
Cross-Coupling Reaction of Allylic Ethers with Aryl Grignard Reagents Catalyzed by a Nickel Pincer Complex


Abstract
: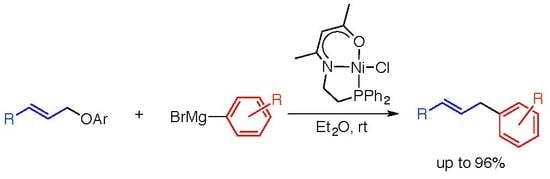
1. Introduction
2. Results and Discussion
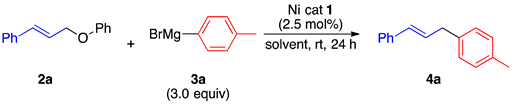
2.1. Optimization of the Reaction Conditions
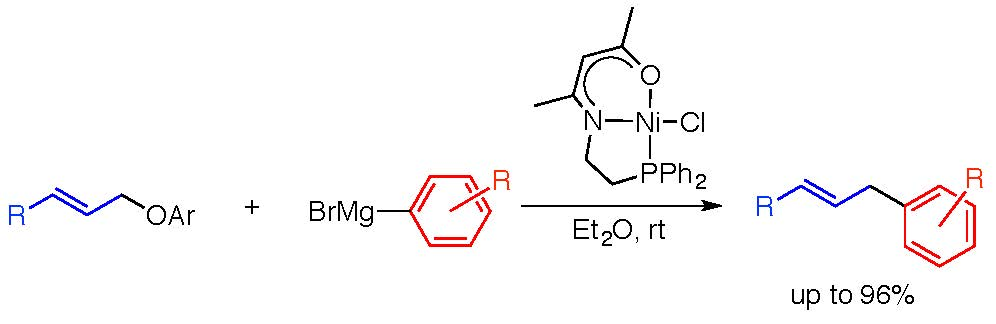
2.1.1. Initial Optimization of the Nickel(II) Complexes
2.1.2. Investigation of the Effect of the Solvent and Nickel(II) Complexes
2.1.3. Investigation of the Effect of the Leaving Group on the Allylic Electrophile
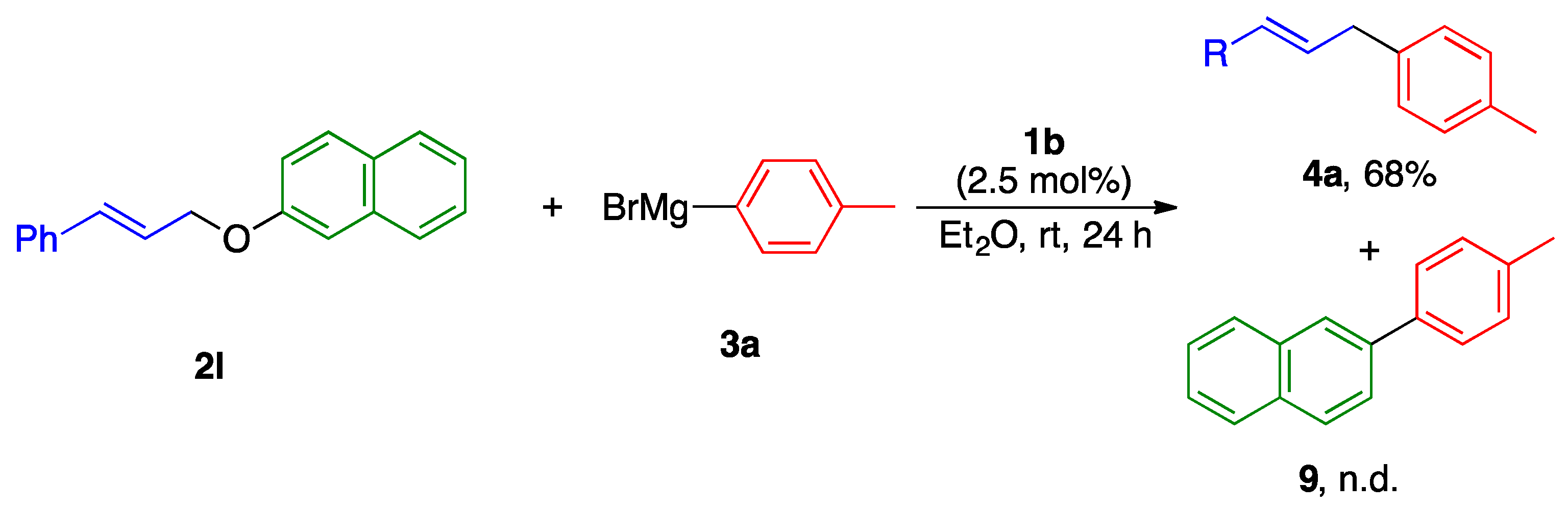
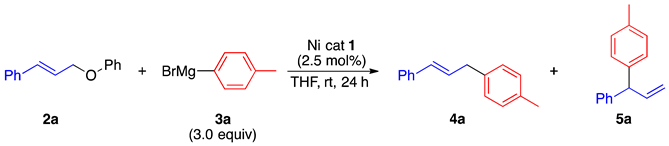
2.2. Substrate Scope
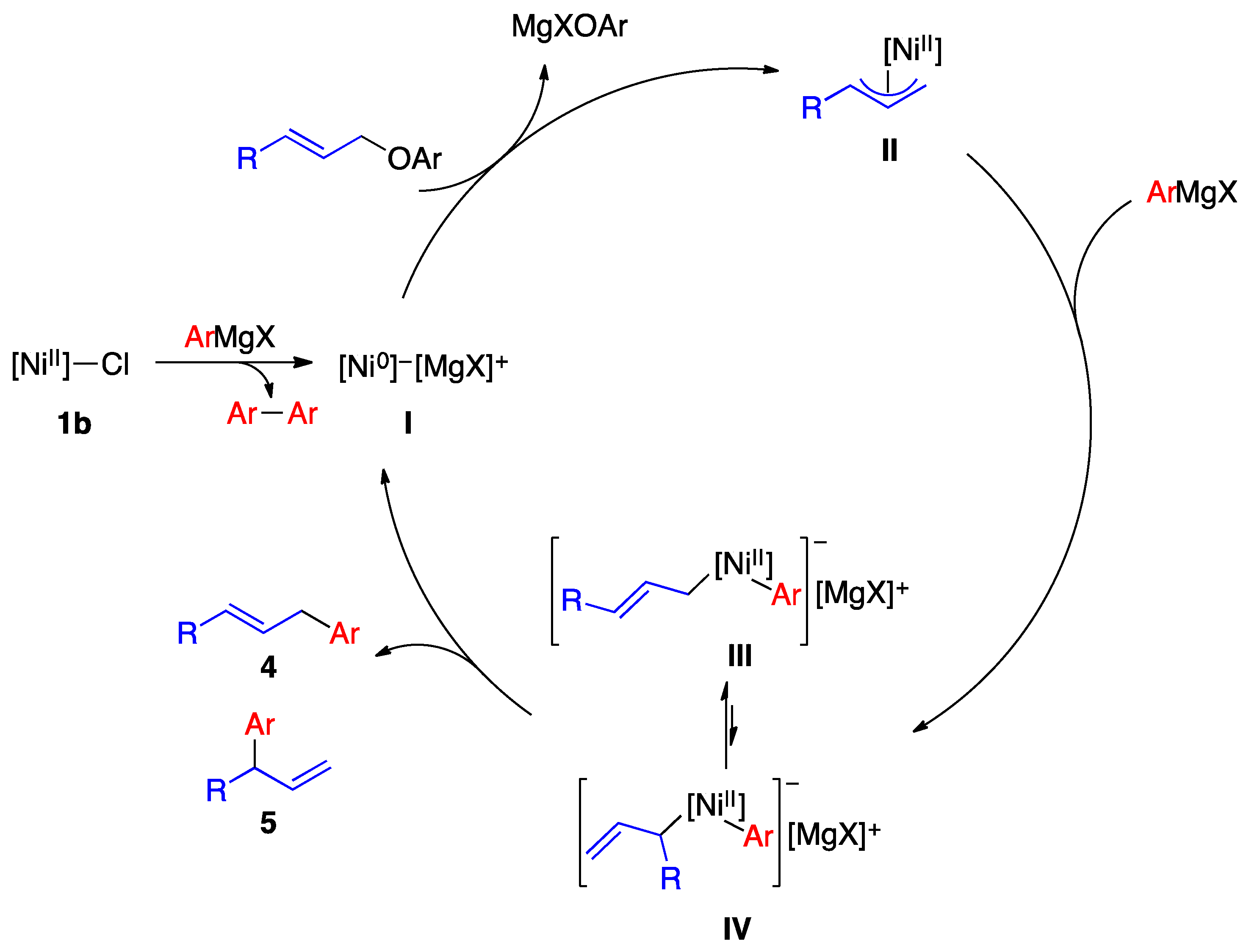
2.3. Reaction Mechanism
3. Materials and Methods
3.1. General Information
3.2. General Procedure for the Cross-Coupling Reaction of Allylic Ethers with Arylmagnesium Reagents
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Meijere, A.; Diederich, F. (Eds.) Metal-Catalyzed Cross-Coupling Reactions, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- Cheng, Q.; Tu, H.-F.; Zhen, C.; Qu, J.-P.; Helmchen, G.; You, S.-L. Iridium-Catalyzed Asymmetric Allylic Substitution Reactions. Chem. Rev. 2019, 119, 1855–1969. [Google Scholar] [CrossRef]
- Butt, N.A.; Zhang, X. Transition metal-catayzed allylic substitution reactions with unactivated allylic substrates. Chem. Soc. Rev. 2015, 44, 7929–7967. [Google Scholar] [CrossRef]
- Sundararaju, B.; Achard, M.; Bruneau, C. Transition Metal Catalyzed Nucleophilic Allylic Substitution: activation of allylic alcohols via π-allylic species. Chem. Soc. Rev. 2012, 41, 4467–4483. [Google Scholar] [CrossRef]
- Harutyunyan, S.R.; den Hartog, T.; Geurts, K.; Minnaard, A.J.; Feringa, B.L. Catalytic Asymmetric Conjugate Addition and Allylic Alkylation with Grignard Reagents. Chem. Rev. 2008, 108, 2824–2852. [Google Scholar] [CrossRef]
- Alexakis, A.; Bäckvall, J.E.; Krause, N.; Pàmies, O.; Diéguez, M. Enantioselective Copper-Catalyzed Conjugate Addition and Allylic Substitution Reactions. Chem. Rev. 2008, 108, 2796–2823. [Google Scholar] [CrossRef]
- Pérez, M.; Fañanás-Mastral, M.; Hornillos, V.; Rudolph, A.; Bos, P.H.; Harutyunyan, S.R.; Feringa, B.L. Asymmetric Allylic Alkylation of Acyclic Allylic Ethers with Organolithium Reagents. Chem. Eur. J. 2012, 18, 11880–11883. [Google Scholar] [CrossRef] [Green Version]
- Yasui, H.; Mizutani, K.; Yorimitsu, H.; Oshima, K. Cobalt- and rhodium-catalyzed cross-coupling reaction of allylic ethers and halides with organometallic reagents. Tetrahedron 2006, 62, 1410–1415. [Google Scholar] [CrossRef]
- Gärtner, D.; Konnerth, H.; von Wangelin, A.J. Highly practical iron-catalyzed C–O cleavage reactions. Catal. Sci. Technol. 2013, 3, 2541–2545. [Google Scholar] [CrossRef]
- Qi, L.; Ma, E.; Jia, F.; Li, Z. Iron-catalyzed allylic substitution reactions of allylic ethers with Grignard reagents. Tetrahedron Lett. 2016, 57, 2211–2214. [Google Scholar] [CrossRef] [Green Version]
- Ma, E.; Jiang, Y.; Chen, Y.; Qi, L.; Yan, X.; Li, Z. Salicylate-Directed C−O Bond Cleavage: Iron-Catalyzed Allylic Substitution with Grignard Reagents. Asian J. Org. Chem. 2018, 7, 914–917. [Google Scholar] [CrossRef]
- Seto, C.; Otsuka, T.; Takeuchi, Y.; Tabuchi, D.; Nagano, T. Iron-Catalyzed Grignard Cross-Couplings with Allylic Methyl Ethers or Allylic Trimethylsilyl Ethers. Synlett 2018, 29, 1211–1214. [Google Scholar] [CrossRef] [Green Version]
- Tao, J.-L.; Yang, B.; Wang, Z.-X. Pincer-Nickel-Catalyzed Allyl-Aryl Coupling between Allyl Methyl Ethers and Arylzinc Chlorides. J. Org. Chem. 2015, 80, 12627–12634. [Google Scholar] [CrossRef]
- Gossage, R.A.; van de Kuil, L.; van Koten, G. Diaminoarylnickel(II) “Pincer” Complexes: Mechanistic Considerations in the Kharasch Addition Reaction, Controlled Polymerization, and Dendrimeric Transition Metal Catalysts. Acc. Chem. Res. 1998, 31, 423–431. [Google Scholar] [CrossRef]
- Albrecht, M.; van Koten, G. Platinum Group Organometallics Based on “Pincer” Complexes: Sensors, Switches, and Catalysts. Angew. Chem. Int. Ed. 2001, 40, 3750–3781. [Google Scholar] [CrossRef]
- Van der Boom, M.E.; Milstein, D. Cyclometalated Phosphine-Based Pincer Complexes: Mechanistic Insight in Catalysis, Coordination, and Bond Activation. Chem. Rev. 2003, 103, 1759–1792. [Google Scholar] [CrossRef]
- Nishiyama, H. Synthesis and use of bisoxazolinyl-phenyl pincers. Chem. Soc. Rev. 2007, 36, 1133–1141. [Google Scholar] [CrossRef]
- Benito-Garagorri, D.; Kirchner, K. Modularly Designed Transition Metal PNP and PCP Pincer Complexes based on Aminophosphines: Synthesis and Catalytic Applications. Acc. Chem. Res. 2008, 41, 201–213. [Google Scholar] [CrossRef]
- Wang, Z.-X.; Liu, N. Nickel-Catalyzed Cross-Coupling with Pincer Ligands. Eur. J. Inorg. Chem. 2012, 901–911. [Google Scholar] [CrossRef]
- Choi, J.; MacArthur, A.H.R.; Brookhart, M.; Goldman, A.S. Dehydrogenation and Related Reactions Catalyzed by Iridium Pincer Complexes. Chem. Rec. 2011, 111, 1761–1779. [Google Scholar] [CrossRef]
- Selander, N.; Szabó, K. Catalysis by Palladium Pincer Complexes. Chem. Rec. 2011, 111, 2048–2076. [Google Scholar]
- Niu, J.-L.; Hao, X.-Q.; Gong, J.-F.; Song, M.-P. Symmetrical and unsymmetrical pincer complexes with group 10 metals: synthesis via aryl C–H activation and some catalytic applications. Dalton Trans. 2011, 40, 5135–5150. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.; Meiners, J.; Askevold, B. Cooperative Aliphatic PNP Amido Pincer Ligands – Versatile Building Blocks for Coordination Chemistry and Catalysis. Eur. J. Inorg. Chem. 2012, 412–429. [Google Scholar] [CrossRef]
- Asay, M.; Morales-Morales, D. Non-symmetric pincer ligands: complexes and applications in catalysis. Dalton Trans. 2015, 44, 17432–17447. [Google Scholar] [CrossRef] [PubMed]
- Murugesan, S.; Kirchner, K. Non-precious metal complexes with an anionic PCP pincer architecture. Dalton Trans. 2016, 45, 416–439. [Google Scholar] [CrossRef]
- Asano, E.; Hatayama, Y.; Kurisu, N.; Ohtani, A.; Hashimoto, T.; Kurihara, Y.; Ueda, K.; Ishihara, S.; Nagano, H.; Yamaguchi, Y. Acetylacetonato-based pincer-type nickel(II) complexes: Synthesis and catalysis in cross-couplings of aryl chlorides with aryl Grignard reagents. Dalton Trans. 2018, 47, 8003–8012. [Google Scholar] [CrossRef] [PubMed]
- Kurisu, N.; Asano, E.; Hatayama, Y.; Kurihara, Y.; Hashimoto, T.; Funatsu, K.; Ueda, K.; Yamaguchi, Y. A β-diketiminato-based pincer-type nickel(II) complex: Synthesis and catalytic performance in the cross-coupling of aryl fluorides with aryl Grignard reagents. Eur. J. Inorg. Chem. 2018, 126–133. [Google Scholar] [CrossRef]
- Yang, B.; Wang, Z.-X. Nickel-Catalyzed Cross-Coupling of Allyl Alcohols with Aryl- or Alkenylzinc Reagents. J. Org. Chem. 2017, 82, 4542–4549. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.-G.; Guo, P.; Duan, J.; Shu, X.-Z. Dual nickel and Lewis acid catalysis for cross-electrophile coupling: the allylation of aryl halides with allylic alcohols. Chem. Sci. 2018, 9, 640–645. [Google Scholar] [CrossRef]
- Cao, H.; Jiang, H.; Feng, H.; Kwan, J.M.C.; Liu, X.; Wu, J. Photo-induced Decarboxylative Heck-Type Coupling of Unactivated Aliphatic Acids and Terminal Alkenes in the Absence of Sacrificial Hydrogen Acceptors. J. Am. Chem. Soc. 2018, 140, 16360–16367. [Google Scholar] [CrossRef]
- Nishikata, T.; Lipshutz, B.H. Allylic Ethers as Educts for Suzuki-Miyaura Couplings in Water at Room Temperature. J. Am. Chem. Soc. 2009, 131, 12103–12105. [Google Scholar] [CrossRef]
- Mino, T.; Kogure, T.; Abe, T.; Koizumi, T.; Fujita, T.; Sakamoto, M. Palladium-Catalyzed Allylic Arylation of Allylic Ethers with Arylboronic Acids Using Hydrazone Ligands. Eur. J. Org. Chem. 2013, 1501–1505. [Google Scholar] [CrossRef]
- Zhang, Y.; Yin, S.-C.; Lu, J.-M. N-Heterocyclic carbene-palladium(II)-1-methylimidazole complex catalyzed allyl-aryl coupling of allylic alcohols with arylboronic acids in neat water. Tetrahedron 2015, 71, 544–549. [Google Scholar] [CrossRef]
- Evans, P.A.; Uraguchi, D. Regio- and Enantiospecific Rhodium-Catalyzed Arylation of Unsymmetrical Fluorinated Acyclic Allylic Carbonates: Inversion of Absolute Configuration. J. Am. Chem. Soc. 2003, 125, 7158–7159. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
| Ni cat 1 Yield of 4a (%) b Recovery of 2a (%) b | 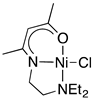 1a 7 43 | 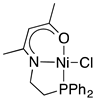 1b 86 0 |  1c 2 83 | 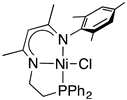 1d 4 90 |
 | |||||||
| Entry | Ni Cat 1 | Solvent | 4a (%) b | Entry | Ni Cat 1 | Solvent | 4a (%) b |
| 1 | 1b | THF | 86 | 7 | 1b | DMF | 0 |
| 2 | 1b | CPME | 89 | 8 | 1b | Et2O | 99(94) c |
| 3 | 1b | 1,4-dioxane | 71 | 9 | 1a | Et2O | 22 |
| 4 | 1b | MTBE | 82 | 10 | 1c | Et2O | 19 |
| 5 | 1b | toluene | 83 | 11 | 1d | Et2O | 30 |
| 6 | 1b | CH2Cl2 | 24 | 12d | 1b | Et2O | 95(91) c |
 | ||
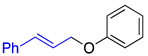 | 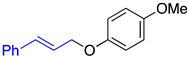 | 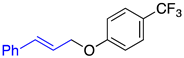 |
| 2a, 91%, 60% b | 2b, 96%, 95% b | 2c, 62% c |
 |  |  |
| 2d, 90% c, 73% b | 7, 40% c,d | 8, 38% c |
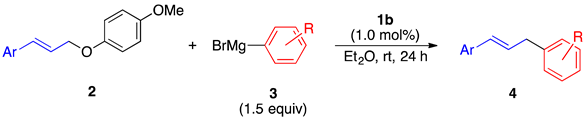 | ||
 |  |  |
| 4b, 94% | 4c, 94% | 4d, 95% b |
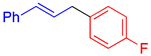 |  |  |
| 4e, 96% | 4f, 85% c | 4g, 79% d |
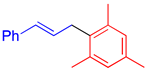 |  |  |
| 4h, 80% e | 4i, 78% b | 4j, 89% f |
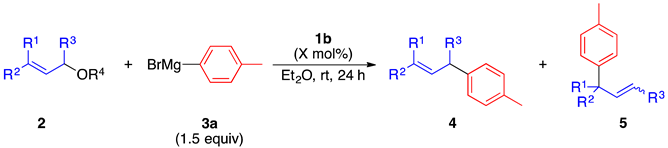 | |||||
| Entry | 2 | X (mol%) | 4/5 | 4 + 5 (%) | |
| 1 |  | 2b′ (cis:trans = 86:14) | 1.0 | 4a:5a = >99:<1 | 88 |
| 2 |  | 2g | 2.5 | 4k:5k = 54:46 | 96 |
| 3 | 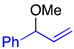 | 2h | 2.5 | 4a:5a = >99:<1 | 94 |
| 4 | 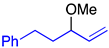 | 2i | 2.5 | 4k:5k = 57:43 | 93 |
| 5 | 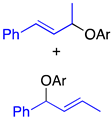 | 2j 2k (2j:2k = 67:33) | 2.5 | 4l:5l = >99:<1 | 90 |
 | ||||
| Entry | X (equiv) | 4a (%) b | 6a (mmol) c | Recovery of 2b (%) b |
| 1 | 1 | N.D. | 0.03 | 83 |
| 2 | 2 | 23 | 0.09 | 40 |
| 3 | 3 | 83 | 0.19 | 0 |
| 4 | 4 | 83 | 0.23 | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hashimoto, T.; Funatsu, K.; Ohtani, A.; Asano, E.; Yamaguchi, Y. Cross-Coupling Reaction of Allylic Ethers with Aryl Grignard Reagents Catalyzed by a Nickel Pincer Complex. Molecules 2019, 24, 2296. https://doi.org/10.3390/molecules24122296
Hashimoto T, Funatsu K, Ohtani A, Asano E, Yamaguchi Y. Cross-Coupling Reaction of Allylic Ethers with Aryl Grignard Reagents Catalyzed by a Nickel Pincer Complex. Molecules. 2019; 24(12):2296. https://doi.org/10.3390/molecules24122296
Chicago/Turabian StyleHashimoto, Toru, Kei Funatsu, Atsufumi Ohtani, Erika Asano, and Yoshitaka Yamaguchi. 2019. "Cross-Coupling Reaction of Allylic Ethers with Aryl Grignard Reagents Catalyzed by a Nickel Pincer Complex" Molecules 24, no. 12: 2296. https://doi.org/10.3390/molecules24122296