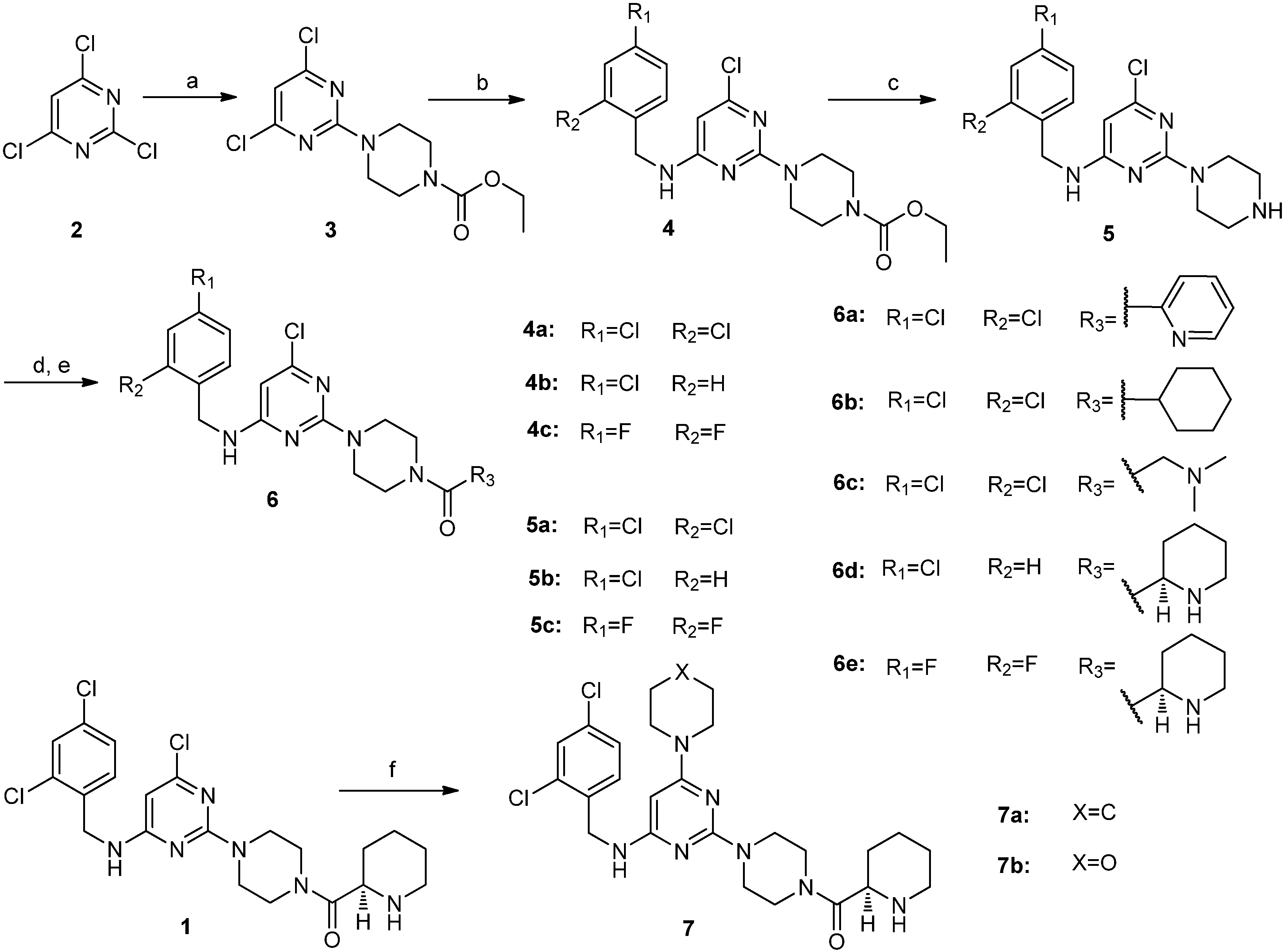
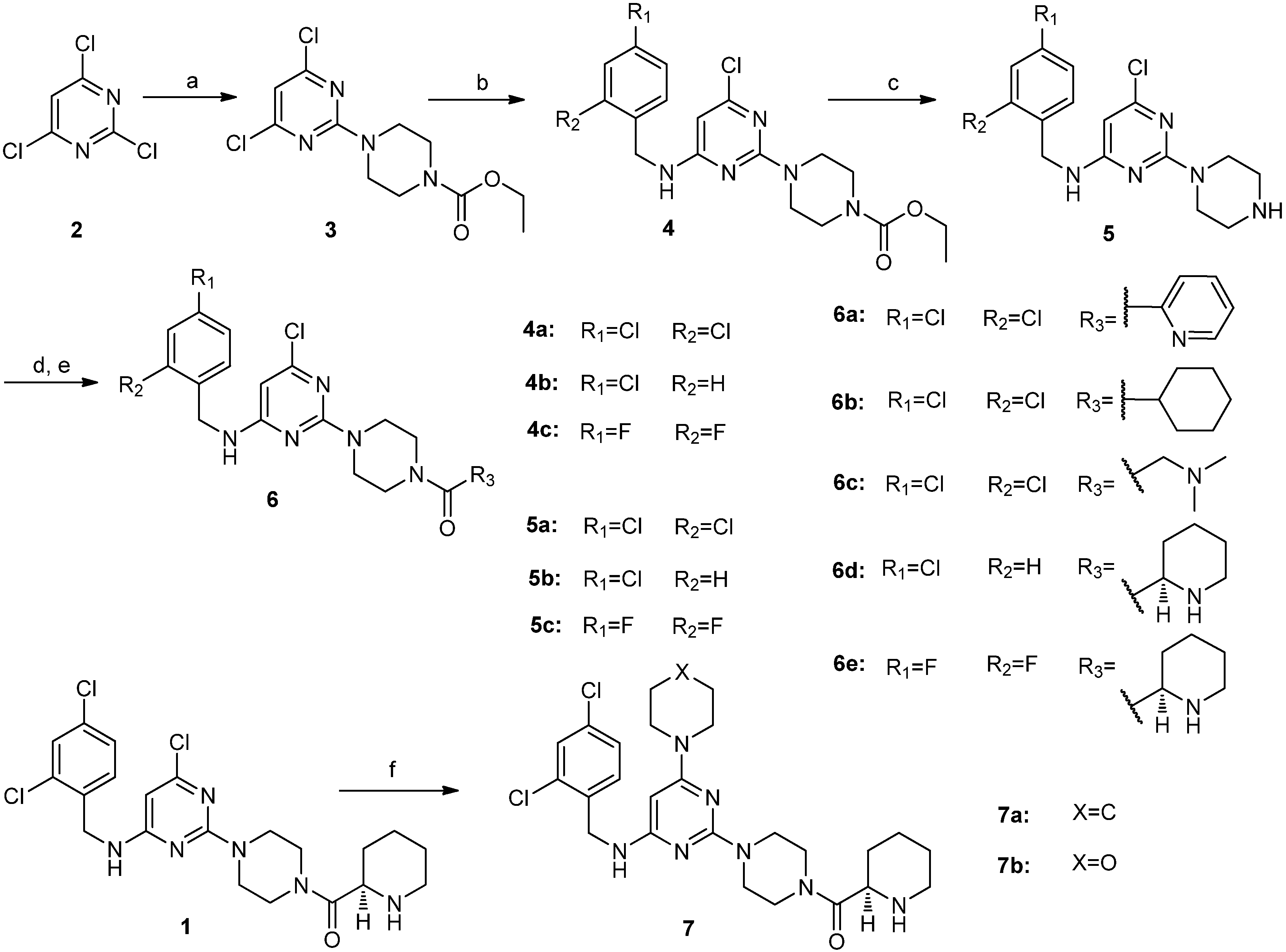
3.2. Chemical Synthesis
3.2.1. Procedure for the Synthesis of Compounds 6a–e
4,6-Dichloro-2-(4-ethoxycarbonylpiperazino)pyrimidine (
3). Compound
3 was synthesized according to a well-established literature procedure [
15].
6-Chloro-2-(4-ethoxycarbonylpiperazino)-N-(2,4-dichlorobenzyl)pyrimidin-4-amine (4a). 2,4-Dichlorobenzylamine (1.76 g, 10.0 mmol) and DIEA (2.60 g, 20.0 mmol) were added to a solution of compound 3 (3.05 g, 10.0 mmol) in NMP (30 mL), and the resulting mixture was stirred at 90 °C for 12 h. The reaction mixture was cooled to room temperature. Water was added, and the resulting solution was extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/ethyl acetate v/v = 5:2) to obtain compound 4a (4.12 g, 92.6%) as a white solid. 1H-NMR (CDCl3) δ ppm: 7.41 (1H, d, J = 1.96 Hz), 7.27 (1H, d, J = 8.6 Hz), 7.23 (1H, d, J = 8.4 Hz), 5.73 (1H, s), 5.10 (1H, brs), 4.57 (2H, s), 4.20–4.14 (2H, q), 3.74 (4H, m), 3.49 (4H, m), 1.30–1.26 (3H, t); ESI-MS: m/z = 444.3 [M+H]+.
6-Chloro-2-(4-ethoxycarbonylpiperazino)-N-(4-chlorobenzyl)pyrimidin-4-amine (4b). Compound 4b was synthesized from compound 3 and 4-chlorobenzylamine according to the procedure described for the synthesis of compound 4a. It was obtained as a white solid. 1H-NMR (CDCl3) δ ppm: 7.32 (2H, d, J = 8.4 Hz), 7.24 (2H, d, J = 8.4 Hz), 5.73 (1H, s), 5.14 (1H, brs), 4.50 (2H, s), 4.19–4.14 (2H, q), 3.75 (4H, m), 3.62 (4H, m), 1.30–1.26 (3H, t); ESI-MS: m/z = 410.4 [M+H]+.
6-Chloro-2-(4-ethoxycarbonylpiperazino)-N-(2,4-difluorobenzyl)pyrimidin-4-amine (4c). Compound 4c was synthesized from Compound 3 and 2,4-difluorobenzylamine according to the procedure described for the synthesis of compound 4a. It was obtained as a white solid. 1H-NMR (CDCl3) δ ppm: 7.30 (1H, m), 6.87–6.80 (2H, m), 5.74 (1H, s), 5.04 (1H, brs), 4.54 (2H, s), 4.20–4.14 (2H, q), 3.76 (4H, m), 3.49 (4H, m), 1.30–1.25 (3H, t); ESI-MS: m/z = 412.4 [M+H]+.
6-Chloro-2-piperazino-N-(2,4-dichlorobenzyl)pyrimidin-4-amine (5a). A mixture of compound 4a (3.78 g, 8.5 mmol), 10% NaOH aqueous solution (40 mL) and ethanol (45 mL) was stirred at reflux for 24 h. The reaction mixture was concentrated in vacuo. Water was added to the residue, and the resulting mixture was extracted with dichloromethane. The organic layer was washed with brine, dried over sodium sulfate, filtered and concentrated in vacuo. The residue was washed with hexane to give compound 5a (3.02 g, 95.5%) as an off-white solid. This solid was used in a subsequent reaction without further purification. 1H-NMR (CDCl3) δ ppm: 7.40 (1H, d, J = 1.96 Hz), 7.27 (1H, d, J = 8.6 Hz), 7.22 (1H, d, J = 8.4 Hz), 5.68 (1H, s), 5.17 (1H, brs), 4.57 (2H, s), 3.72 (4H, m), 2.87 (4H, m), 1.88 (1H, brs); ESI-MS: m/z = 372.0 [M+H]+.
6-Chloro-2-piperazino-N-(4-chlorobenzyl)pyrimidin-4-amine (5b). Compound 5b was obtained as an off-white solid (2.75 g, 95.4%) from compound 4b according to the procedure given for the synthesis of compound 5a. 1H-NMR (CDCl3) δ ppm: 7.30 (2H, d, J = 8.4 Hz), 7.22(2H, d, J = 8.4 Hz), 5.67 (1H, s), 5.14 (1H, brs), 4.50 (2H, s), 3.72 (4H, m), 2.86 (4H, m), 1.88 (3H, brs); ESI-MS: m/z = 338.1 [M+H]+.
6-Chloro-2-piperazino-N-(2,4-difluorobenzyl)pyrimidin-4-amine (5c). Compound 5c was obtained as an off-white solid (2.73 g, 95.0%) from compound 4c according to the procedure described for the synthesis of compound 5a. 1H-NMR (CDCl3) δ ppm: 7.29 (1H, m), 6.84 (2H, m), 5.74 (1H, s), 5.04 (1H, brs), 4.54 (2H, s), 3.73 (4H, m), 2.87 (4H, m), 1.89 (1H, brs); ESI-MS: m/z = 440.1 [M+H]+.
6-Chloro-2-[4-(pyridin-2-yl)carbonylpiperazino]-N-(2,4-dichlorobenzyl)pyrimidin-4-amine (6a). A mixture of compound 5a (0.37 g, 1.0 mmol), EDCI (0.23 g, 1.2 mmol), HOBt (0.16 g, 1.2 mmol), pyridine-2-carboxylic acid (0.13 g, 1.0 mmol), TEA (1 mL) and dichloromethane (20 mL) was stirred at room temperature overnight. The reaction mixture was concentrated in vacuo. Water was added to the residue, and the resulting mixture was extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/ethyl acetate/methanol) to obtain compound 6a (0.38 g, 79.5%) as a white solid. M.p. 175.1–176.9 °C; 1H-NMR (CDCl3) δ ppm: 8.59 (1H, d, J = 4.7 Hz), 7.81 (1H, m, J1= 7.5 Hz, J2 = 1.6 Hz), 7.68 (1H, m), 7.38 (2H, m), 7.25(1H, d, J = 8.4 Hz), 7.21 (1H, dd, J1 = 8.4 Hz, J2 = 1.96 Hz), 5.73 (1H, s), 5.17 (1H, brs), 4.56 (2H, s), 3.87–3.62 (8H, m); 13C-NMR (CDCl3): δ 167.6, 163.3, 160.7, 159.7, 153.7, 148.3, 137.1, 134.2, 133.7, 129.9, 129.3, 127.1, 124.6, 123.9, 92.6, 46.9, 44.1, 43.4, 42.2; ESI-MS: m/z = 477.0 [M+H]+; HRMS (TOF): calcd for C21H19Cl3N6O [M+Na]+: 499.0579, Found: 499.0580.
6-Chloro-2-(4-cyclohexylcarbonylpiperazino)-N-(2,4-dichlorobenzyl)pyrimidin-4-amine (6b). Compound 6b was obtained as a white solid (0.41 g, 85.4%) from compound 5a and cyclohexanecarboxylic acid according to the procedure described for the synthesis of compound 6a. M.p. 175.3–177.1 °C; 1H-NMR (CDCl3) δ ppm: 7.41 (1H, d, J = 1.96 Hz), 7.29 (1H, d, J = 8.4 Hz), 7.23 (1H, dd, J1= 8.4 Hz, J2= 1.96 Hz), 5.74 (1H, s), 5.15(1H,brs), 4.58 (2H, s), 3.76–3.51 (8H, m), 2.48 (1H, m), 1.82–1.25 (10H, m); 13C-NMR (CDCl3): δ 174.8, 163.4, 160.7, 133.8, 129.9, 129.4, 127.2, 92.8, 45.0, 44.1, 43.6, 42.4, 41.3, 40.4, 29.3, 25.7; ESI-MS: m/z = 482.3 [M+H]+; HRMS (TOF): calcd for C22H26Cl3N5O [M+Na]+: 504.1101, Found: 504.1009.
6-Chloro-2-[4-(N,N-dimethylaminomethyl)carbonylpiperazino]-N-(2,4-dichlorobenzyl)pyrimidin-4-amine (6c). Compound 6c was obtained as a white solid (0.30 g, 65.4%) from compound 5a and 2-(dimethylamino)acetic acid according to the procedure given for the synthesis of compound 6a. M.p. 166.5–168.5 °C; 1H-NMR (CDCl3) δ ppm: 7.41 (1H, d, J = 1.96 Hz), 7.27 (1H, d, J = 8.4 Hz), 7.23 (1H, dd, J1= 8.4 Hz, J2= 1.96 Hz), 5.74 (1H, s), 5.15(1H,brs), 4.58 (2H, s), 3.76 (4H, m), 3.63 (4H, m), 3.21 (2H, s), 2.35 (6H, s); 13C-NMR (CDCl3): δ 168.6, 163.3, 160.7, 133.7, 133.6, 129.9, 129.2, 127.1, 92.4, 62.5, 45.4, 45.2, 44.0, 43.5, 42.3, 41.5; ESI-MS: m/z = 457.0 [M+H]+; HRMS (TOF): calcd for C19H23Cl3N6O [M+H]+: 457.1072, Found: 457.1071.
(R)-6-Chloro-2-[4-(piperidin-2-yl)carbonylpiperazino]-N-(4-chlorobenzyl)pyrimidin-4-amine (6d). A mixture of compound 5b (0.51 g, 1.5 mmol), EDCI (0.35 g, 1.8 mmol), HOBt (0.24 g, 1.8 mmol), (R)-N-piperidine-2-carboxylic acid (0.35 g, 1.5 mmol), TEA (1.5 mL) and dichloromethane (30 mL) was stirred at room temperature overnight. The reaction mixture was concentrated in vacuo. Water was added to the residue, and the resulting mixture was extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, filtered, and concentrated in vacuo. The residue (0.74 g) was dissolved in dichloromethane (10) and 4 M HCl in dioxane (0.5 mL) was added dropwise. The reaction mixture was stirred at room temperature for 1 h. Then the pH of reaction mixture was adjusted to 10 with a 10% NaOH aqueous solution. The resulting mixture was extracted with dichloromethane. The organic layer was washed with brine, dried over sodium sulfate, filtered and concentrated in vacuo. Ethyl acetate was added to the crude residue, stirred and filtered. The solid was washed with additional ethyl acetate and dried to obtain compound 6d (0.47 g, 70.6%) as a white solid. M.p. 100.3–101.9 °C; 1H-NMR (CDCl3) δ ppm: 7.30 (2H, d, J = 8.4 Hz), 7.24 (2H, d, J = 8.4 Hz), 5.74 (1H, s), 5.24 (1H, m), 4.48 (2H, s), 3.78–3.53 (8H, m), 3.43 (1H, m), 3.27 (1H, d), 2.80–2.74 (1H, m), 1.91 (1H, d), 1.76 (1H, d), 1.63 (1H, d), 1.55–1.47 (3H, m); 13C-NMR (CDCl3): δ 171.2, 163.4, 160.7, 136.9, 133.0, 128.7, 128.6, 92.7, 56.1, 45.1, 45.0, 44.4, 43.8, 43.4, 41.6, 29.3, 25.5, 23.8; ESI-MS: m/z = 449.5 [M+H]+; HRMS (TOF): calcd for C22H28Cl2N6O [M+H]+: 449.1618, Found: 449.1618.
(R)-6-Chloro-2-[4-(piperidin-2-yl)carbonylpiperazino]-N-((2,4-difluorobenzyl)pyrimidin-4-amine (6e). Compound 6c was obtained as a white solid (0.44 g, 65.4%) from compound 5c according to the procedure described for the synthesis of compound 6d. M.p. 87.2–89.0 °C; 1H-NMR (CDCl3) δ ppm: 7.28 (1H, m), 6.84 (2H, m), 5.77 (1H, s), 5.11 (1H, m), 4.54 (2H, s), 3.79–3.47 (9H, m), 3.16 (1H, d), 2.76 (1H, brs), 2.71–2.65 (1H, m), 1.92 (1H, d), 1.73 (1H, d), 1.58 (1H, d), 1.50–1.45 (3H, m); 13C-NMR (CDCl3): δ 172.1, 163.3, 162.0, 161.0, 160.7, 159.5, 130.3, 111.3, 111.1, 103.8, 92.6, 56.4, 45.9, 45.6, 44.9, 43.8, 43.4, 41.5, 38.4, 30.0, 26.6, 24.3; ESI-MS: m/z = 451.3 [M+H]+; HRMS (TOF): calcd for C21H25ClF2N6O [M+H]+: 451.1819, Found: 451.1816.
3.2.2. Procedure for the Synthesis of Compounds 7a and 7b
(R)-2-[4-(Piperidin-2-yl)carbonylpiperazino]-N-(2,4-dichlorobenzyl)-6-piperidino pyrimidin-4-amine (7a). A mixture of compound 1 (0.48 g, 1.0 mmol) and piperidine (5 mL) was stirred at reflux overnight. Water was added, and the resulting solution was extracted with ethyl acetate. The organic layer was washed with brine, dried over sodium sulfate, filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/ethyl acetate/methanol) to obtain compound 7a (0.28 g, 52.6%) as an off-white solid. M.p. 147.1–149.1 °C; 1H-NMR (CDCl3) δ ppm: 7.38 (1H, d, J = 1.96 Hz), 7.32 (1H, d, J = 8.4 Hz), 7.19 (1H, dd, J1 = 8.4 Hz, J2 = 1.96 Hz), 4.93 (1H, s), 4.50 (2H, d), 3.77–3.55 (8H, m), 3.47 (5H, m), 3.28 (1H, d), 2.77 (1H, t), 1.92 (1H, m), 1.78 (1H, m), 1.63–1.50 (10H, m); 13C-NMR (CDCl3): δ 172.0, 163.9, 163.7, 161.0, 135.4, 133.5, 133.2, 129.9, 129.0, 127.0, 73.0, 56.4, 45.6, 45.2, 44.0, 43.6, 42.6, 41.7, 30.1, 26.6, 25.4, 24.7, 24.4; ESI-MS: m/z = 532.4 [M+H]+; HRMS (TOF): calcd for C26H35Cl2N7O [M+H]+: 532.2358, Found: 532.2356.
(R)-2-[4-(Piperidin-2-yl)carbonylpiperazino]-N-(2,4-dichlorobenzyl)-6-morpholino pyrimidin-4-amine (7b). Compound 7b was obtained as an off-white solid (0.30 g, 55.4%) from compound 1 and morpholine according to the procedure described for the synthesis of compound 7a. M.p. 167.8–169.5 °C; 1H-NMR (CDCl3) δ ppm: 7.39 (1H, d, J = 1.96 Hz), 7.30 (1H, d, J = 8.4 Hz), 7.20 (1H, dd, J1 = 8.4 Hz, J2 = 1.96 Hz), 4.92 (1H, s), 4.90 (1H, brs), 4.52 (2H, d), 3.76–3.54 (12H, m), 3.44 (5H, m), 3.28 (1H, d), 2.76 (1H, t), 1.92 (1H, m), 1.77 (1H, m), 1.63 (1H, d), 1.51–1.43 (3H, m); 13C-NMR (CDCl3): δ 172.1, 164.1, 163.9, 160.9, 135.2, 133.5, 133.2, 129.8, 129.1, 127.0, 73.3, 66.5, 56.4, 45.6, 45.1, 44.5, 44.0, 43.5, 42.5, 41.6, 30.1, 26.6, 24.4; ESI-MS: m/z = 534.3 [M+H]+; HRMS (TOF): calcd for C25H33Cl2N7O2 [M+H]+: 534.2146, Found: 534.2142.
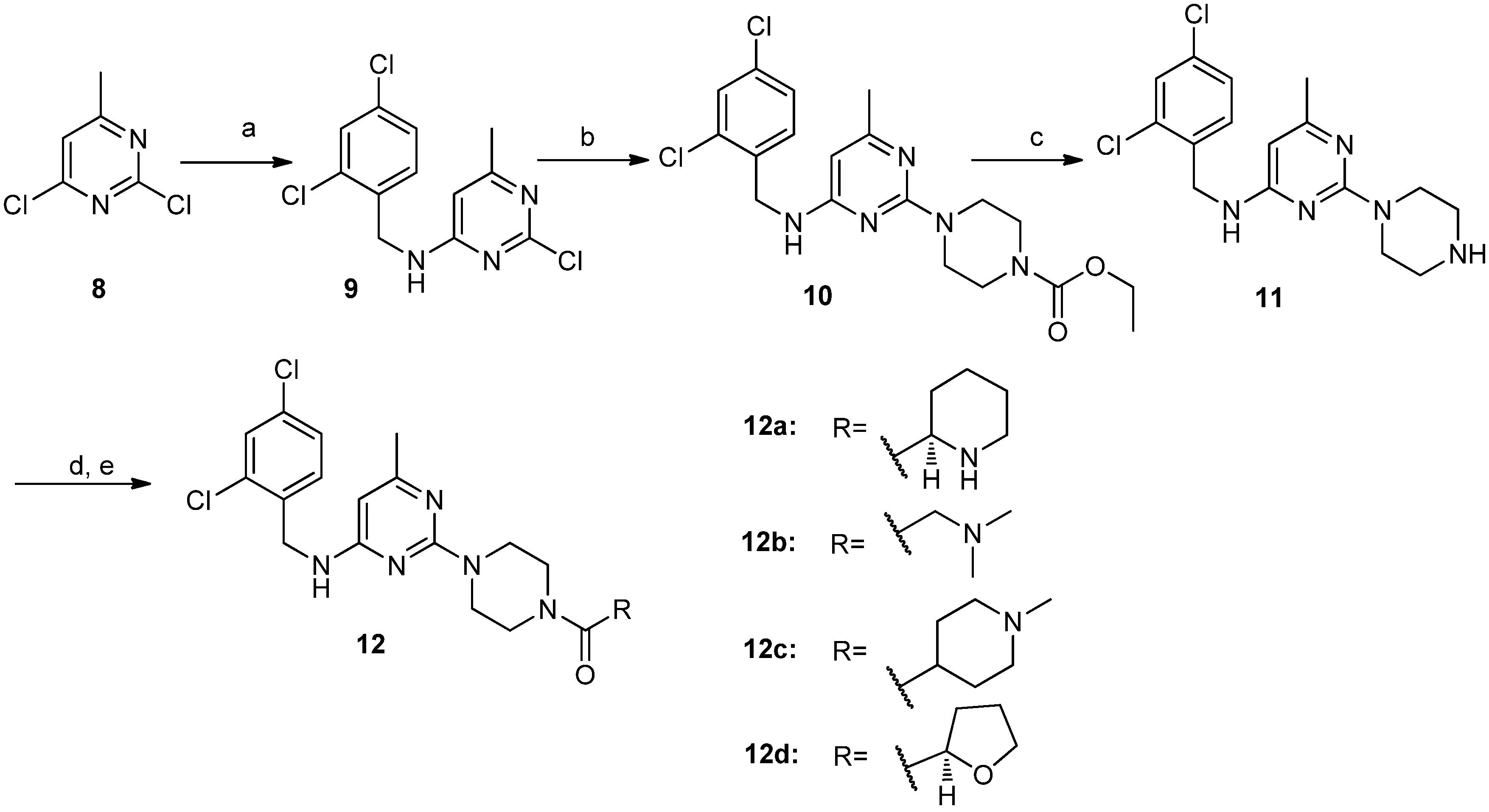
3.2.3. Procedure for the Synthesis of Compounds 12a–d
2-Chloro-N-(2,4-dichlorobenzyl)-6-methylpyrimidin-4-amine (9). 2,4-Dichlorobenzylamine (7.04 g, 40.0 mmol) was dropped into a mixture of compound 8 (6.52 g, 40.0 mmol), DIEA (6.22 g, 48.0 mmol) and dichloromethane (150 mL) at room temperature. The resulting mixture was then stirred at reflux overnight. The reaction mixture was washed with distilled water and brine successively. The obtained organic layer was dried over sodium sulfate, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (hexane/ethyl acetate v/v = 5:1) to give compound 9 (7.56 g, 62.6%) as a white solid. 1H-NMR (DMSO-d6) δ ppm: 8.27 (1H, brs), 7.64 (1H, d, J = 1.96 Hz), 7.43–7.41 (1H, dd, J1 = 8.4 Hz, J2 = 1.96 Hz), 7.34 (1H, d, J = 8.2 Hz), 6.40 (1H, s), 4.52 (2H, s), 2.20 (3H, s); ESI-MS: m/z = 302.5 [M+H]+.
2-(4-Ethoxycarbonylpiperazino)-N-(2,4-dichlorobenzyl)-6-methylpyrimidin-4-amine (10). Compound 10 was obtained as a white solid (5.01 g, 89.4%) from compound 9 and N-ethoxycarbonylpiperidine according to the procedure described for the synthesis of compound 4a. 1H-NMR (DMSO-d6) δ ppm: 7.60 (1H, d, J = 1.96 Hz), 7.54 (1H, brs), 7.41–7.38 (1H, dd, J1 = 8.4 Hz, J2 = 1.96 Hz), 7.34 (1H, d, J = 8.4 Hz), 5.74 (1H, s), 4.48 (2H, s), 4.02–4.06 (2H, q), 3.58 (4H, m), 3.31 (4H, m), 2.06 (3H, s), 1.20–1.17 (3H, t); ESI-MS: m/z = 424.2 [M+H]+.
2-Piperazino-N-(2,4-dichlorobenzyl)-6-methylpyrimidin-4-amine (11). Compound 11 was obtained as an off-white solid (2.81 g, 95.4%) from compound 10 according to the procedure described for the synthesis of compound 5a. 1H-NMR (CDCl3) δ ppm: 7.40 (1H, d, J = 1.96 Hz), 7.27 (1H, d, J = 8.4 Hz), 7.20 (1H, dd, J1= 8.4 Hz, J2= 1.96 Hz), 5.60 (1H, s), 5.04 (1H, brs), 4.57 (2H, s), 3.76 (4H, m), 2.89 (4H, m), 2.19 (3H, s); ESI-MS: m/z = 352.2 [M+H]+.
(R)-2-[4-(Piperidin-2-yl)carbonylpiperazino]-N-(2,4-dichlorobenzyl)-6-methylpyrimidin-4-amine (12a). Compound 12a was obtained as a white solid (0.49 g, 71.4%) from compound 11 and (R)-N-piperidine-2-carboxylic acid according to the procedure described for the synthesis of compound 6d. M.p. 82.6–84.5 °C; 1H-NMR (CDCl3) δ ppm: 7.39 (1H, d, J = 1.96 Hz), 7.27 (1H, d, J = 8.4 Hz), 7.20 (1H, dd, J1 = 8.4 Hz, J2 = 1.96 Hz), 5.59 (1H, s), 5.01 (1H, brs), 4.57 (2H, d), 3.78–3.50 (8H, m), 3.43 (1H, m), 3.23–3.19 (1H, d), 2.73–2.67 (1H, m), 2.18 (3H, s), 1.94–1.91 (1H, d), 1.76 (1H, d), 1.61 (1H, d), 1.51–1.44 (3H, m); 13C-NMR (CDCl3): δ 171.6, 166.1, 163.0, 161.4, 135.0, 133.6, 133.4, 129.9, 129.2, 127.1, 92.9, 55.9, 45.1, 43.9, 43.5, 42.2, 41.7, 29.7, 26.1, 24.1, 22.4; ESI-MS: m/z = 463.5 [M+H]+; HRMS (TOF): calcd for C22H28Cl2N6O [M+H]+: 463.1776, Found: 463.1774.
2-[4-(N,N-dimethylaminomethyl)carbonylpiperazino]-N-(2,4-dichlorobenzyl)-6-methyl pyrimidin-4-amine (12b). Compound 12b was obtained as a white solid (0.28 g, 64.0%) from compound 11 and 2-(dimethylamino)acetic acid according to the procedure described for the synthesis of compound 6a. M.p. 147.5–149.1 °C; 1H-NMR (DMSO-d6) δ ppm: 7.60 (1H, d, J = 1.96 Hz), 7.53 (1H, brs), 7.39 (1H, dd, J1 = 8.4 Hz, J2 = 1.96 Hz), 7.35 (1H, d, J = 8.4 Hz), 5.75 (1H, s), 4.50 (2H, s), 3.58 (4H, m), 3.47–3.40 (4H, m), 3.09 (2H, s), 2.18 (6H, s), 2.07 (3H, s); 13C-NMR (CDCl3): δ 168.5, 166.1, 162.9, 161.5, 135.0, 133.6, 133.4, 129.9, 129.2, 127.0, 92.8, 62.5, 45.4, 44.1, 43.6, 42.2, 41.6, 24.1; ESI-MS: m/z = 437.4 [M+H]+; HRMS (TOF): calcd for C20H26Cl2N6O [M+H]+: 437.1618, Found: 437.1617.
2-[4-(N-methylpiperidin-4-yl)carbonylpiperazino]-N-(2,4-dichlorobenzyl)-6-methylpyrimidin-4-amine (12c). Compound 12c was obtained as a white solid (0.35 g, 73.5%) from compound 11 and 1-methylpiperidine-4-carboxylic acid according to the procedure described for the synthesis of compound 6a. M.p. 146.4–148.0 °C; 1H-NMR (CDCl3) δ ppm: 7.40 (1H, d, J = 1.96 Hz), 7.28 (1H, d, J = 8.4 Hz), 7.19 (1H, dd, J1 = 8.4 Hz, J2 = 1.96 Hz), 5.60 (1H, s), 5.01 (1H, s), 4.57 (2H, d), 3.76 (4H, m), 3.64 (2H, m), 3.49 (2H, m), 2.94 (2H, d), 2.49 (1H, m), 2.31 (3H, s), 2.19 (3H, s), 2.05 (2H, m), 1.93 (2H, m), 1.77 (2H, m); 13C-NMR (CDCl3): δ 173.4, 166.1, 162.9, 161.4, 135.0, 133.6, 133.4, 129.8, 129.1, 127.0, 93.2, 54.9, 46.1, 45.2, 44.1, 43.6, 42.2, 41.5, 37.7, 28.4, 24.1; ESI-MS: m/z = 477.2 [M+H]+; HRMS (TOF): calcd for C23H30Cl2N6O [M+H]+: 477.1931, Found: 477.1934.
(R)-2-[4-(Tetrahydrofuran-2-yl)carbonylpiperazino]-N-(2,4-dichlorobenzyl)-6-methylpyrimidin-4-amine(12d). Compound 12d was obtained as a white solid (0.39 g, 86.7%) from compound 11 and (R)-tetrahydrofuran-2-carboxylic acid according to the procedure described for the synthesis of compound 6a. M.p. 124.3–125.8 °C; 1H-NMR (DMSO-d6) δ ppm: 7.60 (1H, d, J = 1.96 Hz), 7.53 (1H, brs), 7.39 (1H, dd, J1 = 8.4 Hz, J2 = 1.96 Hz), 7.35 (1H, d, J = 8.4 Hz), 5.75 (1H, s), 4.67 (1H, t), 4.50 (2H, s), 3.76 (2H, m), 3.61–3.43 (8H, m), 2.07 (3H, s), 2.00 (2H, m), 1.83 (2H, m); 13C-NMR (CDCl3): δ 170.0, 166.1, 162.9, 161.4, 135.0, 133.6, 133.4, 129.9, 129.1, 127.0, 93.1, 75.8, 69.0, 45.2, 43.9, 43.5, 42.2, 41.9, 28.4, 25.6, 24.1; ESI-MS: m/z = 451.3 [M+H]+; HRMS (TOF): calcd for C21H25Cl2N5O2 [M+H]+: 450.1458, Found: 450.1453.



{kind=link}
{kind=link}
{kind=link}