Diversity of Phage-Displayed Libraries of Peptides during Panning and Amplification

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction—Motivation for Writing This Review
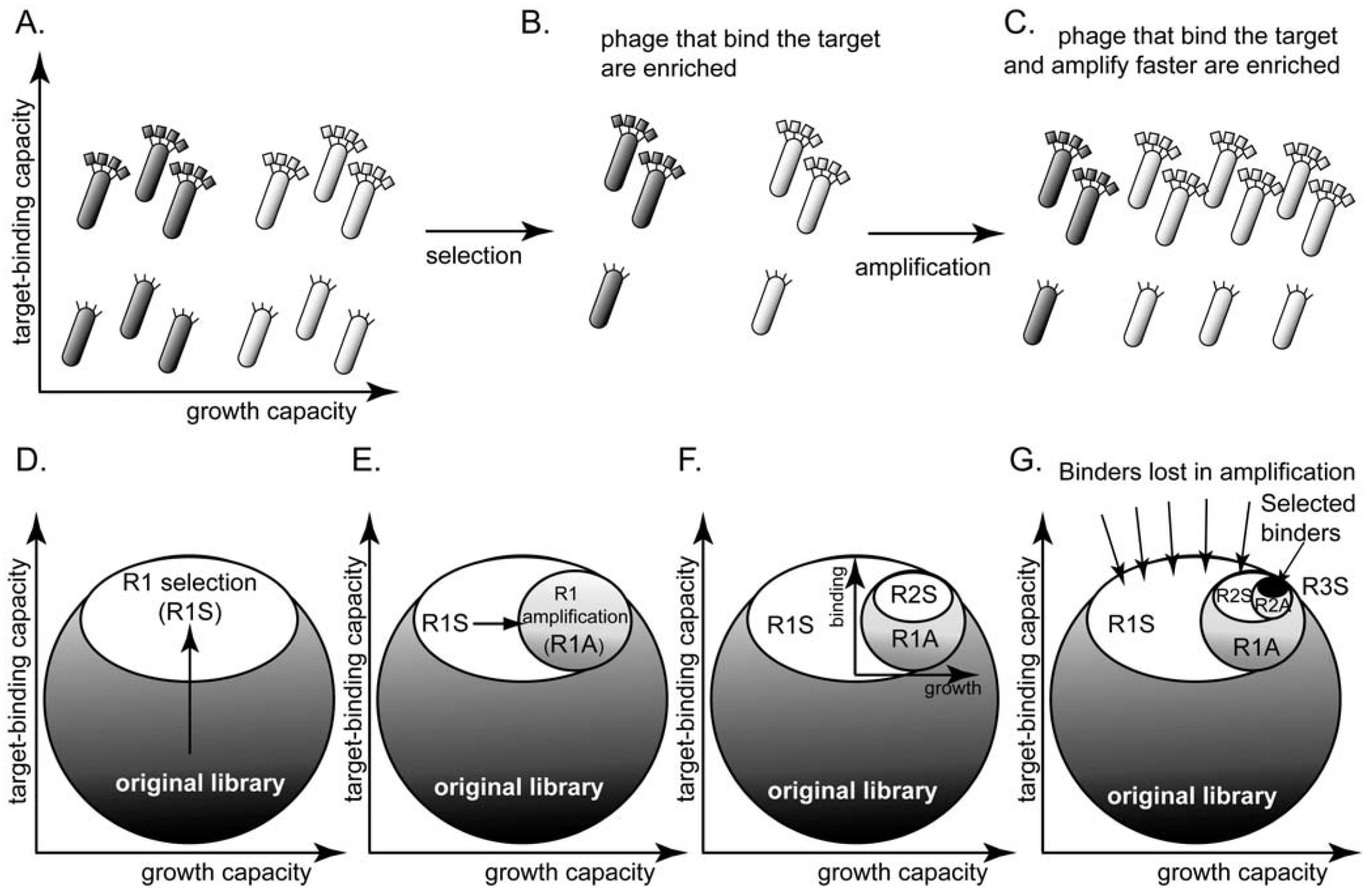
2. The Problem: Panning vs. Rate of Amplification

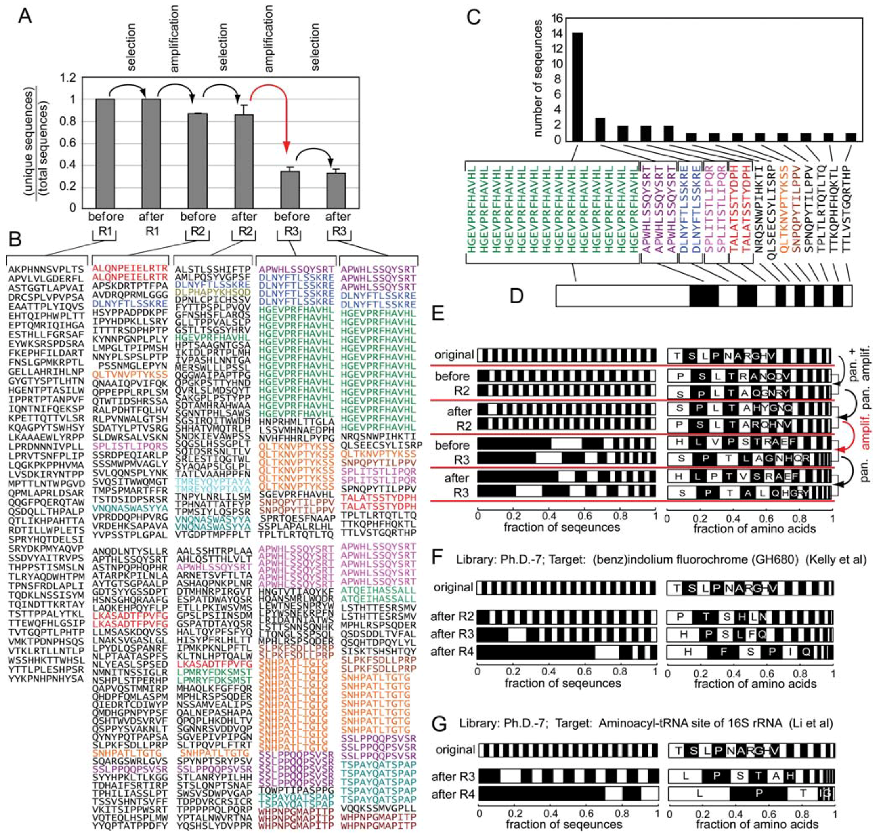
3. Evidence for Amplification-Induced Convergence Based on Comparison of Sequence Diversity at Every Step of the Selection Process

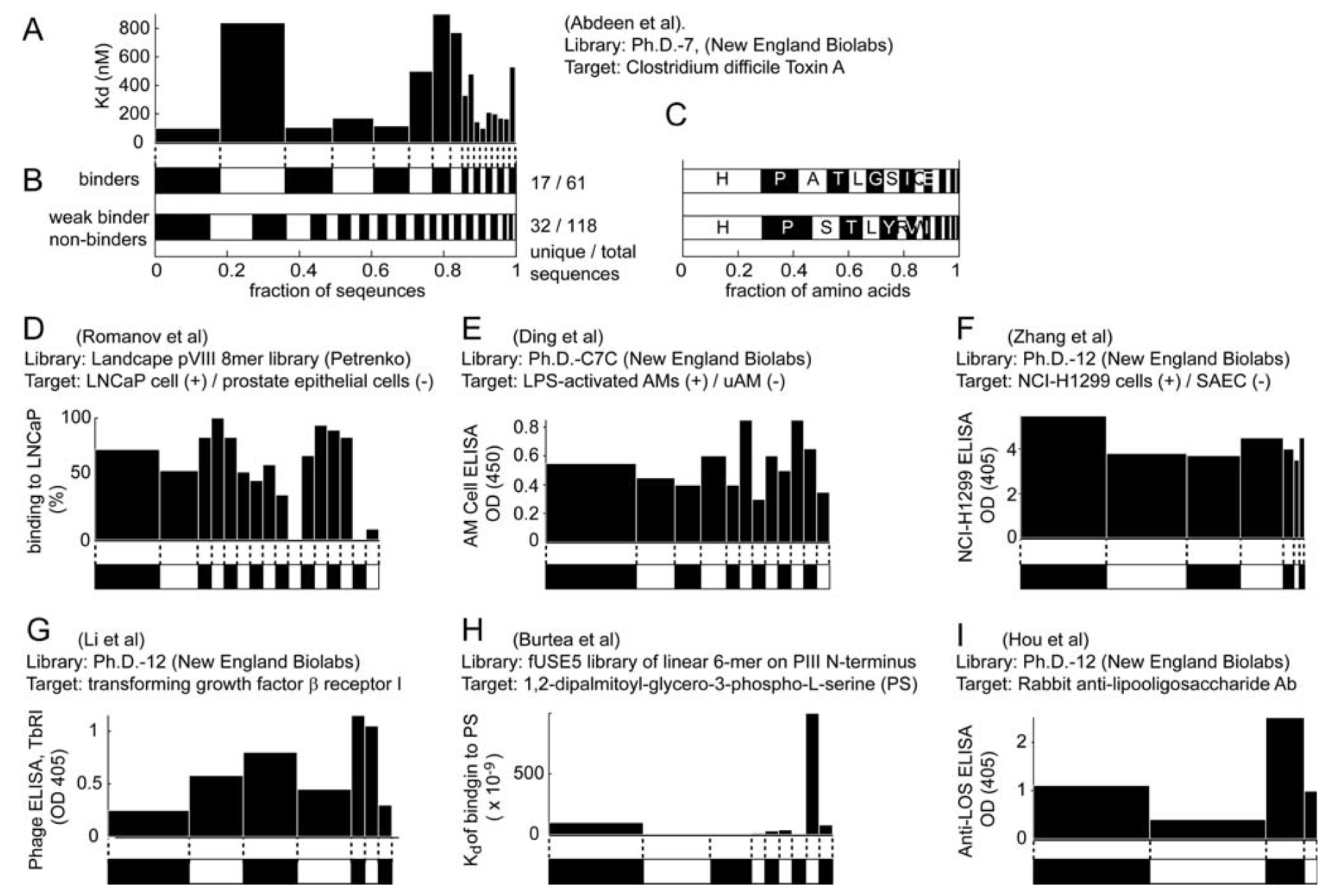
4. Evidence for Amplification-Induced Convergence Based on Comparison of the Diversity of Identified Ligands for Targets with One, Few, Or Many Binding Sites


5. The Relative Abundances of Ligands in the Library Are Not Correlated to Their Binding Strength

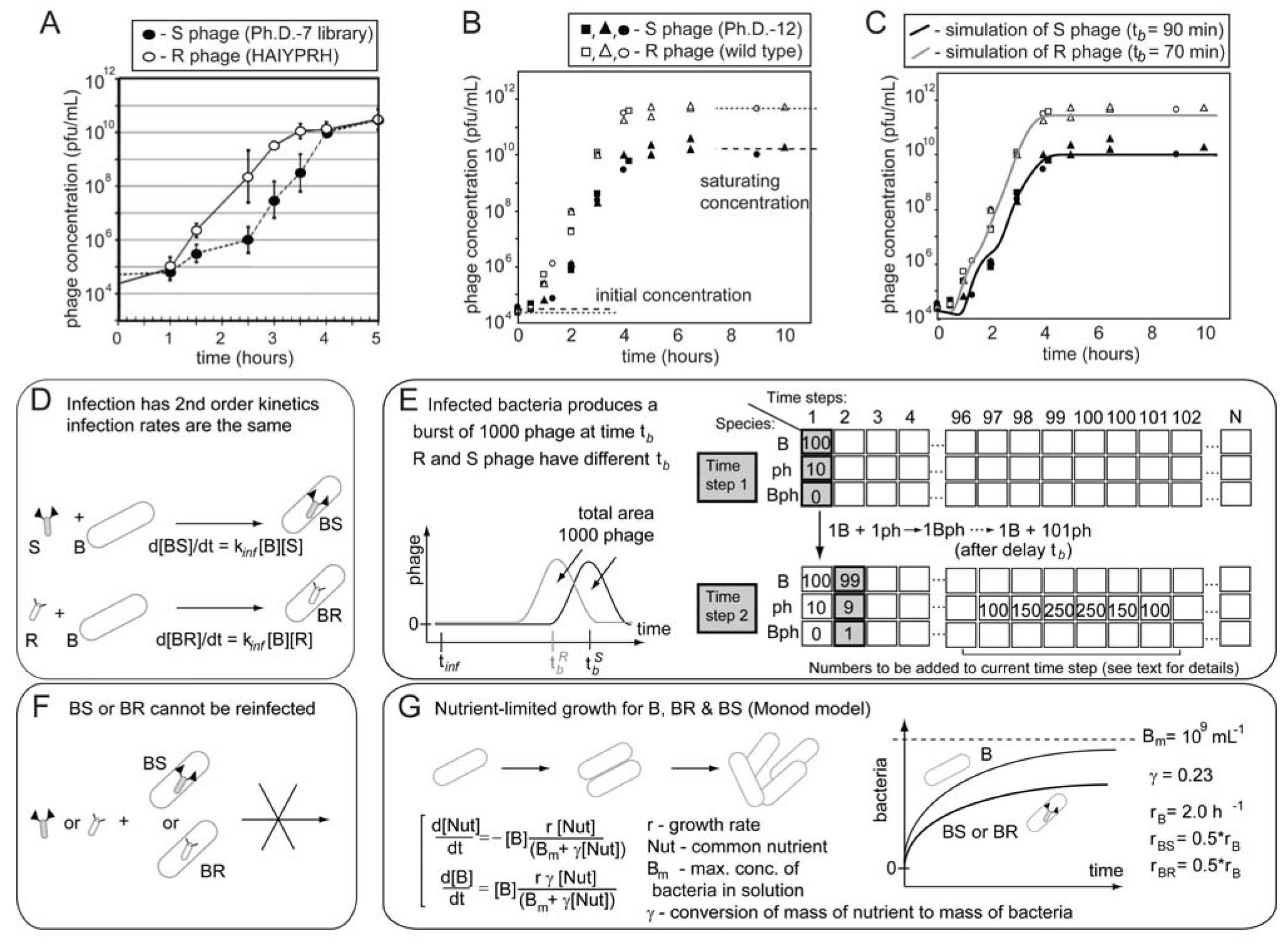
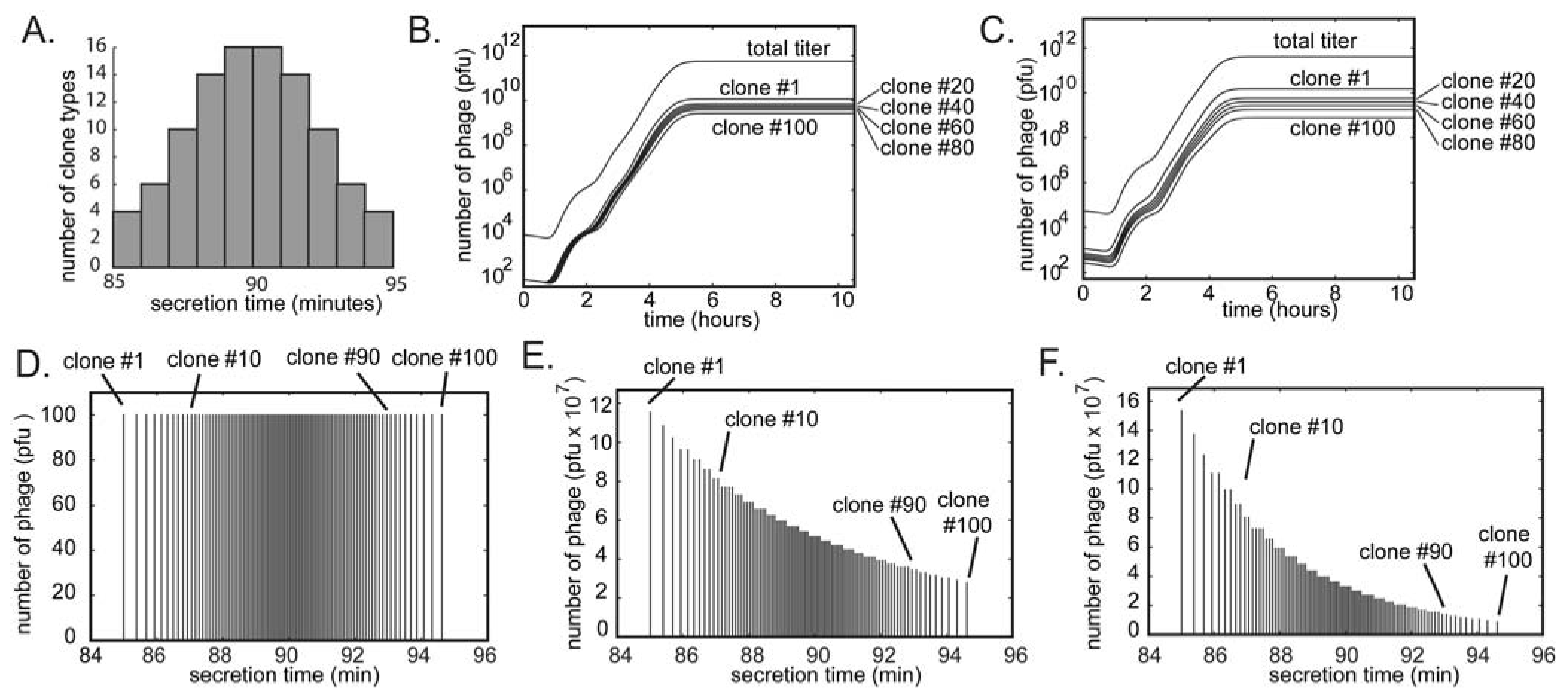
6. Subtle Differences in Growth Rate Yield Drastic Differences in Clone Abundances after Rounds of Amplifications
 103 106 109 upon serial infection in an excess of uninfected bacteria. We used a discrete-step model of phage growth to visualize how small differences in growth rate lead to large differences in the number of phage produced after multiple cycles of re-infection. The model used four parameters: (1) infection rate was described using a simple 2nd order kinetics with respect to concentration of phage and bacteria (Figure 6D); (2) delayed burst of 1,000 phage particles (Figure 6E) [36]; (3) no re-infection of bacteria that were already infected (Figure 6F) [67]; and (4) substrate-limited growth for infected and non-infected bacteria (Figure 6G) [36,68]. The model fits well with the data in Figure 6B and we can therefore attribute the 1:1 to 1:300 increase in R/S ratio to ~30% difference in growth rates of R and S phage (Figure 6C).
103 106 109 upon serial infection in an excess of uninfected bacteria. We used a discrete-step model of phage growth to visualize how small differences in growth rate lead to large differences in the number of phage produced after multiple cycles of re-infection. The model used four parameters: (1) infection rate was described using a simple 2nd order kinetics with respect to concentration of phage and bacteria (Figure 6D); (2) delayed burst of 1,000 phage particles (Figure 6E) [36]; (3) no re-infection of bacteria that were already infected (Figure 6F) [67]; and (4) substrate-limited growth for infected and non-infected bacteria (Figure 6G) [36,68]. The model fits well with the data in Figure 6B and we can therefore attribute the 1:1 to 1:300 increase in R/S ratio to ~30% difference in growth rates of R and S phage (Figure 6C).

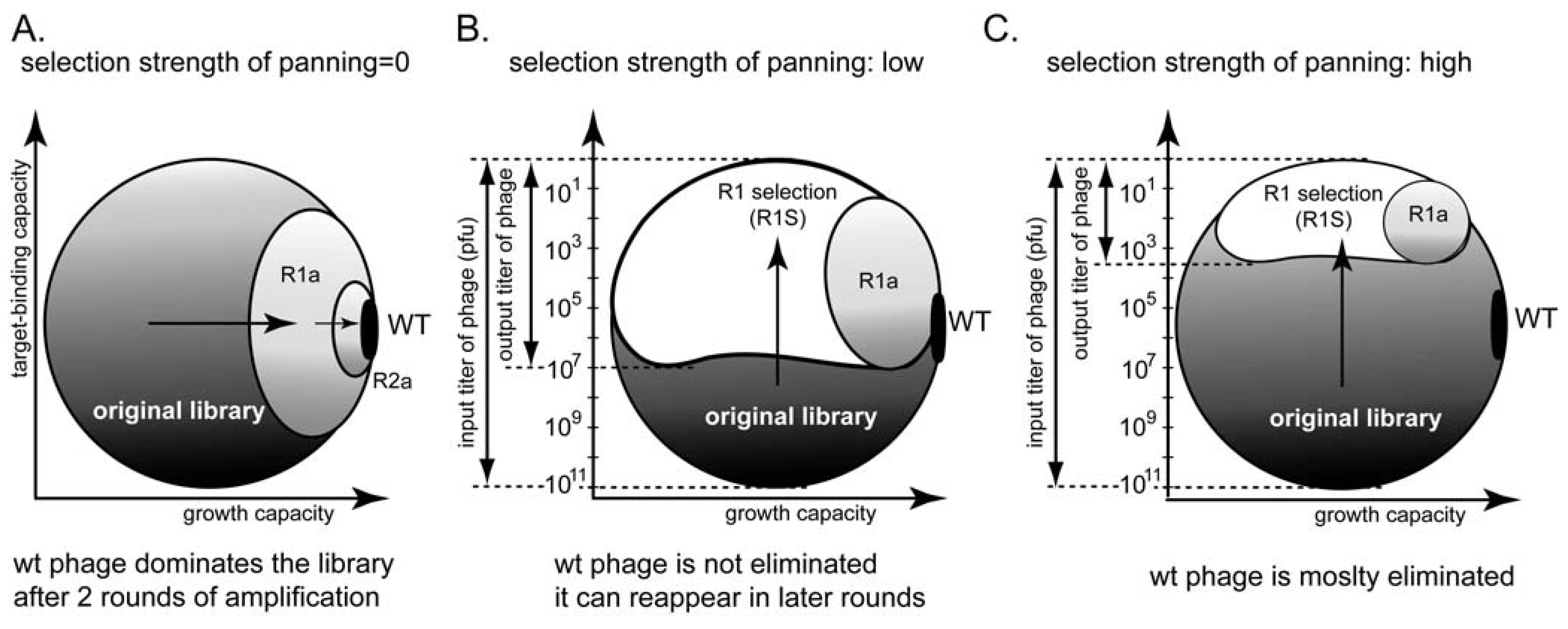
7. Relationships between Panning and Amplification

8. How Many Binding Ligands Are Lost in the Screen?
9. Mitigating Amplification-Induced Convergence: Screens without Amplification
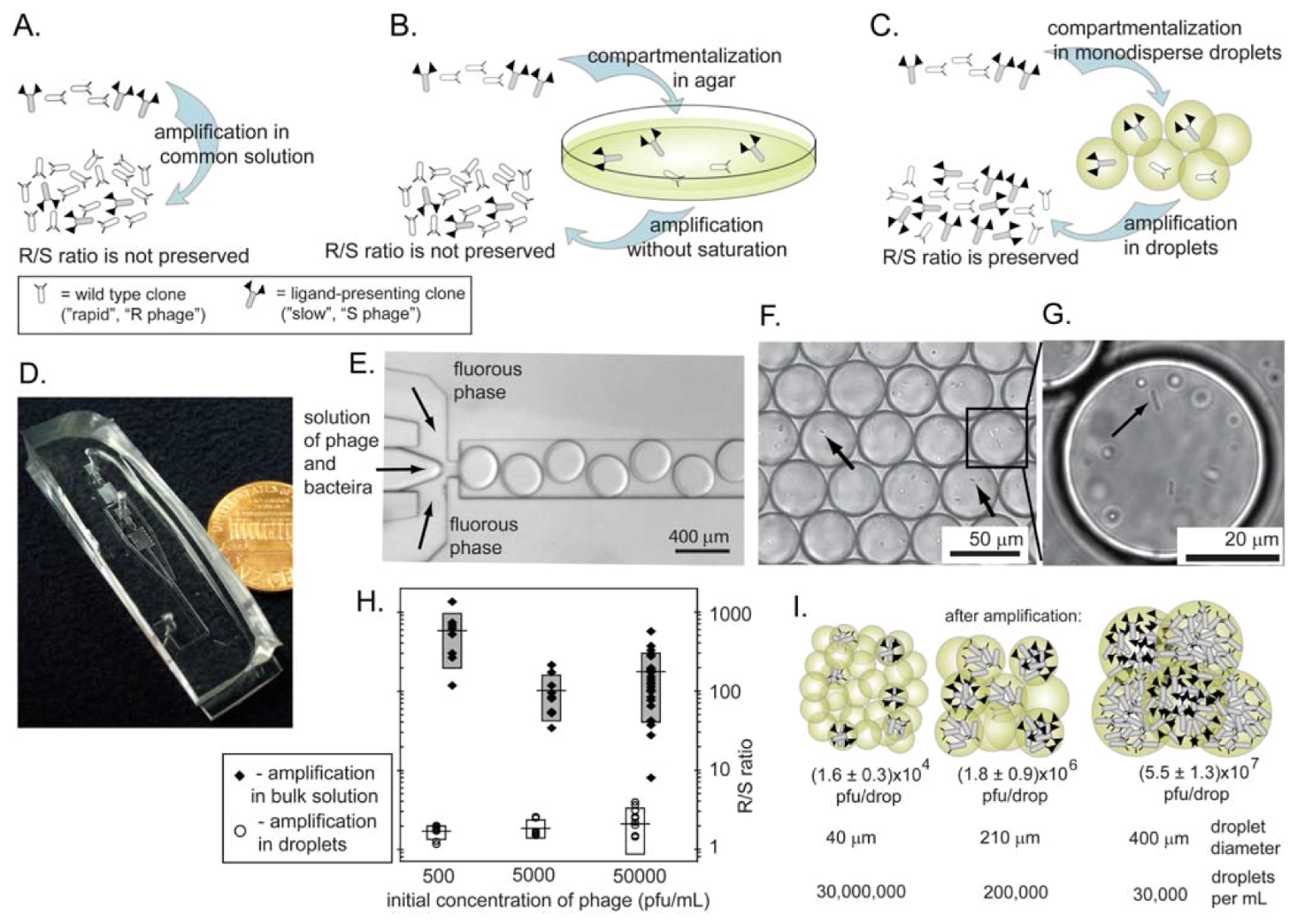
10. Mitigating Amplification-Induced Convergence: Amplification in Isolated Compartments
10.1. Agarose plates as isolated compartments
10.2. Monodisperse droplets as isolated compartments

11. Indirect Mitigation of Amplification-Induced Convergence Using Bioinformatics Analysis
12. Indirect Mitigation of Amplification-Induced Convergence Using Databases
13. Loss of Diversity in Other Display Systems
14. Outlook
Acknowledgements
References and Notes
- Devlin, J.J.; Panganiban, L.C.; Devlin, P.E. Random Peptide Libraries - a Source of Specific Protein-Binding Molecules. Science 1990, 249, 404–406. [Google Scholar]
- McCafferty, J.; Griffiths, A.D.; Winter, G.; Chiswell, D.J. Phage Antibodies - Filamentous Phage Displaying Antibody Variable Domains. Nature 1990, 348, 552–554. [Google Scholar]
- Barbas, C.F.; Bain, J.D.; Hoekstra, D.M.; Lerner, R.A. Semisynthetic Combinatorial Antibody Libraries - a Chemical Solution to the Diversity Problem. Proc. Natl. Acad. Sci. USA 1992, 89, 4457–4461. [Google Scholar] [CrossRef]
- Barbas, C.F.; Kang, A.S.; Lerner, R.A.; Benkovic, S.J. Assembly of Combinatorial Antibody Libraries on Phage Surfaces - the Gene-Iii Site. Proc. Natl. Acad. Sci. USA 1991, 88, 7978–7982. [Google Scholar] [CrossRef]
- Smith, G.P. Filamentous Fusion Phage - Novel Expression Vectors That Display Cloned Antigens on the Virion Surface. Science 1985, 228, 1315–1317. [Google Scholar]
- Smith, G.P.; Petrenko, V.A. Phage display. Chem. Rev. 1997, 97, 391–410. [Google Scholar]
- Scott, J.K.; Smith, G.P. Searching for Peptide Ligands with an Epitope Library. Science 1990, 249, 386–390. [Google Scholar]
- Lam, K.S.; Salmon, S.E.; Hersh, E.M.; Hruby, V.J.; Kazmierski, W.M.; Knapp, R.J. A New Type of Synthetic Peptide Library for Identifying Ligand-Binding Activity. Nature 1991, 354, 82–84. [Google Scholar]
- Blackwell, H.E. Hitting the SPOT: small-molecule macroarrays advance combinatorial synthesis. Curr. Opin. Chem. Biol. 2006, 10, 203–212. [Google Scholar] [CrossRef]
- Thompson, L.A.; Ellman, J.A. Synthesis and applications of small molecule libraries. Chem. Rev. 1996, 96, 555–600. [Google Scholar]
- Schreiber, S.L. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science 2000, 287, 1964–1969. [Google Scholar] [CrossRef]
- Moran, E.J.; Sarshar, S.; Cargill, J.F.; Shahbaz, M.M.; Lio, A.; Mjalli, A.M.M.; Armstrong, R.W. Radio-Frequency Tag Encoded Combinatorial Library Method for the Discovery of Tripeptide-Substituted Cinnamic Acid Inhibitors of the Protein-Tyrosine-Phosphatase Ptp1b. J. Am. Chem. Soc. 1995, 117, 10787–10788. [Google Scholar]
- Vaino, A.R.; Janda, K.D. Euclidean shape-encoded combinatorial chemical libraries. Proc. Natl. Acad. Sci. USA 2000, 97, 7692–7696. [Google Scholar] [CrossRef]
- Kehoe, J.W.; Kay, B.K. Filamentous phage display in the new millennium. Chem. Rev. 2005, 105, 4056–4072. [Google Scholar] [CrossRef]
- Funke, S.A.; Willbold, D. Mirror image phage display-a method to generate D-peptide ligands for use in diagnostic or therapeutical applications. Mol. Biosyst. 2009, 5, 783–786. [Google Scholar] [CrossRef]
- Deutscher, S.L. Phage Display in Molecular Imaging and Diagnosis of Cancer. Chem. Rev. 2010, 110, 3196–3211. [Google Scholar] [CrossRef]
- Kather, I.; Bippes, C.A.; Schmid, F.X. A stable disulfide-free gene-3-protein of phage fd generated by in vitro evolution. J. Mol. Biol. 2005, 354, 666–678. [Google Scholar]
- Dunn, I.S. Phage display of proteins. Curr. Opin. Biotechnol. 1996, 7, 547–553. [Google Scholar] [CrossRef]
- Baneyx, F.; Schwartz, D.T. Selection and analysis of solid-binding peptides. Curr. Opin. Biotechnol. 2007, 18, 312–317. [Google Scholar] [CrossRef]
- Kriplani, U.; Kay, B.K. Selecting peptides for use in nanoscale materials using phagedisplayed combinatorial peptide libraries. Curr. Opin. Biotechnol. 2005, 16, 470–475. [Google Scholar] [CrossRef]
- Kay, B.K.; Kasanov, J.; Knight, S.; Kurakin, A. Convergent evolution with combinatorial peptides. FEBS Lett. 2000, 480, 55–62. [Google Scholar] [CrossRef]
- Kuzmicheva, G.A.; Jayanna, P.K.; Sorokulova, I.B.; Petrenko, V.A. Diversity and censoring of landscape phage libraries. Protein Eng. Des. Sel. 2009, 22, 9–18. [Google Scholar]
- Derda, R.; Musah, S.; Orner, B.P.; Klim, J.R.; Li, L.Y.; Kiessling, L.L. High-Throughput Discovery of Synthetic Surfaces That Support Proliferation of Pluripotent Cells. J. Am. Chem. Soc. 2010, 132, 1289–1295. [Google Scholar]
- Rodi, D.J.; Soares, A.S.; Makowski, L. Quantitative assessment of peptide sequence diversity in M13 combinatorial peptide phage display libraries. J. Mol. Biol. 2002, 322, 1039–1052. [Google Scholar] [CrossRef]
- Iannolo, G.; Minenkova, O.; Gonfloni, S.; Castagnoli, L.; Cesareni, G. Construction, exploitation and evolution of a new peptide library displayed at high density by fusion to the major coat protein of filamentous phage. Biol. Chem. 1997, 378, 517–521. [Google Scholar] [CrossRef]
- Breitling, F.; Dubel, S.; Seehaus, T.; Klewinghaus, I.; Little, M. A Surface Expression Vector for Antibody Screening. Gene 1991, 104, 147–153. [Google Scholar] [CrossRef]
- Bass, S.; Greene, R.; Wells, J.A. Hormone Phage - an Enrichment Method for Variant Proteins with Altered Binding-Properties. Protein.-Struct. Funct. Genet. 1990, 8, 309–314. [Google Scholar] [CrossRef]
- Marks, J.D.; Hoogenboom, H.R.; Bonnert, T.P.; McCafferty, J.; Griffiths, A.D.; Winter, G. By-Passing Immunization - Human-Antibodies from V-Gene Libraries Displayed on Phage. J. Mol. Biol. 1991, 222, 581–597. [Google Scholar] [CrossRef]
- Barbas, C.F.; Lerner, R.A. Combinatorial immunoglobulin libraries on the surface of phage: Rapid selection of antigen-specific Fabs. Methods Companion Methods Enzymol. 1991, 2, 119–124. [Google Scholar] [CrossRef]
- Rader, C.; Barbas, C.F. Phage display of combinatorial antibody libraries. Curr. Opin. Biotechnol. 1997, 8, 503–508. [Google Scholar] [CrossRef]
- Lofblom, J.; Feldwisch, J.; Tolmachev, V.; Carlsson, J.; Stahl, S.; Frejd, F.Y. Affibody molecules: Engineered proteins for therapeutic, diagnostic and biotechnological applications. FEBS Lett. 2010, 584, 2670–2680. [Google Scholar] [CrossRef]
- Sidhu, S.S.; Koide, S. Phage display for engineering and analyzing protein interaction interfaces. Curr. Opin. Struct. Biol. 2007, 17, 481–487. [Google Scholar] [CrossRef]
- Rebar, E.J.; Pabo, C.O. Zinc-Finger Phage - Affinity Selection of Fingers with New DNA-Binding Specificities. Science 1994, 263, 671–673. [Google Scholar]
- Adey, N.B.; Mataragnon, A.H.; Rider, J.E.; Carter, J.M.; Kay, B.K. Characterization of Phage That Bind Plastic from Phage-Displayed Random Peptide Libraries. Gene 1995, 156, 27–31. [Google Scholar] [CrossRef]
- Menendez, A.; Scott, J.K. The nature of target-unrelated peptides recovered in the screening of phage-displayed random peptide libraries with antibodies. Anal. Biochem. 2005, 336, 145–157. [Google Scholar]
- Barbas, C.F.I.; Burton, D.R.; Scott, J.; Silverman, G.J. Phage Display: A Laboratory Manual; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001; pp. 1.2–1.36. [Google Scholar]
- Rodi, D.J.; Makowski, L. In Structure and Function of Macromolecular Assembly; Namba, K., Ed.; Matsushita Electric Industrial Co.Ltd: Matsushita, Japan, 1997; pp. 155–164. [Google Scholar]
- Thomas, W.D.; Golomb, M.; Smith, G.P. Corruption of phage display libraries by target-unrelated clones: Diagnosis and countermeasures. Anal. Biochem. 2010, 407, 237–240. [Google Scholar]
- Brammer, L.A.; Bolduc, B.; Kass, J.L.; Felice, K.M.; Noren, C.J.; Hall, M.F. A target-unrelated peptide in an M13 phage display library traced to an advantageous mutation in the gene II ribosome-binding site. Anal. Biochem. 2008, 373, 88–98. [Google Scholar]
- Mandecki, W.; Chen, Y.C.J.; Grihalde, N. A Mathematical-Model for Biopanning (Affinity Selection) Using Peptide Libraries on Filamentous Phage. J. Theor. Biol. 1995, 176, 523–530. [Google Scholar] [CrossRef]
- Zhuang, G.Q.; Katakura, Y.; Furuta, T.; Omasa, T.; Kishimoto, M.; Suga, K. A kinetic model for a biopanning process considering antigen desorption and effective antigen concentration on a solid phase. J. Biosci. Bioeng. 2001, 91, 474–481. [Google Scholar]
- Levitan, B. Stochastic modeling and optimization of phage display. J. Mol. Biol. 1998, 277, 893–916. [Google Scholar] [CrossRef]
- Zhao, S.J.; Zhao, W.X.; Ma, L. Novel peptide ligands that bind specifically to mouse embryonic stem cells. Peptides 2010, 31, 2027–2034. [Google Scholar] [CrossRef]
- Lu, S.; Xu, X.; Zhao, W.; Wu, W.; Yuan, H.; Shen, H.; Zhou, C.; Li, L.S.; Ma, L. Targeting of embryonic stem cells by peptide-conjugated quantum dots. PLoS One 2010, 5, e12075(1)–e12075(10). [Google Scholar]
- Zhao, W.X.; Yuan, H.; Xu, X.; Ma, L. Isolation and Initial Application of a Novel Peptide That Specifically Recognizes the Neural Stem Cells Derived from Rhesus Monkey Embryonic Stem Cells. J. Biomol. Screen 2010, 15, 687–694. [Google Scholar] [CrossRef]
- Kelly, K.A.; Carson, J.; McCarthy, J.R.; Weissleder, R. Novel Peptide Sequence ("IQ-tag'') with High Affinity for NIR Fluorochromes Allows Protein and Cell Specific Labeling for In Vivo Imaging. PLoS One 2007, 2, e665(1)–e665(9). [Google Scholar]
- Li, M.; Duc, A.C.E.; Klosi, E.; Pattabiraman, S.; Spaller, M.R.; Chow, C.S. Selection of Peptides That Target the Aminoacyl-tRNA Site of Bacterial 16S Ribosomal RNA. Biochemistry 2009, 48, 8299–8311. [Google Scholar]
- Zimmermann, B.; Gesell, T.; Chen, D.; Lorenz, C.; Schroeder, R. Monitoring Genomic Sequences during SELEX Using High-Throughput Sequencing: Neutral SELEX. PLoS One 2010, 5, e9169(1)–e9169(7). [Google Scholar]
- Ru, B.; Huang, J.; Dai, P.; Li, S.; Xia, Z.; Ding, H.; Lin, H.; Guo, F.; Wang, X. MimoDB: a new repository for mimotope data derived from phage display technology. Molecules 2010, 15, 8279–8288. [Google Scholar]
- Abdeen, S.J.; Swett, R.J.; Feig, A.L. Peptide Inhibitors Targeting Clostridium difficile Toxins A and B. ACS Chem. Biol. 2010, 5, 1097–1103. [Google Scholar] [CrossRef]
- Kini, R.M.; Evans, H.J. A Hypothetical Structural Role for Proline Residues in the Flanking Segments of Protein-Protein Interaction Sites. Biochem. Biophys. Res. Commun. 1995, 212, 1115–1124. [Google Scholar] [CrossRef]
- Kay, B.K.; Williamson, M.P.; Sudol, P. The importance of being proline: the interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J. 2000, 14, 231–241. [Google Scholar]
- Romanov, V.I.; Durand, D.B.; Petrenko, V.A. Phage display selection of peptides that affect prostate carcinoma cells attachment and invasion. Prostate 2001, 47, 239–251. [Google Scholar] [CrossRef]
- Hou, Y.C.; Gu, X.X. Development of peptide mimotopes of lipooligosaccharide from nontypeable Haemophilus influenzae as vaccine candidates. J. Immunol. 2003, 170, 4373–4379. [Google Scholar]
- Li, L.Y.; Orner, B.P.; Huang, T.; Hinck, A.P.; Kiessling, L.L. Peptide ligands that use a novel binding site to target both TGF-beta receptors. Mol. Biosyst. 2010, 6, 2392–2402. [Google Scholar] [CrossRef]
- Zang, L.Q.; Shi, L.; Guo, J.; Pan, Q.; Wu, W.; Pan, X.D.; Wang, J.Y. Screening and Identification of a peptide specifically targeted to NCI-H1299 from a phage display peptide library. Cancer Lett. 2009, 281, 64–70. [Google Scholar] [CrossRef]
- Ding, N.; Xiao, H.; Wang, F.; Xu, L.X.; She, S.Z. A FQHPSFI peptide selectively binds to LPS-activated alveolar macrophages and inhibits LPS-induced MIP-2 production. Inflamm. Res. 2010, 59, 627–634. [Google Scholar] [CrossRef]
- Burtea, C.; Laurent, S.; Lancelot, E.; Ballet, S.; Murariu, O.; Rousseaux, O.; Port, M.; Elst, L.V.; Corot, C.; Muller, R.N. Peptidic Targeting of Phosphatidylserine for the MRI Detection of Apoptosis in Atherosclerotic Plaques. Mol. Pharm. 2009, 6, 1903–1919. [Google Scholar] [CrossRef]
- Iannolo, G.; Minenkova, O.; Petruzzelli, R.; Cesareni, G. Modifying Filamentous Phage Capsid - Limits in the Size of the Major Capsid Protein. J. Mol. Biol. 1995, 248, 835–844. [Google Scholar] [CrossRef]
- Legendre, D.; Fastrez, J. Construction and exploitation in model experiments of functional selection of a landscape library expressed from a phagemid. Gene 2002, 290, 203–215. [Google Scholar] [CrossRef]
- Petrenko, V.A.; Smith, G.P.; Gong, X.; Quinn, T. A library of organic landscapes on filamentous phage. Protein Eng. 1996, 9, 797–801. [Google Scholar] [CrossRef]
- Li, Z.P.; Koch, H.; Dubel, S. Mutations in the N-terminus of the major coat protein (pVIII, gp8) of filamentous bacteriophage affect infectivity. J. Mol. Microbiol. Biotechnol. 2003, 6, 57–66. [Google Scholar] [CrossRef]
- Petrenko, V.A.; Smith, G.P.; Mazooji, M.M.; Quinn, T. alpha-helically constrained phage display library. Protein Eng. 2002, 15, 943–950. [Google Scholar] [CrossRef]
- Held, H.A.; Sidhu, S.S. Comprehensive mutational analysis of the M13 major coat protein: Improved scaffolds for C-terminal phage display. J. Mol. Biol. 2004, 340, 587–597. [Google Scholar] [CrossRef]
- Roth, T.A.; Weiss, G.A.; Eigenbrot, C.; Sidhu, S.S. A minimized M13 coat protein defines the requirements for assembly into the bacteriophage particle. J. Mol. Biol. 2002, 322, 357–367. [Google Scholar] [CrossRef]
- Derda, R.; Tang, S.K.Y.; Whitesides, G.M. Uniform Amplification of Phage with Different Growth Characteristics in Individual Compartments Consisting of Monodisperse Droplets. Angew. Chem.-Int. Edit. 2010, 49, 5301–5304. [Google Scholar]
- Marvin, D.A.; Hohn, B. Filamentous Bacterial Viruses. Bacteriol. Rev. 1969, 33, 172–209. [Google Scholar]
- Ray, D.S.; Bscheide, H.P.; Hofschne, P.H. Replication of Single-Stranded DNA of Male-Specific Bacteriophage M13. J. Mol. Biol. 1966, 21, 473–483. [Google Scholar] [CrossRef]
- Malik, P.; Tarry, T.D.; Gowda, L.R.; Langara, A.; Petukhov, S.A.; Symmons, M.F.; Welsh, L.C.; Marvin, D.A.; Perham, R.N. Role of capsid structure and membrane protein processing in determining the size and copy number of peptides displayed on the major coat protein of filamentous bacteriophage. J. Mol. Biol. 1996, 260, 9–21. [Google Scholar] [CrossRef]
- Rodi, D.J.; Makowski, L.; Kay, B.K. One from column A and two from column B: the benefits of phage display in molecular-recognition studies. Curr. Opin. Chem. Biol. 2002, 6, 92–96. [Google Scholar] [CrossRef]
- A 5-mer of defined sequence ABCDE can be shifted 8 times in a random 12mer X12 (ABCDE-X7, X-ABCDE-X6, X2-ABCDE-X5, …, X7-ABCDE). The number of ABCDE-X7 sequences is (20)^7. The total number, thus, is 8x(20)^7 = 10^10.
- Montgomery-Smith, S.J.; Schmidt, F.J. Statistical methods for estimating complexity from competition experiments between two populations. J. Theor. Biol. 2010, 264, 1043–1046. [Google Scholar] [CrossRef]
- Birtalan, S.; Fisher, R.D.; Sidhu, S.S. The functional capacity of the natural amino acids for molecular recognition. Mol. Biosyst. 2010, 6, 1186–1194. [Google Scholar] [CrossRef]
- Koide, S.; Sidhu, S.S. The Importance of Being Tyrosine: Lessons in Molecular Recognition from Minimalist Synthetic Binding Proteins. ACS Chem. Biol. 2009, 4, 325–334. [Google Scholar] [CrossRef]
- Liu, J.W.; Cao, Z.H.; Lu, Y. Functional Nucleic Acid Sensors. Chem. Rev. 2009, 109, 1948–1998. [Google Scholar] [CrossRef]
- Famulok, M.; Hartig, J.S.; Mayer, G. Functional aptamers and aptazymes in biotechnology, diagnostics, and therapy. Chem. Rev. 2007, 107, 3715–3743. [Google Scholar] [CrossRef]
- Wulff, G. Enzyme-like catalysis by molecularly imprinted polymers. Chem. Rev. 2002, 102, 1–27. [Google Scholar] [CrossRef]
- Williams, B.R.; Sharon, J. Polyclonal anti-colorectal cancer Fab phage display library selected in one round using density gradient: centrifugation to separate antigen-bound and free phage. Immunol. Lett. 2002, 81, 141–148. [Google Scholar] [CrossRef]
- Chen, L.Y.; Williams, B.R.; Yang, C.Y.; Cevallos, A.M.; Bhat, N.; Ward, H.; Sharon, J. Polyclonal Fab phage display libraries with a high percentage of diverse clones to Cryptosporidium parvum glycoproteins. Int. J. Parasitol. 2003, 33, 281–291. [Google Scholar] [CrossRef]
- Derda, R.; Wherritt, D.J.; Kiessling, L.L. Solid-phase synthesis of alkanethiols for the preparation of self-assembled monolayers. Langmuir 2007, 23, 11164–11167. [Google Scholar] [CrossRef]
- Orner, B.P.; Derda, R.; Lewis, R.L.; Thomson, J.A.; Kiessling, L.L. Arrays for the combinatorial exploration of cell adhesion. J. Am. Chem. Soc. 2004, 126, 10808–10809. [Google Scholar]
- Arap, W.; Kolonin, M.G.; Trepel, M.; Lahdenranta, J.; Cardo-Vila, M.; Giordano, R.J.; Mintz, P.J.; Ardelt, P.U.; Yao, V.J.; Vidal, C.I.; Chen, L.; Flamm, A.; Valtanen, H.; Weavind, L.M.; Hicks, M.E.; Pollock, R.E.; Botz, G.H.; Bucana, C.D.; Koivunen, E.; Cahill, D.; Troncoso, P.; Baggerly, K.A.; Pentz, R.D.; Do, K.A.; Logothetis, C.J.; Pasqualini, R. Steps toward mapping the human vasculature by phage display. Nat. Med. 2002, 8, 121–127. [Google Scholar]
- Krag, D.N.; Fuller, S.P.; Oligino, L.; Pero, S.C.; Weaver, D.L.; Soden, A.L.; Hebert, C.; Mills, S.; Liu, C.; Peterson, D. Phage-displayed random peptide libraries in mice: Toxicity after serial panning. Cancer Chemother. Pharmacol. 2002, 50, 325–332. [Google Scholar] [CrossRef]
- Krag, D.N.; Shukla, G.S.; Shen, G.P.; Pero, S.; Ashikaga, T.; Fuller, S.; Weaver, D.L.; Burdette-Radoux, S.; Thomas, C. Selection of tumor-binding ligands in cancer patients with phage display libraries. Cancer Res. 2006, 66, 7724–7733. [Google Scholar]
- Brown, K.C. Peptidic Tumor Targeting Agents: The Road from Phage Display Peptide Selections to Clinical Applications. Curr. Pharm. Des. 2010, 16, 1040–1054. [Google Scholar] [CrossRef]
- Kridel, S.J.; Chen, E.; Kotra, L.P.; Howard, E.W.; Mobashery, S.; Smith, J.M. Substrate hydrolysis by matrix metalloproteinase-9. J. Biol. Chem. 2001, 276, 20572–20578. [Google Scholar]
- McConnell, S.J.; Uveges, A.J.; Spinella, D.G. Comparison of Plate Versus Liquid Amplification of M13 Phage Display Libraries. Biotechniques 1995, 18, 803–806. [Google Scholar]
- Rodi, D.J.; Janes, R.W.; Sanganee, H.J.; Holton, R.A.; Wallace, B.A.; Makowski, L. Screening of a library of phage-displayed peptides identifies human Bcl-2 as a taxol binding protein. J. Mol. Biol. 1999, 285, 197–203. [Google Scholar] [CrossRef]
- Garufi, G.; Minenkova, O.; Lo Passo, C.; Pernice, I.; Felici, F. In Biotechnology Annual Review; El-Gewely, M.R., Ed.; Elsevier Science Bv: Amsterdam, Netherlands, 2005; Volume 11, pp. 153–190. [Google Scholar]
- Tawfik, D.S.; Griffiths, A.D. Man-made cell-like compartments for molecular evolution. Nat. Biotechnol. 1998, 16, 652–656. [Google Scholar] [CrossRef]
- Ganan-Calvo, A.M.; Gordillo, J.M. Perfectly monodisperse microbubbling by capillary flow focusing. Phys. Rev. Lett. 2001, 87, 274501–274504. [Google Scholar] [CrossRef]
- Garstecki, P.; Stone, H.A.; Whitesides, G.M. Mechanism for flow-rate controlled breakup in confined geometries: A route to monodisperse emulsions. Phys. Rev. Lett. 2005, 94, 1645011–1645014. [Google Scholar]
- Garstecki, P.; Fuerstman, M.J.; Stone, H.A.; Whitesides, G.M. Formation of droplets and bubbles in a microfluidic T-junction - scaling and mechanism of break-up. Lab Chip 2006, 6, 437–446. [Google Scholar] [CrossRef]
- Clausell-Tormos, J.; Lieber, D.; Baret, J.C.; El-Harrak, A.; Miller, O.J.; Frenz, L.; Blouwolff, J.; Humphry, K.J.; Koster, S.; Duan, H.; Holtze, C.; Weitz, D.A.; Griffiths, A.D.; Merten, C.A. Droplet-based microfluidic platforms for the encapsulation and screening of mammalian cells and multicellular organisms. Chem. Biol. 2008, 15, 427–437. [Google Scholar] [CrossRef]
- Holtze, C.; Rowat, A.C.; Agresti, J.J.; Hutchison, J.B.; Angile, F.E.; Schmitz, C.H.J.; Koster, S.; Duan, H.; Humphry, K.J.; Scanga, R.A.; Johnson, J.S.; Pisignano, D.; Weitz, D.A. Biocompatible surfactants for water-in-fluorocarbon emulsions. Lab Chip 2008, 8, 1632–1639. [Google Scholar] [CrossRef]
- Liu, W.S.; Kim, H.J.; Lucchetta, E.M.; Du, W.B.; Ismagilov, R.F. Isolation, incubation, and parallel functional testing and identification by FISH of rare microbial single-copy cells from multi-species mixtures using the combination of chemistrode and stochastic confinement. Lab Chip 2009, 9, 2153–2162. [Google Scholar] [CrossRef]
- Vincent, M.E.; Liu, W.S.; Haney, E.B.; Ismagilov, R.F. Microfluidic stochastic confinement enhances analysis of rare cells by isolating cells and creating high density environments for control of diffusible signals. Chem. Soc. Rev. 2010, 39, 974–984. [Google Scholar] [CrossRef]
- Makowski, L.; Soares, A. Estimating the diversity of peptide populations from limited sequence data. Bioinformatics 2003, 19, 483–489. [Google Scholar] [CrossRef]
- Mandava, S.; Makowski, L.; Devarapalli, S.; Uzubell, J.; Rodi, D.J. RELIC - A bioinformatics server for combinatorial peptide analysis and identification of protein-ligand interaction sites. Proteomics 2004, 4, 1439–1460. [Google Scholar] [CrossRef]
- Huang, J.; Ru, B.; Dai, P. Bioinformatics Resources and Tools for Phage Display. Molecules 2010, 16, 694–709. [Google Scholar] [CrossRef]
- Huang, J.; Ru, B.; Li, S.; Lin, H.; Guo, F.-B. SAROTUP: Scanner and reporter of target-unrelated peptides. J. Biomed. Biotechnol. 2010, 2010, 101932. [Google Scholar]
- Breaker, R.R.; Joyce, G.F. Emergence of a Replicating Species from an in-Vitro Rna Evolution Reaction. Proc. Natl. Acad. Sci. USA 1994, 91, 6093–6097. [Google Scholar] [CrossRef]
- Meyers, L.A.; Lee, J.F.; Cowperthwaite, M.; Ellington, A.D. The robustness of naturally and artificially selected nucleic acid secondary structures. J. Mol. Evol. 2004, 58, 681–691. [Google Scholar] [CrossRef]
- Halpin, D.R.; Harbury, P.B. DNA display I. Sequence-encoded routing of DNA populations. Plos Biol. 2004, 2, 1015–1021. [Google Scholar]
- Gartner, Z.J.; Kanan, M.W.; Liu, D.R. Multistep small-molecule synthesis programmed by DNA templates. J. Am. Chem. Soc. 2002, 124, 10304–10306. [Google Scholar] [CrossRef]
- Wilson, D.S.; Keefe, A.D.; Szostak, J.W. The use of mRNA display to select high-affinity protein-binding peptides. Proc. Natl. Acad. Sci. USA 2001, 98, 3750–3755. [Google Scholar] [CrossRef]
- Hanes, J.; Pluckthun, A. In vitro selection and evolution of functional proteins by using ribosome display. Proc. Natl. Acad. Sci. USA 1997, 94, 4937–4942. [Google Scholar] [CrossRef]
- Amstutz, P.; Forrer, P.; Zahnd, C.; Pluckthun, A. In vitro display technologies: novel developments and applications. Curr. Opin. Biotechnol. 2001, 12, 400–405. [Google Scholar] [CrossRef]
- Griffiths, A.D.; Tawfik, D.S. Miniaturising the laboratory in emulsion droplets. Trends Biotechnol. 2006, 24, 395–402. [Google Scholar] [CrossRef]
- Williams, R.; Peisajovich, S.G.; Miller, O.J.; Magdassi, S.; Tawfik, D.S.; Griffiths, A.D. Amplification of complex gene libraries by emulsion PCR. Nat. Methods 2006, 3, 545–550. [Google Scholar] [CrossRef]
- Krumpe, L.R.H.; Atkinson, A.J.; Smythers, G.W.; Kandel, A.; Schumacher, K.M.; McMahon, J.B.; Makowski, L.; Mori, T. T7 lytic phage-displayed peptide libraries exhibit less sequence bias than M13 filamentous phage-displayed peptide libraries. Proteomics 2006, 6, 4210–4222. [Google Scholar]
- Kim, J.; Schumann, W. Display of proteins on Bacillus subtilis endospores. Cell. Mol. Life Sci. 2009, 66, 3127–3136. [Google Scholar] [CrossRef]
- Rockberg, J.; Lofblom, J.; Hjelm, B.; Uhlen, M.; Stahl, S. Epitope mapping of antibodies using bacterial surface display. Nat. Methods 2008, 5, 1039–1045. [Google Scholar] [CrossRef]
- Gunneriusson, E.; Samuelson, P.; Ringdahl, J.; Gronlund, H.; Nygren, P.A.; Stahl, S. Staphylococcal surface display of immunoglobulin A (IgA)- and IgE-specific in vitro-selected binding proteins (affibodies) based on Staphylococcus aureus protein A. Appl. Environ. Microbiol. 1999, 65, 4134–4140. [Google Scholar]
- Boder, E.T.; Wittrup, K.D. Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 1997, 15, 553–557. [Google Scholar] [CrossRef]
- Lee, S.Y.; Choi, J.H.; Xu, Z.H. Microbial cell-surface display. Trends Biotechnol. 2003, 21, 45–52. [Google Scholar] [CrossRef]
- Sergeeva, A.; Kolonin, M.G.; Molldrem, J.J.; Pasqualini, R.; Arap, W. Display technologies: Application for the discovery of drug and gene delivery agents. Advan. Drug Delivery Rev. 2006, 58, 1622–1654. [Google Scholar] [CrossRef]
- Metzker, M.L. Sequencing technologies - the next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef]
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; Boutell, J.M.; Bryant, J.; Carter, R.J.; Cheetham, R.K.; Cox, A.J.; Ellis, D.J.; Flatbush, M.R.; Gormley, N.A.; Humphray, S.J.; Irving, L.J.; Karbelashvili, M.S.; Kirk, S.M.; Li, H.; Liu, X.H.; Maisinger, K.S.; Murray, L.J.; Obradovic, B.; Ost, T.; Parkinson, M.L.; Pratt, M.R.; Rasolonjatovo, I.M.J.; Reed, M.T.; Rigatti, R.; Rodighiero, C.; Ross, M.T.; Sabot, A.; Sankar, S.V.; Scally, A.; Schroth, G.P.; Smith, M.E.; Smith, V.P.; Spiridou, A.; Torrance, P.E.; Tzonev, S.S.; Vermaas, E.H.; Walter, K.; Wu, X.L.; Zhang, L.; Alam, M.D.; Anastasi, C.; Aniebo, I.C.; Bailey, D.M.D.; Bancarz, I.R.; Banerjee, S.; Barbour, S.G.; Baybayan, P.A.; Benoit, V.A.; Benson, K.F.; Bevis, C.; Black, P.J.; Boodhun, A.; Brennan, J.S.; Bridgham, J.A.; Brown, R.C.; Brown, A.A.; Buermann, D.H.; Bundu, A.A.; Burrows, J.C.; Carter, N.P.; Castillo, N.; Catenazzi, M.C.E.; Chang, S.; Cooley, R.N.; Crake, N.R.; Dada, O.O.; Diakoumakos, K.D.; Dominguez-Fernandez, B.; Earnshaw, D.J.; Egbujor, U.C.; Elmore, D.W.; Etchin, S.S.; Ewan, M.R.; Fedurco, M.; Fraser, L.J.; Fajardo, K.V.F.; Furey, W.S.; George, D.; Gietzen, K.J.; Goddard, C.P.; Golda, G.S.; Granieri, P.A.; Green, D.E.; Gustafson, D.L.; Hansen, N.F.; Harnish, K.; Haudenschild, C.D.; Heyer, N.I.; Hims, M.M.; Ho, J.T.; Horgan, A.M.; Hoschler, K.; Hurwitz, S.; Ivanov, D.V.; Johnson, M.Q.; James, T.; Jones, T.A.H.; Kang, G.D.; Kerelska, T.H.; Kersey, A.D.; Khrebtukova, I.; Kindwall, A.P.; Kingsbury, Z.; Kokko-Gonzales, P.I.; Kumar, A.; Laurent, M.A.; Lawley, C.T.; Lee, S.E.; Lee, X.; Liao, A.K.; Loch, J.A.; Lok, M.; Luo, S.J.; Mammen, R.M.; Martin, J.W.; McCauley, P.G.; McNitt, P.; Mehta, P.; Moon, K.W.; Mullens, J.W.; Newington, T.; Ning, Z.M.; Ng, B.L.; Novo, S.M.; O'Neill, M.J.; Osborne, M.A.; Osnowski, A.; Ostadan, O.; Paraschos, L.L.; Pickering, L.; Pike, A.C.; Pike, A.C.; Pinkard, D.C.; Pliskin, D.P.; Podhasky, J.; Quijano, V.J.; Raczy, C.; Rae, V.H.; Rawlings, S.R.; Rodriguez, A.C.; Roe, P.M.; Rogers, J.; Bacigalupo, M.C.R.; Romanov, N.; Romieu, A.; Roth, R.K.; Rourke, N.J.; Ruediger, S.T.; Rusman, E.; Sanches-Kuiper, R.M.; Schenker, M.R.; Seoane, J.M.; Shaw, R.J.; Shiver, M.K.; Short, S.W.; Sizto, N.L.; Sluis, J.P.; Smith, M.A.; Sohna, J.E.S.; Spence, E.J.; Stevens, K.; Sutton, N.; Szajkowski, L.; Tregidgo, C.L.; Turcatti, G.; vandeVondele, S.; Verhovsky, Y.; Virk, S.M.; Wakelin, S.; Walcott, G.C.; Wang, J.W.; Worsley, G.J.; Yan, J.Y.; Yau, L.; Zuerlein, M.; Rogers, J.; Mullikin, J.C.; Hurles, M.E.; McCooke, N.J.; West, J.S.; Oaks, F.L.; Lundberg, P.L.; Klenerman, D.; Durbin, R.; Smith, A.J. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008, 456, 53–59. [Google Scholar]
- Shendure, J.; Porreca, G.J.; Reppas, N.B.; Lin, X.X.; McCutcheon, J.P.; Rosenbaum, A.M.; Wang, M.D.; Zhang, K.; Mitra, R.D.; Church, G.M. Accurate multiplex polony sequencing of an evolved bacterial genome. Science 2005, 309, 1728–1732. [Google Scholar]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.J.; Chen, Z.T.; Dewell, S.B.; Du, L.; Fierro, J.M.; Gomes, X.V.; Godwin, B.C.; He, W.; Helgesen, S.; Ho, C.H.; Irzyk, G.P.; Jando, S.C.; Alenquer, M.L.I.; Jarvie, T.P.; Jirage, K.B.; Kim, J.B.; Knight, J.R.; Lanza, J.R.; Leamon, J.H.; Lefkowitz, S.M.; Lei, M.; Li, J.; Lohman, K.L.; Lu, H.; Makhijani, V.B.; McDade, K.E.; McKenna, M.P.; Myers, E.W.; Nickerson, E.; Nobile, J.R.; Plant, R.; Puc, B.P.; Ronan, M.T.; Roth, G.T.; Sarkis, G.J.; Simons, J.F.; Simpson, J.W.; Srinivasan, M.; Tartaro, K.R.; Tomasz, A.; Vogt, K.A.; Volkmer, G.A.; Wang, S.H.; Wang, Y.; Weiner, M.P.; Yu, P.G.; Begley, R.F.; Rothberg, J.M. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376–380. [Google Scholar]
- Dias-Neto, E.; Nunes, D.N.; Giordano, R.J.; Sun, J.; Botz, G.H.; Yang, K.; Setubal, J.C.; Pasqualini, R.; Arap, W. Next-Generation Phage Display: Integrating and Comparing Available Molecular Tools to Enable Cost-Effective High-Throughput Analysis. PLoS One 2009, 4, e8338(1)–e8338(11). [Google Scholar]
- Stockwell, B.R. Chemical genetics: Ligand-based discovery of gene function. Nat. Rev. Genet. 2000, 1, 116–125. [Google Scholar] [CrossRef]
- Schreiber, S.L. Chemical genetics resulting from a passion for synthetic organic chemistry. Bioorg. Med. Chem. 1998, 6, 1127–1152. [Google Scholar] [CrossRef]
- Little, L.E.; Dane, K.Y.; Daugherty, P.S.; Healy, K.E.; Schaffer, D.V. Exploiting bacterial peptide display technology to engineer biomaterials for neural stem cell culture. Biomaterials 2010, 32, 1484–1494. [Google Scholar]
- Blackwell, H.E.; Perez, L.; Stavenger, R.A.; Tallarico, J.A.; Eatough, E.C.; Foley, M.A.; Schreiber, S.L. A one-bead, one-stock solution approach to chemical genetics: part 1. Chem. Biol. 2001, 8, 1167–1182. [Google Scholar] [CrossRef]
- Lehar, J.; Stockwell, B.R.; Giaever, G.; Nislow, C. Combination chemical genetics. Nat. Chem. Biol. 2008, 4, 674–681. [Google Scholar] [CrossRef]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Derda, R.; Tang, S.K.Y.; Li, S.C.; Ng, S.; Matochko, W.; Jafari, M.R. Diversity of Phage-Displayed Libraries of Peptides during Panning and Amplification. Molecules 2011, 16, 1776-1803. https://doi.org/10.3390/molecules16021776
Derda R, Tang SKY, Li SC, Ng S, Matochko W, Jafari MR. Diversity of Phage-Displayed Libraries of Peptides during Panning and Amplification. Molecules. 2011; 16(2):1776-1803. https://doi.org/10.3390/molecules16021776
Chicago/Turabian StyleDerda, Ratmir, Sindy K.Y. Tang, S. Cory Li, Simon Ng, Wadim Matochko, and Mohammad R. Jafari. 2011. "Diversity of Phage-Displayed Libraries of Peptides during Panning and Amplification" Molecules 16, no. 2: 1776-1803. https://doi.org/10.3390/molecules16021776