Synthesis and Evaluation of Pyrimidine Steroids as Antiproliferative Agents

 ,
,  and
and
Abstract
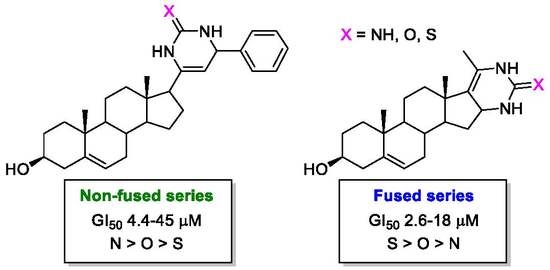
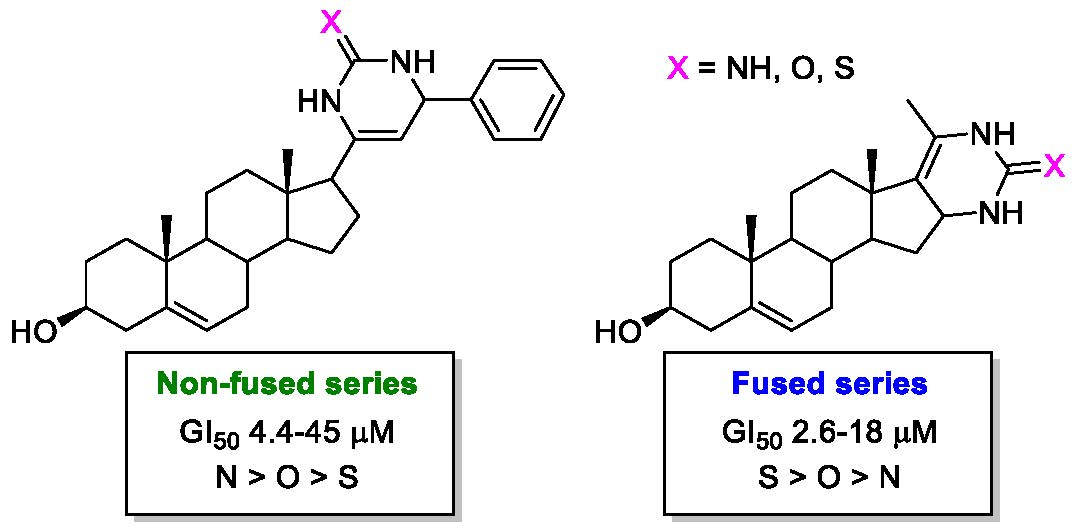
:
1. Introduction
2. Results and Discussion
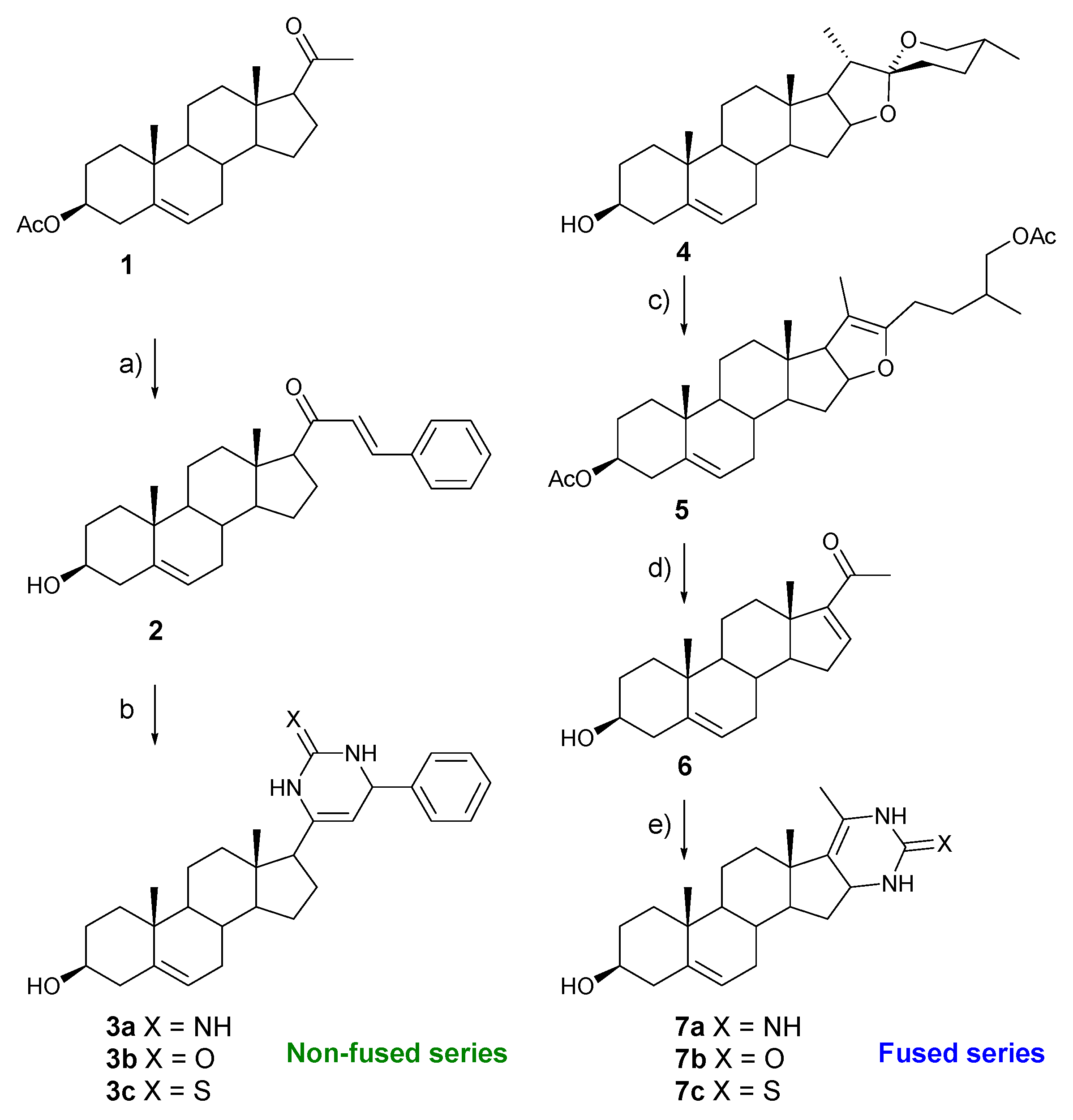
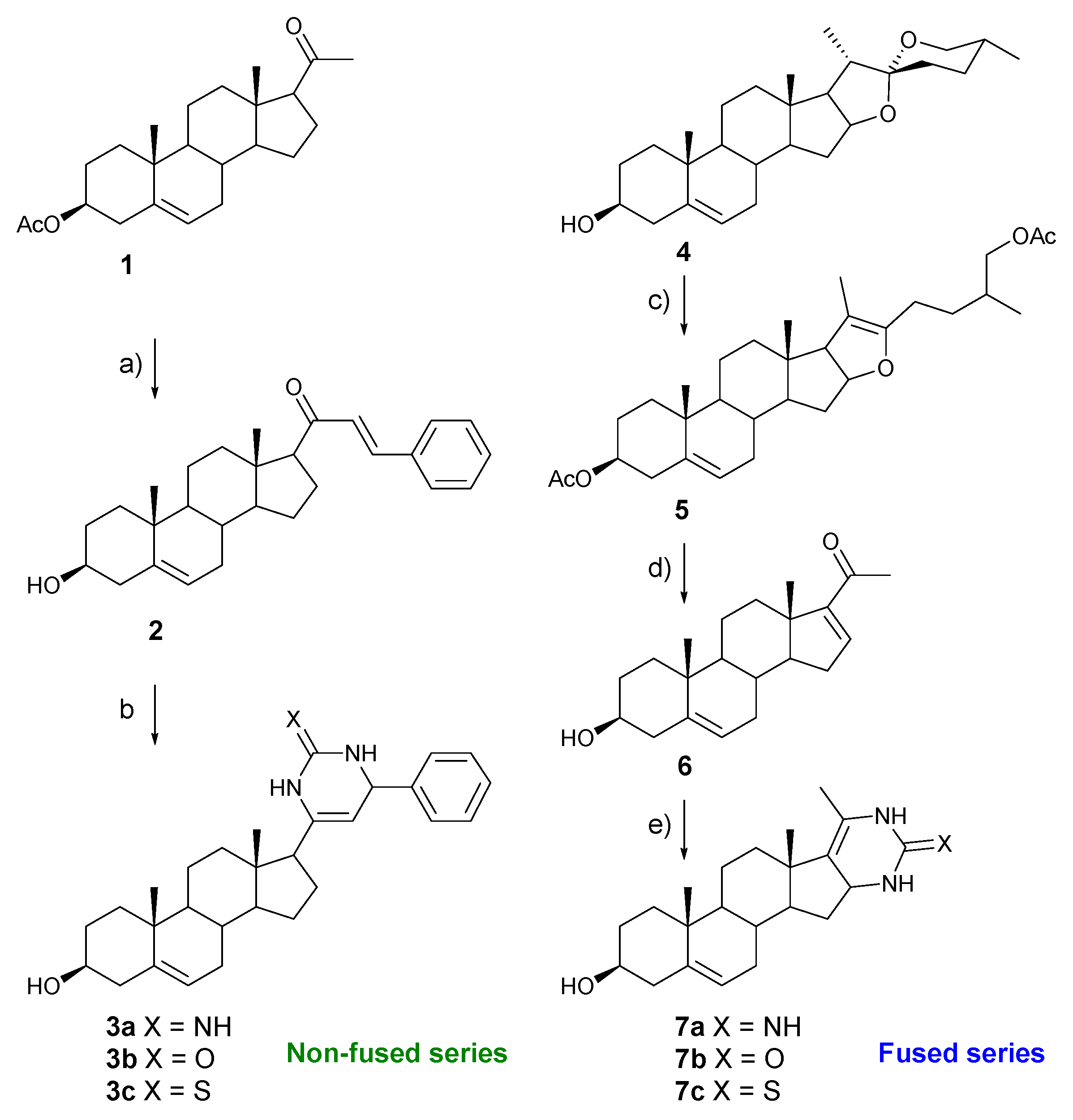
2.1. Chemistry
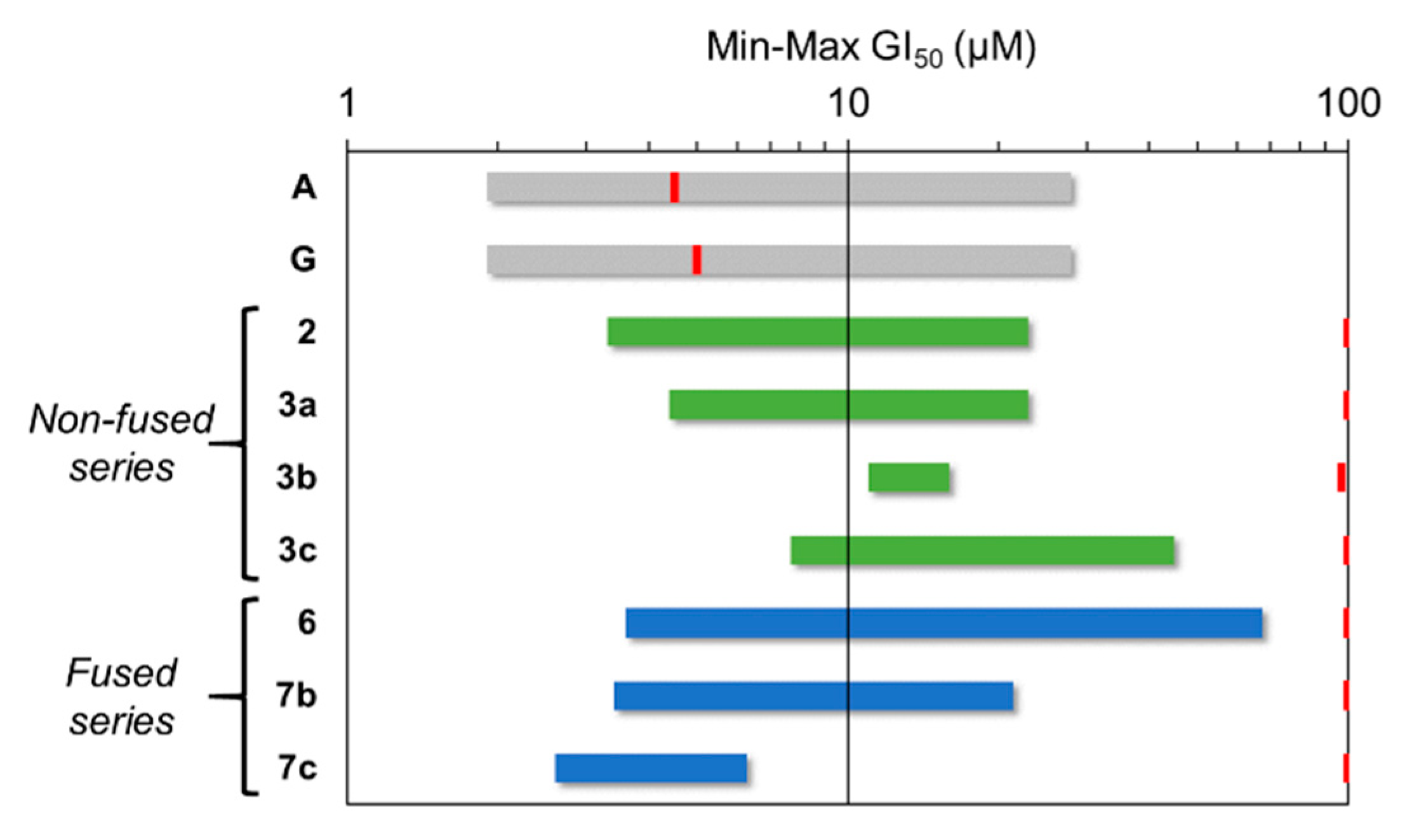
2.2. Antiproliferative Activity
2.3. P-Glycoprotein Assay
3. Materials and Methods
3.1. Chemistry
3.1.1. General Methods
3.1.2. (E)-21-benzyliden-3β-hydroxypregn-5-en-20-one (2)
3.1.3. General Procedure for the Preparation of 3a–c
3.1.4. (25R)-furosta-5,20(22)-diene-3β,26-diyl Diacetate (5)
3.1.5. 20-Oxopregna-5,16-dien-3β-yl Acetate (6)
3.1.6. General Procedure for the Preparation of 7a–c
3.2. Biological Tests
3.2.1. Cell Lines and Growth Conditions
3.2.2. Growth Inhibition Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
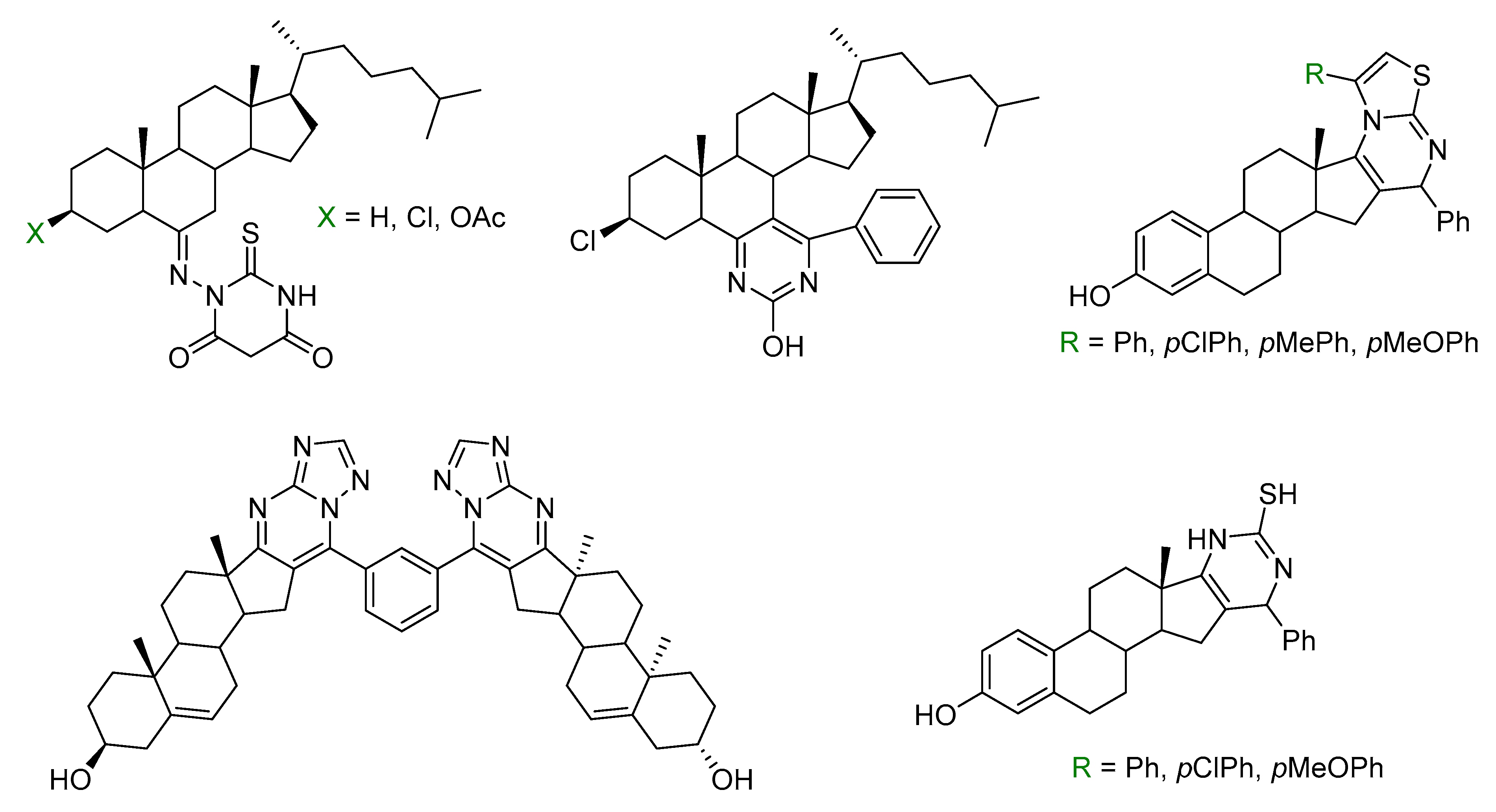
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbone, A.; Parrino, B.; Cusimano, M.G.; Spanò, V.; Montalbano, A.; Barraja, P.; Schillaci, D.; Cirrincione, G.; Diana, P.; Cascioferro, S. New thiazole nortopsentin analogues inhibit bacterial biofilm formation. Mar. Drugs 2018, 16, 274. [Google Scholar] [CrossRef] [PubMed]
- Spanò, V.; Attanzio, A.; Cascioferro, S.; Carbone, A.; Montalbano, A.; Barraja, P.; Tesoriere, L.; Cirrincione, G.; Diana, P.; Parrino, B. Synthesis and antitumor activity of new thiazole nortopsentin analogs. Mar. Drugs 2016, 14, 226. [Google Scholar] [CrossRef] [PubMed]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef] [PubMed]
- Shamsuzzaman; Dar, A.M.; Yaseen, Z.; Alam, K.; Hussain, A.; Gatoo, M.A. Steroidal pyrimidines; Synthesis, characterization, molecular docking studies with DNA and vitro cytotoxicity. J. Mol. Struct. 2013, 1045, 62–71. [Google Scholar]
- Shamsuzzaman; Dar, A.M.; Tabassum, S.; Zaki, M.; Khan, Y.; Sohail, A.; Gatoo, M.A. DNA binding, docking studies, artificial nuclease activity and in vitro cytotoxicity of newly synthesized steroidal 1H-pyrimidines. C. R. Chimie 2014, 17, 359–369. [Google Scholar] [CrossRef]
- Dar, A.M.; Rah, B.; Mir, S.; Nabi, R.; Shamsuzzaman; Gatoo, M.A.; Mashrai, A.; Khand, Y. DNA binding, artificial nuclease activity and cytotoxic studies of newly synthesized steroidal pyrimidines. Int. J. Biol. Macromol. 2018, 111, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Mohareb, R.M.; Al-Omran, F.; Azzam, R.A. Heterocyclic ring extension of estrone: Synthesis and cytotoxicity of fused pyran, pyrimidine and thiazole derivatives. Steroids 2014, 84, 46–56. [Google Scholar] [CrossRef]
- Yu, B.; Shi, X.-J.; Zheng, Y.-F.; Fang, Y.; Zhang, E.; Yu, D.-Q.; Liu, H.-M. A novel [1,2,4] triazolo [1,5-a] pyrimidine-based phenyl-linked steroid dimer: Synthesis and its cytotoxic activity. Eur. J. Med. Chem. 2013, 69, 323–330. [Google Scholar] [CrossRef]
- Ali, A.; Asif, M.; Alam, P.; Alam, J.M.; Sherwani, A.M.; Khan, H.R.; Ahmad, S.; Shamsuzzaman. DFT/B3LYP calculations, in vitro cytotoxicity and antioxidant activities of steroidal pyrimidines and their interaction with HSA using molecular docking and multispectroscopic techniques. Bioorg. Chem. 2017, 73, 83–99. [Google Scholar] [CrossRef]
- Abdalla, M.M.; Al-Omar, M.A.; Bhat, M.A.; Amr, A.E.; Al-Mohizea, A.M. Steroidal pyrazolines evaluated as aromatase and quinone reductase-2-inhibitors for chemoprevention of cancer. Int. J. Biol. Macromol. 2012, 50, 1127–1132. [Google Scholar] [CrossRef] [PubMed]
- Abdelhalim, M.M.; El-Saidi, M.M.T.; Rabie, S.T.; Elmegeed, G.A. Synthesis of novel steroidal heterocyclic derivatives as antibacterial agents. Steroids 2007, 72, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Arenas-González, A.; Mendez-Delgado, L.A.; Merino-Montiel, P.; Padrón, J.M.; Montiel-Smith, S.; Vega-Báez, J.L.; Meza-Reyes, S. Synthesis of monomeric and dimeric steroids containing [1,2,4]triazolo[1,5-a]pyrimidines. Steroids 2016, 116, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Romero-Hernández, L.L.; Merino-Montiel, P.; Meza-Reyes, S.; Vega-Baez, J.L.; López, Ó.; Padrón, J.M.; Montiel-Smith, S. Synthesis of unprecedented steroidal spiro heterocycles as potential antiproliferative drugs. Eur. J. Med. Chem. 2018, 143, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Romero-López, A.; Montiel-Smith, S.; Meza-Reyes, S.; Merino-Montiel, P.; Vega-Baez, J.L. Synthesis of steroidal derivatives containing substituted, fused and spiro pyrazolines. Steroids 2014, 87, 86–92. [Google Scholar]
- Viñas-Bravo, O.; Martínez-Pascual, R.; Vega-Báez, J.L.; Gómez-Calvario, V.; Sandoval-Ramírez, J.; Montiel Smith, S.; Meza-Reyes, S.; López-De la Rosa, A.; Martínez-Montiel, M.; Reyes, M.; et al. Rapid conversion of spirostans into furostan skeletons at room temperatura. Steroids 2012, 77, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Monks, A.; Scudiero, D.; Skehan, P.; Shoemaker, R.; Paull, K.; Vistica, D.; Hose, C.; Langley, J.; Cronise, P.; Vaigro-Wolff, A.; et al. Feasibility of a High-Flux Anticancer Drug Screen Using a Diverse Panel of Cultured Human Tumor Cell Lines. J. Natl. Cancer Inst. 1991, 83, 757–766. [Google Scholar] [CrossRef]
- Chang, C.; Bahadduri, P.M.; Polli, J.E.; Swaan, P.W.; Ekins, S. Rapid Identification of P-glycoprotein Substrates and Inhibitors. Drug Metab. Dispos. 2006, 34, 1976–1984. [Google Scholar] [CrossRef] [Green Version]
- Bergman, A.M.; Pinedo, H.M.; Talianidis, I.; Veerman, G.; Loves, W.J.; Van der Wilt, C.L.; Peters, G.J. Increased sensitivity to gemcitabine of P-glycoprotein and multidrug resistance-associated protein-overexpressing human cancer cell lines. Br. J. Cancer 2003, 88, 1963–1970. [Google Scholar] [CrossRef] [Green Version]
- Castaing, M.; Loiseau, A.; Cornish-Bowden, A. Synergy between verapamil and other multidrug-resistance modulators in model membranes. J. Biosci. 2007, 32, 737–746. [Google Scholar] [CrossRef]
- Miranda, P.O.; Padrón, J.M.; Padrón, J.I.; Villar, J.; Martín, V.S. Prins-type synthesis and SAR study of cytotoxic alkyl chloro dihydropyrans. ChemMedChem 2006, 1, 323–329. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Series | Compound | X | Time (h) | Yield (%) |
|---|---|---|---|---|
| Non-fused | 3a | N | 7 | 68 |
| 3b | O | 12 | 56 | |
| 3c | S | 10 | 62 | |
| Fused | 7a | N | 9 | 58 |
| 7b | O | 14 | 52 | |
| 7c | S | 8 | 65 |
| Compound | Cell Line (Origin) | |||||
|---|---|---|---|---|---|---|
| A549 (Lung) | HBL-100 (Breast) | HeLa (Cervix) | T-47D (Breast) | WiDr (Colon) | BJ-hTERT (Fibroblasts) | |
| 2 | 23 ± 1.6 | 19 ± 3.7 | 3.3 ± 0.3 | 9.9 ± 3.5 | 6.3 ± 0.5 | >100 |
| 3a | 4.4 ± 1.0 | 23 ± 2.8 | 6.1 ± 1.2 | 8.8 ± 2.2 | 8.3 ± 1.1 | >100 |
| 3b | 11 ± 2.9 | 16 ± 1.3 | 14 ± 3.6 | 15 ± 1.3 | 15 ± 3.5 | 97 ± 5.9 |
| 3c | 7.7 ± 1.0 | 45 ± 7.9 | 11 ± 1.5 | 13 ± 2.3 | 9.2 ± 1.6 | >100 |
| 6 | 26 ± 5.2 | 64 ± 1.6 | 3.6 ± 0.4 | 29 ± 4.5 | 37 ± 2.1 | >100 |
| 7b | 13 ± 3.7 | 15 ± 3.2 | 3.4 ± 0.2 | 15 ± 1.9 | 18 ± 1.8 | >100 |
| 7c | 3.4 ± 0.5 | 3.7 ± 1.0 | 2.6 ± 0.7 | 3.0 ± 0.5 | 3.1 ± 0.4 | >100 |
| Abiraterone | 95 ± 0.8 | >100 | 7.9 ± 0.5 | 24 ± 4.5 | 42 ± 7.7 | 4.5 ± 1.0 |
| Galeterone | 3.9 ± 1.3 | 10 ± 0.9 | 5.3 ± 0.4 | 2.1 ± 0.1 | 2.7 ± 0.2 | 5.0 ± 1.3 |
| Compound | w/o Verapamil | w Verapamil | ||||
|---|---|---|---|---|---|---|
| SW1573 | SW1573/PGP | Rf | SW1573 | SW1573/PGP | Rf | |
| 2 | 23 ± 0.2 | 29 ± 5.5 | 1.2 | 20 ± 2.9 | 30 ± 5.9 | 1.5 |
| 3a | 6.9 ± 0.5 | 10 ± 4.2 | 1.5 | 8.1 ± 0.7 | 5.8 ± 0.1 | 0.7 |
| 3b | 18 ± 0.9 | 34 ± 13 | 1.9 | 18 ± 3.4 | 28 ± 5.7 | 1.6 |
| 3c | 12 ± 2.7 | 38 ± 6.1 | 3.2 | 14 ± 4.4 | 25 ± 1.1 | 1.8 |
| 6 | 72 ± 10 | >100.0 | 1.4 | 71 ± 9.1 | 90 ± 19 | 1.3 |
| 7b | 34 ± 4.7 | 49 ± 4.4 | 1.5 | 24 ± 3.0 | 33 ± 10 | 1.4 |
| 7c | 9.4 ± 1.0 | 16 ± 1.3 | 1.7 | 8.1 ± 0.5 | 8.5 ± 1.8 | 1.1 |
| PTX | 1.5 ± 0.5 | 196 ± 53 | 128 | 1.6 ± 0.2 | 4.2 ± 0.9 | 2.6 |
| VB | 0.9 ± 0.3 | 2051 ± 682 | 2388 | 0.8 ± 0.2 | 1.0 ± 0.5 | 1.3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cortés-Percino, A.; Vega-Báez, J.L.; Romero-López, A.; Puerta, A.; Merino-Montiel, P.; Meza-Reyes, S.; Padrón, J.M.; Montiel-Smith, S. Synthesis and Evaluation of Pyrimidine Steroids as Antiproliferative Agents. Molecules 2019, 24, 3676. https://doi.org/10.3390/molecules24203676
Cortés-Percino A, Vega-Báez JL, Romero-López A, Puerta A, Merino-Montiel P, Meza-Reyes S, Padrón JM, Montiel-Smith S. Synthesis and Evaluation of Pyrimidine Steroids as Antiproliferative Agents. Molecules. 2019; 24(20):3676. https://doi.org/10.3390/molecules24203676
Chicago/Turabian StyleCortés-Percino, Alejandra, José Luis Vega-Báez, Anabel Romero-López, Adrián Puerta, Penélope Merino-Montiel, Socorro Meza-Reyes, José M. Padrón, and Sara Montiel-Smith. 2019. "Synthesis and Evaluation of Pyrimidine Steroids as Antiproliferative Agents" Molecules 24, no. 20: 3676. https://doi.org/10.3390/molecules24203676