Placental Adaptations in Growth Restriction

 and
and
Abstract
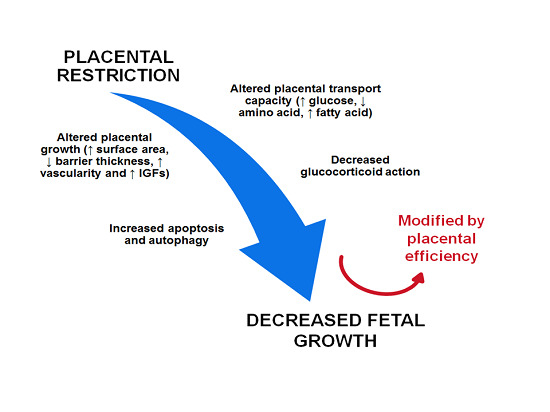
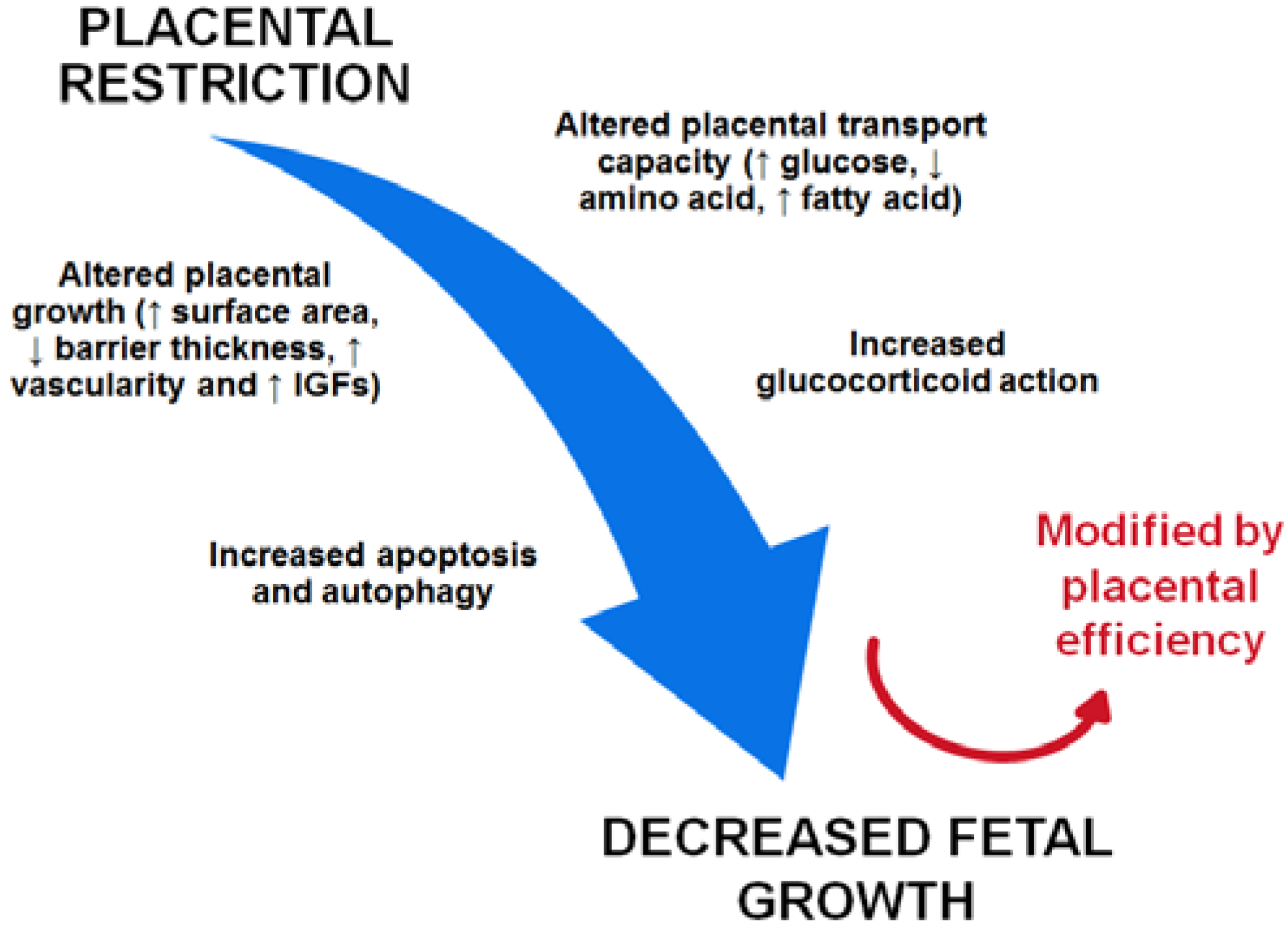
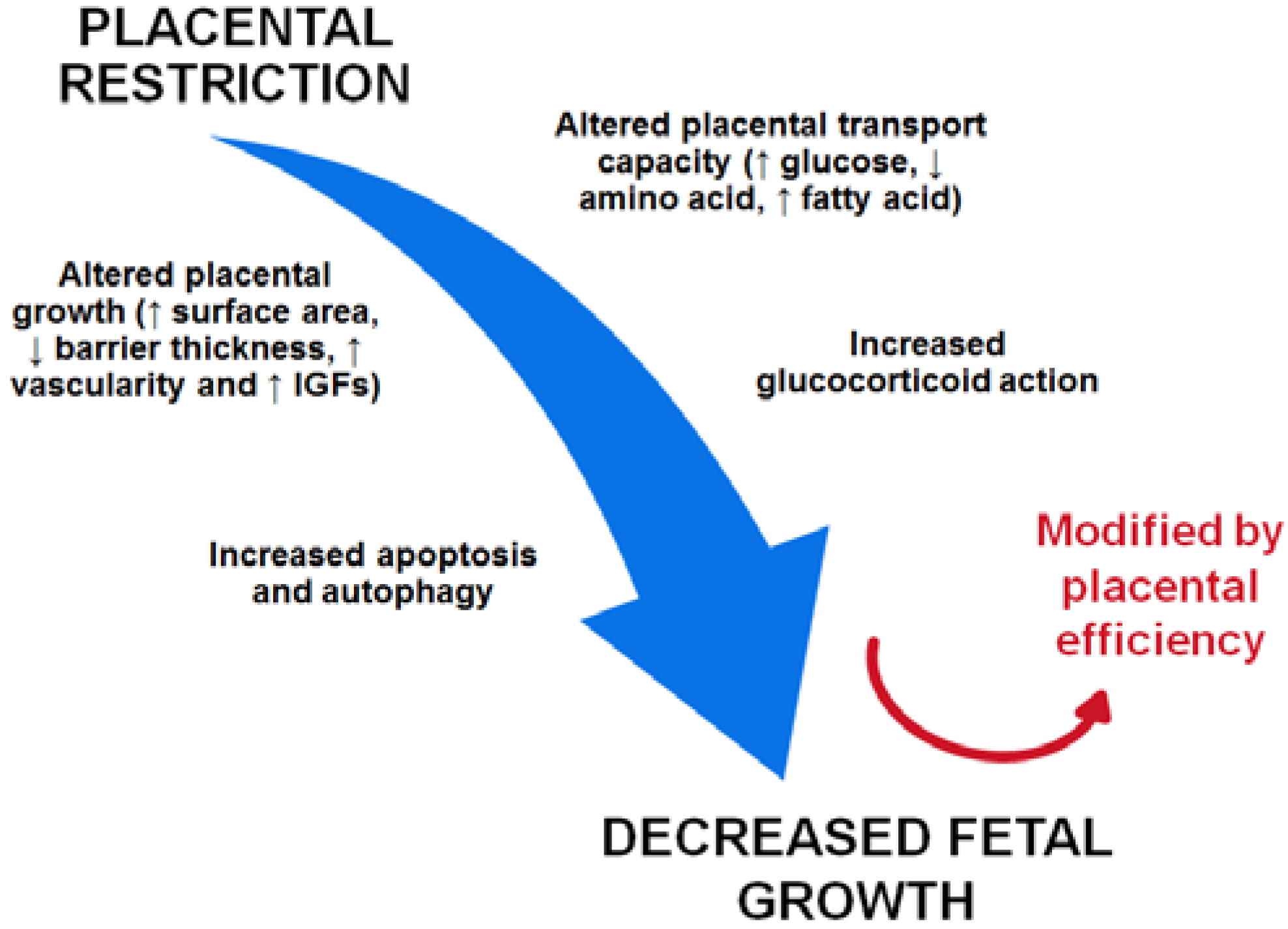
:
1. Introduction
2. Placental Structure and Function

{kind=link}
{kind=link}
| Species | Placental Shape | Placental Structure |
|---|---|---|
| Humans | Discoid | Hemochorial |
| Ruminants (Sheep, cattle, goats) | Cotyledonary | Epitheliochorial |
| Rodents (rats, mice) | Discoid | Hemochorial |
| Pigs | Diffuse | Epitheliochorial |
| Horses | Diffuse | Epitheliochorial |
| Carnivores (cats, dogs) | Zonary | Epitheliochorial |
| Primates | Discoid | Hemochorial |
3. Factors Affecting Placental Substrate Transport Capacity
3.1. Fetal Oxygenation
3.2. Placental Size and Morphology
3.3. Blood Flow and Vascularity
3.4. Insulin-Like Growth Factors
3.5. Placental Apoptosis, Autophagy and Glucocorticoid Actions
3.6. Placental Transporter Abundance
3.6.1. Placental Glucose Transport Systems
3.6.2. Placental Amino Acid Transport Systems
3.6.3. Placental Fatty Acid Transporter Systems
4. The Placenta and Development of Intrauterine Growth Restriction
| Experimental Intervention | Impact on the Placenta | Impact on the Fetus | |
|---|---|---|---|
| Surgical Umbilical Artery Ligation (SUAL) | Isolation and ligation of one umbilical artery close to the fetal abdomen | placental infarction causing ↓ umbilical blood flow and ↓ placental substrate transfer [118,119] | Hypoxemia IUGR |
| Maternal Hyperthermia | Exposure of pregnant ewes to an environment with an increased ambient temperature | ↓ uterine artery flow and ↓ placental weight due to ↑ maternal temperature [2,30] | IUGR |
| Placental Embolism | Repeated injection of microspheres (15μm) into the placenta via the umbilical artery through a catheter implanted in the descending aorta or fetal umbilical vein | block placental capillaries causing ↓ placental surface area [120] | Hypoxemia Hypoglycemia IUGR |
| Uterine Carunclectomy | Surgical removal of the majority of the endometrial caruncles from the uterus of non-pregnant ewes prior to conception | ↓ placental weight due to ↓ placentomes [116] | Hypoxemia Hypoglycemia IUGR |
5. Animal Models of Placental Insufficiency
5.1. Single Umbilical Artery Ligation (SUAL)
5.2. Maternal Hyperthermia
5.3. Placental Embolism
5.4. Endometrial Carunclectomy
6. Regulation of Placental Growth and Substrate Transport in IUGR Pregnancy
6.1. Placental Size and Morphology
6.2. Oxygen Supply/Uptake, Blood Flow and Vascularity
6.3. IGFs
6.4. Apoptosis, Autophagy and Glucocorticoid Action
6.5. Glucose Transport Systems
| Models of IUGR | Impact on Glucose Transporters | Impact on Amino Acid Transporters | Impact on Fatty Acid Transporters |
|---|---|---|---|
| Human IUGR | ↓ GLUT1 mRNA, ↔ GLUT1 protein [169] ↑ GLUT4 mRNA, ↔ GLUT1 protein [169] ↑ GLUT3 mRNA & protein [169] ↓ GLUT1 protein [171] ↓ GLUT1 protein ↔ GLUT3 protein [170] ↔ GLUT1 protein [172] | ↓ System A transporter activity [173,174] ↓ SNAT2 mRNA & protein [175] | NS |
| Sheep | |||
| Surgical Umbilical Artery Ligation (SUAL) | NS | NS | NS |
| Maternal Hyperthermia | ↓ GLUT8 mRNA & protein | ↑ LAT-1 & LAT-2 mRNA [19] | NS |
| Placental Embolism | NS | NS | NS |
| Uterine Carunclectomy | NS | NS | NS |
6.6. Amino Acid Transport Systems
6.7. Fatty Acid Transport Systems
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Li, Z.; Zeki, R.; Hilder, L.; Sullivan, E.A. Australia’s Mothers and Babies 2010, Ca. no. PER 57 ed.; AIHW National Perinatal Epidemiology and Statistics: Canberra, Australia, 2012. [Google Scholar]
- Limesand, S.W.; Rozance, P.J.; Zerbe, G.O.; Hutton, J.C.; Hay, W.W., Jr. Attenuated insulin release and storage in fetal sheep pancreatic islets with intrauterine growth restriction. Endocrinology 2006, 147, 1488–1497. [Google Scholar] [CrossRef]
- Bamfo, J.E.; Odibo, A.O. Diagnosis and management of fetal growth restriction. J. Pregnancy 2011, 2011. [Google Scholar] [CrossRef]
- Barker, D.J. Developmental origins or chronic disease. Public Healh 2012, 126, 185–189. [Google Scholar] [CrossRef]
- Barker, D.J.; Gluckman, P.D.; Godfrey, K.M.; Harding, J.E.; Owens, J.A.; Robinson, J.S. Fetal nutrition and cardiovascular disease in adult life. Lancet 1993, 341, 938–941. [Google Scholar] [CrossRef] [PubMed]
- Li, C.C.Y.; Maloney, C.A.; Cropley, J.E.; Suter, C.M. Epigenetic programming by maternal nutritions: Shaping future generations. Epigenomics 2010, 2, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J.; Gelow, J.; Thornburg, K.; Osmond, C.; Kajantie, E.; Eriksson, J.G. The early origins of chronic heart failure: Impaired placental growth and initiation of insulin resistance in childhood. Eur. J. Heart Fail. 2010, 12, 819–825. [Google Scholar] [CrossRef] [PubMed]
- Jansson, T.; Powell, T.L. Role of the placenta in fetal programming: Underlying mechanisms and potential interventional approaches. Clin. Sci. (Lond.) 2007, 113, 1–13. [Google Scholar] [CrossRef]
- Jones, H.N.; Powell, T.L.; Jansson, T. Regulation of placental nutrient transport—A review. Placenta 2007, 28, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Marconi, A.M.; Paolini, C.L. Nutrient transport across the intrauterine growth-restricted placenta. Semin. Perinatol. 2008, 32, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Gude, N.M.; Roberts, C.T.; Kalionis, B.; King, R.G. Growth and function of the normal human placenta. Thromb. Res. 2004, 114, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Sibley, C.P.; Brownbill, P.; Dilworth, M.; Glazier, J.D. Review: Adaption in placental nutrient supply to meet fetal growth demand: Implications for programmings. Placenta 2010, 24, 70–74. [Google Scholar] [CrossRef]
- Lager, S.; Powell, T.L. Regulation of nutrient transport across the placenta. J. Pregnancy 2012, 2012, 1–14. [Google Scholar] [CrossRef]
- Barry, J.S.; Anthony, R.V. The pregnant sheep as a model for human pregnancy. Theriogenology 2008, 69, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Fowden, A.L.; Ward, J.W.; Wooding, F.P.B.; Forhead, A.J.; Constancia, M. Programming placental nutrient transfer capacity. J. Physiol. 2006, 572, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.W.; Forhead, A.J.; Wooding, F.P.B.; Fowden, A.L. Functional significance and cortisol dependence of the gross morphology of ovine placentomes during late gestation. Biol. Reprod. 2006, 74, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Ahokas, R.A.; McKinney, E.T. Development and Physiology of the Placenta and Membrane; The Global Library of Women’s Medicine: London, UK, 2008. [Google Scholar]
- Fowden, A.L.; Sferruzzi-Perri, A.N.; Coan, P.M.; Burton, G. Placental efficiency and adaptation: Endocrine regulation. J. Physiol. 2009, 587, 3459–3472. [Google Scholar] [CrossRef] [PubMed]
- Regnault, T.R.; Marconi, A.M.; Smith, C.H.; Glazier, J.D.; Novak, D.A.; Sibley, C.P.; Jansson, T. Placental amino acid transport systems and fetal growth restriction—A workshop report. Placenta 2005, 26, 76–80. [Google Scholar] [CrossRef]
- Roberts, C.T.; Sohlstrom, A.; Kind, K.L.; Grant, P.A.; Earl, R.A.; Robinson, J.S.; Khong, T.Y.; Owens, P.C.; Owens, J.A. Altered placental structure induced by maternal food restriction in guinea pigs: A role for circulating IGF-II and IGFBP-2 in the mother? Placenta 2001, 22, 77–82. [Google Scholar] [CrossRef]
- Anthony, R.V.; Scheaffer, A.N.; Wright, C.D.; Regnault, T.R. Ruminant models of prenatal growth restriction. Reprod. Suppl. 2003, 61, 183–194. [Google Scholar] [PubMed]
- Regnault, T.R.; de Vrijer, B.; Galan, H.L.; Wilkening, R.B.; Battaglia, F.C.; Meschia, G. Umbilical uptakes and transplacental concentration ratios of amino acids in severe fetal growth restriction. Pediatr. Res. 2013, 73, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Morrison, J.L. Sheep models of intrauterine growth restriction: Fetal adaptations and consequences. Clin. Exp. Pharmacol. Physiol. 2008, 35, 730–743. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, R.; Langridge, J.; Inchley, K.; Murotsuki, J.; Possmayer, F. Changes in surfactant-associated protein mRNA profile in growth-restricted fetal sheep. Am. J. Physiol. 1999, 276, 459–465. [Google Scholar]
- Jones, C.T.; Roebuck, M.M.; Walker, D.W.; Lagercrantz, H.; Johnston, B.M. Cardiovascular, metabolic and endocrine effects of chemical sympathectomy and of adrenal demedullation in fetal sheep. J. Dev. Physiol. 1987, 9, 347–367. [Google Scholar] [PubMed]
- Miller, S.L.; Chai, M.; Loose, J.; Castillo-Melendez, M.; Walker, D.W.; Jenkin, G.; Wallace, E.M. The effects of maternal betamethasone administration on the intrauterine growth-restricted fetus. Endocrinology 2007, 148, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Mallard, E.C.; Rees, S.; Stringer, M.; Cock, M.L.; Harding, R. Effects of chronic placental insufficiency on brain development in fetal sheep. Pediatr. Res. 1998, 43, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.S.; Hart, I.C.; Kingston, E.J.; Jones, C.T.; Thorburn, G.D. Studies on the growth of the fetal sheep. The effects of reduction of placental size on hormone concentration in fetal plasma. J. Dev. Physiol. 1980, 2, 239–248. [Google Scholar] [PubMed]
- Dobbing, J.; Sands, J. Comparative aspects of the brain growth spurt. Early Hum. Dev. 1979, 3, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Regnault, T.R.H.; de Vrijer, B.; Galan, H.L.; Wilkening, R.B.; Battaglia, F.C.; Meschia, G. Development and mechanisms of fetal hypoxia in severe fetal growth restriction. Placenta 2007, 28, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.E. Role of placental function in mediating conceptus growth and survival. J. Anim. Sci. 2002, 80, 195–201. [Google Scholar]
- Vonnahme, K.A.; Arndt, W.J.; Johnson, M.L.; Borowicz, P.P.; Reynolds, L.P. Effect of morphology on placentome size, vascularity and vasoreactivity in late pregnant sheep. Biol. Reprod. 2008, 79, 976–982. [Google Scholar] [CrossRef] [PubMed]
- Ehrhardt, R.A.; Bell, A.W. Growth and metabolism of the ovine placenta during mid-gestation. Placenta 1995, 16, 727–741. [Google Scholar] [CrossRef] [PubMed]
- Vatnick, I.; Schoknecht, P.A.; Darrigrand, R.; Bell, A.W. Growth and metabolism of the placenta after unilateral fetectomy in twin pregnant ewes. J. Dev. Physiol. 1991, 15, 351–356. [Google Scholar] [PubMed]
- Robinson, J.S.; Hartwich, K.M.; Walker, S.K.; Erwich, J.J.; Owens, J.A. Early influences on embryonic and placental growth. Acta Paediatr. Suppl. 1997, 423, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Alexander, G. Studies on the placenta of the sheep (Ovis aries L.). Effect of surgical reduction in the number of caruncles. J. Reprod. Fertil. 1964, 30, 307–322. [Google Scholar] [CrossRef]
- Wilson, M.E.; Ford, S.P. Comparative aspects of placental efficiency. Reprod. Suppl. 2001, 58, 223–232. [Google Scholar] [PubMed]
- Barker, D.J.P.; Thornburg, K.L. Placental programming of chronic diseases, cancer and lifespan: A review. Placenta 2013, 34, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J.; Bull, A.R.; Osmond, C.; Simmonds, S.J. Fetal and placental size and risk of hypertension in adult life. BMJ 1990, 301, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Sibai, B.M.; Frangieh, A. Maternal adaptation to pregnancy. Curr. Opin. Obstet. Gynecol. 1995, 7, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Carter, A.M. Factors affecting gas transfer across the placenta and the oxygen supply to the fetus. J. Dev. Physiol. 1989, 12, 305–322. [Google Scholar] [PubMed]
- Bell, A.W.; Kennaugh, J.M.; Battaglia, F.C.; Makowski, E.L.; Meschia, G. Metabolic and circulatory studies of fetal lamb at midgestation. Am. J. Physiol. 1986, 250, 538–544. [Google Scholar]
- Bonds, D.R.; Crosby, L.O.; Cheek, T.G.; Hagerdal, M.; Gutsche, B.B.; Gabbe, S.G. Estimation of human fetal-placental unit metabolic rate by application of the bohr principle. J. Dev. Physiol. 1986, 8, 49–54. [Google Scholar] [PubMed]
- Campbell, A.G.; Dawes, G.S.; Fishman, A.P.; Hyman, A.I.; James, G.B. The oxygen consumption of the placenta and foetal membranes in the sheep. J. Physiol. 1966, 182, 439–464. [Google Scholar] [CrossRef] [PubMed]
- Coan, P.M.; Ferguson-Smith, A.C.; Burton, G. Developmental dynamics of the definitive mouse placenta assessed by stereology. Biol. Reprod. 2004, 70, 1806–1813. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, L.P.; Borawicz, P.P.; Vonnahme, K.A.; Johnson, M.L.; Grazul-Bilska, A.T.; Wallace, J.M.; Caton, J.S.; Redmer, D.A. Animal models of placental angiogenesis. Placenta 2005, 26, 689–708. [Google Scholar] [CrossRef] [PubMed]
- Demir, R.; Seval, Y.; Huppertz, B. Vasculogenesis and angiogenesis in the early human placenta. Acta Histochem. 2007, 109, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Charnock-Jones, D.S.; Kaufmann, P.; Mayhew, T.M. Aspects of human fetoplacental vasculogenesis and angiogenesis. I. Molecular regulation. Placenta 2004, 25, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Perkins, J. Angiogenesis and intrauterine growth restriction. Baillieres Best Pract. Res. Clin. Obstet. Gynaecol. 2000, 14, 981–998. [Google Scholar] [CrossRef] [PubMed]
- Maisonpierre, P.C.; Suri, C.; Jones, P.F.; Bartunkova, S.; Wiegand, S.J.; Radziejewski, C.; Compton, D.; McClain, J.; Aldrich, T.H.; Papadopoulos, N.; et al. Angiopoietin-2, a natural antagonist for tie2 that disrupts in vivo angiogenesis. Science 1997, 277, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Plaisier, M.; Rodrigues, S.; Willems, F.; Koolwijk, P.; van Hinsbergh, V.W.; Helmerhorst, F.M. Different degrees of vascularization and their relationship to the expression of vascular endothelial growth factor, placental growth factor, angiopoietins, and their receptors in first-trimester decidual tissues. Fertil. Steril. 2007, 88, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Cross, J.C.; Hemberger, M.; Lu, Y.; Nozaki, T.; Whiteley, K.; Masutani, M.; Adamson, S.L. Trophoblast functions, angiogenesis and remodeling of the maternal vasculature in the placenta. Mol. Cell. Endocrinol 2002, 187, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Regnault, T.R.; de Vrijer, B.; Galan, H.L.; Davidsen, M.L.; Trembler, K.A.; Battaglia, F.C.; Wilkening, R.B.; Anthony, R.V. The relationship between transplacental O2 diffusion and placental expression of PLGF, VEGF and their receptors in a placental insufficiency model of fetal growth restriction. J. Physiol. 2003, 550, 641–656. [Google Scholar] [CrossRef] [PubMed]
- Hagen, A.S.; Orbus, R.J.; Wilkening, R.B.; Regnault, T.R.; Anthony, R.V. Placental expression of angiopoietin-1, angiopoietin-2 and tie-2 during placental development in an ovine model of placental insufficiency-fetal growth restriction. Pediatr. Res. 2005, 58, 1228–1232. [Google Scholar] [CrossRef] [PubMed]
- Fowden, A.L.; Forhead, A.J.; Coan, P.M.; Burton, G. The placenta and intrauterine programming. J. Neuroendocrinol. 2008, 20, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Sferruzzi-Perri, A.N.; Owens, J.A.; Pringle, K.G.; Roberts, C.T. The neglected role of insulin-like growth factors in the maternal circulation regulating fetal growth. J. Physiol. 2011, 589, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Roos, S.; Lagerlöf, O.; Wennergren, M.; Powell, T.L.; Jansson, T. Regulation of amino acid transporters by glucose and growth factors in cultured primary human trophoblast cells is mediated by mtor signalling. Am. J. Physiol. Cell Physiol. 2009, 297, 723–731. [Google Scholar] [CrossRef]
- Harding, J.E.; Liu, L.; Evans, P.C.; Gluckman, P.D. Insulin-like growth factor 1 alters feto-placental protein and carbohydrate metabolism in fetal sheep. Endocrinology. 1994, 134, 1509–1514. [Google Scholar] [PubMed]
- Constancia, M.; Angiolini, E.; Sandovic, I.; Smith, P.; Smith, R.; Kelsey, G.; Dean, W.; Ferguson-Smith, A.C.; Sibley, C.P.; Reik, W.; et al. Adaption of nutrient supply to fetal demand in the mouse involves interaction between the Igf2 gene and placental transporter proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 19219–19224. [Google Scholar] [CrossRef] [PubMed]
- Sibley, C.P.; Coan, P.M.; Ferguson-Smith, A.C.; Dean, W.; Hughes, J.; Smith, P.; Reik, W.; Burton, G.J.; Fowden, A.L.; Constancia, M. Placental-specific insulin-like growth factor 2 (Igf2) regulates the diffusional exchange characteristics of the mouse placenta. Proc. Natl. Acad. Sci. USA 2004, 101, 8204–8208. [Google Scholar] [CrossRef] [PubMed]
- Constancia, M.; Hemberger, M.; Hughes, J.; Dean, W.; Ferguson-Smith, A.; Fundele, R.; Stewart, F.; Kelsey, G.; Fowden, A.; Sibley, C.; et al. Placental-specific IGF-II is a major modulator of placental and fetal growth. Nature 2002, 417, 945–948. [Google Scholar] [CrossRef] [PubMed]
- Huppertz, B.; Kadyrov, M.; Kingdom, J.C. Apoptosis and its role in the trophoblast. Am. J. Obstet. Gynecol. 2006, 195, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Signorelli, P.; Avagliano, L.; Virgili, E.; Gagliostro, V.; Doi, P.; Braidotti, P.; Bulfamante, G.P.; Ghidoni, R.; Marconi, A.M. Autophagy in term normal human placentas. Placenta 2011, 32, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.H.; Hsieh, T.T.; Chen, S.F.; Li, M.J.; Yeh, Y.L. Autophagy in the human placenta throughout gestation. PLoS One 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar] [CrossRef] [PubMed]
- Crighton, D.; Wilkinson, S.; Ryan, K.M. Dram links autophagy to p53 and programmed cell death. Autophagy 2007, 3, 72–74. [Google Scholar] [CrossRef]
- Michael, A.E.; Thurston, L.M.; Rae, M.T. Glucocorticoid metabolism and reproduction: A tale of two enzymes. Reproduction 2003, 126, 425–441. [Google Scholar] [CrossRef] [PubMed]
- Korgun, E.T.; Ozmen, A.; Unek, G.; Mendilcioglu, I. The Effects of Glucocorticoids on Fetal and Placental Development; InTech: Rijeka, Croatia, 2012. [Google Scholar]
- Alfaidy, N.; Li, W.; MacIntosh, T.; Yang, K.; Challis, J. Late gestation increase in 11beta-hydroxysteroid dehydrogenase 1 expression in human fetal membranes: A novel intrauterine source of cortisol. J. Clin. Endocrinol. Metab. 2003, 88, 5033–5038. [Google Scholar] [CrossRef] [PubMed]
- Shams, M.; Kilby, M.D.; Somerset, D.A.; Howie, A.J.; Gupta, A.; Wood, P.J.; Afnan, M.; Stewart, P.M. 11beta-hydroxysteroid dehydrogenase type 2 in human pregnancy and reduced expression in intrauterine growth restriction. Hum. Reprod. 1998, 13, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Whorwood, C.B.; Firth, K.M.; Budge, H.; Symonds, M.E. Maternal undernutrition during early to midgestation programs tissue-specific alterations in the expression of the glucocorticoid receptor, 11beta-hydroxysteroid dehydrogenase isoforms, and type 1 angiotensin ii receptor in neonatal sheep. Endocrinology 2001, 142, 2854–2864. [Google Scholar] [PubMed]
- Sibley, C.P.; Turner, M.A.; Cetin, I.; Ayuk, P.; Boyd, R.; D’Souza, S.W.; Glazier, J.D.; Greenwood, S.L.; Jansson, T.; Powell, T.L. Placental phenotypes of intrauterine growth. Pediatr. Res. 2005, 58, 827–832. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.; Heller, D.S.; Zamudio, S.; Illsley, N.P. Glucose transporter 3 (GLUT3) protein expression in human placenta across gestation. Placenta 2011, 32, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, A.L.; Powell, T.L.; Wennergren, M.; Jansson, T. Glucose metabolism in the the human preterm and term placenta of IUGR fetuses. Placenta 2004, 25, 337–346. [Google Scholar] [CrossRef] [PubMed]
- DiGiacomo, J.E.; Hay, W.W., Jr. Placental-fetal glucose exchange and placental glucose consumption in pregnant sheep. Am. J. Physiol. 1990, 258, 360–367. [Google Scholar]
- Hay, W.W., Jr. Placental-fetal glucose exchange and fetal glucose metabolism. Trans. Am. Clin. Climatol. Assoc. 2006, 117, 321–340. [Google Scholar] [PubMed]
- Novakovic, B.; Gordon, L.; Robinson, W.P.; Desoye, G.; Saffery, R. Glucose as a fetal nutrients: Dynamic regulation of several glucose transporter genes by DNA methylation in the human placenta across gestation. J. Nutr. Biochem. 2013, 24, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Dandrea, J.; Wilson, V.; Gopalakrishnan, G.; Heasman, L.; Budge, H.; Stephenson, T.; Symonds, M.E. Maternal nutritional manipulation of placental growth and glucose transporter 1 (GLUT-1) abundance in sheep. Reproduction 2001, 122, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Larqué, E.; Ruiz-Palacios, M.R.; Koletzko, B. Placental regulation of fetal nutrient supply. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Wooding, F.B.; Fowden, A.L.; Bell, A.W.; Ehrhardt, R.A.; Limesand, S.W.; Hay, W.W., Jr. Localisation of glucose transport in the ruminant placenta: Implications for sequential use of transporter isoforms. Placenta 2005, 26, 626–640. [Google Scholar] [CrossRef] [PubMed]
- Wolf, H.J.; Desoye, G. Immunohistochemical localization of glucose transporters and insulin receptors in human fetal membranes at term. Histochemistry 1993, 100, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Xing, A.Y.; Challier, J.C.; Lepercq, J.; Cauzac, M.; Charron, M.J.; Girard, J.; Hauguel-de Mouzon, S. Unexpected expression of glucose transporter 4 in villous stromal cells of human placenta. J. Clin. Endocrinol. Metab. 1998, 83, 4097–4101. [Google Scholar] [PubMed]
- Limesand, S.W.; Regnault, T.R.; Hay, W.W., Jr. Characterization of glucose transporter 8 (GLUT8) in the ovine placenta of normal and growth restricted fetuses. Placenta 2004, 25, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Carayannopoulos, M.O.; Chi, M.M.; Cui, Y.; Pingsterhaus, J.M.; McKnight, R.A.; Mueckler, M.; Devaskar, S.U.; Moley, K.H. GLUT8 is a glucose transporter responsible for insulin-stimulated glucose uptake in the blastocyst. Proc. Natl. Acad. Sci. USA 2000, 97, 7313–7318. [Google Scholar] [CrossRef] [PubMed]
- Regnault, T.R.H.; Friedman, J.E.; Wilkening, R.B.; Anthony, R.V.; Hay, W.W., Jr. Fetoplacental transport and utilization of amino acids in IUGR. A review. Placenta 2005, 26, 52–62. [Google Scholar] [CrossRef]
- Grillo, M.A.; Lanza, A.; Colombatto, S. Transport of amino acids through the placenta and their role. Amino Acids 2008, 34, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Desforges, M.; Sibley, C.P. Placental nutrient supply and fetal growth. Int. J. Dev. Biol. 2010, 54, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Desforges, M.; Lacey, H.A.; Glazier, J.D.; Greenwood, S.L.; Mynett, K.J.; Speake, P.F.; Sibley, C.P. Snat4 isoform of system A amino acid transporter is expressed in human placenta. Am. J. Physiol. Cell Physiol. 2006, 290, 305–312. [Google Scholar] [CrossRef]
- Regnault, T.R.H.; de Vrijer, B.; Battaglia, F.C. Transport and metabolism of amino acids in placenta. Endocrine 2002, 19, 23–41. [Google Scholar] [CrossRef] [PubMed]
- Desforges, M.; Greenwood, S.L.; Glazier, J.D.; Westwood, M.; Sibley, C.P. The contribution of snat1 to system A amino acid transporter activity in human placental trophoblast. Biochem. Biophys. Res. Commun. 2010, 398, 130–134. [Google Scholar] [CrossRef] [PubMed]
- Verrey, F. System l: Heteromeric exchangers of large, neutral amino acids involved in directional transport. Pflugers Arch. 2003, 445, 529–533. [Google Scholar] [PubMed]
- Okamoto, Y.; Sakata, M.; Ogura, K.; Yamamoto, T.; Yamaguchi, M.; Tasaka, K.; Kurachi, H.; Tsurudome, M.; Murata, Y. Expression and regulation of 4f2hc and hlat1 in human trophoblasts. Am. J. Physiol. 2002, 282, 196–204. [Google Scholar] [CrossRef]
- Gaccioli, F.; Roos, S.; Powell, T.L.; Jansson, T. Isoforms of the system l-amino acid transporter are differentially polarised to the syncytiotrophoblast plasma membrane in human placenta. Reprod. Sci. 2011, 18, 200. [Google Scholar]
- Lewis, R.M.; Glazier, J.D.; Greenwood, S.L.; Bennett, E.J.; Godfrey, K.M.; Jackson, A.A.; Sibley, C.P.; Cameron, I.T.; Hanson, M.A. L-serine uptake by human placental microvillous membrane vesicles. Placenta 2007, 28, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Kudo, Y.; Boyd, C.A.R. Characterisation of l-tryptophan transporters in human placenta: A comparison of brush border and basal membrane vesicles. J. Physiol. 2001, 531, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Bodoy, S.; Martin, L.; Zorzano, A.; Palacin, M.; Estevez, R.; Bertran, J. Identification of lat4, a novel amino acid transporter with system l activity. J. Biol. Chem. 2005, 280, 12002–12011. [Google Scholar] [CrossRef] [PubMed]
- Babu, E.; Kanai, Y.; Chairoungdua, A.; Kim, D.K.; Iribe, Y.; Tangtrongsup, S.; Jutabha, P.; Li, Y.; Ahmed, N.; Sakamoto, S.; et al. Identification of a novel system l amino acid transporter structurally distinct from heterodimeric amino acid transporters. J. Biol. Chem. 2003, 278, 43838–43845. [Google Scholar] [CrossRef] [PubMed]
- Cleal, J.K.; Glazier, J.D.; Ntani, G.; Crozier, S.R.; Day, P.E.; Harvey, N.C.; Robinson, S.M.; Cooper, C.; Godfrey, K.M.; Hanson, M.A.; et al. Facilitated transporters mediate net efflux of amino acids to the fetus across the basal membrane of the placental syncytiotrophoblast. J. Physiol. 2011, 589, 987–997. [Google Scholar] [CrossRef] [PubMed]
- Cleal, J.K.; Brownbill, P.; Godfrey, K.M.; Jackson, J.M.; Jackson, A.A.; Sibley, C.P.; Hanson, M.A.; Lewis, R.M. Modification of fetal plasma amino acid composition by placental amino acid exchangers in vitro. J. Physiol. 2007, 582, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Ayuk, P.T.; Sibley, C.P.; Donnai, P.; D’Souza, S.; Glazier, J.D. Development and polarization of cationic amino acid transporters and regulators in the human placenta. Am. J. Physiol. Cell Physiol. 2000, 278, 1162–1171. [Google Scholar]
- Kamath, S.G.; Furesz, T.C.; Way, B.A.; Smith, C.H. Identification of three cationic amino acid transporters in placental trophoblast: Cloning, expression, and characterization of hcat-1. J. Membr. Biol. 1999, 171, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Arriza, J.L.; Fairman, W.A.; Wadiche, J.I.; Murdoch, G.H.; Kavanaugh, M.P.; Amara, S.G. Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J. Neurosci. 1994, 14, 5559–5569. [Google Scholar] [PubMed]
- Fairman, W.A.; Vandenberg, R.J.; Arriza, J.L.; Kavanaugh, M.P.; Amara, S.G. An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature 1995, 375, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Matthews, J.C.; Beveridge, M.J.; Malandro, M.S.; Rothstein, J.D.; Campbell-Thompson, M.; Verlander, J.W.; Kilberg, M.S.; Novak, D.A. Activity and protein localization of multiple glutamate transporters in gestation day 14 vs. Day 20 rat placenta. Am. J. Physiol. 1998, 274, 603–614. [Google Scholar]
- Jansson, T. Amino acid transporters in the human placenta. Pediatr. Res. 2001, 49, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Haggarty, P. Fatty acid supply to the human fetus. Annu. Rev. Nutr. 2010, 30, 237–255. [Google Scholar] [CrossRef] [PubMed]
- Kazantzis, M.; Stahl, A. Fatty acid transport proteins, implications in physiology and disease. Biochim. Biophys. Acta 2012, 1821, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Schaiff, W.T.; Bildirici, I.; Cheong, M.; Chern, P.L.; Nelson, D.M.; Sadovsky, Y. Peroxisome proliferator-activated receptor-γ and retinoid X receptor signaling regulate fatty acid uptake by primary human placental trophoblasts. J. Clin. Endocrinol. Metab. 2005, 90, 4267–4275. [Google Scholar] [CrossRef] [PubMed]
- Campbell, F.M.; Bush, P.G.; Veerkamp, J.H.; Dutta-Roy, A.K. Detection and cellular localization of plasma membrane-associated and cytoplasmic fatty acid-binding proteins in human placenta. Placenta 1998, 19, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Biron-Shental, T.; Schaiff, W.T.; Ratajczak, C.K.; Bildirici, I.; Nelson, D.M.; Sadovsky, Y. Hypoxia regulates the expression of fatty acid-binding proteins in primary term human trophoblasts. Am. J. Obstet. Gynecol. 2007, 197, 1–6. [Google Scholar] [CrossRef] [PubMed]
- McMillen, I.C.; Robinson, J.S. Developmental origins of the metabolic syndrome: Prediction, plasticity, and programming. Physiol. Rev. 2005, 85, 571–633. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J. Fetal programming of coronary heart disease. Trends Endocrinol. Metab. 2002, 13, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J.; Osmond, C.; Forsen, T.J.; Kajantie, E.; Eriksson, J.G. Trajectories of growth among children who have coronary events as adults. N. Engl. J. Med. 2005, 353, 1802–1809. [Google Scholar] [CrossRef] [PubMed]
- Moore, V.M.; Miller, A.G.; Boulton, T.J.C.; Cockington, R.A.; Craig, I.H.; Magarey, A.M.; Robinson, J.S. Placental weight, birth measurements and blood pressure at age 8 years. Arch. Dis. Child. 1996, 74, 538–541. [Google Scholar] [CrossRef] [PubMed]
- McMillen, I.C.; Adams, M.B.; Ross, J.T.; Coulter, C.L.; Simonetta, G.; Owens, J.A.; Robinson, J.S.; Edwards, L.J. Fetal growth restriction: Adaptations and consequences. Reproduction 2001, 122, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Morrison, J.L.; Duffield, J.A.; Muhulhausler, B.S.; Gentili, S.; McMillen, I.C. Fetal growth restriction, catch-up growth and the early origins of insulin resistance and visceral obesity. Pediatr. Nephrol. 2009, 25, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Emmanouilides, G.C.; Townsend, D.E.; Bauer, R.A. Effects of single umbilical artery ligation in the lamb fetus. Pediatrics 1968, 42, 919–927. [Google Scholar] [PubMed]
- Oh, W.; Omori, K.; Erenberg, A.; Emmanouilides, G.C. Umbilical blood flow and glucose uptake in lamb fetus following single umbilical artery ligation. Biol. Neonate 1975, 26, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Cheung, C.Y.; Bogic, L.; Gagnon, R.; Harding, R.; Brace, R.A. Morphologic alterations in ovine placenta and fetal liver following induced severe placental insufficiency. J. Soc. Gynecol. Investig. 2004, 11, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, A.E.; Crossley, K.J.; Allison, B.J.; Jenkin, G.; Wallace, E.M.; Miller, S.L. The effects of intrauterine growth restriction and antenatal glucocorticoids on ovine fetal lung development. Pediatr. Res. 2012, 76, 689–696. [Google Scholar] [CrossRef]
- Early, R.J.; McBride, B.W.; Vatnick, I.; Bell, A.W. Chronic heat stress and prenatal development in sheep: Ii. Placental cellularity and metabolism. J. Anim. Sci. 1991, 69, 3610–3616. [Google Scholar] [PubMed]
- Regnault, T.R.; Orbus, R.J.; de Vrijer, B.; Davidsen, M.L.; Galan, H.L.; Wilkening, R.B.; Anthony, R.V. Placental expression of VEGF, PLGF and their receptors in a model of placental insufficiency-intrauterine growth restriction (PI-IUGR). Placenta 2002, 23, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Clapp, J.F., 3rd; Szeto, H.H.; Larrow, R.; Hewitt, J.; Mann, L.I. Umbilical blood flow response to embolization of the uterine circulation. Am. J. Obstet. Gynecol. 1980, 138, 60–67. [Google Scholar] [PubMed]
- Gagnon, R.; Johnston, L.; Murotsuki, J. Fetal placental embolization in the late-gestation ovine fetus: Alterations in umbilical blood FLW and fetal heart rate patterns. Am. J. Obstet. Gynecol. 1996, 175, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Louey, S.; Cock, M.L.; Stevenson, K.M.; Harding, R. Placental insufficiency and fetal growth restriction lead to postnatal hypotension and alterd postnatal growth in sheep. Pediatr. Res. 2000, 48, 808–814. [Google Scholar] [CrossRef] [PubMed]
- Owens, J.A.; Kind, K.L.; Carbone, F.; Robinson, J.S.; Owens, P.C. Circulating insulin-like growth factors-I and -II and substrates in fetal sheep following restriction of placental growth. J. Endocrinol. 1994, 140, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Danielson, L.; IMcMillen, I.C.; Dyer, J.L.; Morrison, J.L. Restriction of placental growth results in greater hypotensive response to α-adrenergic blockade in sheep during late gestation. J. Physiol. 2005, 563, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Soo, P.S.; Hiscock, J.; Botting, K.J.; Roberts, C.T.; Davey, A.K.; Morrison, J.L. Maternal undernutrition reduces P-glycoprotein in guinea pigs placenta and developing brain in late gestation. Reprod. Toxicol. 2012, 33, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.S.; Kingston, E.J.; Jones, C.T.; Thornburn, G.D. Studies on experimental growth retardation in sheep. The effect of removal of endometrial caruncles on fetal size and metabolism. J. Dev. Physiol. 1979, 1, 379–398. [Google Scholar] [PubMed]
- Chen, C.P.; Bajoria, R.; Aplin, J.D. Decreased vascularization and cell proliferation in placentas of intrauterine growth-restricted fetuses with abnormal umbilical artery flow velocity waveforms. Am. J. Obstet. Gynecol. 2002, 187, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Mayhew, T.M.; Ohadike, C.; Baker, P.N.; Crocker, I.P.; Mitchell, C.; Ong, S.S. Stereological investigation of placental morphology in pregnancies complicated by pre-eclampsia with and without intrauterine growth restriction. Placenta 2003, 24, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Krebs, C.; Macara, L.M.; Leiser, R.; Bowman, A.W.; Greer, I.A.; Kingdom, J.C. Intrauterine growth restriction with absent end-diastolic flow velocity in the umbilical artery is associated with maldevelopment of the placental terminal villous tree. Am. J. Obstet. Gynecol. 1996, 175, 1534–1542. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.T.; Owens, P.C.; Owens, J.A. Maternal food restriction reduces the exchange surface area and increases the barrier thickness of the placenta in the guinea pig. Placenta 2001, 22, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Bacon, B.J.; Gilbert, R.D.; Kaufmann, P.; Smith, A.D.; Trevino, F.T.; Longo, L.D. Placental anatomy and diffusing capacity in guinea pigs following long-term maternal hypoxia. Placenta 1984, 5, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Reshetnikova, O.S.; Burton, G.J.; Milovanov, A.P. Effects of hypobaric hypoxia on the fetoplacental unit: The morphometric diffusing capacity of the villous membrane at high altitude. Am. J. Obstet. Gynecol. 1994, 171, 1560–1565. [Google Scholar] [CrossRef] [PubMed]
- Parraguez, V.H.; Atlagich, M.; Diaz, R.; Cepeda, R.; Gonzalez, C.; de los Reyes, M.; Bruzzone, M.E.; Behn, C.; Raggi, L.A. Ovine placenta at high altitudes: Comparison of animals with different times of adaptation to hypoxic environment. Anim. Reprod. Sci. 2006, 95, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Penninga, L.; Longo, L.D. Ovine placentome morphology: Effect of high altitude, long-term hypoxia. Placenta 1998, 19, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Steyn, C.; Hawkins, P.; Saito, T.; Noakes, D.E.; Kingdom, J.C.P.; Hanson, M.A. Undernutrition during the first half of gestation increases the predominance of fetal tissue in late-gestation ovine placentomes. Eur. J. Obstet. Gynecol. Reprod. Biol. 2001, 98, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Gardner, D.S.; Ward, J.W.; Giussani, D.A.; Fowden, A.L. The effect of a reversible period of adverse intrauterine conditions during late gestation on fetal and placental weight and placentome distribution in sheep. Placenta 2002, 23, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hif-1: Mediator of physiological and pathophysiological responses to hypoxia. J. Appl. Physiol. 2000, 88, 1474–1480. [Google Scholar] [PubMed]
- Caniggia, I.; Winter, J.L. Adriana and Luisa Castellucci award lecture 2001. Hypoxia inducible factor-1: Oxygen regulation of trophoblast differentiation in normal and pre-eclamptic pregnancies—A review. Placenta 2002, 23, 47–57. [Google Scholar] [CrossRef]
- Pringle, K.G.; Kind, K.L.; Thompson, J.G.; Roberts, C.T. Complex interactions between hypoxia inducible factors, insulin-like growth factor-ii and oxygen in early murine trophoblasts. Placenta 2007, 28, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, T.; Elcock, C.L.; Anthony, F.W. Angiogenesis and the placental environment. Placenta 1995, 16, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Zhang, E.G.; Smith, S.K.; Baker, P.N.; Charnock-Jones, D.S. The regulation and localization of angiopoietin-1, -2, and their receptor tie2 in normal and pathologic human placentae. Mol. Med. 2001, 7, 624–635. [Google Scholar] [PubMed]
- Toal, M.; Chan, C.; Fallah, S.; Alkazalen, F.; Chaddha, V.; Winderm, R.C.; Kingdom, J.C. Usefulness of a placental profile in high-risk pregnancies. Am. J. Obstet. Gynecol. 2007, 196, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Pijnenborg, R.; Vercruysse, L.; Hanssens, M. The uterine spiral arteries in human pregnancy: Facts and controversies. Placenta 2006, 27, 939–958. [Google Scholar] [CrossRef] [PubMed]
- Lyall, F.; Young, A.; Boswell, F.; Kingdom, J.C.; Greer, I.A. Placental expression of vascular endothelial growth factor in placentae from pregnancies complicated by pre-eclampsia and intrauterine growth restriction does not support placental hypoxia at delivery. Placenta 1997, 18, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Barut, F.; Barut, A.; Gun, B.D.; Kandemir, N.O.; Harma, M.I.; Harma, M.; Aktunc, E.; Ozdamar, S.O. Intrauterine growth restriction and placental angiogenesis. Diagn. Pathol. 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Akram, S.K.; Sahlin, L.; Ostlund, E.; Hagenas, L.; Fried, G.; Soder, O. Placental IGF-I, estrogen receptor, and progesterone receptor expression, and maternal anthropometry in growth-restricted pregnancies in the swedish population. Horm. Res. Paediatr. 2011, 75, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Koutsaki, M.; Sifakis, S.; Zaravinos, A.; Koutroulakis, D.; Koukoura, O.; Spandidos, D.A. Decreased placental expression of hPGH, IGF-I and IGFBP-1 in pregnancies complicated by fetal growth restriction. Growth Horm. IGF Res. 2011, 21, 31–36. [Google Scholar] [CrossRef] [PubMed]
- De Vrijer, B.; Davidsen, M.L.; Wilkening, R.B.; Anthony, R.V.; Regnault, T.R. Altered placental and fetal expression of IGFS and IGF-binding proteins associated with intrauterine growth restriction in fetal sheep during early and mid-pregnancy. Pediatr. Res. 2006, 60, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Cuffe, J.S.; Walton, S.L.; Singh, R.R.; Spiers, J.G.; Bielefeldt-Ohmann, H.; Wilkinson, L.; Little, M.H.; Moritz, K.M. Mid- to late term hypoxia in the mouse alters placental morphology, glucocorticoid regulatory pathways and nutrient transporters in a sex-specific manner. J. Physiol. 2014, 592, 3127–3141. [Google Scholar] [CrossRef] [PubMed]
- Erel, C.T.; Dane, B.; Calay, Z.; Kaleli, S.; Aydinli, K. Apoptosis in the placenta of pregnancies complicated with iugr. Int. J. Gynaecol. Obstet. 2001, 73, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.C.; Baker, P.N.; Symonds, E.M. Increased placental apoptosis in intrauterine growth restriction. Am. J. Obstet. Gynecol. 1997, 177, 1395–1401. [Google Scholar] [CrossRef] [PubMed]
- Levy, R.; Smith, S.D.; Chandler, K.; Sadovsky, Y.; Nelson, D.M. Apoptosis in human cultured trophoblasts is enhanced by hypoxia and diminished by epidermal growth factor. Am. J. Physiol. Cell Physiol. 2000, 278, 982–988. [Google Scholar]
- Hung, T.H.; Chen, S.F.; Liou, J.D.; Hsu, J.J.; Li, M.J.; Yeh, Y.L.; Hsieh, T.T. Bax, Bak and mitochondrial oxidants are involved in hypoxia-reoxygenation-induced apoptosis in human placenta. Placenta 2008, 29, 565–583. [Google Scholar] [CrossRef] [PubMed]
- Levy, R.; Smith, S.D.; Yusuf, K.; Huettner, P.C.; Kraus, F.T.; Sadovsky, Y.; Nelson, D.M. Trophoblast apoptosis from pregnancies complicated by fetal growth restriction is associated with enhanced p53 expression. Am. J. Obstet. Gynecol. 2002, 186, 1056–1061. [Google Scholar] [CrossRef] [PubMed]
- Endo, H.; Okamoto, A.; Yamada, K.; Nikaido, T.; Tanaka, T. Frequent apoptosis in placental villi from pregnancies complicated with intrauterine growth restriction and without maternal symptoms. Int. J. Mol. Med. 2005, 16, 79–84. [Google Scholar] [PubMed]
- Heazell, A.E.; Sharp, A.N.; Baker, P.N.; Crocker, I.P. Intra-uterine growth restriction is associated with increased apoptosis and altered expression of proteins in the p53 pathway in villous trophoblast. Apoptosis 2011, 16, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Hung, T.H.; Chen, S.F.; Lo, L.M.; Li, M.J.; Yeh, Y.L.; Hsieh, T.T. Increased autophagy in placentas of intrauterine growth-restricted pregnancies. PLoS One 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Longtine, M.S.; Nelson, D.M. Hypoxia induces autophagy in primary human trophoblasts. Endocrinology 2012, 153, 4946–4954. [Google Scholar] [CrossRef] [PubMed]
- Jaquiery, A.L.; Oliver, M.H.; Bloomfield, F.H.; Connor, K.L.; Challis, J.R.; Harding, J.E. Fetal exposure to excess glucocorticoid is unlikely to explain the effects of periconceptional undernutrition in sheep. J. Physiol. 2006, 572, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Mericq, V.; Medina, P.; Kakarieka, E.; Marquez, L.; Johnson, M.C.; Iniguez, G. Differences in expression and activity of 11beta-hydroxysteroid dehydrogenase type 1 and 2 in human placentas of term pregnancies according to birth weight and gender. Eur. J. Endocrinol. 2009, 161, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Zamudio, S.; Torricos, T.; Fik, E.; Oyala, M.; Echalar, L.; Pullockaran, J.; Tutino, E.; Martin, B.; Belliappa, S.; Balanza, E.; et al. Hypoglycemia and the origin of hypoxia-induced reduction in human fetal growth. PLoS One 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Owens, J.A.; Falconer, J.; Robinson, J.S. Restriction of placental size in sheep enhances efficiency of placental transfer of antipyrine, 3-o-methyl-d-glucose but not of urea. J. Dev. Physiol. 1987, 9, 457–464. [Google Scholar] [PubMed]
- Owens, J.A.; Falconer, J.; Robinson, J.S. Effect of restriction of placental growth on fetal and utero-placental metabolism. J. Dev. Physiol. 1987, 9, 225–238. [Google Scholar] [PubMed]
- Baumann, M.U.; Zamudio, S.; Illsley, N.P. Hypoxic upregulation of glucose transporters in BeWo choriocarcinoma cells is mediated by hypoxia-inducible factor-1. Am. J. Physiol. Cell Physiol. 2007, 293, 477–485. [Google Scholar] [CrossRef]
- Janzen, C.; Lei, M.Y.; Cho, J.; Sullivan, P.; Shin, B.C.; Devaskar, S.U. Placental glucose transporter 3 (GLUT3) is up-regulated in human pregnancies complicated by late-onset intrauterine growth restriction. Placenta 2013, 34, 1072–1078. [Google Scholar] [CrossRef] [PubMed]
- Dubova, E.A.; Pavlov, K.A.; Kulikova, G.V.; Shchegolev, A.I.; Sukhikh, G.T. Glucose transporters expression in the placental terminal villi of preeclampsia and intrauterine growth retardation complicated pregnancies. Health 2013, 5, 100–104. [Google Scholar] [CrossRef]
- Zamudio, S.; Baumann, M.U.; Illsley, N.P. Effects of chronic hypoxia in vivo on the expression of human placental glucose transporter. Placenta 2006, 27, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Jansson, T.; Wennergren, M.; Illsley, N.P. Glucose transporter protein expression in human placenta throughout gestation and in intrauterine growth retardation. J. Clin. Endocrinol. Metab. 1993, 77, 1554–1562. [Google Scholar] [PubMed]
- Jansson, T.; Ylven, K.; Wennergren, M.; Powell, T.L. Glucose transport and system A activity in syncytiotrophoblast microvillous and basal plasma membranes in intrauterine growth restriction. Placenta 2002, 23, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Glazier, J.D.; Cetin, I.; Perugino, G.; Ronzoni, S.; Grey, A.M.; Mahendran, D.; Marconi, A.M.; Pardi, G.; Sibley, C.P. Association between the activity of the system A amino acid transporter in the microvillous plasma membrane of the human placenta and severity of fetal compromise in intrauterine growth restriction. Pediatr. Res. 1997, 42, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Mando, C.; Tabano, S.; Pileri, P.; Colapietro, P.; Marino, M.A.; Avagliano, L.; Doi, P.; Bulfamante, G.; Miozzo, M.; Cetin, I. Snat2 expression and regulation in human growth-restricted placentas. Pediatr. Res. 2013, 74, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Jansson, T.; Scholtbach, V.; Powell, T.L. Placental transport of leucine and lysine is reduced in intrauterine growth restriction. Pediatr. Res. 1998, 44, 532–537. [Google Scholar] [CrossRef] [PubMed]
- Paolini, C.L.; Marconi, A.M.; Ronzoni, S.; di Noio, M.; Fennessey, P.V.; Pardi, G.; Battaglia, F.C. Placental transport of leucine, phenylalanine, glycine, and proline in intrauterine growth-restricted pregnancies. J. Clin. Endocrinol. Metab. 2001, 86, 5427–5432. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.C.; Fennessey, P.V.; Wilkening, R.B.; Battaglia, F.C.; Meschia, G. Placental transport and fetal utilization of leucine in a model of fetal growth retardation. Am. J. Physiol. 1996, 270, 491–503. [Google Scholar]
- Anderson, A.H.; Fennessey, P.V.; Meschia, G.; Wilkening, R.B.; Battaglia, F.C. Placental transport of threonine and its utilization in the normal and growth-restricted fetus. Am. J. Physiol. 1997, 272, 892–900. [Google Scholar]
- De Vrijer, B.; Regnault, T.R.; Wilkening, R.B.; Meschia, G.; Battaglia, F.C. Placental uptake and transport of ACP, a neutral nonmetabolizable amino acid, in an ovine model of fetal growth restriction. Am. J. Physiol. Endocrinol. Metab. 2004, 287, 1114–1124. [Google Scholar] [CrossRef]
- Godfrey, K.M.; Matthews, N.; Glazier, J.; Jackson, A.; Wilman, C.; Sibley, C.P. Neutral amino acid uptake by the microvillous plasma membrane of the human placenta is inversely related to fetal size at birth in normal pregnancy. J. Clin. Endocrinol. Metab. 1998, 83, 3320–3326. [Google Scholar] [PubMed]
- Nelson, D.M.; Smith, S.D.; Furesz, T.C.; Sadovsky, Y.; Ganapathy, V.; Parvin, C.A.; Smith, C.H. Hypoxia reduces expression and function of system A amino acid transporters in cultured term human trophoblasts. Am. J. Physiol. Cell Physiol. 2003, 284, 310–315. [Google Scholar] [CrossRef]
- Mishima, T.; Miner, J.H.; Morizane, M.; Stahl, A.; Sadovsky, Y. The expression and function of fatty acid transport protein-2 and -4 in the murine placenta. PLoS One 2011, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laivuori, H.; Gallaher, M.J.; Collura, L.; Crombleholme, W.R.; Markovic, N.; Rajakumar, A.; Hubel, C.A.; Roberts, J.M.; Powers, R.W. Relationships between maternal plasma leptin, placental leptin mRNA and protein in normal pregnancy, pre-eclampsia and intrauterine growth restriction without pre-eclampsia. Mol. Hum. Reprod. 2006, 12, 551–556. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, S.; Regnault, T.R.H.; Barker, P.L.; Botting, K.J.; McMillen, I.C.; McMillan, C.M.; Roberts, C.T.; Morrison, J.L. Placental Adaptations in Growth Restriction. Nutrients 2015, 7, 360-389. https://doi.org/10.3390/nu7010360
Zhang S, Regnault TRH, Barker PL, Botting KJ, McMillen IC, McMillan CM, Roberts CT, Morrison JL. Placental Adaptations in Growth Restriction. Nutrients. 2015; 7(1):360-389. https://doi.org/10.3390/nu7010360
Chicago/Turabian StyleZhang, Song, Timothy R.H. Regnault, Paige L. Barker, Kimberley J. Botting, Isabella C. McMillen, Christine M. McMillan, Claire T. Roberts, and Janna L. Morrison. 2015. "Placental Adaptations in Growth Restriction" Nutrients 7, no. 1: 360-389. https://doi.org/10.3390/nu7010360