APOE Promoter Polymorphism-219T/G is an Effect Modifier of the Influence of APOE ε4 on Alzheimer’s Disease Risk in a Multiracial Sample
 ,
,  , , , , , , , , , , and add
Show full author list
, , , , , , , , , , and add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Participants
2.2. Data Generation and Analysis
2.2.1. SNP Genotyping
2.2.2. Quality Control of Genome-Wide Data
2.3. Statistical Genetic Analysis
2.3.1. Association of AD Risk with SNPs in the APOE Region
2.3.2. Follow-up Association Analyses with rs405509
2.4. APOE Reporter Gene Assays
2.4.1. APOE Promoter Construct
2.4.2. Luciferase Assay
2.5. Human Postmortem Brain Tissues
3. Results
3.1. Ethnic Variability in the Association of APOE ε4 with AD
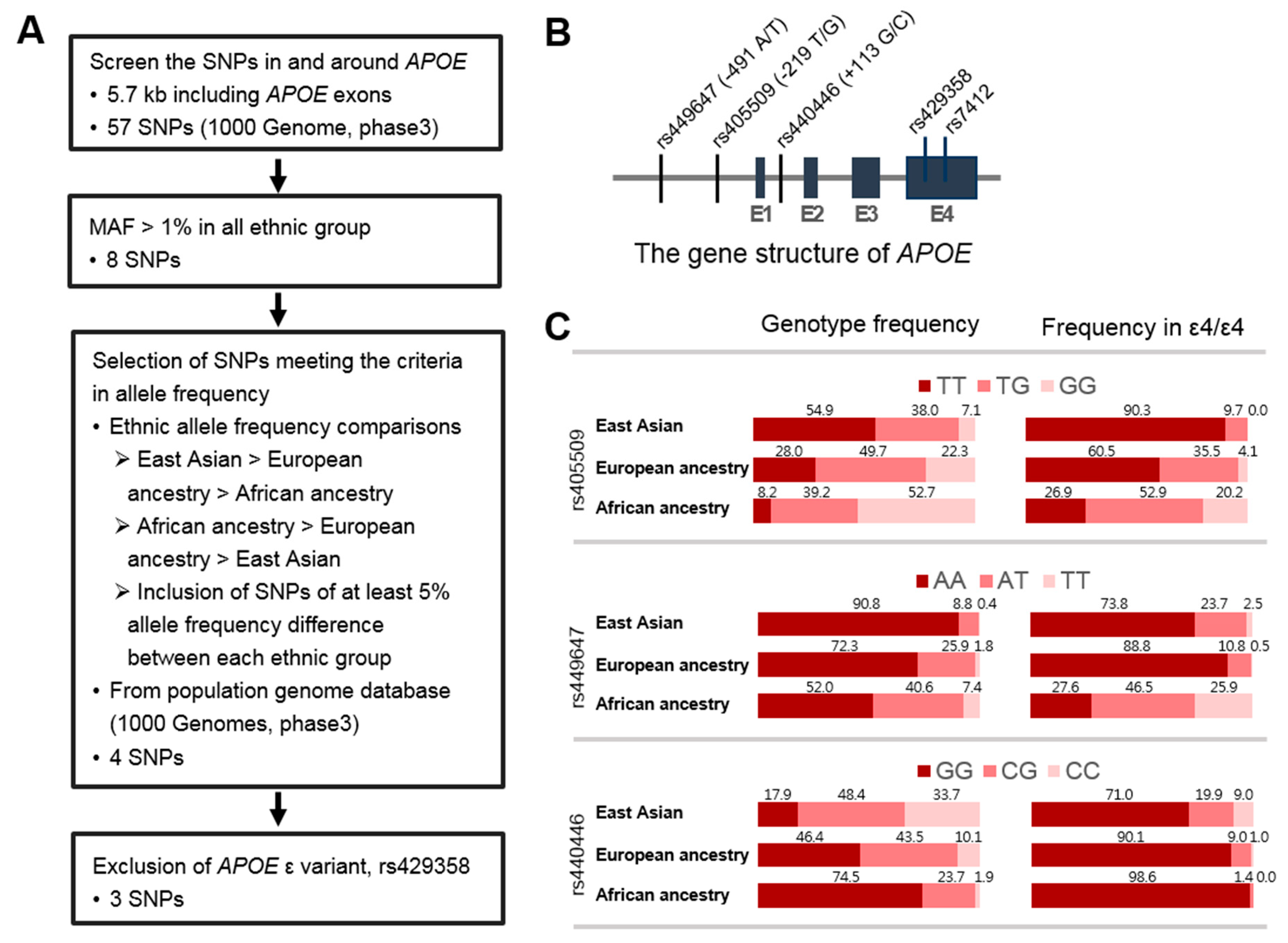
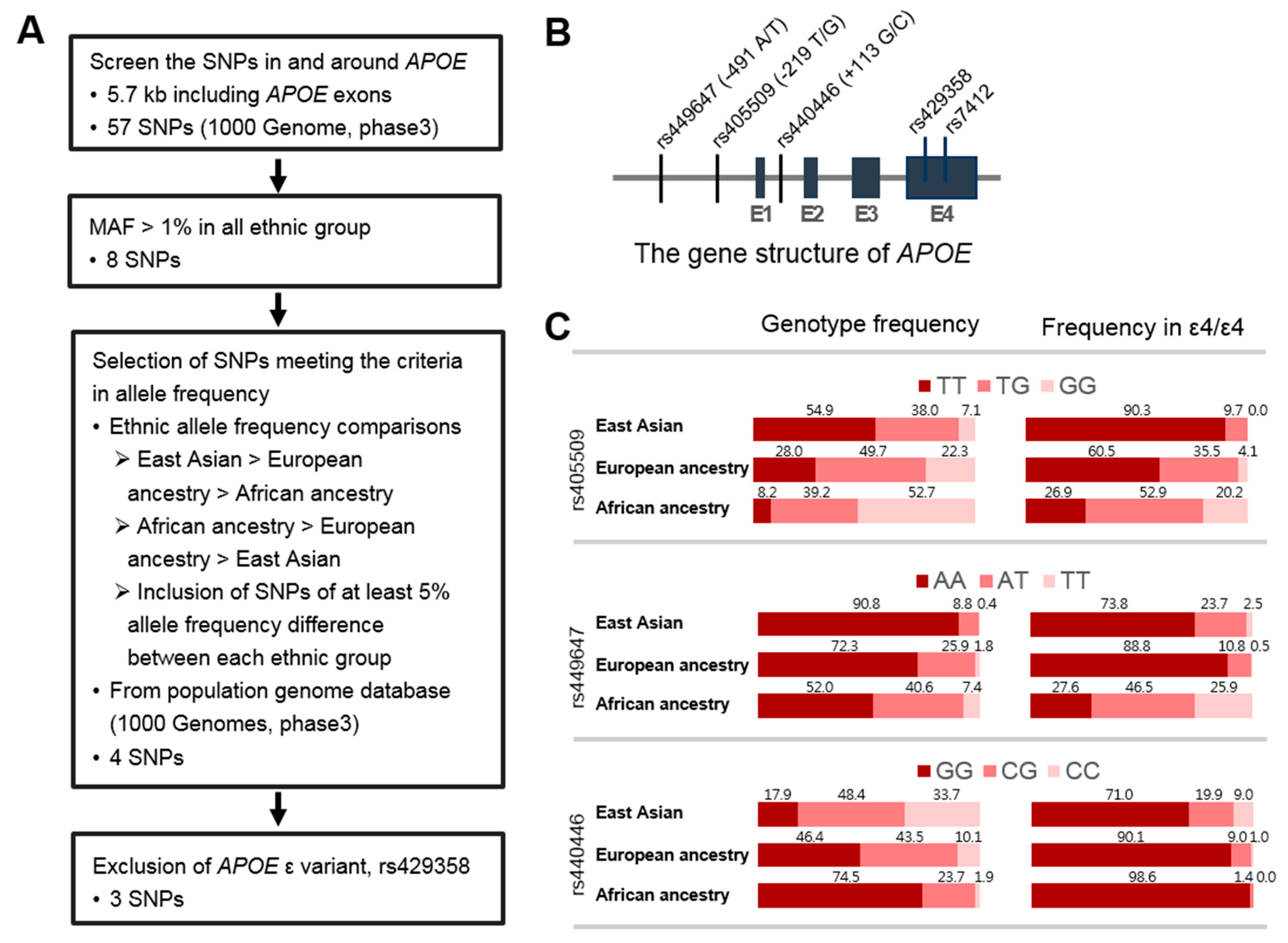
3.2. Identification of SNPs in the APOE Region Contributing to Ethnic Variability in AD Risk
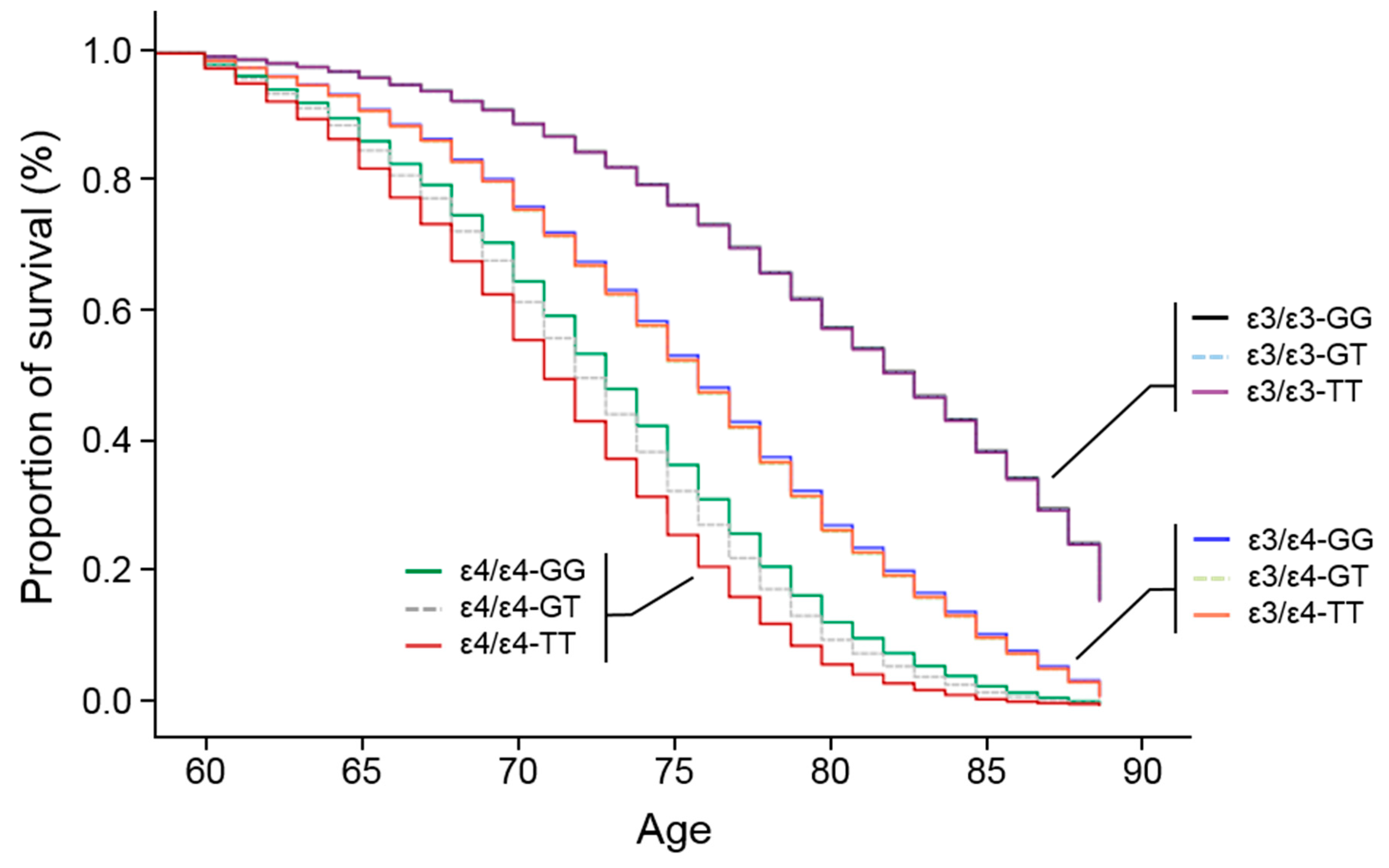
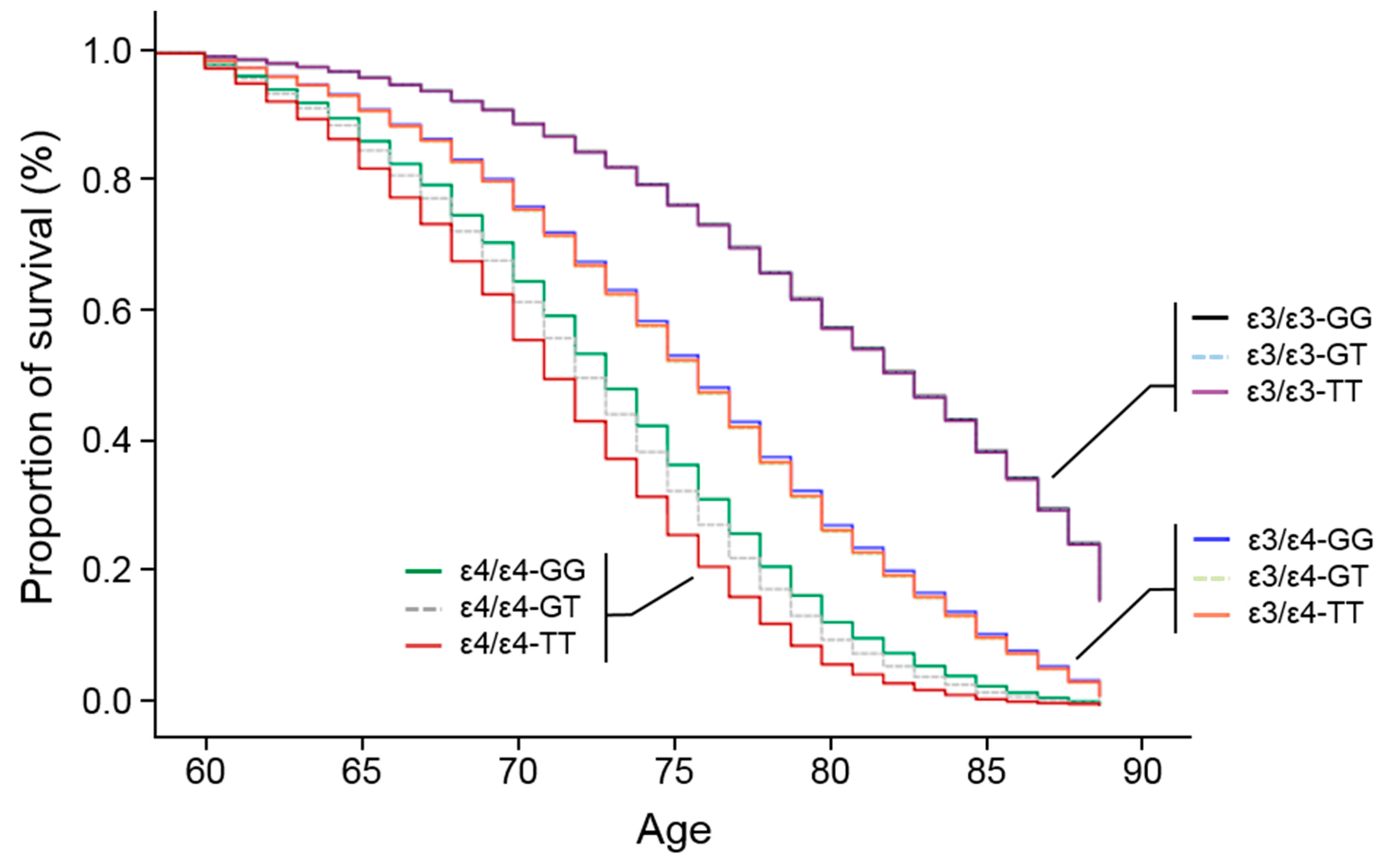
3.3. APOE Promoter SNP, rs405509, Modulates the ε4 Association for AD
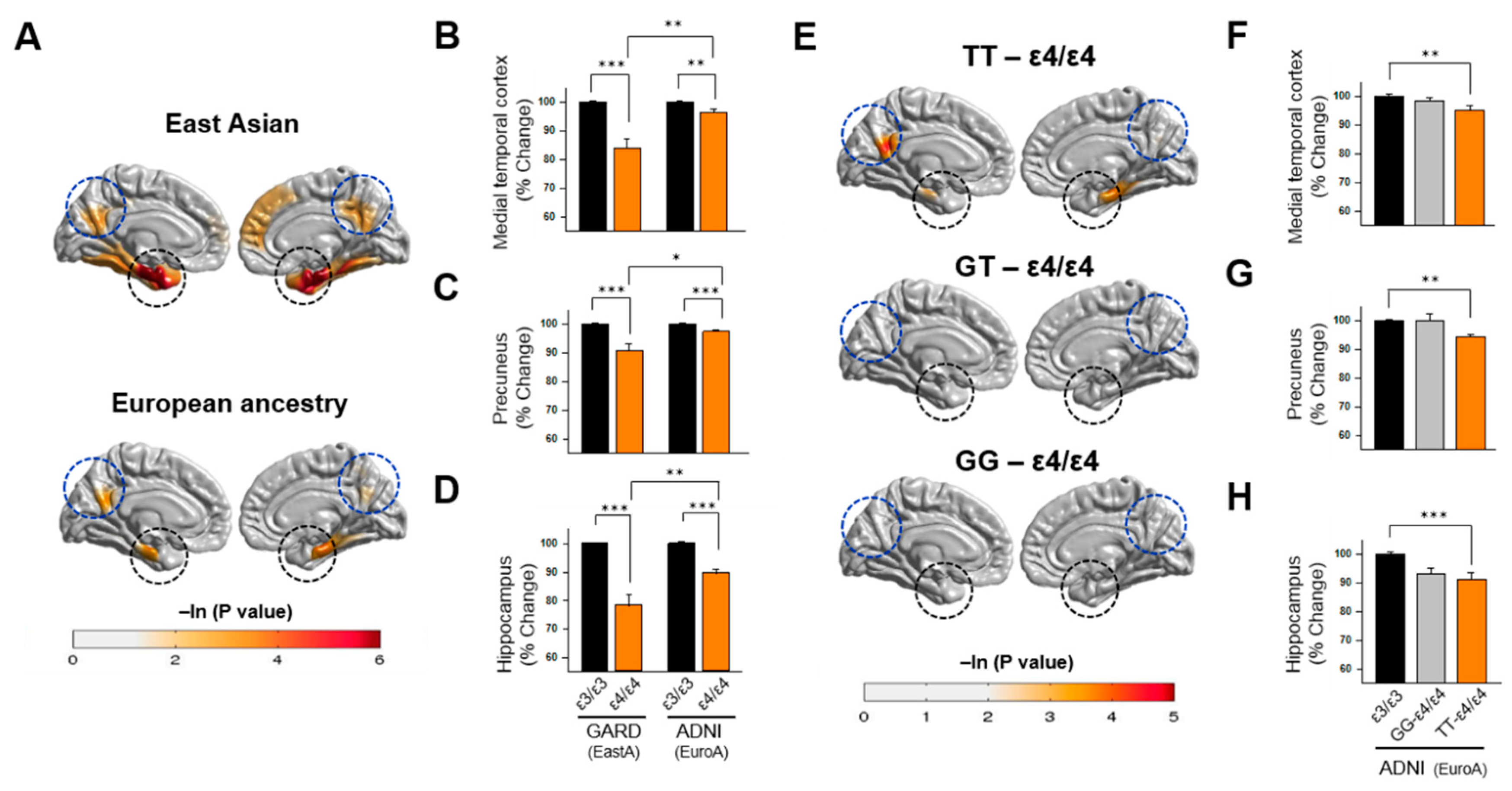
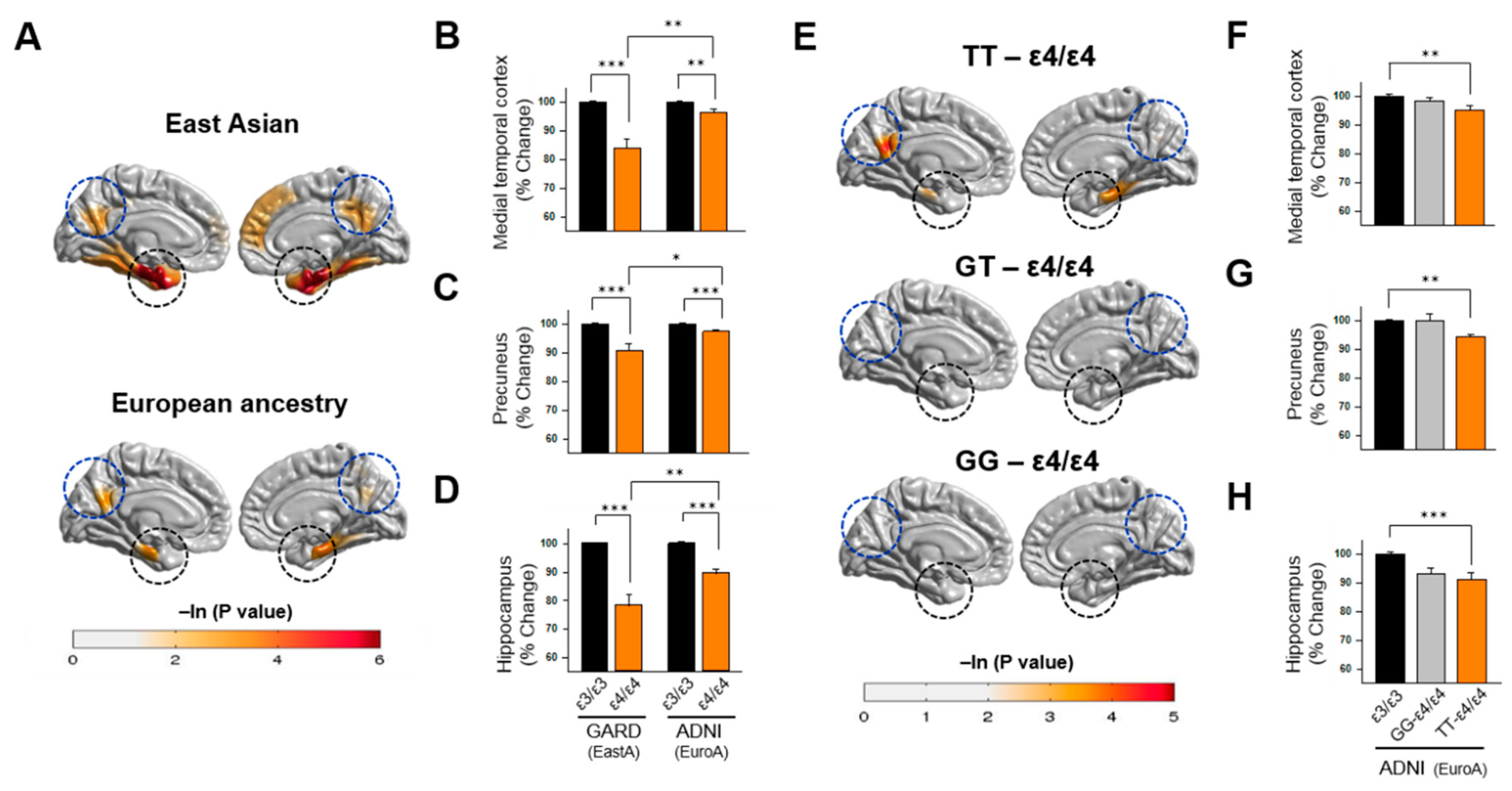
3.4. Association of APOE Polymorphisms with Brain Atrophy
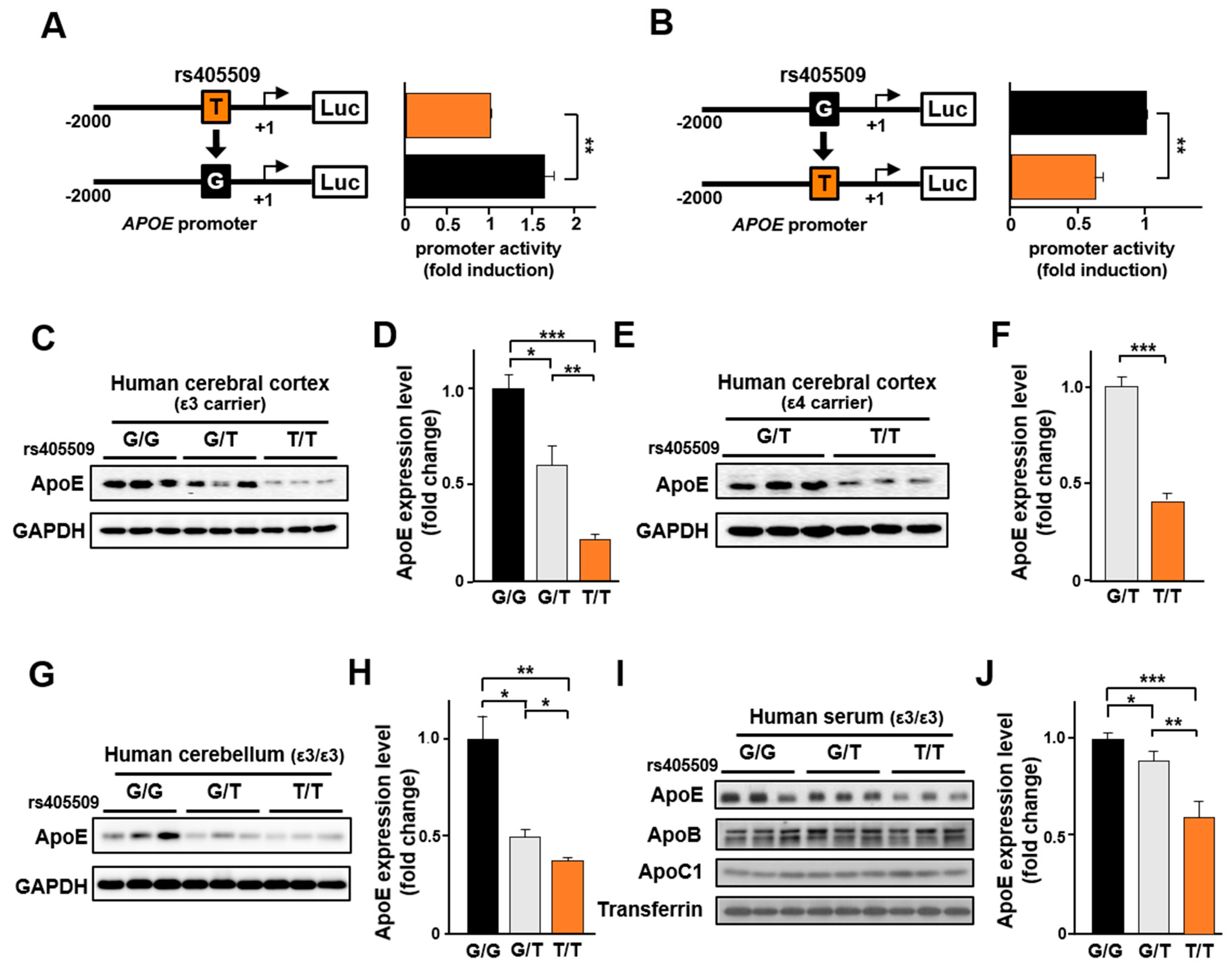
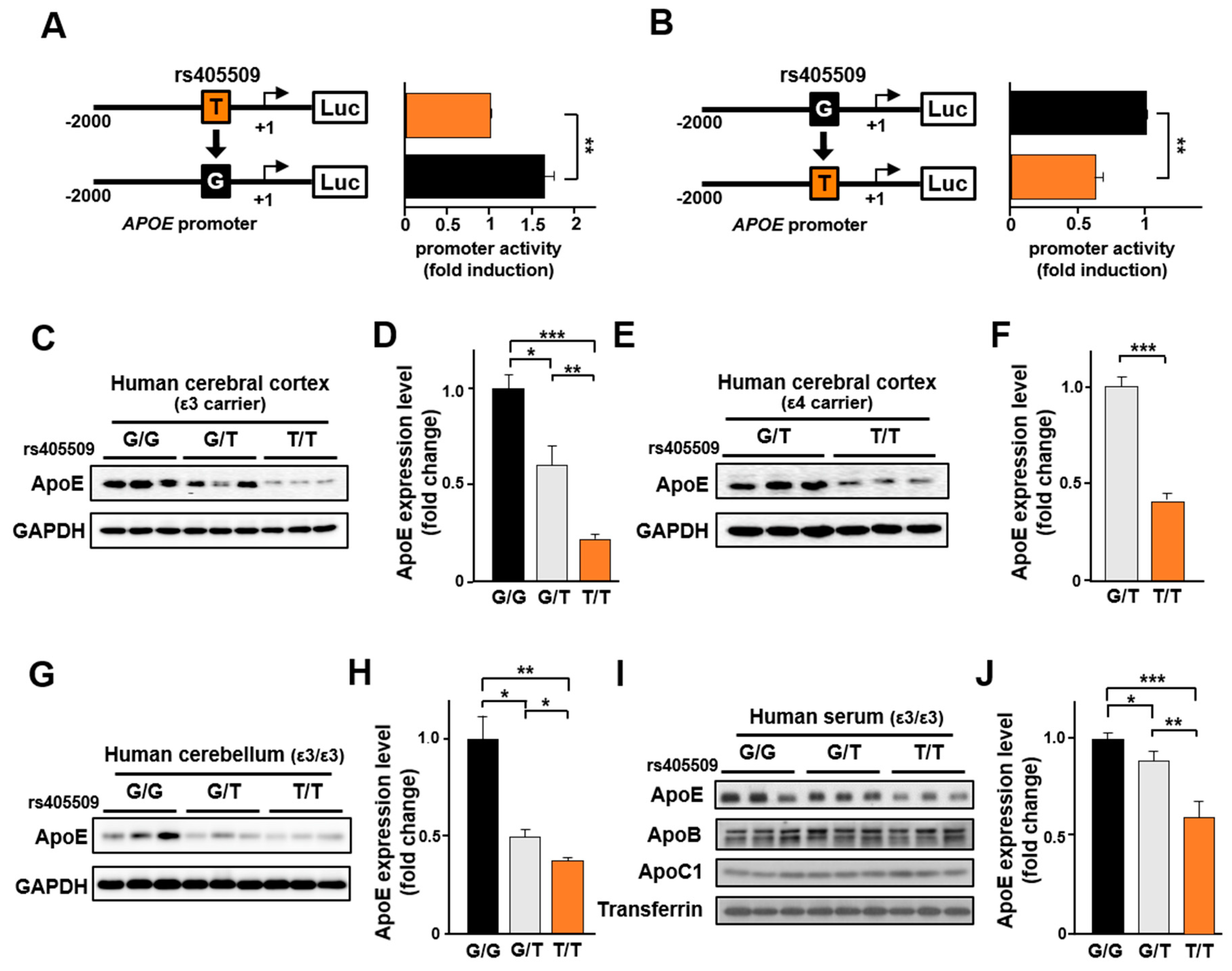
3.5. Rs405509 Regulates APOE Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Barnes, D.E.; Yaffe, K. The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011, 10, 819–828. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2012 Alzheimer’s disease facts and figures. Alzheimers Dement. 2012, 8, 131–168. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr. Alzheimer disease: New concepts on its neurobiology and the clinical role imaging will play. Radiology 2012, 263, 344–361. [Google Scholar] [CrossRef] [PubMed]
- Trojanowski, J.Q.; Vandeerstichele, H.; Korecka, M.; Clark, C.M.; Aisen, P.S.; Petersen, R.C.; Blennow, K.; Soares, H.; Simon, A.; Lewczuk, P.; et al. Update on the biomarker core of the Alzheimer’s Disease Neuroimaging Initiative subjects. Alzheimers Dement. 2010, 6, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Rocchi, A.; Pellegrini, S.; Siciliano, G.; Murri, L. Causative and susceptibility genes for Alzheimer’s disease: A review. Brain Res. Bull. 2003, 61, 1–24. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef] [PubMed]
- Myers, R.H.; Schaefer, E.J.; Wilson, P.W.; D’Agostino, R.; Ordovas, J.M.; Espino, A.; Au, R.; White, R.F.; Knoefel, J.E.; Cobb, J.L.; et al. Apolipoprotein E epsilon4 association with dementia in a population-based study: The Framingham study. Neurology 1996, 46, 673–677. [Google Scholar] [CrossRef]
- Gomez-Tortosa, E.; Barquero, M.S.; Baron, M.; Sainz, M.J.; Manzano, S.; Payno, M.; Ros, R.; Almaraz, C.; Gomez-Garre, P.; Jimenez-Escrig, A. Variability of age at onset in siblings with familial Alzheimer disease. Arch. Neurol. 2007, 64, 1743–1748. [Google Scholar] [CrossRef]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: A meta-analysis. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Risch, N.J.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C., Jr.; Rimmler, J.B.; Locke, P.A.; Conneally, P.M.; Schmader, K.E.; et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat. Genet. 1994, 7, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Anstey, K.; Christensen, H. Education, activity, health, blood pressure and apolipoprotein E as predictors of cognitive change in old age: A review. Gerontology 2000, 46, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Caselli, R.J.; Dueck, A.C.; Osborne, D.; Sabbagh, M.N.; Connor, D.J.; Ahern, G.L.; Baxter, L.C.; Rapcsak, S.Z.; Shi, J.; Woodruff, B.K.; et al. Longitudinal modeling of age-related memory decline and the APOE epsilon4 effect. New Engl. J. Med. 2009, 361, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Craft, S.; Teri, L.; Edland, S.D.; Kukull, W.A.; Schellenberg, G.; McCormick, W.C.; Bowen, J.D.; Larson, E.B. Accelerated decline in apolipoprotein E-epsilon4 homozygotes with Alzheimer’s disease. Neurology 1998, 51, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Mayeux, R.; Small, S.A.; Tang, M.; Tycko, B.; Stern, Y. Memory performance in healthy elderly without Alzheimer’s disease: Effects of time and apolipoprotein-E. Neurobiol. Aging 2001, 22, 683–689. [Google Scholar] [CrossRef]
- Van Gerven, P.W.; Van Boxtel, M.P.; Ausems, E.E.; Bekers, O.; Jolles, J. Do apolipoprotein E genotype and educational attainment predict the rate of cognitive decline in normal aging? A 12-year follow-up of the Maastricht Aging Study. Neuropsychology 2012, 26, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Beaudreau, S.A.; Kaci Fairchild, J.; Spira, A.P.; Lazzeroni, L.C.; O’Hara, R. Neuropsychiatric symptoms, apolipoprotein E gene, and risk of progression to cognitive impairment, no dementia and dementia: The Aging, Demographics, and Memory Study (ADAMS). Int. J. Geriatr. Psychiatry 2013, 28, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Jorm, A.F.; Mather, K.A.; Butterworth, P.; Anstey, K.J.; Christensen, H.; Easteal, S. APOE genotype and cognitive functioning in a large age-stratified population sample. Neuropsychology 2007, 21, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kleiman, T.; Zdanys, K.; Black, B.; Rightmer, T.; Grey, M.; Garman, K.; Macavoy, M.; Gelernter, J.; van Dyck, C. Apolipoprotein E epsilon4 allele is unrelated to cognitive or functional decline in Alzheimer’s disease: Retrospective and prospective analysis. Dement. Geriatr. Cogn. Disord. 2006, 22, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Deary, I.J.; Whiteman, M.C.; Pattie, A.; Starr, J.M.; Hayward, C.; Wright, A.F.; Carothers, A.; Whalley, L.J. Cognitive change and the APOE epsilon 4 allele. Nature 2002, 418, 932. [Google Scholar] [CrossRef] [PubMed]
- Farlow, M.R.; He, Y.; Tekin, S.; Xu, J.; Lane, R.; Charles, H.C. Impact of APOE in mild cognitive impairment. Neurology 2004, 63, 1898–1901. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, H.; Crivello, F.; Dufouil, C.; Grassiot, B.; Tzourio, C.; Alperovitch, A.; Mazoyer, B. No epsilon4 gene dose effect on hippocampal atrophy in a large MRI database of healthy elderly subjects. Neuroimage 2005, 24, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Crivello, F.; Lemaitre, H.; Dufouil, C.; Grassiot, B.; Delcroix, N.; Tzourio-Mazoyer, N.; Tzourio, C.; Mazoyer, B. Effects of ApoE-epsilon4 allele load and age on the rates of grey matter and hippocampal volumes loss in a longitudinal cohort of 1186 healthy elderly persons. Neuroimage 2010, 53, 1064–1069. [Google Scholar] [CrossRef]
- Gureje, O.; Ogunniyi, A.; Baiyewu, O.; Price, B.; Unverzagt, F.W.; Evans, R.M.; Smith-Gamble, V.; Lane, K.A.; Gao, S.; Hall, K.S.; et al. APOE epsilon4 is not associated with Alzheimer’s disease in elderly Nigerians. Ann. Neurol. 2006, 59, 182–185. [Google Scholar] [CrossRef] [PubMed]
- Rassas, A.A.; Mrabet Khiari, H.; Hadj Fredj, S.; Sahnoun, S.; Batti, H.; Zakraoui, N.O.; Cherif, A.; Anane, N.; Ben Ali, N.; Messaoud, T.; et al. High APOE epsilon 4 allele frequencies associated with Alzheimer disease in a Tunisian population. Neurol. Sci. 2012, 33, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.X.; Stern, Y.; Marder, K.; Bell, K.; Gurland, B.; Lantigua, R.; Andrews, H.; Feng, L.; Tycko, B.; Mayeux, R. The APOE-epsilon4 allele and the risk of Alzheimer disease among African Americans, whites, and Hispanics. JAMA 1998, 279, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C.; Jun, G.; Naj, A.; Rajbhandary, R.; Vardarajan, B.N.; Wang, L.S.; Valladares, O.; Lin, C.F.; Larson, E.B.; Graff-Radford, N.R.; et al. Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E 4,and the risk of late-onset Alzheimer disease in African Americans. JAMA 2013, 309, 1483–1492. [Google Scholar] [CrossRef]
- Singh, P.P.; Singh, M.; Mastana, S.S. APOE distribution in world populations with new data from India and the UK. Ann. Hum. Biol. 2006, 33, 279–308. [Google Scholar] [CrossRef] [PubMed]
- Eto, M.; Watanabe, K.; Ishii, K. A racial difference in apolipoprotein E allele frequencies between the Japanese and Caucasian populations. Clin. Genet. 1986, 30, 422–427. [Google Scholar] [CrossRef]
- Hendrie, H.C.; Murrell, J.; Gao, S.; Unverzagt, F.W.; Ogunniyi, A.; Hall, K.S. International studies in dementia with particular emphasis on populations of African origin. Alzheimer Dis. Assoc. Disord. 2006, 20, S42–S46. [Google Scholar] [CrossRef]
- Farrer, L.A.; Friedland, R.P.; Bowirrat, A.; Waraska, K.; Korczyn, A.; Baldwin, C.T. Genetic and environmental epidemiology of Alzheimer’s disease in arabs residing in Israel. J. Mol. Neurosci. 2003, 20, 207–212. [Google Scholar] [CrossRef]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984, 34, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Go, M.J.; Hu, C.; Hong, C.B.; Kim, Y.K.; Lee, J.Y.; Hwang, J.Y.; Oh, J.H.; Kim, D.J.; Kim, N.H.; et al. Large-scale genome-wide association studies in East Asians identify new genetic loci influencing metabolic traits. Nat. Genet. 2011, 43, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Han, B.G.; Ko, G.E.S.G. Cohort Profile: The Korean Genome and Epidemiology Study (KoGES) Consortium. Int. J. Epidemiol. 2017, 46, 1350. [Google Scholar] [CrossRef] [PubMed]
- Jee, Y.H.; Lee, S.J.; Jung, K.J.; Jee, S.H. Alcohol Intake and Serum Glucose Levels from the Perspective of a Mendelian Randomization Design: The KCPS-II Biobank. PLoS ONE 2016, 11, e0162930. [Google Scholar] [CrossRef] [PubMed]
- Jun, G.R.; Chung, J.; Mez, J.; Barber, R.; Beecham, G.W.; Bennett, D.A.; Buxbaum, J.D.; Byrd, G.S.; Carrasquillo, M.M.; Crane, P.K.; et al. Transethnic genome-wide scan identifies novel Alzheimer’s disease loci. Alzheimers Dement. 2017, 13, 727–738. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, S.; Das, S.; Kretzschmar, W.; Delaneau, O.; Wood, A.R.; Teumer, A.; Kang, H.M.; Fuchsberger, C.; Danecek, P.; Sharp, K.; et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet. 2016, 48, 1279–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, R.; Boutin, T.S.; Marten, J.; Huffman, J.E.; Kerr, S.M.; Campbell, A.; Evenden, L.; Gibson, J.; Amador, C.; Howard, D.M.; et al. Exploration of haplotype research consortium imputation for genome-wide association studies in 20,032 Generation Scotland participants. Genome Med. 2017, 9, 23. [Google Scholar] [CrossRef]
- Surakka, I.; Sarin, A.P.; Ruotsalainen, S.E.; Durbin, R.; Salomaa, V.; Daly, M.; Palotie, A.; Ripatti, S. The rate of false polymorphisms introduced when imputing genotypes from global imputation panels. BioRxiv 2016. [Google Scholar] [CrossRef]
- Patterson, N.; Price, A.L.; Reich, D. Population structure and eigenanalysis. PLoS Genet. 2006, 2, e190. [Google Scholar] [CrossRef]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Viechtbauer, W. Conducting Meta-Analyses in R with the metafor Package. J. Stat. Softw. 2010, 36, 1–48. [Google Scholar] [CrossRef]
- Zubenko, G.S.; Hughes, H.B.; Zubenko, W.N. D10S1423 Identifies a Susceptibility Locus for Alzheimer’s Disease (AD7) in a Prospective, Longitudinal, Double-Blind Study of Asymptomatic Individuals: Results at 14 Years. Am. J. Med Genet. Part B Neuropsychiatr. Genet. 2010, 153B, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Fan, M.; Liu, B.; Zhou, Y.; Zhen, X.; Xu, C.; Jiang, T. Cortical thickness is associated with different apolipoprotein E genotypes in healthy elderly adults. Neurosci. Lett. 2010, 479, 332–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, J.E.; Adler, R.J. Euler Characteristics for Gaussian Fields on Manifolds. Ann. Probab. 2003, 31, 533–563. [Google Scholar] [CrossRef]
- Hickie, I.; Naismith, S.; Ward, P.B.; Turner, K.; Scott, E.; Mitchell, P.; Wilhelm, K.; Parker, G. Reduced hippocampal volumes and memory loss in patients with early- and late-onset depression. Br. J. Psychiatry 2005, 186, 197. [Google Scholar] [CrossRef]
- Zierhut, K.C.; Graßmann, R.; Kaufmann, J.; Steiner, J.; Bogerts, B.; Schiltz, K. Hippocampal CA1 deformity is related to symptom severity and antipsychotic dosage in schizophrenia. Brain 2013, 136, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Fleisher, A.; Grundman, M.; Jack, C.R.; Petersen, R.C.; Taylor, C.; Kim, H.T.; Schiller, D.H.; Bagwell, V.; Sencakova, D.; Weiner, M.F.; et al. Sex, Apolipoprotein E ε4 status, and hippocampal volume in mild cognitive impairment. Arch. Neurol. 2005, 62, 953–957. [Google Scholar] [CrossRef]
- Michaelson, D.M. APOE epsilon4: The most prevalent yet understudied risk factor for Alzheimer’s disease. Alzheimers Dement. 2014, 10, 861–868. [Google Scholar] [CrossRef]
- Tosto, G.; Fu, H.; Vardarajan, B.N.; Lee, J.H.; Cheng, R.; Reyes-Dumeyer, D.; Lantigua, R.; Medrano, M.; Jimenez-Velazquez, I.Z.; Elkind, M.S.; et al. F-box/LRR-repeat protein 7 is genetically associated with Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2015, 2, 810–820. [Google Scholar] [CrossRef]
- Agarwal, R.; Tripathi, C.B. Association of apolipoprotein E genetic variation in Alzheimer’s disease in Indian population: A meta-analysis. Am. J. Alzheimers Dis. Other Dement. 2014, 29, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Li, H.L.; Liu, Z.J.; Tao, Q.Q.; Xu, M.; Guo, Q.H.; Hong, Z.; Sun, Y.M. Associations between apolipoprotein E gene polymorphisms and Alzheimer’s disease risk in a large Chinese Han population. Clin. Interv. Aging 2015, 10, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Bowirrat, A.; Friedland, R.P.; Chapman, J.; Korczyn, A.D. The very high prevalence of AD in an Arab population is not explained by APOE epsilon4 allele frequency. Neurology 2000, 55, 731. [Google Scholar] [CrossRef] [PubMed]
- Farrer, L.A. Intercontinental epidemiology of Alzheimer disease: A global approach to bad gene hunting. JAMA 2001, 285, 796–798. [Google Scholar] [CrossRef] [PubMed]
- Bachman, D.L.; Green, R.C.; Benke, K.S.; Cupples, L.A.; Farrer, L.A.; Group, M.S. Comparison of Alzheimer’s disease risk factors in white and African American families. Neurology 2003, 60, 1372–1374. [Google Scholar] [CrossRef] [PubMed]
- Donix, M.; Burggren, A.C.; Suthana, N.A.; Siddarth, P.; Ekstrom, A.D.; Krupa, A.K.; Jones, M.; Rao, A.; Martin-Harris, L.; Ercoli, L.M.; et al. Longitudinal changes in medial temporal cortical thickness in normal subjects with the APOE-4 polymorphism. Neuroimage 2010, 53, 37–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, W.K.; Hallmayer, J.; O’Hara, R.; Alzheimer’s Disease Neuroimaging Initiative. Design considerations for characterizing psychiatric trajectories across the lifespan: Application to effects of APOE-epsilon4 on cerebral cortical thickness in Alzheimer’s disease. Am. J. Psychiatry 2011, 168, 894–903. [Google Scholar] [CrossRef]
- Foley, J.M.; Salat, D.H.; Stricker, N.H.; McGlinchey, R.E.; Milberg, W.P.; Grande, L.J.; Leritz, E.C. Glucose Dysregulation Interacts With APOE-4 to Potentiate Temporoparietal Cortical Thinning. Am. J. Alzheimers Dis. Other Dement. 2016, 31, 76–86. [Google Scholar] [CrossRef]
- Lambert, J.C.; Pasquier, F.; Cottel, D.; Frigard, B.; Amouyel, P.; Chartier-Harlin, M.C. A new polymorphism in the APOE promoter associated with risk of developing Alzheimer’s disease. Hum. Mol. Genet. 1998, 7, 533–540. [Google Scholar] [CrossRef]
- Lambert, J.C.; Araria-Goumidi, L.; Myllykangas, L.; Ellis, C.; Wang, J.C.; Bullido, M.J.; Harris, J.M.; Artiga, M.J.; Hernandez, D.; Kwon, J.M.; et al. Contribution of APOE promoter polymorphisms to Alzheimer’s disease risk. Neurology 2002, 59, 59–66. [Google Scholar] [CrossRef]
- Bertram, L.; McQueen, M.B.; Mullin, K.; Blacker, D.; Tanzi, R.E. Systematic meta-analyses of Alzheimer disease genetic association studies: The AlzGene database. Nat. Genet. 2007, 39, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Lescai, F.; Chiamenti, A.M.; Codemo, A.; Pirazzini, C.; D’Agostino, G.; Ruaro, C.; Ghidoni, R.; Benussi, L.; Galimberti, D.; Esposito, F.; et al. An APOE haplotype associated with decreased epsilon4 expression increases the risk of late onset Alzheimer’s disease. J. Alzheimers Dis. JAD 2011, 24, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.Q.; Rogaeva, E.; Premkumar, S.; Brindle, N.; Kawarai, T.; Orlacchio, A.; Yu, G.; Levesque, G.; Nishimura, M.; Ikeda, M.; et al. Absence of association between Alzheimer disease and the −491 regulatory region polymorphism of APOE. Neurosci. Lett. 1998, 250, 189–192. [Google Scholar] [CrossRef]
- Rebeck, G.W.; Cheung, B.S.; Growdon, W.B.; Deng, A.; Akuthota, P.; Locascio, J.; Greenberg, S.M.; Hyman, B.T. Lack of independent associations of apolipoprotein E promoter and intron 1 polymorphisms with Alzheimer’s disease. Neurosci. Lett. 1999, 272, 155–158. [Google Scholar] [CrossRef]
- Toji, H.; Maruyama, H.; Sasaki, K.; Nakamura, S.; Kawakami, H. Apolipoprotein E promoter polymorphism and sporadic Alzheimer’s disease in a Japanese population. Neurosci. Lett. 1999, 259, 56–58. [Google Scholar] [CrossRef]
- Wang, J.C.; Kwon, J.M.; Shah, P.; Morris, J.C.; Goate, A. Effect of APOE genotype and promoter polymorphism on risk of Alzheimer’s disease. Neurology 2000, 55, 1644–1649. [Google Scholar] [CrossRef]
- Zurutuza, L.; Verpillat, P.; Raux, G.; Hannequin, D.; Puel, M.; Belliard, S.; Michon, A.; Pothin, Y.; Camuzat, A.; Penet, C.; et al. APOE promoter polymorphisms do not confer independent risk for Alzheimer’s disease in a French population. Eur. J. Hum. Genet. 2000, 8, 713–716. [Google Scholar] [CrossRef]
- Xiao, H.; Gao, Y.; Liu, L.; Li, Y. Association between polymorphisms in the promoter region of the apolipoprotein E (APOE) gene and Alzheimer’s disease: A meta-analysis. EXCLI J. 2017, 16, 921–938. [Google Scholar] [CrossRef]
- Ma, C.; Zhang, Y.; Li, X.; Chen, Y.; Zhang, J.; Liu, Z.; Chen, K.; Zhang, Z. The TT allele of rs405509 synergizes with APOE epsilon4 in the impairment of cognition and its underlying default mode network in non-demented elderly. Curr. Alzheimer Res. 2016, 13, 708–717. [Google Scholar] [CrossRef]
- Tycko, B.; Lee, J.H.; Ciappa, A.; Saxena, A.; Li, C.M.; Feng, L.; Arriaga, A.; Stern, Y.; Lantigua, R.; Shachter, N.; et al. APOE and APOC1 promoter polymorphisms and the risk of Alzheimer disease in African American and Caribbean Hispanic individuals. Arch. Neurol. 2004, 61, 1434–1439. [Google Scholar] [CrossRef]
- Rantalainen, V.; Lahti, J.; Kajantie, E.; Tienari, P.; Eriksson, J.G.; Raikkonen, K. APOE ɛ4, rs405509, and rs440446 promoter and intron-1 polymorphisms and dementia risk in a cohort of elderly Finns—Helsinki Birth Cohort Study. Neurobiol. Aging 2019, 73, 230.e235–230.e238. [Google Scholar] [CrossRef] [PubMed]
- Xin, X.Y.; Ding, J.Q.; Chen, S.D. Apolipoprotein E Promoter Polymorphisms and Risk of Alzheimer’s Disease: Evidence From Meta-Analysis. J. Alzheimers Dis. JAD 2010, 19, 1283–1294. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Zhang, Y.; Li, X.; Zhang, J.; Chen, K.; Liang, Y.; Chen, Y.; Liu, Z.; Zhang, Z. Is there a significant interaction effect between apolipoprotein E rs405509 T/T and ε4 genotypes on cognitive impairment and gray matter volume? Eur. J. Neurol. 2016, 23, 1415–1425. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, P.; Gu, B.; Liu, Z.; Li, X.; Evans, A.C.; Gong, G.; Zhang, Z. The effects of an APOE promoter polymorphism on human cortical morphology during nondemented aging. J. Neurosci. 2015, 35, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, K.L.; Tybjaerg-Hansen, A.; Nordestgaard, B.G.; Frikke-Schmidt, R. Plasma levels of apolipoprotein E and risk of dementia in the general population. Ann. Neurol. 2015, 77, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, K.L.; Tybjaerg-Hansen, A.; Nordestgaard, B.G.; Frikke-Schmidt, R. Plasma apolipoprotein E levels and risk of dementia: A Mendelian randomization study of 106,562 individuals. Alzheimers Dement. 2018, 14, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.C.; Mann, D.; Richard, F.; Tian, J.; Shi, J.; Thaker, U.; Merrot, S.; Harris, J.; Frigard, B.; Iwatsubo, T.; et al. Is there a relation between APOE expression and brain amyloid load in Alzheimer’s disease? J. Neurol. Neurosurg. Psychiatry 2005, 76, 928–933. [Google Scholar] [CrossRef] [PubMed]
- Rezeli, M.; Zetterberg, H.; Blennow, K.; Brinkmalm, A.; Laurell, T.; Hansson, O.; Marko-Varga, G. Quantification of total apolipoprotein E and its specific isoforms in cerebrospinal fluid and blood in Alzheimer’s disease and other neurodegenerative diseases. EuPA Open Proteom. 2015, 8, 137–143. [Google Scholar] [CrossRef]
- Cruchaga, C.; Kauwe, J.S.; Nowotny, P.; Bales, K.; Pickering, E.H.; Mayo, K.; Bertelsen, S.; Hinrichs, A.; Alzheimer’s Disease Neuroimaging Initiative; Fagan, A.M.; et al. Cerebrospinal fluid APOE levels: An endophenotype for genetic studies for Alzheimer’s disease. Hum. Mol. Genet. 2012, 21, 4558–4571. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | N | Control/AD | ε3/ε4 | ε4/ε4 | ||
|---|---|---|---|---|---|---|
| OR (95% CI) a | p | OR (95% CI) a | p | |||
| East Asian b | 19,398 | 17,096/2302 | 4.98 (4.4–5.6) | 2.6 × 10−152 | 25.12 (19.0–33.5) | 2.8 × 10−109 |
| European ancestry (ADGC) | 15,836 | 7417/8419 | 3.83 (3.6–4.1) | 2.0 × 10−270 | 14.35 (12.0–17.1) | 2.3 × 10−187 |
| African ancestry (ADGC) | 4985 | 3462/1523 | 2.49 (2.2–2.9) | 1.3 × 10−35 | 8.17 (6.3–10.7) | 3.0 × 10−54 |
| Population | rs405509 | n | ε3/ε3 | ε3/ε4 | ε4/ε4 | ||
|---|---|---|---|---|---|---|---|
| Odds Ratio (95% CI) | p | Odds Ratio (95% CI) | p | ||||
| East Asian | TT | 9770 | Ref | 5.13 (4.40–5.98) | 5.10 × 10−98 | 27.02 (19.81–37.18) | 8.80 × 10−94 |
| GT | 7941 | Ref | 4.55 (3.69–5.61) | 1.09 × 10−45 | 15.87 (6.32–39.49) | 2.62 × 10−9 | |
| GG | 1681 | Ref | 3.55 (1.38–8.60) | 0.006 | NAa | - | |
| European ancestry (ADGC) | TT | 4713 | Ref | 4.25 (3.71–4.88) | 1.55 × 10−94 | 18.13 (14.02–23.44) | 2.69 × 10−108 |
| GT | 7510 | Ref | 3.89 (3.49–4.34) | 2.55 × 10−134 | 12.63 (9.41–16.94) | 3.44× 10−64 | |
| GG | 3385 | Ref | 3.39 (2.81–4.09) | 4.87 × 10−37 | 8.35 (4.58–15.21) | 4.07 × 10−12 | |
| Total | TT | 14,483 | Ref | 4.62 (4.17–5.11) | 2.60 × 10−187 | 20.96 (17.07–25.73) | 7.69 × 10−186 |
| GT | 15,451 | Ref | 4.10 (3.55–4.74) | 4.53 × 10−176 | 12.90 (9.75–17.07) | 1.05 × 10−71 | |
| GG | 5066 | Ref | 3.40 (2.83–4.08) | 7.16 × 10−39 | 8.44 (4.66–15.27) | 1.84 × 10−12 | |
| Region of Interest | rs405509-TT | rs405509-GG | ||||||
|---|---|---|---|---|---|---|---|---|
| ε4/ε4 vs. ε3/ε3 (n = 175) | ε3/ε4 vs. ε3/ε3 (n = 239) | ε4/ε4 vs. ε3/ε3 (n = 128) | ε3/ε4 vs. ε3/ε3 (n = 161) | |||||
| F-value | p | F-value | p | F-value | p | F-value | p | |
| Medial temporal cortex * | 6.33 | 0.013 | 1.95 | 0.16 | 0.04 | 0.85 | 0.24 | 0.62 |
| Precuneus | 8.27 | 0.005 | 4.58 | 0.03 | 0.01 | 0.92 | 0.10 | 0.75 |
| Hippocampal Volume | 18.13 | 3.4 × 10−5 | 7.40 | 0.04 | 1.43 | 2.65 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, K.Y.; Lee, J.J.; Gunasekaran, T.I.; Kang, S.; Lee, W.; Jeong, J.; Lim, H.J.; Zhang, X.; Zhu, C.; Won, S.-Y.; et al. APOE Promoter Polymorphism-219T/G is an Effect Modifier of the Influence of APOE ε4 on Alzheimer’s Disease Risk in a Multiracial Sample. J. Clin. Med. 2019, 8, 1236. https://doi.org/10.3390/jcm8081236
Choi KY, Lee JJ, Gunasekaran TI, Kang S, Lee W, Jeong J, Lim HJ, Zhang X, Zhu C, Won S-Y, et al. APOE Promoter Polymorphism-219T/G is an Effect Modifier of the Influence of APOE ε4 on Alzheimer’s Disease Risk in a Multiracial Sample. Journal of Clinical Medicine. 2019; 8(8):1236. https://doi.org/10.3390/jcm8081236
Chicago/Turabian StyleChoi, Kyu Yeong, Jang Jae Lee, Tamil Iniyan Gunasekaran, Sarang Kang, Wooje Lee, Jangho Jeong, Ho Jae Lim, Xiaoling Zhang, Congcong Zhu, So-Yoon Won, and et al. 2019. "APOE Promoter Polymorphism-219T/G is an Effect Modifier of the Influence of APOE ε4 on Alzheimer’s Disease Risk in a Multiracial Sample" Journal of Clinical Medicine 8, no. 8: 1236. https://doi.org/10.3390/jcm8081236