Synthesis of Disaccharides Containing 6-Deoxy-a-L-talose as Potential Heparan Sulfate Mimetics


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
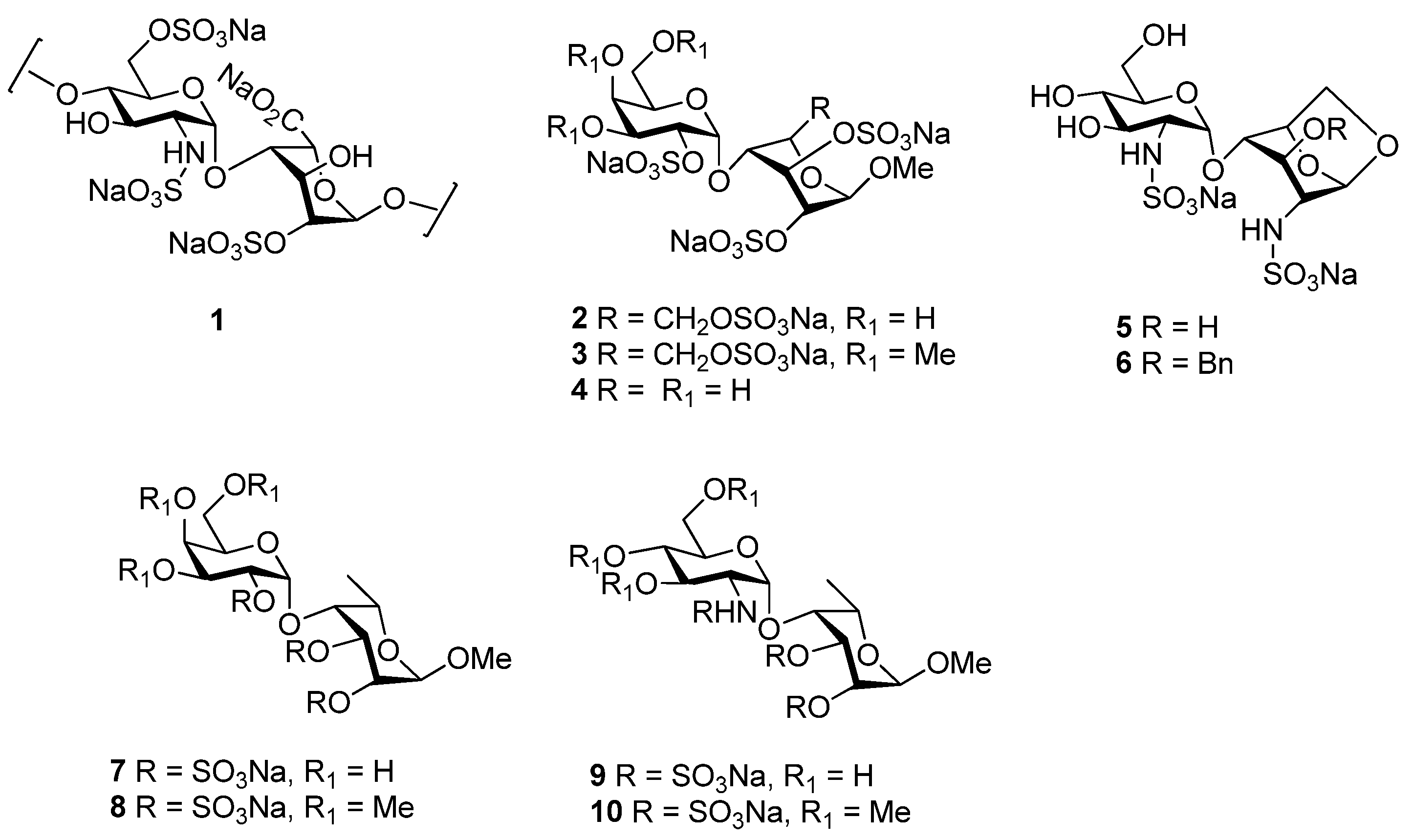
:1. Introduction
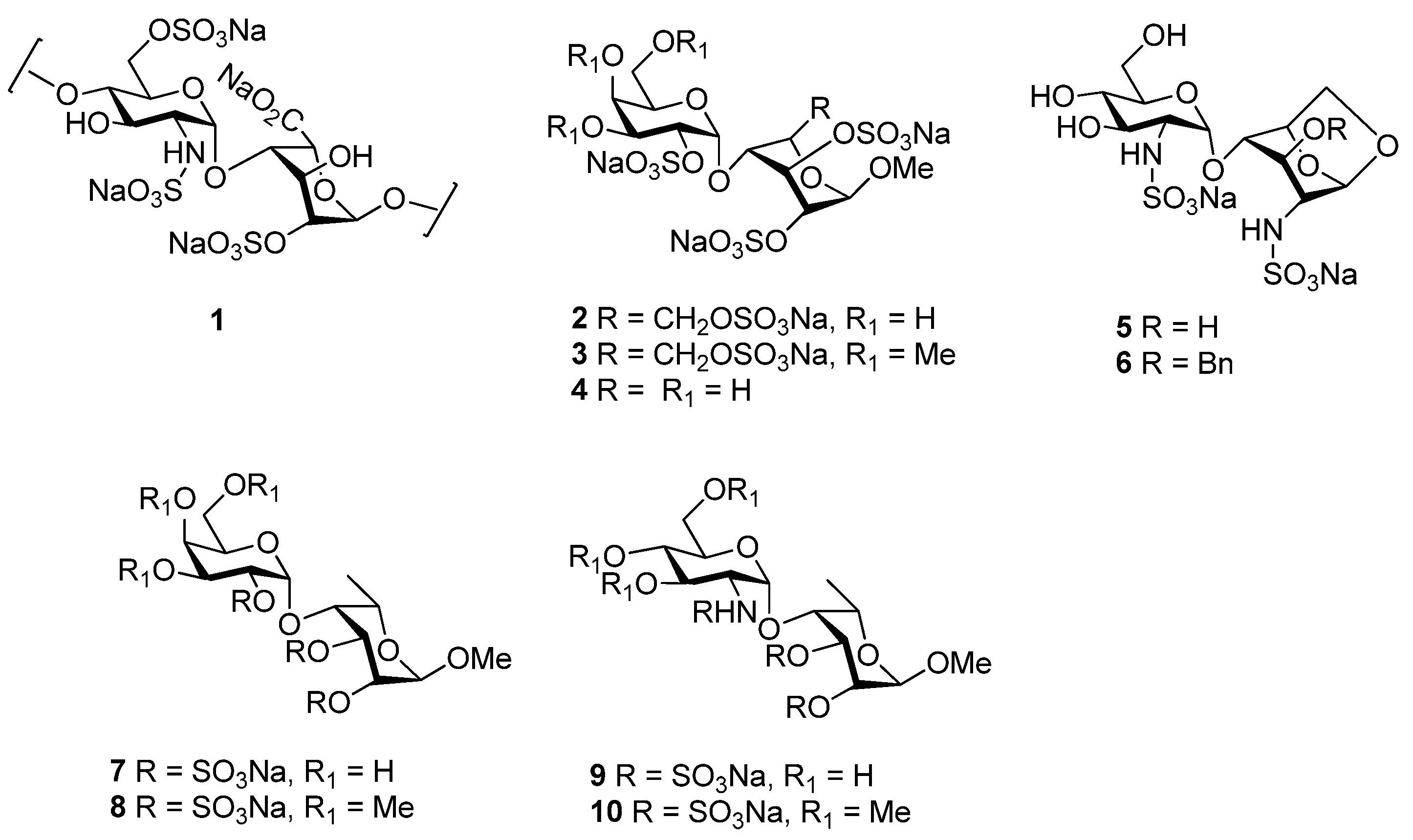
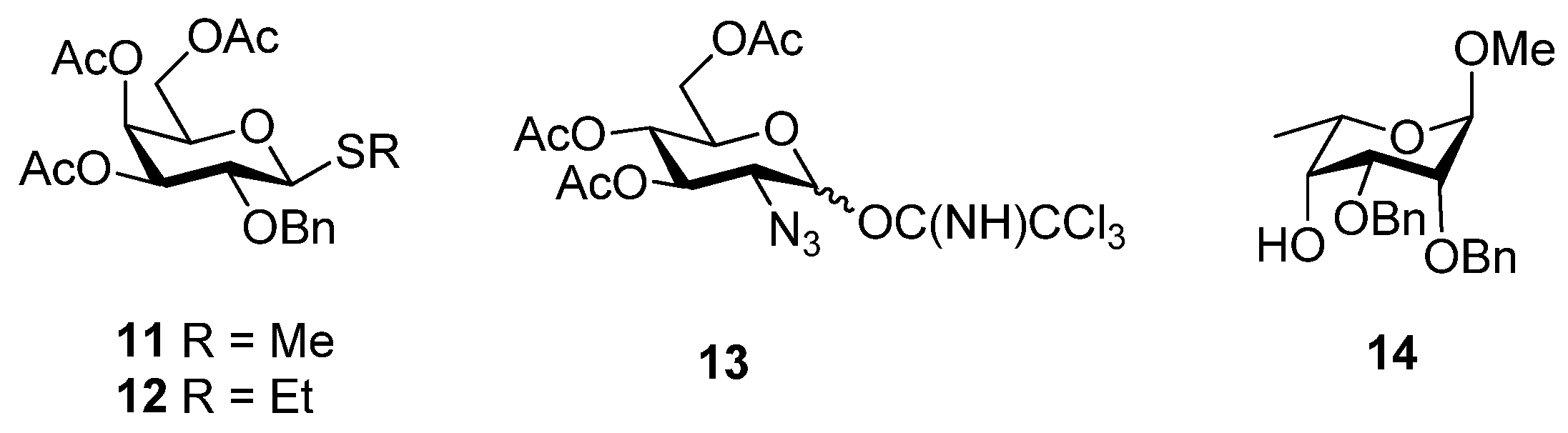
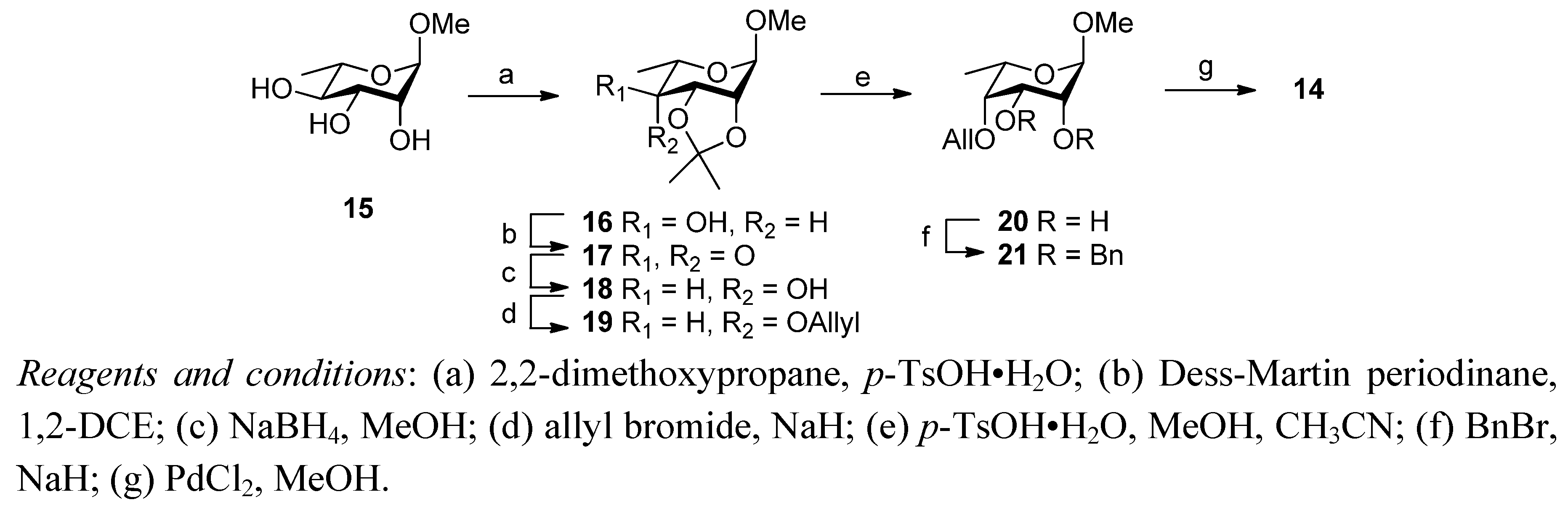
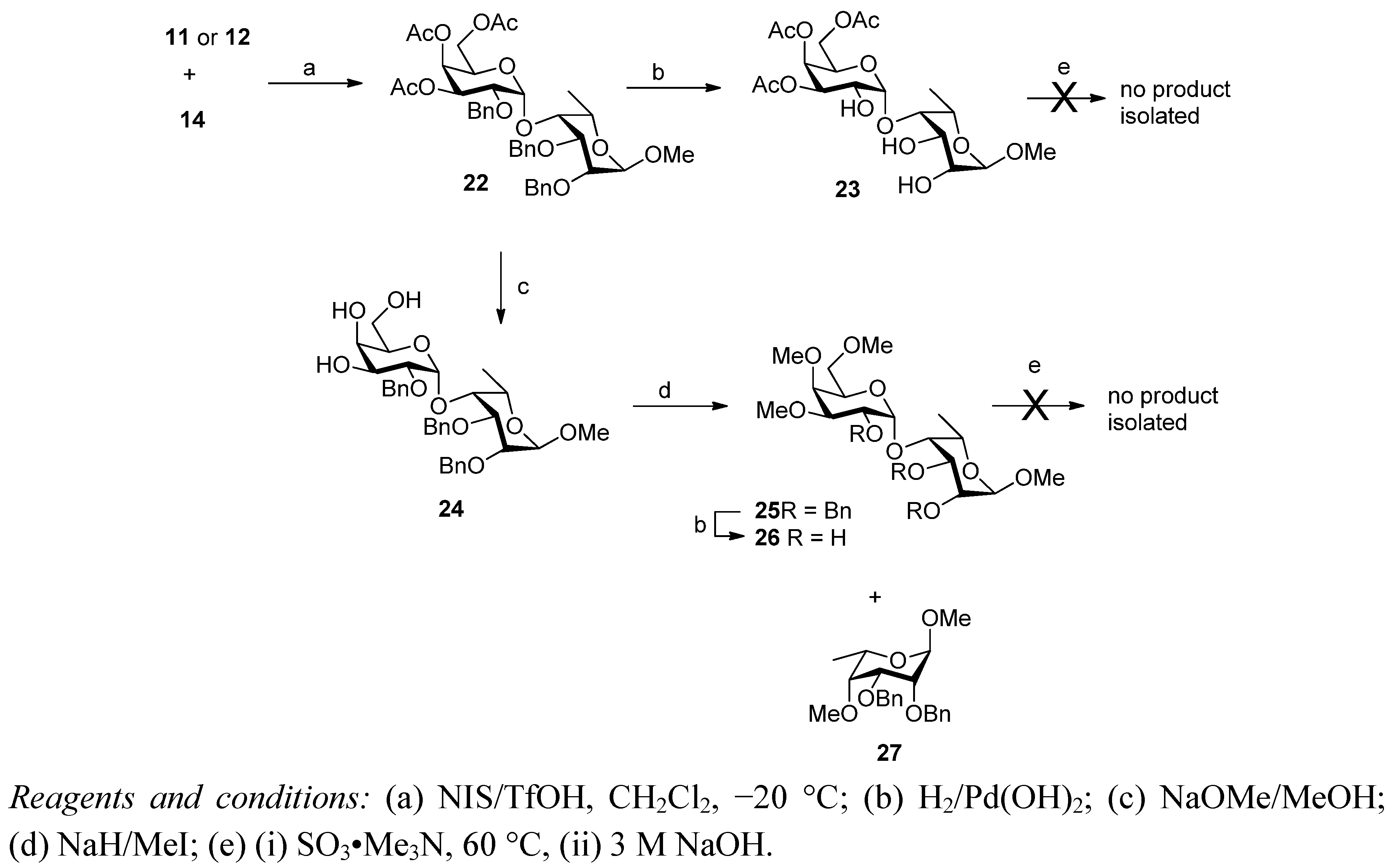
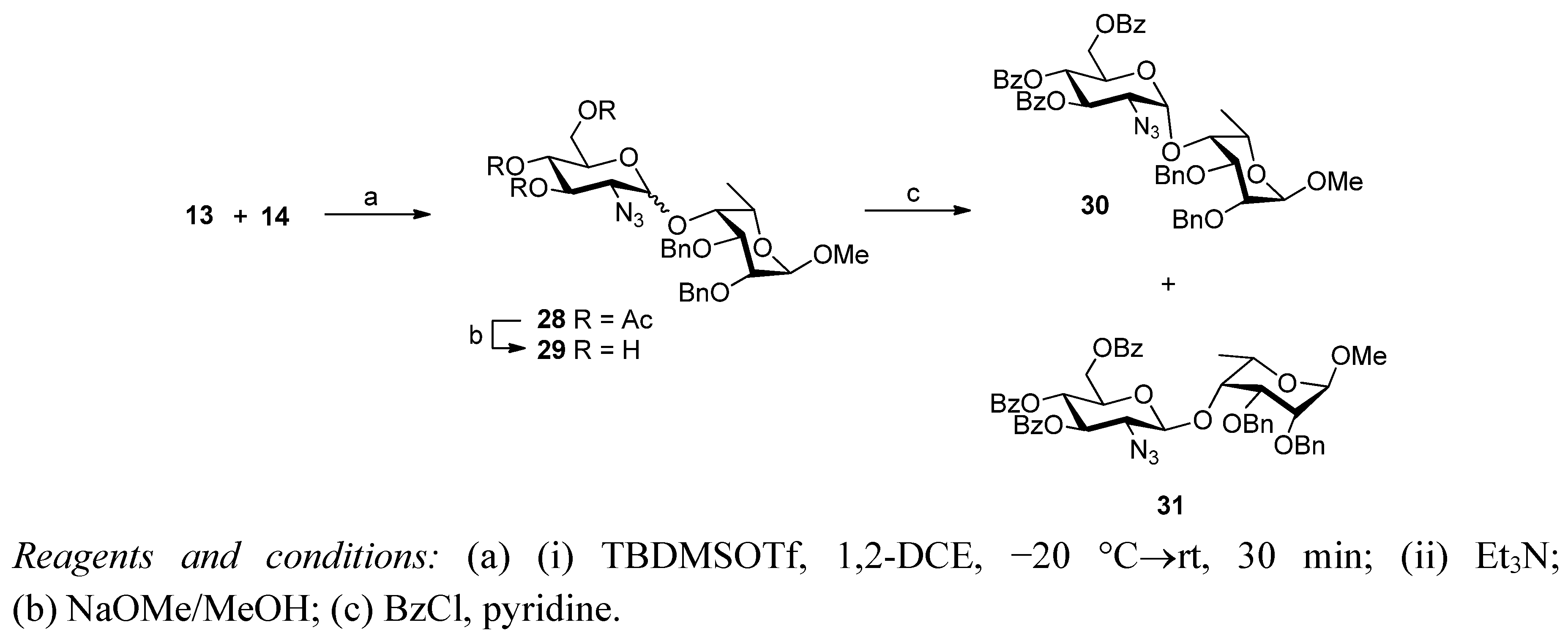
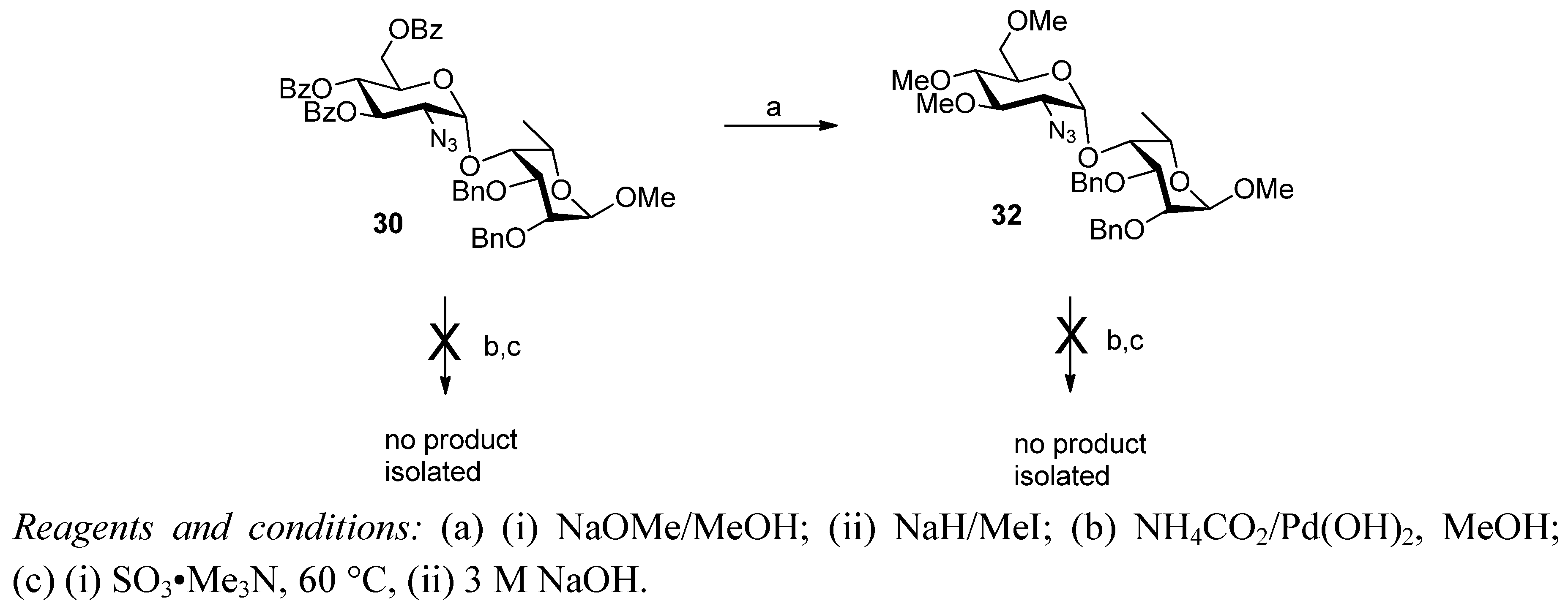
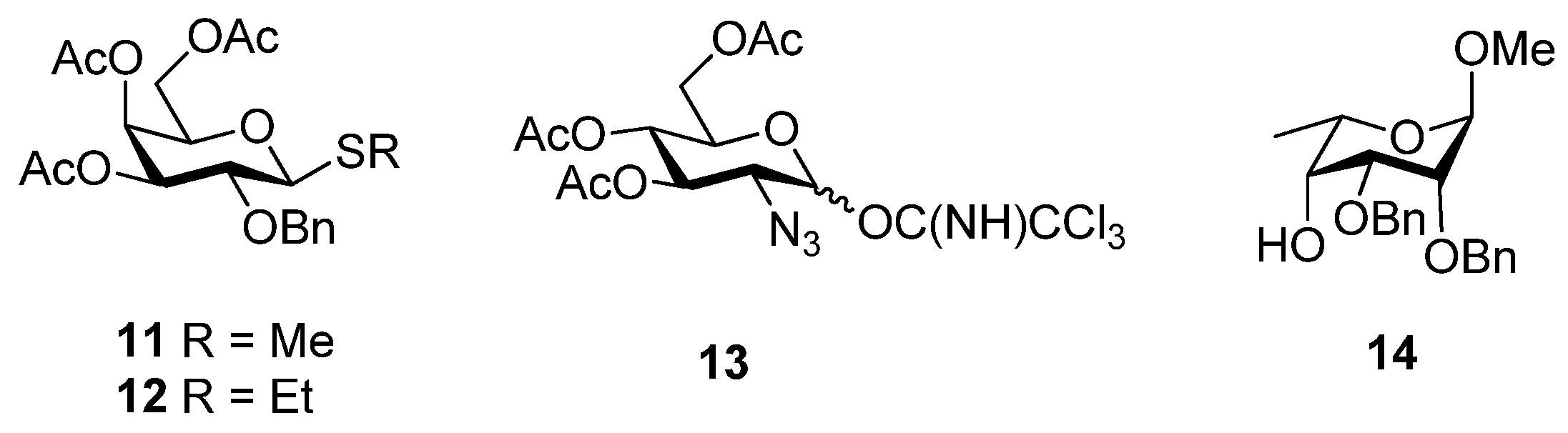
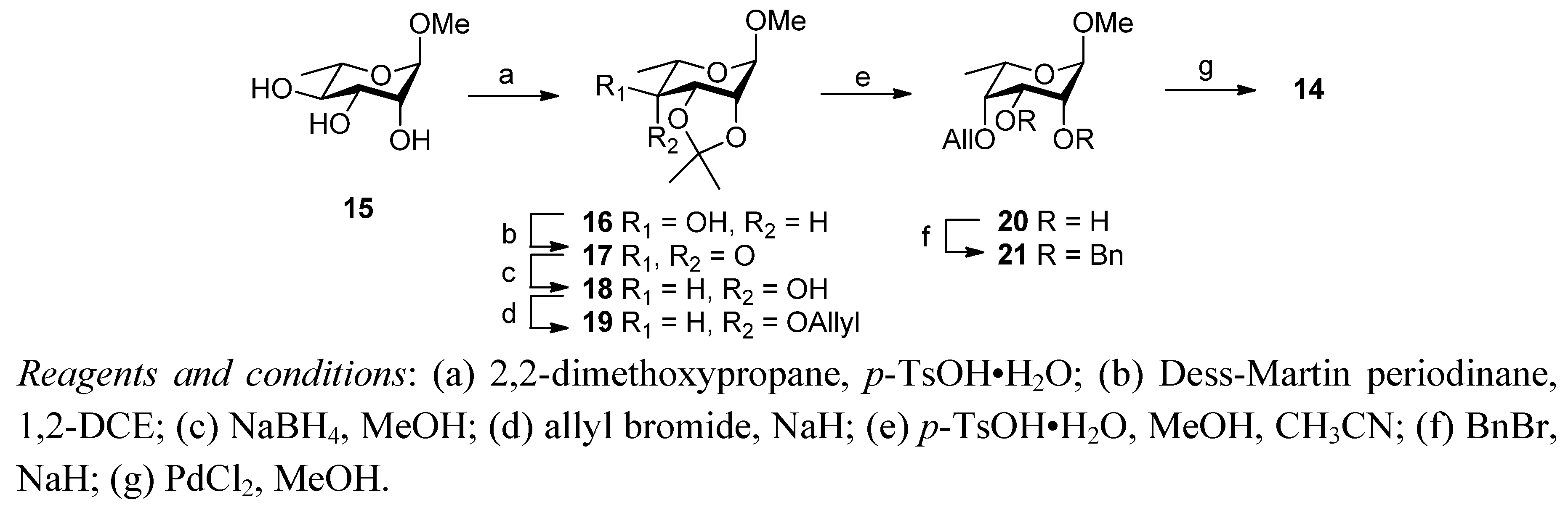
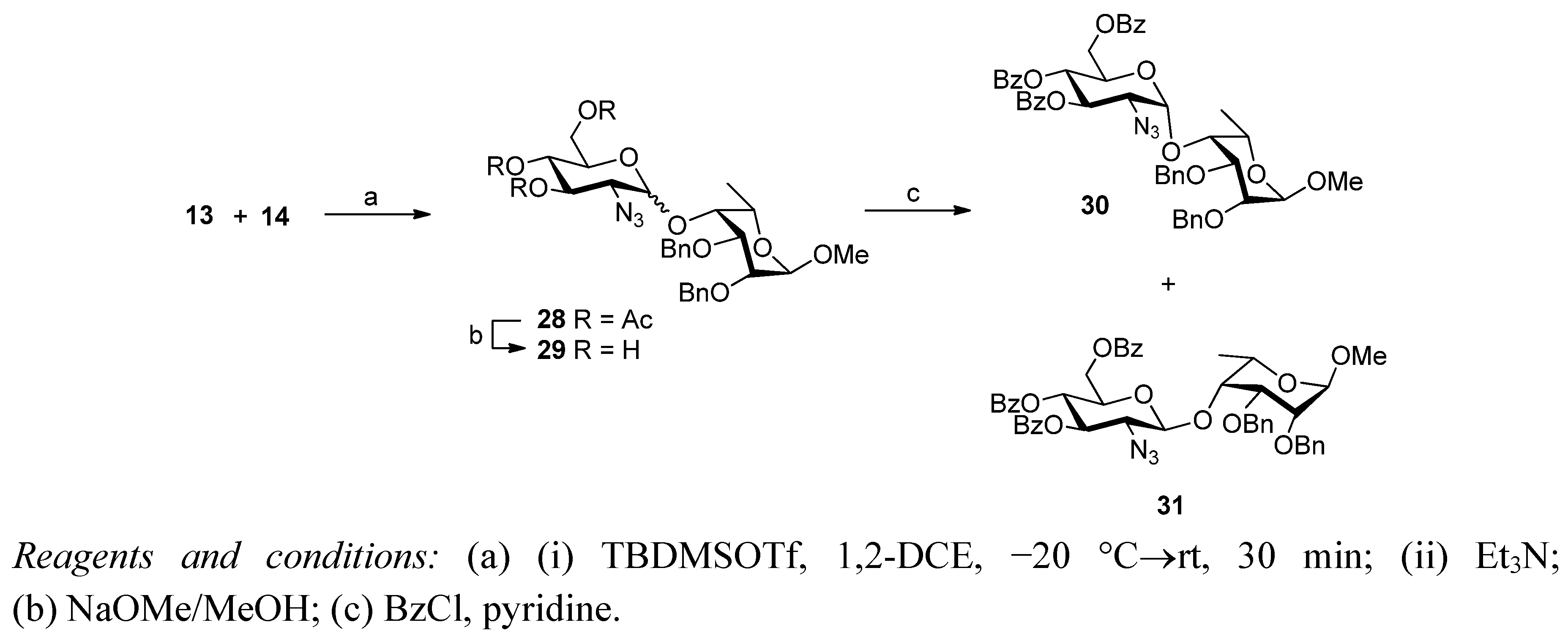
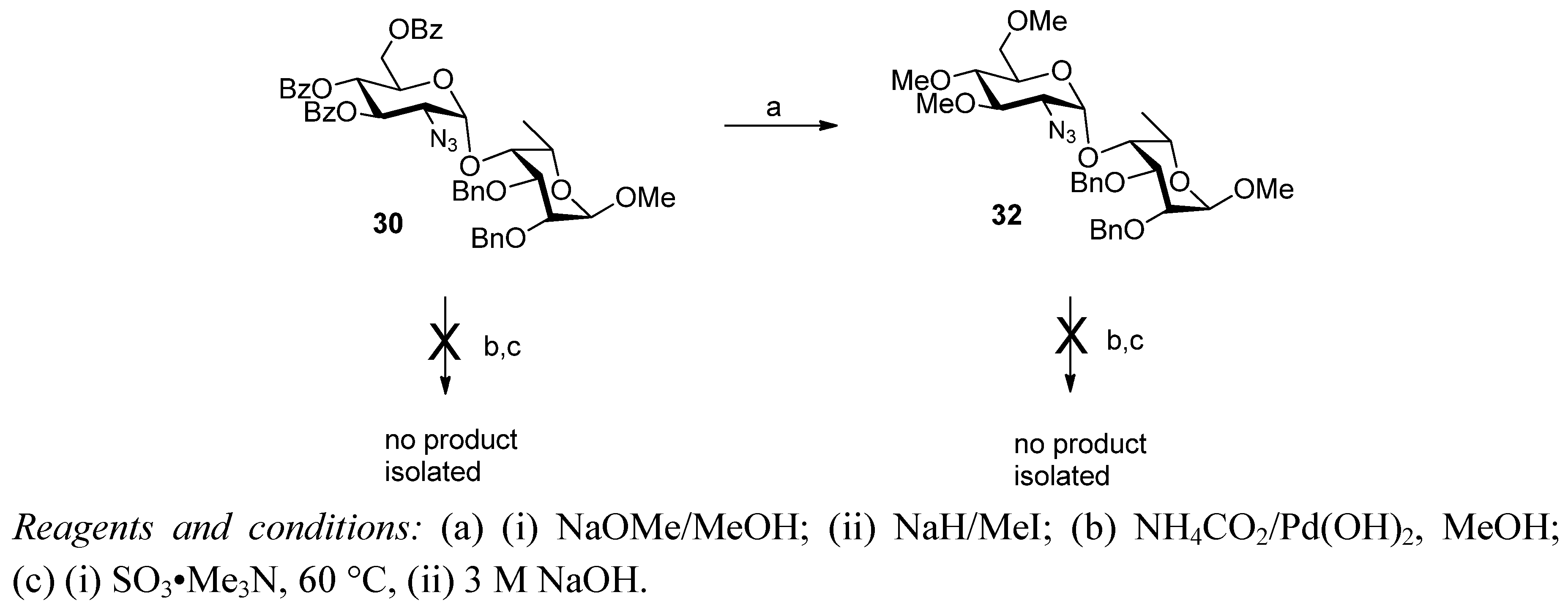
2. Results and Discussion

3. Experimental
General
4. Conclusions
Supplementary Materials
Acknowledgments
References
- Presta, M.; Dell’Era, P.; Mitola, S.; Moroni, E.; Ronca, R.; Rusnati, M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005, 16, 159–178. [Google Scholar] [CrossRef] [Green Version]
- Korc, M.; Friesel, R.E. The role of fibroblast growth factors in tumor growth. Curr. Cancer Drug Targets 2009, 9, 639–651. [Google Scholar] [CrossRef]
- Eswarakumar, V.P.; Lax, I.; Schlessinger, J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005, 16, 139–149. [Google Scholar] [CrossRef]
- Parish, C.R.; Freeman, C.; Brown, K.J.; Francis, D.J.; Cowden, W.B. Identification of sulfated oligosaccharide-based inhibitors of tumor growth and metastasis using novel in vitro assays for angiogenesis and heparanase activity. Cancer Res. 1999, 59, 3433–3441. [Google Scholar]
- Ferro, V.; Liu, L.; Johnstone, K.D.; Wimmer, N.; Karoli, T.; Handley, P.; Rowley, J.; Dredge, K.; Li, C.P.; Hammond, E.; et al. Discovery of PG545: a highly potent and simultaneous inhibitor of angiogenesis, tumor growth, and metastasis. J. Med. Chem. 2012, 55, 3804–3813. [Google Scholar]
- Casu, B.; Naggi, A.; Torri, G. Heparin-derived heparan sulfate mimics to modulate heparan sulfate-protein interaction in inflammation and cancer. Matrix Biol. 2010, 29, 442–452. [Google Scholar] [CrossRef]
- Force, T.; Kolaja, K.L. Cardiotoxicity of kinase inhibitors: The prediction and translation of preclinical models to clinical outcomes. Nat. Rev. Drug Discov. 2011, 10, 111–126. [Google Scholar] [CrossRef]
- Angulo, J.; Ojeda, R.; de Paz, J.L.; Lucas, R.; Nieto, P.M.; Lozano, R.M.; Redondo-Horcajo, M.; Giménez-Gallego, G.; Martín-Lomas, M. The activation of fibroblast growth factors (FGFs) by glycosaminoglycans: influence of the sulfation pattern on the biological activity of FGF-1. ChemBioChem 2004, 5, 55–61. [Google Scholar] [CrossRef]
- Poletti, L.; Lay, L. Chemical contributions to understanding heparin activity: synthesis of related sulfated oligosaccharides. Eur. J. Org. Chem. 2003, 2999–3024. [Google Scholar] [CrossRef]
- O’Brien, A.; Lynch, C.; O’Boyle, K.M.; Murphy, P.V. Synthesis of disaccharides derived from heparin and evaluation of effects on endothelial cell growth and on binding of heparin to FGF-2. Carbohydr. Res. 2004, 339, 2343–2354. [Google Scholar]
- Canales, A.; Angulo, J.; Ojeda, R.; Bruix, M.; Fayos, R.; Lozano, R.; Gimenez-Gallego, G.; Martin-Lomas, M.; Nieto, P.M.; Jimenez-Barbero, J. Conformational flexibility of a synthetic glycosylaminoglycan bound to a fibroblast growth factor. FGF-1 recognizes both the (1)C(4) and (2)S(O) conformations of a bioactive heparin-like hexasaccharide. J. Am. Chem. Soc. 2005, 127, 5778–5779. [Google Scholar]
- Hung, S.C.; Lu, X.A.; Lee, J.C.; Chang, M.D.; Fang, S.L.; Fan, T.C.; Zulueta, M.M.; Zhong, Y.Q. Synthesis of heparin oligosaccharides and their interaction with eosinophil-derived neurotoxin. Org. Biomol. Chem. 2012, 10, 760–772. [Google Scholar] [CrossRef]
- Tiruchinapally, G.; Yin, Z.J.; El-Dakdouki, M.; Wang, Z.; Huang, X.F. Divergent heparin oligosaccharide synthesis with preinstalled sulfate esters. Chem.-Eur. J. 2011, 17, 10106–10112. [Google Scholar]
- Maza, S.; Macchione, G.; Ojeda, R.; Lopez-Prados, J.; Angulo, J.; de Paz, J.L.; Nieto, P.M. Synthesis of amine-functionalized heparin oligosaccharides for the investigation of carbohydrate-protein interactions in microtiter plates. Org. Biomol. Chem. 2012, 10, 2146–2163. [Google Scholar]
- Murphy, P.V.; Pitt, N.; O’Brien, A.; Enright, P.M.; Dunne, A.; Wilson, S.J.; Duane, R.M.; O’Boyle, K.M. Identification of novel inhibitors of fibroblast growth factor (FGF-2) binding to heparin and endothelial cell survival from a structurally diverse carbohybrid library. Bioorg. Med. Chem. Lett. 2002, 12, 3287–3290. [Google Scholar] [CrossRef]
- Liu, L.; Li, C.P.; Cochran, S.; Ferro, V. Application of the four-component Ugi condensation for the preparation of sulfated glycoconjugate libraries. Bioorg. Med. Chem. Lett. 2004, 14, 2221–2226. [Google Scholar]
- Zhang, J.; Riverst, G.; Zhu, Y.; Jacobson, A.; Peyers, J.; Grundstrom, G.; Burch, P.; Hussein, S.; Marolewski, A.; Herlihy, W.; Rusche, J. Identification of inhibitors of heparin-growth factor interactions from combinatorial libraries of four-component condensation reactions. Bioorg. Med. Chem. 2001, 9, 825–836. [Google Scholar]
- Liu, L.; Bytheway, I.; Karoli, T.; Fairweather, J.K.; Cochran, S.; Li, C.; Ferro, V. Design, synthesis, FGF-1 binding, and molecular modeling studies of conformationally flexible heparin mimetic disaccharides. Bioorg. Med. Chem. Lett. 2008, 18, 344–349. [Google Scholar] [CrossRef]
- Fairweather, J.K.; Karoli, T.; Liu, L.; Bytheway, I.; Ferro, V. Synthesis of a heparan sulfate mimetic disaccharide with a conformationally locked residue from a common intermediate. Carbohydr. Res. 2009, 344, 2394–2398. [Google Scholar] [Green Version]
- Pellegrini, L. Role of heparan sulfate in fibroblast growth factor signalling: A structural view. Curr. Opin. Struct. Biol. 2001, 11, 629–634. [Google Scholar] [CrossRef]
- Ferro, D.R.; Provasoli, A.; Ragazzi, M.; Torri, G.; Casu, B.; Gatti, G.; Jacquinet, J.C.; Sinaÿ, P.; Petitou, M.; Choay, J. Evidence for conformational equilibrium of the sulfated L-iduronate residue in heparin and in synthetic heparin mono- and oligosaccharides: NMR and force-field studies. J. Am. Chem. Soc. 1986, 108, 6773–6778. [Google Scholar]
- Ferro, D.R.; Provasoli, A.; Ragazzi, M.; Casu, B.; Torri, G.; Bossennec, V.; Perly, B.; Sinaÿ, P.; Petitou, M.; Choay, J. Conformer populations of L-iduronic acid residues in glycosaminoglycan sequences. Carbohydr. Res. 1990, 195, 157–167. [Google Scholar] [CrossRef]
- Probst, K.C.; Wessel, H.P. Synthesis and conformational investigations of sulfated carbohydrates. J. Carbohydr. Chem. 2001, 20, 549–560. [Google Scholar]
- Wessel, H.P.; Bartsch, S. Conformational flexibility in highly sulfated -β-D-glucopyranoside derivatives. Carbohydr. Res. 1995, 274, 1–9. [Google Scholar] [CrossRef]
- DiGabriele, A.D.; Lax, I.; Chen, D.I.; Svahn, C.M.; Jaye, M.; Schlessinger, J.; Hendrickson, W.A. Structure of a heparin-linked biologically active dimer of fibroblast growth factor. Nature 1998, 393, 812–817. [Google Scholar]
- Faham, S.; Hileman, R.E.; Fromm, J.R.; Linhardt, R.J.; Rees, D.C. Heparin structure and interactions with basic fibroblast growth factor. Science 1996, 271, 1116–1120. [Google Scholar]
- Clode, D.M.; Horton, D.; Weckerle, W. Reaction of derivatives of methyl 2,3-O-benzylidene-6-deoxy-α-L-mannopyranoside with butyllithium: Synthesis of methyl 2,6-dideoxy-4-O-methyl-α-L-erythro-hexopyranosid-3-ulose. Carbohydr. Res. 1976, 49, 305–314. [Google Scholar] [CrossRef]
- Petit, E.; Papy-Garcia, D.; Muller, G.; Courtois, B.; Caruelle, J.-P.; Courtois, J. Controlled sulfatation of natural anionic bacterial polysaccharides can yield agents with specific regenerating activity in vivo. Biomacromolecules 2004, 5, 445–452. [Google Scholar]
- Cui, J.-F.; Byström, S.; Eneroth, P.; Björkhem, I. Novel mechanism for oxidative cleavage of glycosidic bonds: evidence for an oxygen dependent reaction. J. Org. Chem. 1994, 59, 8251–8255. [Google Scholar]
- Lai, Y.Z. Kinetic evidence of anionic intermediates in the base-catalyzed cleavage of glycosidic bonds in the methyl D-glucopyranosides. Carbohydr. Res. 1972, 24, 57–65. [Google Scholar] [CrossRef]
- Orgueira, H.A.; Bartolozzi, A.; Schell, P.; Litjens, R.E.J.N.; Palmacci, E.R.; Seeberger, P.H. Modular synthesis of heparin oligosaccharides. Chem. Eur. J. 2003, 9, 140–169. [Google Scholar] [CrossRef]
- Farndale, R.W.; Buttle, D.J.; Barrett, A.J. Improved quantitation and discrimination of sulphated glycosaminoglycans by use of dimethylmethylene blue. Biochim. Biophys. Acta 1986, 883, 173–177. [Google Scholar] [CrossRef]
- Yu, G.; Gunay, N.S.; Linhardt, R.J.; Toida, T.; Fareed, J.; Hoppensteadt, D.A.; Shadid, H.; Ferro, V.; Li, C.; Fewings, K.; et al. Preparation and anticoagulant activity of the phosphosulfomannan PI-88. Eur. J. Med. Chem. 2002, 37, 783–791. [Google Scholar]
- Pozsgay, V.; Trinh, L.; Shiloach, J.; Robbins, J.B.; Donohue-Rolfe, A.; Calderwood, S.B. Purification of subunit B of Shiga toxin using a synthetic trisaccharide-based affinity matrix. Bioconjug. Chem. 1996, 7, 45–55. [Google Scholar]
- Lipták, A.; Imre, J.; Nánási, P. Preparation of carbohydrate isopropylidene derivatives with 2,2-dimethoxypropane in the presence of toluene-p-sulphonic acid. Carbohydr. Res. 1981, 92, 154–156. [Google Scholar] [CrossRef]
- Bajza, I.; Kövér, K.E.; Lipták, A. Synthesis of p-trifluoroacetamidophenyl (4,6-dideoxy-4-formamido-3-C-methyl-2-O-methyl-α-L-mannopyranosyl)- (1→3)-(2-O-methyl-α-D-rhamnopyranosyl)-(1→3)- (2-O-methyl-α-L-fucopyranosyl)-(1→3)-(α-L-rhamnopyranosyl)- (1→2)-6-deoxy-α-L-talopyranoside: A spacer-armed pentasaccharide glycopeptidolipid antigen of Mycobacterium avium serovar 14. Carbohydr. Res. 1998, 308, 247–258. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fairweather, J.K.; Liu, L.; Karoli, T.; Ferro, V. Synthesis of Disaccharides Containing 6-Deoxy-a-L-talose as Potential Heparan Sulfate Mimetics. Molecules 2012, 17, 9790-9802. https://doi.org/10.3390/molecules17089790
Fairweather JK, Liu L, Karoli T, Ferro V. Synthesis of Disaccharides Containing 6-Deoxy-a-L-talose as Potential Heparan Sulfate Mimetics. Molecules. 2012; 17(8):9790-9802. https://doi.org/10.3390/molecules17089790
Chicago/Turabian StyleFairweather, Jon K., Ligong Liu, Tomislav Karoli, and Vito Ferro. 2012. "Synthesis of Disaccharides Containing 6-Deoxy-a-L-talose as Potential Heparan Sulfate Mimetics" Molecules 17, no. 8: 9790-9802. https://doi.org/10.3390/molecules17089790