired bacteriophage-mediated immunity. Bacteriophage 2013, 3, e25857. [Google Scholar] [CrossRef]
- Gorski, A.; Dabrowska, K.; Hodyra-Stefaniak, K.; Borysowski, J.; Miedzybrodzki, R.; Weber-Dabrowska, B. Phages targeting infected tissues: novel approach to phage therapy. Future Microbiol. 2015, 10, 199–204. [Google Scholar] [CrossRef]
- Abedon, S.T. Bacteriophage secondary infection. Virol. Sin. 2015, 30, 3–10. [Google Scholar] [CrossRef]
- Stewart, P.S.; Franklin, M.J. Physiological heterogeneity in biofilms. Nat. Rev. Microbiol. 2008, 6, 199–210. [Google Scholar] [CrossRef]
- Abedon, S.T. Disambiguating bacteriophage pseudolysogeny: An historical analysis of lysogeny, pseudolysogeny, and the phage carrier state. In Contemporary Trends in Bacteriophage Research; Adams, H.T., Ed.; Nova Science Publishers: New York, NY, USA, 2009; pp. 285–307. [Google Scholar]
- Moons, P.; Faster, D.; Aertsen, A. Lysogenic conversion and phage resistance development in phage exposed Escherichia coli biofilms. Viruses 2013, 5, 150–161. [Google Scholar] [CrossRef]
- Weitz, J.S.; Mileyko, Y.; Joh, R.I.; Voit, E.O. Collective decision making in bacterial viruses. Biophys. J. 2008, 95, 2673–2680. [Google Scholar] [CrossRef]
- Abedon, S.T.; Yin, J. Impact of spatial structure on phage population growth. In Bacteriophage Ecology; Abedon, S.T., Ed.; Cambridge University Press: Cambridge, UK, 2008; pp. 94–113. [Google Scholar]
- Abedon, S.T. Communication among phages, bacteria, and soil environments. In Biocommunication of Soil Microorganisms; Witzany, G., Ed.; Springer: New York, NY, USA, 2011; pp. 37–65. [Google Scholar]
- Kutter, E.; Kellenberger, E.; Carlson, K.; Eddy, S.; Neitzel, J.; Messinger, L.; North, J.; Guttman, B. Effects of bacterial growth conditions and physiology on T4 infection. In The Molecular Biology of Bacteriophage T4; Karam, J.D., Kutter, E., Carlson, K., Guttman, B., Eds.; ASM Press: Washington, DC, USA, 1994; pp. 406–418. [Google Scholar]
- Miller, R.V.; Day, M. Contribution of lysogeny, pseudolysogeny, and starvation to phage ecology. In Bacteriophage Ecology; Abedon, S.T., Ed.; Cambridge University Press: Cambridge, UK, 2008; pp. 114–143. [Google Scholar]
- Golec, P.; Karczewska-Golec, J.; Łoś, M.; Węgrzyn, G. Bacteriophage T4 can produce progeny virions in extremely slowly growing Escherichia coli host: Comparison of a mathematical model with the experimental data. FEMS Microbiol. Lett. 2014, 351, 156–161. [Google Scholar] [CrossRef]
- Heilmann, S.; Sneppen, K.; Krishna, S. Coexistence of phage and bacteria on the boundary of self-organized refuges. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 12828–12833. [Google Scholar] [CrossRef]
- Borer, E.T.; Halpern, B.S.; Seabloom, E.W. Asymmetry in community regulation: Effects of predators and productivity. Ecology 2006, 87, 2813–2820. [Google Scholar] [CrossRef]
- Kutter, E.; De Vos, D.; Gvasalia, G.; Alavidze, Z.; Gogokhia, L.; Kuhl, S.; Abedon, S.T. Phage therapy in clinical practice: Treatment of human infections. Curr. Pharm. Biotechnol. 2010, 11, 69–86. [Google Scholar] [CrossRef]
- Abedon, S.T.; Kuhl, S.J.; Blasdel, B.G.; Kutter, E.M. Phage treatment of human infections. Bacteriophage 2011, 1, 66–85. [Google Scholar] [CrossRef]
- Abedon, S.T. Phage therapy of pulmonary infections. Bacteriophage 2015, 5, e1020260-1–e1020260-13. [Google Scholar] [CrossRef]
- Ślopek, S.; Weber-Dąbrowska, B.; Dąbrowski, M.; Kucharewicz-Krukowska, A. Results of bacteriophage treatment of suppurative bacterial infections in the years 1981–1986. Arch. Immunol. Ther. Exp. 1987, 35, 569–583. [Google Scholar]
- Weber-Dąbrowska, B.; Mulczyk, M.; Górski, A. Bacteriophage therapy of bacterial infections: An update of our institute’s experience. Arch. Immunol. Ther. Exp. 2000, 48, 547–551. [Google Scholar]
- Thawal, N.D.; Yele, A.B.; Sahu, P.K.; Chopade, B.A. Effect of a novel podophage AB7-IBB2 on Acinetobacter baumannii biofilm. Curr. Microbiol. 2012, 65, 66–72. [Google Scholar] [CrossRef]
- Mendes, J.J.; Leandro, C.; Mottola, C.; Barbosa, R.; Silva, F.A.; Oliveira, M.; Vilela, C.L.; Melo-Cristino, J.; Gorski, A.; Pimentel, M.; Sao-Jose, C.; Cavaco-Silva, P.; Garcia, M. In vitro design of a novel lytic bacteriophage cocktail with therapeutic potential against organisms causing diabetic foot infections. J. Med. Microbiol. 2014, 63, 1055–1065. [Google Scholar] [CrossRef]
- Goldman, G.; Starosvetsky, J.; Armon, R. Inhibition of biofilm formation on UF membrane by use of specific bacteriophages. J. Membr. Sci. 2009, 342, 145–152. [Google Scholar] [CrossRef]
- Castillo-Ruiz, M.; Vines, E.D.; Montt, C.; Fernandez, J.; Delgado, J.M.; Hormazabal, J.C.; Bittner, M. Isolation of a novel Aggregatibacter actinomycetemcomitans serotype b bacteriophage capable of lysing bacteria within a biofilm. Appl. Environ. Microbiol. 2011, 77, 3157–3159. [Google Scholar] [CrossRef]
- Belgini, D.R.; Dias, R.S.; Siqueira, V.M.; Valadares, L.A.; Albanese, J.M.; Souza, R.S.; Torres, A.P.; Sousa, M.P.; Silva, C.C.; De Paula, S.O.; et al. Culturable bacterial diversity from a feed water of a reverse osmosis system, evaluation of biofilm formation and biocontrol using phages. World J. Microbiol. Biotechnol. 2014, 30, 2689–2700. [Google Scholar] [CrossRef]
- Siringan, P.; Connerton, P.L.; Payne, R.J.; Connerton, I.F. Bacteriophage-mediated dispersal of Campylobacter jejuni biofilms. Appl. Environ. Microbiol. 2011, 77, 3320–3326. [Google Scholar] [CrossRef]
- Gong, C.; Jiang, X. Application of bacteriophages to reduce biofilms formed by hydrogen sulfide producing bacteria on surfaces in a rendering plant. Can. J. Microbiol. 2015, 61, 539–544. [Google Scholar] [CrossRef]
- Bhattacharjee, A.S.; Choi, J.D.; Motlagh, A.M.; Mukherji, S.T.; Goel, R. Bacteriophage therapy for membrane biofouling in membrane bioreactors and antibiotic-resistant bacterial biofilms. Biotech. Bioeng. 2015, 112, 1644–1654. [Google Scholar] [CrossRef]
- Hughes, K.A.; Sutherland, I.W.; Jones, M.V. Biofilm susceptibility to bacteriophage attack: The role of phage-borne polysaccharide depolymerase. Microbiology 1998, 144, 3039–3047. [Google Scholar] [CrossRef]
- Tait, K.; Skilman, L.C.; Sutherland, I.W. The efficacy of bacteriophage as a method of biofilm eradication. Biofouling 2002, 18, 305–311. [Google Scholar] [CrossRef]
- Khalifa, L.; Brosh, Y.; Gelman, D.; Coppenhagen-Glazer, S.; Beyth, S.; Poraduso-Cohen, R.; Que, Y.A.; Beyth, N.; Hazan, R. Targeting Enterococcus faecalis biofilm using phage therapy. Appl. Environ. Microbiol. 2015, 81, 2696–2705. [Google Scholar] [CrossRef]
- Doolittle, M.M.; Cooney, J.J.; Caldwell, D.E. Lytic infection of Escherichia coli biofilms by bacteriophage T4. Can. J. Microbiol. 1995, 41, 12–18. [Google Scholar] [CrossRef]
- Doolittle, M.M.; Cooney, J.J.; Caldwell, D.E. Tracing the interaction of bacteriophage with bacterial biofilms using fluorescent and chromogenic probes. J. Indust. Microbiol. 1996, 16, 331–341. [Google Scholar] [CrossRef]
- Corbin, B.D.; McLean, R.J. C.; Aron, G.M. Bacteriophage T4 multiplication in a glucose-limited Escherichia coli biofilm. Can. J. Microbiol. 2001, 47, 680–684. [Google Scholar] [CrossRef]
- Sharma, M.; Ryu, J.H.; Beuchat, L.R. Inactivation of Escherichia coli O157:H7 in biofilm on stainless steel by treatment with an alkaline cleaner and a bacteriophage. J. Appl. Microbiol. 2005, 99, 449–459. [Google Scholar] [CrossRef]
- Moons, P.; Werckx, W.; Van Houdt, R.; Aertsen, A.; Michiels, C.W. Resistance development of bacterial biofilms against bacteriophage attack. Commun. Agric. Appl. Biol. Sci. 2006, 71, 297–300. [Google Scholar]
- Lu, T.K.; Collins, J.J. Dispersing biofilms with engineered enzymatic bacteriophage. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 11197–11202. [Google Scholar] [CrossRef]
- Lu, T.K.; Collins, J.J. Engineered bacteriophage targeting gene networks as adjuvants for antibiotic therapy. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 4629–4634. [Google Scholar] [CrossRef]
- Carson, L.; Gorman, S.P.; Gilmore, B.F. The use of lytic bacteriophages in the prevention and eradication of biofilms of Proteus mirabilis and Escherichia coli. FEMS Immunol. Med. Microbiol. 2010, 59, 447–455. [Google Scholar] [CrossRef]
- Kay, M.K.; Erwin, T.C.; McLean, R.J.; Aron, G.M. Bacteriophage ecology in Escherichia coli and Pseudomonas aeruginosa mixed biofilm communities. Appl. Environ. Microbiol. 2011, 77, 821–829. [Google Scholar] [CrossRef]
- Chibeu, A.; Lingohr, E.J.; Masson, L.; Manges, A.; Harel, J.; Ackermann, H.W.; Kropinski, A.M.; Boerlin, P. Bacteriophages with the ability to degrade uropathogenic Escherichia coli biofilms. Viruses 2012, 4, 471–487. [Google Scholar] [CrossRef]
- Ryan, E.M.; Alkawareek, M.Y.; Donnelly, R.F.; Gilmore, B.F. Synergistic phage-antibiotic combinations for the control of Escherichia coli biofilms in vitro. FEMS Immunol. Med. Microbiol. 2012, 65, 395–398. [Google Scholar] [CrossRef]
- Hosseinidoust, Z.; Tufenkji, N.; van de Ven, T.G. Formation of biofilms under phage predation: Considerations concerning a biofilm increase. Biofouling 2013, 29, 457–468. [Google Scholar] [CrossRef]
- Coulter, L.B.; McLean, R.J.; Rohde, R.E.; Aron, G.M. Effect of bacteriophage infection in combination with tobramycin on the emergence of resistance in Escherichia coli and Pseudomonas aeruginosa biofilms. Viruses 2014, 6, 3778–3786. [Google Scholar] [CrossRef]
- Pei, R.; Lamas-Samanamud, G.R. Inhibition of biofilm formation by T7 bacteriophages producing quorum quenching enzymes. Appl. Environ. Microbiol. 2014, 80, 5340–5348. [Google Scholar] [CrossRef]
- Schmerer, M.; Molineux, I.J.; Ally, D.; Tyerman, J.; Cecchini, N.; Bull, J.J. Challenges in predicting the evolutionary maintenance of a phage transgene. J. Biol. Eng. 2014, 8, 21. [Google Scholar] [CrossRef]
- Bedi, M.S.; Verma, V.; Chhibber, S. Amoxicillin and specific bacteriophage can be used together for eradication of biofilm of Klebsiella pneumoniae B5055. World J. Microbiol. Biotechnol. 2009, 25, 1145–1151. [Google Scholar] [CrossRef]
- Verma, V.; Harjai, K.; Chhibber, S. Restricting ciprofloxacin-induced resistant variant formation in biofilm of Klebsiella pneumoniae B5055 by complementary bacteriophage treatment. J. Antimicrob. Chemother. 2009, 64, 1212–1218. [Google Scholar] [CrossRef]
- Verma, V.; Harjai, K.; Chhibber, S. Structural changes induced by a lytic bacteriophage make ciprofloxacin effective against older biofilm of Klebsiella pneumoniae. Biofouling 2010, 26, 729–737. [Google Scholar] [CrossRef]
- Chhibber, S.; Nag, D.; Bansal, S. Inhibiting biofilm formation by Klebsiella pneumoniae B5055 using an iron antagonizing molecule and a bacteriophage. Bmc Microbiol. 2013, 13, 174. [Google Scholar] [CrossRef]
- Chhibber, S.; Bansal, S.; Kaur, S. Disrupting the mixed species biofilm of Klebsiella pneumoniae B5055 and Pseudomonas aeruginosa PAO using bacteriophages alone or in combination with xylitol. Microbiology 2015, 161, 1369–1377. [Google Scholar] [CrossRef]
- Jamal, M.; Hussain, T.; Das, C.R.; Andleeb, S. Characterization of Siphoviridae phage Z and studying its efficacy against multidrug-resistant Klebsiella pneumoniae planktonic cells and biofilm. J. Med. Microbiol. 2015, 64, 454–462. [Google Scholar] [CrossRef]
- Roy, B.; Ackermann, H.-W.; Pandian, S.; Picard, G.; Goulet, J. Biological inactivation of adhering Listeria monocytogenes by listeriaphages and a quaternary ammonium compound. Appl. Environ. Microbiol. 1993, 59, 2914–2917. [Google Scholar]
- Hibma, A.M.; Jassim, S.A.; Griffiths, M.W. Infection and removal of L-forms of Listeria monocytogenes with bred bacteriophage. Int. J. Food Microbiol. 1997, 34, 197–207. [Google Scholar] [CrossRef]
- Soni, K.A.; Nannapaneni, R. Removal of Listeria monocytogenes biofilms with bacteriophage P100. J. Food Prot. 2010, 73, 1519–1524. [Google Scholar]
- Montanez-Izquierdo, V.Y.; Salas-Vazquez, D.I.; Rodriguez-Jerez, J.J. Use of epifluorescence microscopy to assess the effectiveness of phage P100 in controlling Listeria monocytogenes biofilms on stainless steel surfaces. Food Control 2012, 23, 470–477. [Google Scholar] [CrossRef]
- Ganegama Arachchi, G.J.; Cridge, A.G.; Dias-Wanigasekera, B.M.; Cruz, C.D.; McIntyre, L.; Liu, R.; Flint, S.H.; Mutukumira, A.N. Effectiveness of phages in the decontamination of Listeria monocytogenes adhered to clean stainless steel, stainless steel coated with fish protein, and as a biofilm. J. Ind. Microbiol. Biotechnol. 2013, 40, 1105–1116. [Google Scholar] [CrossRef]
- Chaitiemwong, N.; Hazeleger, W.C.; Beumer, R.R. Inactivation of Listeria monocytogenes by disinfectants and bacteriophages in suspension and stainless steel carrier tests. J. Food Prot. 2014, 77, 2012–2020. [Google Scholar] [CrossRef]
- Lehman, S.M.; Donlan, R.M. Bacteriophage-mediated control of a two-species biofilm formed by microorganisms causing catheter-associated urinary tract infections in an in vitro urinary catheter model. Antimicrob. Agents Chemother. 2015, 59, 1127–1137. [Google Scholar] [CrossRef]
- Hanlon, G.W.; Denyer, S.P.; Olliff, C.J.; Ibrahim, L.J. Reduction in exopolysaccharide viscosity as an aid to bacteriophage penetration through Pseudomonas aeruginosa biofilms. Appl. Environ. Microbiol. 2001, 67, 2746–2753. [Google Scholar] [CrossRef]
- Knezevic, P.; Petrovic, O. A colorimetric microtiter plate method for assessment of phage effect on Pseudomonas aeruginosa biofilm. J. Microbiol. Meth. 2008, 74, 114–118. [Google Scholar] [CrossRef]
- Fu, W.; Forster, T.; Mayer, O.; Curtin, J.J.; Lehman, S.M.; Donlan, R.M. Bacteriophage cocktail for the prevention of biofilm formation by Pseudomonas aeruginosa on catheters in an in vitro model system. Antimicrob. Agents Chemother. 2010, 54, 397–404. [Google Scholar] [CrossRef]
- Ahiwale, S.; Tamboli, N.; Thorat, K.; Kulkarni, R.; Ackermann, H.; Kapadnis, B. In vitro management of hospital Pseudomonas aeruginosa biofilm using indigenous T7-like lytic phage. Curr. Microbiol. 2011, 62, 335–340. [Google Scholar] [CrossRef]
- Knezevic, P.; Obreht, D.; Curcin, S.; Petrusic, M.; Aleksic, V.; Kostanjsek, R.; Petrovic, O. Phages of Pseudomonas aeruginosa: Response to environmental factors and in vitro ability to inhibit bacterial growth and biofilm formation. J. Appl. Microbiol. 2011, 111, 245–254. [Google Scholar] [CrossRef]
- Pires, D.; Sillankorva, S.; Faustino, A.; Azeredo, J. Use of newly isolated phages for control of Pseudomonas aeruginosa PAO1 and ATCC 10145 biofilms. Res. Microbiol. 2011, 162, 798–806. [Google Scholar] [CrossRef] [Green Version]
- Alemayehu, D.; Casey, P.G.; McAuliffe, O.; Guinane, C.M.; Martin, J.G.; Shanahan, F.; Coffey, A.; Ross, R.P.; Hill, C. Bacteriophages ϕMR299-2 and ϕNH-4 can eliminate Pseudomonas aeruginosa in the murine lung and on cystic fibrosis lung airway cells. MBio 2012, 3, e00029-12. [Google Scholar] [CrossRef]
- Liao, K.S.; Lehman, S.M.; Tweardy, D.J.; Donlan, R.M.; Trautner, B.W. Bacteriophages are synergistic with bacterial interference for the prevention of Pseudomonas aeruginosa biofilm formation on urinary catheters. J. Appl. Microbiol. 2012, 113, 1530–1539. [Google Scholar] [CrossRef]
- Phee, A.; Bondy-Denomy, J.; Kishen, A.; Basrani, B.; Azarpazhooh, A.; Maxwell, K. Efficacy of bacteriophage treatment on Pseudomonas aeruginosa biofilms. J Endod. 2013, 39, 364–369. [Google Scholar] [CrossRef]
- Yilmaz, C.; Colak, M.; Yilmaz, B.C.; Ersoz, G.; Kutateladze, M.; Gozlugol, M. Bacteriophage therapy in implant-related infections: an experimental study. J. Bone Joint Surg. Am. 2013, 95, 117–125. [Google Scholar] [CrossRef]
- Zhang, Y.; Hu, Z. Combined treatment of Pseudomonas aeruginosa biofilms with bacteriophages and chlorine. Biotechnol. Bioeng. 2013, 110, 286–295. [Google Scholar] [CrossRef]
- Zhang, Y.; Hunt, H.K.; Hu, Z. Application of bacteriophages to selectively remove Pseudomonas aeruginosa in water and wastewater filtration systems. Water Res. 2013, 47, 4507–4518. [Google Scholar] [CrossRef]
- Danis-Wlodarczyk, K.; Olszak, T.; Arabski, M.; Wasik, S.; Majkowska-Skrobek, G.; Augustyniak, D.; Gula, G.; Briers, Y.; Jang, H.B.; Vandenheuvel, D.; et al. Characterization of the newly isolated lytic bacteriophages KTN6 and KT28 and their efficacy against Pseudomonas aeruginosa biofilm. PLoS ONE 2015, 10, e0127603. [Google Scholar] [CrossRef] [Green Version]
- Sillankorva, S.; Oliveira, R.; Vieira, M.J.; Sutherland, I.W.; Azeredo, J. Bacteriophage Φ S1 infection of Pseudomonas fluorescens planktonic cells versus biofilms. Biofouling 2004, 20, 133–138. [Google Scholar] [CrossRef]
- Sillankorva, S.; Oliveira, R.; Vieira, M.J.; Azeredo, J. Real-time quantification of Pseudomonas fluorescens cell removal from glass surfaces due to bacteriophage φS1 application. J. Appl. Microbiol. 2008, 105, 196–202. [Google Scholar] [CrossRef]
- Sillankorva, S.; Neubauer, P.; Azeredo, J. Pseudomonas fluorescens biofilms subjected to phage phiIBB-PF7A. Bmc Biotechnol. 2008, 8, 79. [Google Scholar] [CrossRef] [Green Version]
- Sillankorva, S.; Neubauer, P.; Azeredo, J. Phage control of dual species biofilms of Pseudomonas fluorescens and Staphylococcus lentus. Biofouling 2010, 26, 567–575. [Google Scholar] [CrossRef] [Green Version]
- Cornelissen, A.; Ceyssens, P.J.; T'Syen, J.; Van, P.H.; Noben, J.P.; Shaburova, O.V.; Krylov, V.N.; Volckaert, G.; Lavigne, R. The T7-related Pseudomonas putida phage φ15 displays virion-associated biofilm degradation properties. PLoS ONE 2011, 6, e18597. [Google Scholar] [CrossRef]
- Gino, E.; Starosvetsky, J.; Kurzbaum, E.; Armon, R. Combined chemical-biological treatment for prevention/rehabilitation of clogged wells by an iron-oxidizing bacterium. Environ. Sci. Technol. 2010, 44, 3123–3129. [Google Scholar] [CrossRef]
- Del Pozo, J.L.; Alonso, M.; Arciola, C.R.; Gonzalez, R.; Leiva, J.; Lasa, I.; Penades, J. Biotechnological war against biofilms. Could phages mean the end of device-related infections? Int. J. Artif. Organs 2007, 30, 805–812. [Google Scholar]
- Son, J.S.; Lee, S.J.; Jun, S.Y.; Yoon, S.J.; Kang, S.H.; Paik, H.R.; Kang, J.O.; Choi, Y.J. Antibacterial and biofilm removal activity of a podoviridae Staphylococcus aureus bacteriophage SAP-2 and a derived recombinant cell-wall-degrading enzyme. Appl. Microbiol. Biotechnol. 2010, 86, 1439–1449. [Google Scholar] [CrossRef]
- Rahman, M.; Kim, S.; Kim, S.M.; Seol, S.Y.; Kim, J. Characterization of induced Staphylococcus aureus bacteriophage SAP-26 and its anti-biofilm activity with rifampicin. Biofouling 2011, 27, 1087–1093. [Google Scholar] [CrossRef]
- Kelly, D.; McAuliffe, O.; Ross, R.P.; Coffey, A. Prevention of Staphylococcus aureus biofilm formation and reduction in established biofilm density using a combination of phage K and modified derivatives. Lett. Appl. Microbiol. 2012, 54, 286–291. [Google Scholar] [CrossRef]
- Seth, A.K.; Geringer, M.R.; Nguyen, K.T.; Agnew, S.P.; Dumanian, Z.; Galiano, R.D.; Leung, K.P.; Mustoe, T.A.; Hong, S.J. Bacteriophage therapy for Staphylococcus aureus biofilm-infected wounds: a new approach to chronic wound care. Plast. Reconstr. Surg. 2013, 131, 225–234. [Google Scholar] [CrossRef]
- Lungren, M.P.; Christensen, D.; Kankotia, R.; Falk, I.; Paxton, B.E.; Kim, C.Y. Bacteriophage K for reduction of Staphylococcus aureus biofilm on central venous catheter material. Bacteriophage 2013, 3, e26825. [Google Scholar] [CrossRef]
- Alves, D.R.; Gaudion, A.; Bean, J.E.; Perez-Esteban, P.; Arnot, T.; Harper, D.R.; Kot, W.; Hansen, L.H.; Enright, M.C.; Jenkins, A.T. Combined use of bacteriophage K and a novel bacteriophage to reduce Staphylococcus aureus biofilm. Appl. Environ. Microbiol. 2014, 80, 6694–6703. [Google Scholar] [CrossRef]
- Drilling, A.; Morales, S.; Boase, S.; Jervis-Bardy, J.; James, C.; Jardeleza, C.; Tan, N.C.; Cleland, E.; Speck, P.; Vreugde, S.; et al. Safety and efficacy of topical bacteriophage and ethylenediaminetetraacetic acid treatment of Staphylococcus aureus infection in a sheep model of sinusitis. Int. Forum Allergy Rhinol. 2014, 4, 176–186. [Google Scholar] [CrossRef]
- Drilling, A.; Morales, S.; Jardeleza, C.; Vreugde, S.; Speck, P.; Wormald, P.J. Bacteriophage reduces biofilm of Staphylococcus aureus ex vivo isolates from chronic rhinosinusitis patients. Am. J. Rhinol. Allergy 2014, 28, 3–11. [Google Scholar] [CrossRef]
- Lungren, M.P.; Donlan, R.M.; Kankotia, R.; Paxton, B.E.; Falk, I.; Christensen, D.; Kim, C.Y. Bacteriophage K antimicrobial-lock technique for treatment of Staphylococcus aureus central venous catheter-related infection: a leporine model efficacy analysis. J. Vasc. Interv. Radiol. 2014, 25, 1627–1632. [Google Scholar] [CrossRef]
- Gutierrez, D.; Vandenheuvel, D.; Martinez, B.; Rodriguez, A.; Lavigne, R.; Garcia, P. Two phages, phiIPLA-RODI and phiIPLA-C1C, lyse mono- and dual-species staphylococcal biofilms. Appl. Environ. Microbiol. 2015, 81, 3336–3348. [Google Scholar] [CrossRef]
- Wood, H.L.; Holden, S.R.; Bayston, R. Susceptibility of Staphylococcus epidermidis biofilm in CSF shunts to bacteriophage attack. Eur. J. Ped. Surgery 2001, 11, S56–S57. [Google Scholar]
- Curtin, J.J.; Donlan, R.M. Using bacteriophages to reduce formation of catheter-associated biofilms by Staphylococcus epidermidis. Antimicrob. Agents Chemother. 2006, 50, 1268–1275. [Google Scholar] [CrossRef]
- Cerca, N.; Oliveria, R.; Azeredo, J. Susceptibility of Staphylococcus epidermidis planktonic cells and biofilms to the lytic action of staphylococcus bacteriophage K. Lett. Appl. Microbiol. 2007, 45, 313–317. [Google Scholar] [CrossRef]
- Tan, D.; Dahl, A.; Middelboe, M. Vibriophages differentially influence biofilm formation by Vibrio anguillarum strains. Appl. Environ. Microbiol. 2015, 81, 4489–4497. [Google Scholar] [CrossRef]
- Abedon, S.T. Phage therapy best practices. In Bacteriophages in Health and Disease; Hyman, P., Abedon, S.T., Eds.; CABI Press: Wallingford, UK, 2012; pp. 256–272. [Google Scholar]
- Abedon, S.T. Bacteriophages as drugs: The pharmacology of phage therapy. In Phage Therapy: Current Research and Applications; Borysowski, J., Miedzybrodzki, R., Górski, A., Eds.; Caister Academic Press: Norfolk, UK, 2014; pp. 69–100. [Google Scholar]
- Abedon, S.T. Phage therapy: Eco-physiological pharmacology. Scientifica 2014, 2014, 581639. [Google Scholar] [CrossRef]
- Wright, A.; Hawkins, C.H.; Änggård, E.E.; Harper, D.R. A controlled clinical trial of a therapeutic bacteriophage preparation in chronic otitis due to antibiotic-resistant Pseudomonas aeruginosa; a preliminary report of efficacy. Clin. Otolaryng. 2009, 34, 349–357. [Google Scholar] [CrossRef]
- Hawkins, C.; Harper, D.; Burch, D.; Anggard, E.; Soothill, J. Topical treatment of Pseudomonas aeruginosa otitis of dogs with a bacteriophage mixture: A before/after clinical trial. Vet. Microbiol. 2010, 145, 309–313. [Google Scholar] [CrossRef]
- Pirnay, J.P.; De, V.D.; Verbeken, G.; Merabishvili, M.; Chanishvili, N.; Vaneechoutte, M.; Zizi, M.; Laire, G.; Lavigne, R.; Huys, I.; et al. The phage therapy paradigm: prêt-à-porter or sur-mesure? Pharm. Res. 2011, 28, 934–937. [Google Scholar] [CrossRef]
- Chan, B.K.; Abedon, S.T. Phage therapy pharmacology: Phage cocktails. Adv. Appl. Microbiol. 2012, 78, 1–23. [Google Scholar]
- Chan, B.K.; Abedon, S.T.; Loc-Carrillo, C. Phage cocktails and the future of phage therapy. Future Microbiol. 2013, 8, 769–783. [Google Scholar] [CrossRef]
- Schmerer, M.; Molineux, I.J.; Bull, J.J. Synergy as a rationale for phage therapy using phage cocktails. PeerJ 2014, 2, e590. [Google Scholar] [CrossRef]
- Abedon, S.T. Lysis from without. Bacteriophage 2011, 1, 46–49. [Google Scholar] [CrossRef]
- Blackledge, M.S.; Worthington, R.J.; Melander, C. Biologically inspired strategies for combating bacterial biofilms. Curr. Opin. Pharmacol. 2013, 13, 699–706. [Google Scholar] [CrossRef]
- Thallinger, B.; Prasetyo, E.N.; Nyanhongo, G.S.; Guebitz, G.M. Antimicrobial enzymes: An emerging strategy to fight microbes and microbial biofilms. Biotechnol. J. 2013, 8, 97–109. [Google Scholar] [CrossRef]
- Shen, Y.; Mitchell, M.S.; Donovan, D.M.; Nelson, D.C. Phage-based enzybiotics. In Bacteriophages in Health and Disease; Hyman, P., Abedon, S.T., Eds.; CABI Press: Wallingford, UK, 2012; pp. 217–239. [Google Scholar]
- Yan, J.; Mao, J.; Xie, J. Bacteriophage polysaccharide depolymerases and biomedical applications. BioDrugs 2014, 28, 265–274. [Google Scholar] [CrossRef]
- Hwang, I.Y.; Tan, M.H.; Koh, E.; Ho, C.L.; Poh, C.L.; Chang, M.W. Reprogramming microbes to be pathogen-seeking killers. ACS Synth. Biol. 2014, 3, 228–237. [Google Scholar] [CrossRef]
- Curtright, A.J.; Abedon, S.T. Phage therapy: Emergent property pharmacology. J. Bioanal. Biomed. 2011, S6. [Google Scholar] [CrossRef]
- Loc-Carrillo, C.; Abedon, S.T. Pros and cons of phage therapy. Bacteriophage 2011, 1, 111–114. [Google Scholar] [CrossRef]
- Viruses vs. Superbugs. Available online: http://www.bacteriophagetherapy.info/ECF40946-8E2F-4890-9CA6-D390A26E39C1/Pros and cons of phage therapy.html (accessed on 6 September 2015).
- Phage Therapy. Available online: phage-therapy.org (accessed on 6 September 2015).
- Kutter, E.M.; Kuhl, S.J.; Abedon, S.T. Re-establishing a place for phage therapy in western medicine. Future Microbiol. 2015, 10, 685–688. [Google Scholar] [CrossRef]
- Hargreaves, K.R.; Clokie, M.R. Clostridium difficile phages: Still difficult? Front Microbiol. 2014, 5, 184. [Google Scholar] [CrossRef]
- Balogh, B.; Jones, J.B.; Iriarte, F.B.; Momol, M.T. Phage therapy for plant disease control. Curr. Pharm. Biotechnol. 2010, 11, 48–57. [Google Scholar] [CrossRef]
- Bull, J.J.; Otto, G.; Molineux, I.J. In vivo growth rates are poorly correlated with phage therapy success in a mouse infection model. Antimicrob. Agents Chemother. 2012, 56, 949–954. [Google Scholar] [CrossRef]
- Henry, M.; Lavigne, R.; Debarbieux, L. Predicting in vivo efficacy of therapeutic bacteriophages used to treat pulmonary infections. Antimicrob. Agents Chemother. 2013, 57, 5961–5968. [Google Scholar] [CrossRef]
- Bull, J.J.; Gill, J.J. The habits of highly effective phages: Population dynamics as a framework for identifying therapeutic phages. Front Microbiol. 2014, 5, 61. [Google Scholar] [CrossRef]
- Lindberg, H.M.; McKean, K.A.; Wang, I.-N. Phage fitness may help predict phage therapy efficacy. Bacteriophage 2014, 4, e964081. [Google Scholar] [CrossRef]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abedon, S.T. Ecology of Anti-Biofilm Agents II: Bacteriophage Exploitation and Biocontrol of Biofilm Bacteria. Pharmaceuticals 2015, 8, 559-589. https://doi.org/10.3390/ph8030559
Abedon ST. Ecology of Anti-Biofilm Agents II: Bacteriophage Exploitation and Biocontrol of Biofilm Bacteria. Pharmaceuticals. 2015; 8(3):559-589. https://doi.org/10.3390/ph8030559
Chicago/Turabian StyleAbedon, Stephen T. 2015. "Ecology of Anti-Biofilm Agents II: Bacteriophage Exploitation and Biocontrol of Biofilm Bacteria" Pharmaceuticals 8, no. 3: 559-589. https://doi.org/10.3390/ph8030559