Appropriate Assignment of Fossil Calibration Information Minimizes the Difference between Phylogenetic and Pedigree Mutation Rates in Humans


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sequences and Alignments
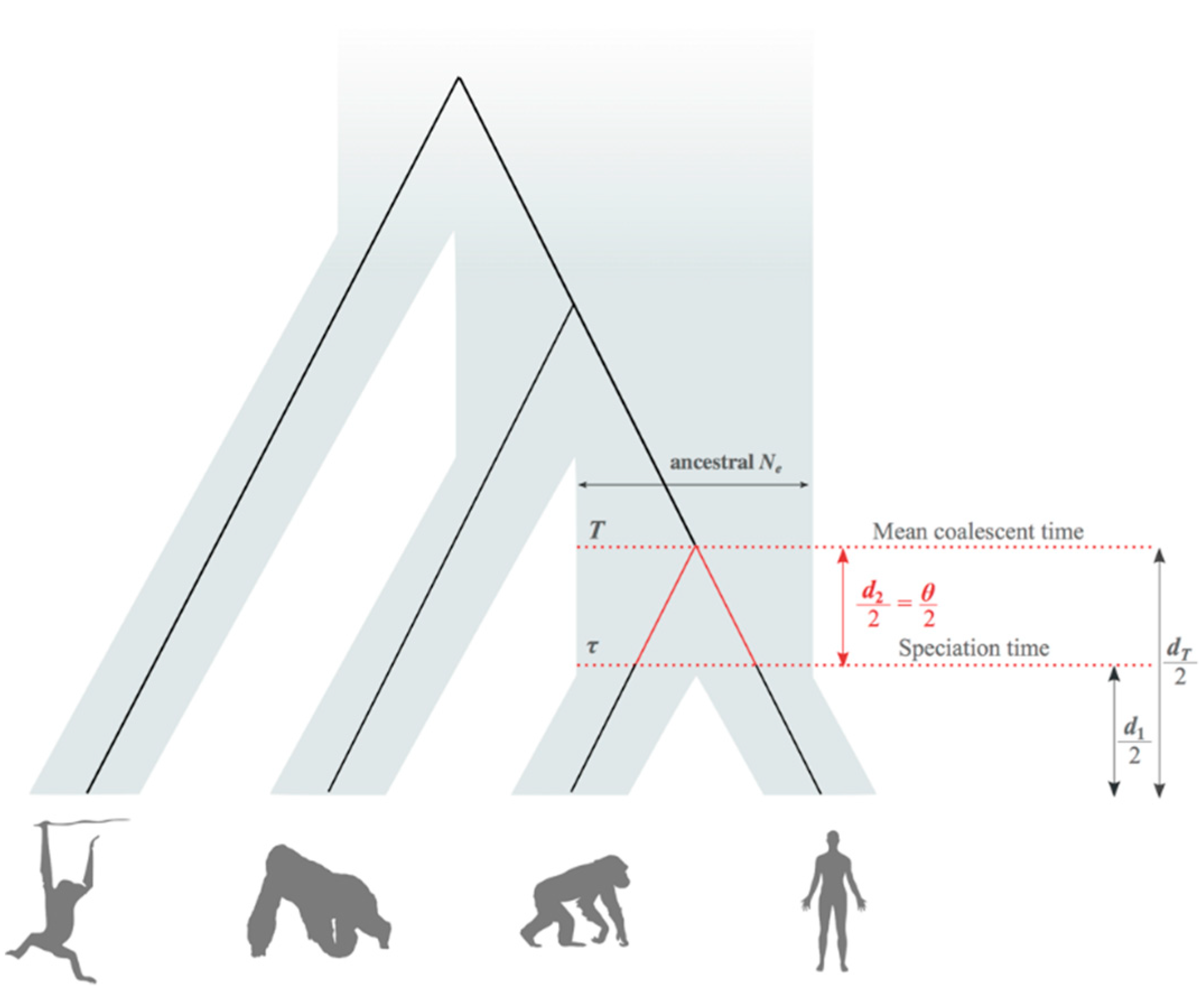
2.2. Average Coalescent Times and Speciation Times
2.3. Estimation of Mean Speciation Times τ
2.4. Fossil African Great Apes and Humans
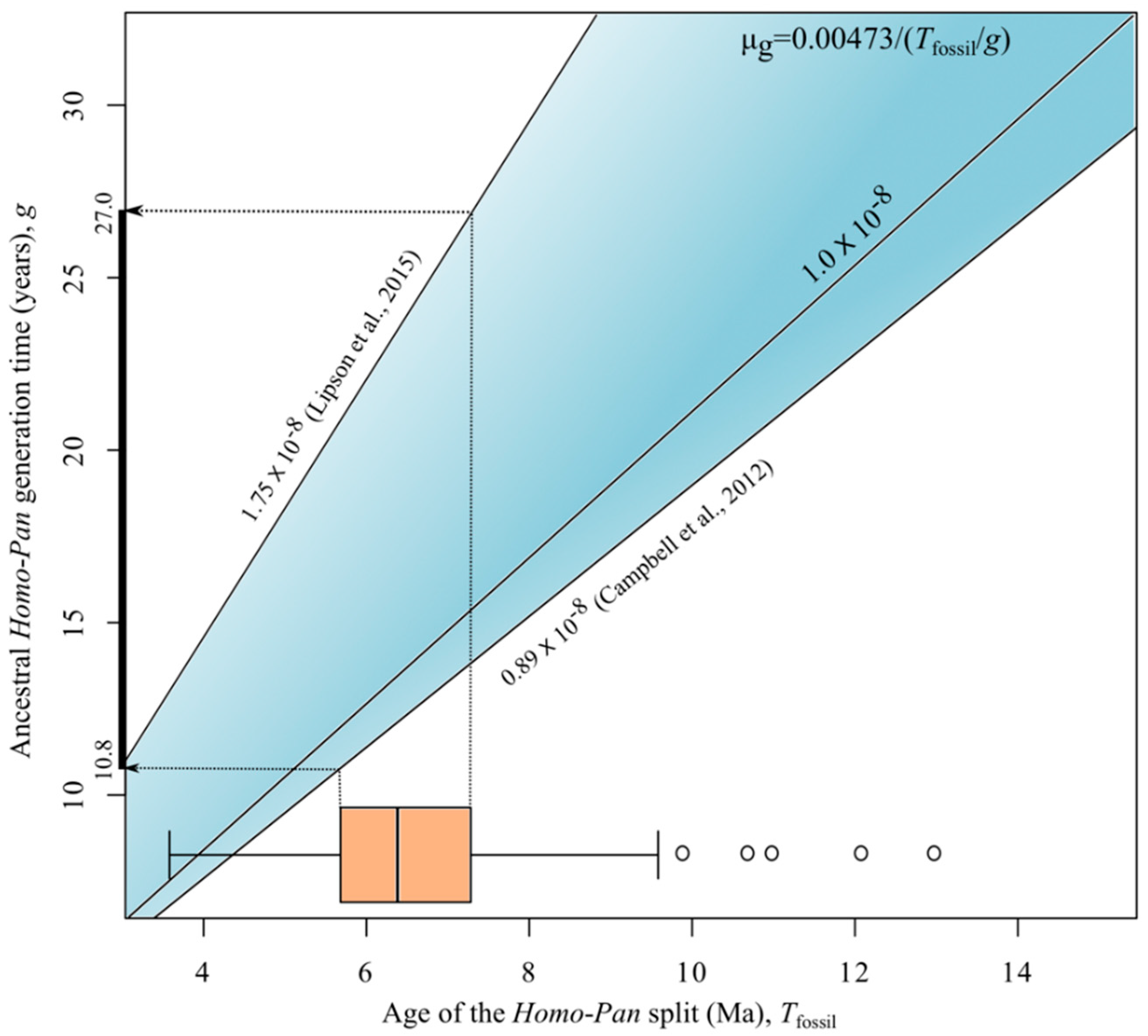
2.5. Generation Times
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kimura, M. The Neutral Theory of Molecular Evolution; Cambridge University Press: New York, CA, USA, 1983. [Google Scholar]
- Nei, M. Mutation-Driven Evolution; Oxford University Press: New York, CA, USA, 2013. [Google Scholar]
- Haldane, J.B.S. The rate of spontaneous mutation of a human gene. J. Genet. 1935, 31, 317–326. [Google Scholar] [CrossRef]
- Drake, J.W.; Charlesworth, B.; Charlesworth, D.; Crow, J.F. Rates of spontaneous mutation. Genetics 1998, 148, 1667–1686. [Google Scholar] [PubMed]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: New York, CA, USA, 2000. [Google Scholar]
- Benton, M.J.; Donoghue, P.C.J.; Asher, R.J.; Friedman, M.; Near, T.J.; Vinther, J. Constraints on the timescale of animal evolutionary history. Palaeontol. Electron. 2015, 18, 1–107. [Google Scholar] [CrossRef]
- The Chimpanzee Sequencing and Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 2005, 437, 69–87. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.C.; Li, W.H. Genomic divergences between humans and other hominoids and the effective population size of the common ancestor of humans and chimpanzees. Am. J. Hum. Genet. 2001, 68, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Ebersberger, I.; Metzler, D.; Schwarz, C.; Paabo, S. Genomewide comparison of DNA sequences between humans and chimpanzees. Am. J. Hum. Genet. 2002, 70, 1490–1497. [Google Scholar] [CrossRef] [PubMed]
- Brunet, M.; Guy, F.; Pilbeam, D.; Mackaye, H.T.; Likius, A.; Ahounta, D.; Beauvilain, A.; Blondel, C.; Bocherens, H.; Boisserie, J.R.; et al. A new hominid from the Upper Miocene of Chad, Central Africa. Nature 2002, 418, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.D.; Eichler, E.E. Properties and rates of germline mutations in humans. Trends Genet. 2013, 29, 575–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venn, O.; Turner, I.; Mathieson, I.; de Groot, N.; Bontrop, R.; McVean, G. Strong male bias drives germline mutation in chimpanzees. Science 2014, 344, 1272–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrago, C.G. The effective population sizes of the anthropoid ancestors of the human-chimpanzee lineage provide insights on the historical biogeography of the great apes. Mol. Biol. Evol. 2014, 31, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Rannala, B.; Yang, Z. Bayes estimation of species divergence times and ancestral population sizes using DNA sequences from multiple loci. Genetics 2003, 164, 1645–1656. [Google Scholar] [PubMed]
- Scally, A.; Durbin, R. Revising the human mutation rate: Implications for understanding human evolution. Nat. Rev. Genet. 2012, 13, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Goodman, M. Rates of Molecular Evolution: The Hominoid Slowdown. Bioessays 1985, 3, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Li, W.H.; Tanimura, M. The Molecular Clock Runs More Slowly in Man Than in Apes and Monkeys. Nature 1987, 326, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Bailey, W.J.; Fitch, D.H.; Tagle, D.A.; Czelusniak, J.; Slightom, J.L.; Goodman, M. Molecular evolution of the psi eta-globin gene locus: Gibbon phylogeny and the hominoid slowdown. Mol. Biol. Evol. 1991, 8, 155–184. [Google Scholar] [CrossRef] [PubMed]
- Koop, B.F.; Goodman, M.; Xu, P.; Chan, K.; Slightom, J.L. Primate Eta-Globin DNA-Sequences and Mans Place among the Great Apes. Nature 1986, 319, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Elango, N.; Warden, C.; Vigoda, E.; Yi, S.V. Heterogeneous genomic molecular clocks in primates. PLoS Genet. 2006, 2, 1527–1534. [Google Scholar] [CrossRef] [PubMed]
- Scally, A.; Dutheil, J.Y.; Hillier, L.W.; Jordan, G.E.; Goodhead, I.; Herrero, J.; Hobolth, A.; Lappalainen, T.; Mailund, T.; Marques-Bonet, T.; et al. Insights into hominid evolution from the gorilla genome sequence. Nature 2012, 483, 169–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, S.Y.W.; Phillips, M.J.; Cooper, A.; Drummond, A.J. Time dependency of molecular rate estimates and systematic overestimation of recent divergence times. Mol. Biol. Evol. 2005, 22, 1561–1568. [Google Scholar] [CrossRef] [PubMed]
- Emerson, B.C. Alarm bells for the molecular clock? No support for Ho et al.’s model of time-dependent molecular rate estimates. Syst. Biol. 2007, 56, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.; Beerli, P. Perspective: Gene divergence, population divergence, and the variance in coalescence time in phylogeographic studies. Evolution 2000, 54, 1839–1854. [Google Scholar] [CrossRef] [PubMed]
- Peterson, G.I.; Masel, J. Quantitative Prediction of Molecular Clock and K-a/K-s at Short Timescales. Mol. Biol. Evol. 2009, 26, 2595–2603. [Google Scholar] [CrossRef] [PubMed]
- Ho, S.Y.W.; Lanfear, R.; Bromham, L.; Phillips, M.J.; Soubrier, J.; Rodrigo, A.G.; Cooper, A. Time-dependent rates of molecular evolution. Mol. Ecol. 2011, 20, 3087–3101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roach, J.C.; Glusman, G.; Smit, A.F.A.; Huff, C.D.; Hubley, R.; Shannon, P.T.; Rowen, L.; Pant, K.P.; Goodman, N.; Bamshad, M.; et al. Analysis of Genetic Inheritance in a Family Quartet by Whole-Genome Sequencing. Science 2010, 328, 636–639. [Google Scholar] [CrossRef] [PubMed]
- Conrad, D.F.; Keebler, J.E.M.; DePristo, M.A.; Lindsay, S.J.; Zhang, Y.J.; Casals, F.; Idaghdour, Y.; Hartl, C.L.; Torroja, C.; Garimella, K.V.; et al. Awadalla for the 1000 genomes project. Variation in genome-wide mutation rates within and between human families. Nat. Genet. 2011, 43, 712–715. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.D.; Chong, J.X.; Malig, M.; Ko, A.; Dumont, B.L.; Han, L.; Vives, L.; O’Roak, B.J.; Sudmant, P.H.; Shendure, J.; et al. Estimating the human mutation rate using autozygosity in a founder population. Nat. Genet. 2012, 44, 1277–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, A.; Frigge, M.L.; Masson, G.; Besenbacher, S.; Sulem, P.; Magnusson, G.; Gudjonsson, S.A.; Sigurdsson, A.; Jonasdottir, A.; Wong, W.S.; et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature 2012, 488, 471–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaelson, J.J.; Shi, Y.J.; Gujral, M.; Zheng, H.C.; Malhotra, D.; Jin, X.; Jian, M.H.; Liu, G.M.; Greer, D.; Bhandari, A.; et al. Whole-Genome Sequencing in Autism Identifies Hot Spots for De Novo Germline Mutation. Cell 2012, 151, 1431–1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Q.; Mittnik, A.; Johnson, P.L.; Bos, K.; Lari, M.; Bollongino, R.; Sun, C.; Giemsch, L.; Schmitz, R.; Burger, J.; et al. A revised timescale for human evolution based on ancient mitochondrial genomes. Curr. Biol. 2013, 23, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Lipson, M.; Loh, P.R.; Sankararaman, S.; Patterson, N.; Berger, B.; Reich, D. Calibrating the Human Mutation Rate via Ancestral Recombination Density in Diploid Genomes. PLoS Genet. 2015, 11, E1005550. [Google Scholar] [CrossRef] [PubMed]
- Besenbacher, S.; Liu, S.; Izarzugaza, J.M.G.; Grove, J.; Belling, K.; Bork-Jensen, J.; Huang, S.; Als, T.D.; Li, S.; Yadav, R.; et al. Novel variation and de novo mutation rates in population-wide de novo assembled Danish trios. Nat. Commun. 2015, 6, 5969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahbari, R.; Wuster, A.; Lindsay, S.J.; Hardwick, R.J.; Alexandrov, L.B.; Al Turki, S.; Dominiczak, A.; Morris, A.; Porteous, D.; Smith, B.; et al. Timing, rates and spectra of human germline mutation. Nat. Genet. 2015, 48, 126–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amster, G.; Sella, G. Life history effects on the molecular clock of autosomes and sex chromosomes. Proc. Natl. Acad. Sci. USA 2016, 113, 1588–1593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, W.S.W.; Solomon, B.D.; Bodian, D.L.; Kothiyal, P.; Eley, G.; Huddleston, K.C.; Baker, R.; Thach, D.C.; Iyer, R.K.; Vockley, J.G.; et al. New observations on maternal age effect on germline de novo mutations. Nat. Commun. 2016, 7, 10486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.H. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.; Rannala, B. Bayesian species delimitation using multilocus sequence data. Proc. Natl. Acad. Sci. USA 2010, 107, 9264–9269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahata, N.; Satta, Y.; Klein, J. Divergence time and population size in the lineage leading to modern humans. Theor. Popul. Biol. 1995, 48, 198–221. [Google Scholar] [CrossRef] [PubMed]
- Hobolth, A.; Dutheil, J.Y.; Hawks, J.; Schierup, M.H.; Mailund, T. Incomplete lineage sorting patterns among human, chimpanzee, and orangutan suggest recent orangutan speciation and widespread selection. Genome Res. 2011, 21, 349–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langergraber, K.E.; Prufer, K.; Rowney, C.; Boesch, C.; Crockford, C.; Fawcett, K.; Inoue, E.; Inoue-Muruyama, M.; Mitani, J.C.; Muller, M.N.; et al. Generation times in wild chimpanzees and gorillas suggest earlier divergence times in great ape and human evolution. Proc. Natl. Acad. Sci. USA 2012, 109, 15716–15721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrago, C.G. The limiting distribution of the effective population size of the ancestor of humans and chimpanzees. J. Theor. Biol. 2014, 357, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Hedges, S.B.; Marin, J.; Suleski, M.; Paymer, M.; Kumar, S. Tree of Life Reveals Clock-Like Speciation and Diversification. Mol. Biol. Evol. 2015, 32, 835–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurst, L.D.; Ellegren, H. Sex biases in the mutation rate. Trends Genet. 1998, 14, 446–452. [Google Scholar] [CrossRef]
- Sun, J.X.; Helgason, A.; Masson, G.; Ebenesersdottir, S.S.; Li, H.; Mallick, S.; Gnerre, S.; Patterson, N.; Kong, A.; Reich, D.; et al. A direct characterization of human mutation based on microsatellites. Nat. Genet. 2012, 44, 1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Manuel, M.; Kuhlwilm, M.; Frandsen, P.; Sousa, V.C.; Desai, T.; Prado-Martinez, J.; Hernandez-Rodriguez, J.; Dupanloup, I.; Lao, O.; Hallast, P.; et al. Chimpanzee genomic diversity reveals ancient admixture with bonobos. Science 2016, 354, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Nater, A.; Mattle-Greminger, M.P.; Nurcahyo, A.; Nowak, M.G.; de Manuel, M.; Desai, T.; Groves, C.; Pybus, M.; Sonay, T.B.; Roos, C.; et al. Morphometric, behavioral, and genomic evidence for a new orangutan species. Curr. Biol. 2017, 27, 3487–3498. [Google Scholar] [CrossRef] [PubMed]
- Hey, J.; Nielsen, R. Integration within the Felsenstein equation for improved Markov chain Monte Carlo methods in population genetics. Proc. Natl. Acad. Sci. USA 2007, 104, 2785–2790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, D.; Yu, Y.; Nakhleh, L. Bayesian inference of reticulate phylogenies under the multispecies network coalescent. PLoS Genet. 2016, 12, e1006006. [Google Scholar] [CrossRef] [PubMed]
- Plouviez, S.; Le Guen, D.; Lecompte, O.; Lallier, F.H.; Jollivet, D. Determining gene flow and the influence of selection across the equatorial barrier of the East Pacific Rise in the tube-dwelling polychaete Alvinella pompejana. BMC Evol. Biol. 2010, 10, 220. [Google Scholar] [CrossRef] [PubMed]
- Etter, R.J.; Boyle, E.E.; Glazier, A.; Jennings, R.M.; Dutra, E.; Chase, M.R. Phylogeography of a pan-Atlantic abyssal protobranch bivalve: Implications for evolution in the Deep Atlantic. Mol. Ecol. 2011, 20, 829–843. [Google Scholar] [CrossRef] [PubMed]
- Oliver, J.C. Microevolutionary processes generate phylogenomic discordance at ancient divergences. Evolution 2013, 67, 1823–1830. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Study | Mean Rate (s/s/g) |
|---|---|
| 1000 Genomes Project Consortium [27] | 1.0–1.2 × 10−8 |
| Roach et al. [28] | 1.1 × 10−8 |
| Conrad et al. [29] | 0.97–1.17 × 10−8 |
| Cambpell et al. [30] | 0.89–1.43 × 10−8 |
| Kong et al. [31] | 1.20 × 10−8 |
| Michaelson et al. [32] | 1.0 × 10−8 |
| Fu et al. [33] * | 1.10–1.42 × 10−8 |
| Lipson et al. [34] | 1.55–1.75 × 10−8 |
| Besenbacher et al. [35] | 1.16–1.38 × 10−8 |
| Rahbari et al. [36] | 1.13–1.43 × 10−8 |
| Amster and Seela [37] | 1.2 × 10−8 |
| Wong et al. [38] | 1.05 × 10−8 |
| Mean Coalescent Time (T) | Speciation Time (τ) | ||
|---|---|---|---|
| Genetic Distance (dT/2) 1 | Yearly Evolutionary Rate 2 | Genetic Distance (d1/2) 1 | Yearly Evolutionary Rate 2 |
| 0.00625 * | 0.089 × 10−8 | 0.00473 ± 0.000040 | 0.067–0.068 × 10−8 |
| Ancestral Homo–Pan Generation Time (Years) | Evolutionary Rate |
|---|---|
| 15 | 0.99 × 10−8–1.03 × 10−8 |
| 20 | 1.33 × 10−8–1.37 × 10−8 |
| 26.3 | 1.75 × 10−8–1.81 × 10−8 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capellão, R.T.; Costa-Paiva, E.M.; Schrago, C.G. Appropriate Assignment of Fossil Calibration Information Minimizes the Difference between Phylogenetic and Pedigree Mutation Rates in Humans. Life 2018, 8, 49. https://doi.org/10.3390/life8040049
Capellão RT, Costa-Paiva EM, Schrago CG. Appropriate Assignment of Fossil Calibration Information Minimizes the Difference between Phylogenetic and Pedigree Mutation Rates in Humans. Life. 2018; 8(4):49. https://doi.org/10.3390/life8040049
Chicago/Turabian StyleCapellão, Renata T., Elisa M. Costa-Paiva, and Carlos G. Schrago. 2018. "Appropriate Assignment of Fossil Calibration Information Minimizes the Difference between Phylogenetic and Pedigree Mutation Rates in Humans" Life 8, no. 4: 49. https://doi.org/10.3390/life8040049