Evidence for the Desmosomal Cadherin Desmoglein-3 in Regulating YAP and Phospho-YAP in Keratinocyte Responses to Mechanical Forces

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
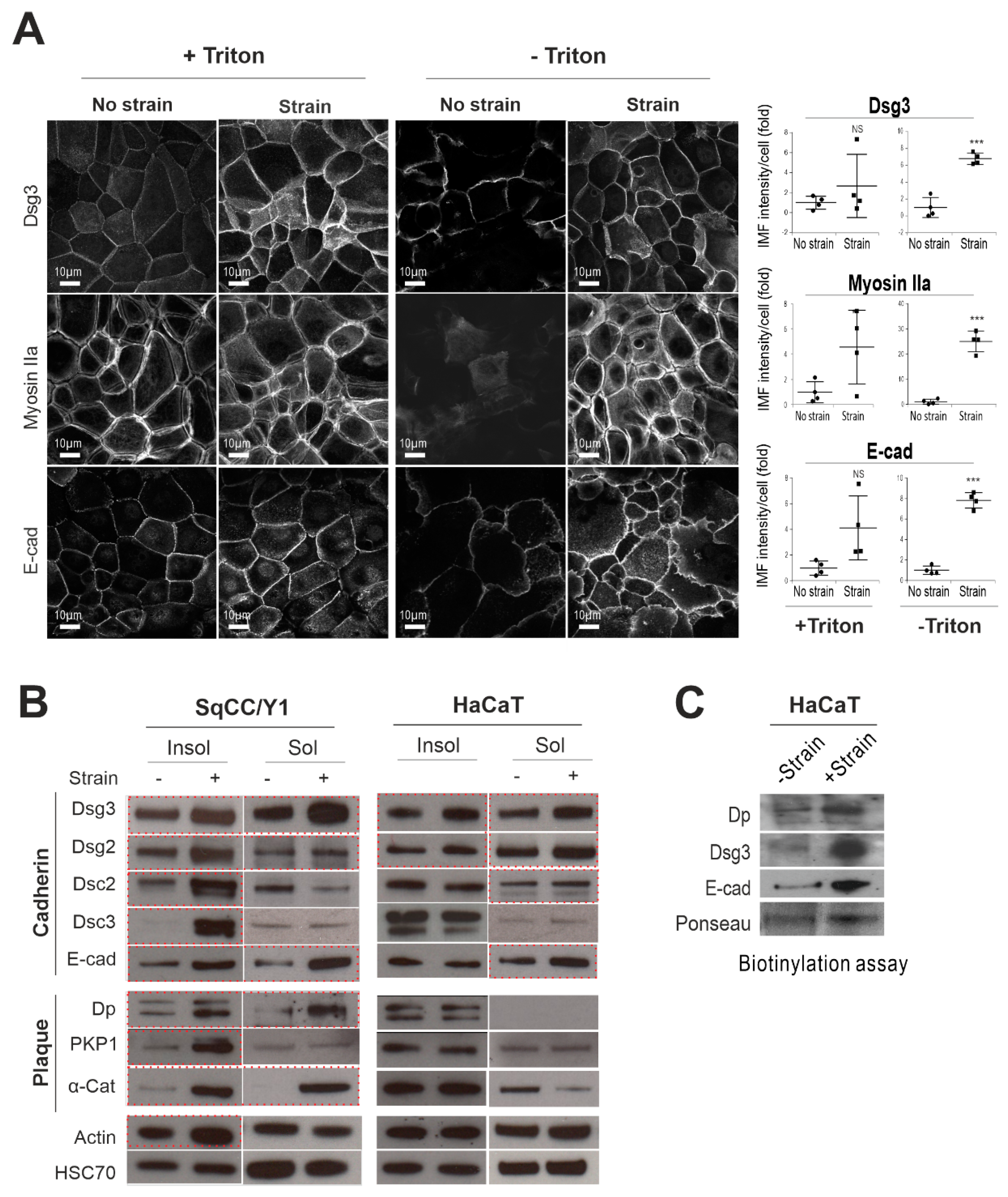
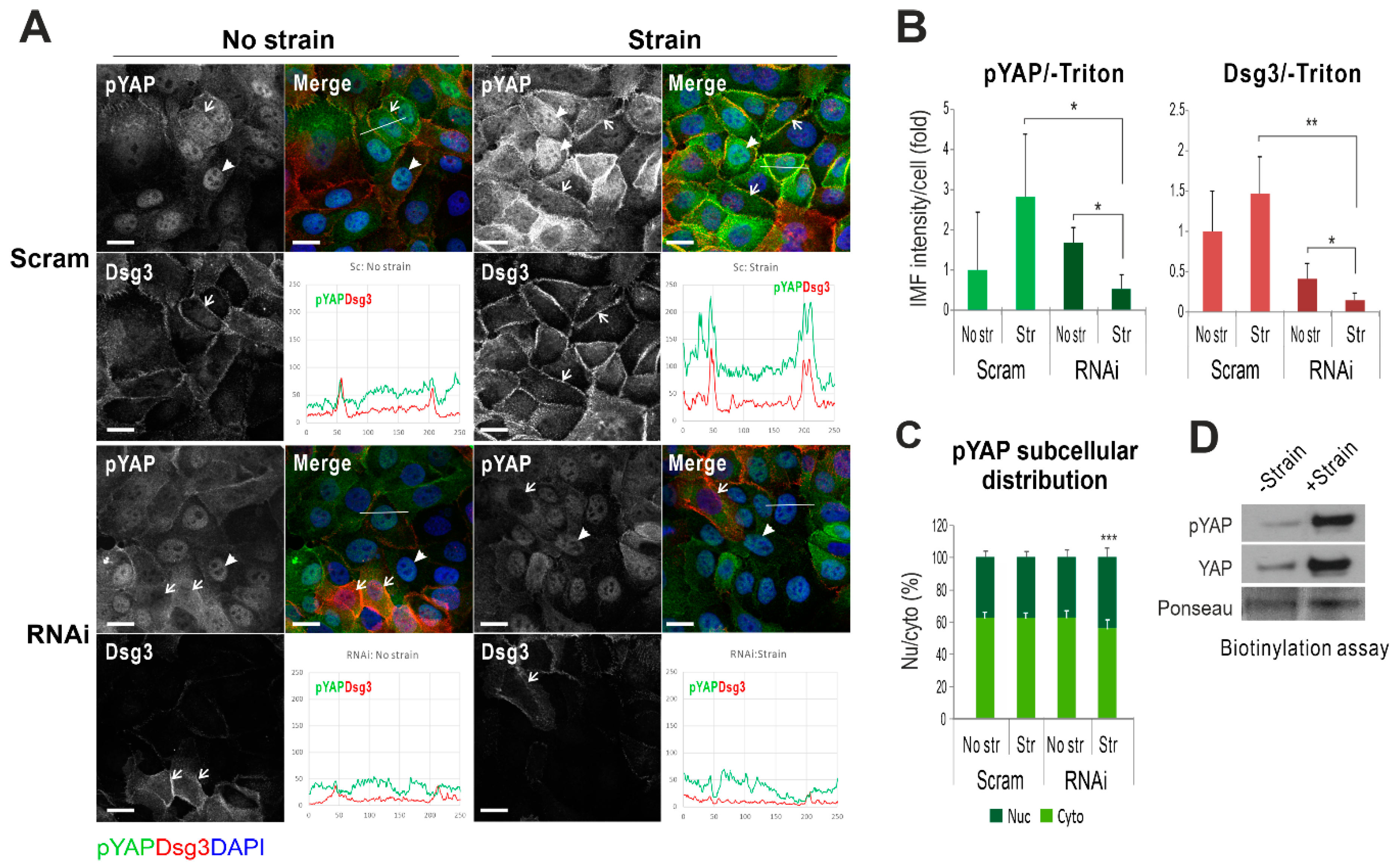
2.1. Mechanical Cyclic Strain Alters Junction Protein Expression and Distribution in Keratinocytes
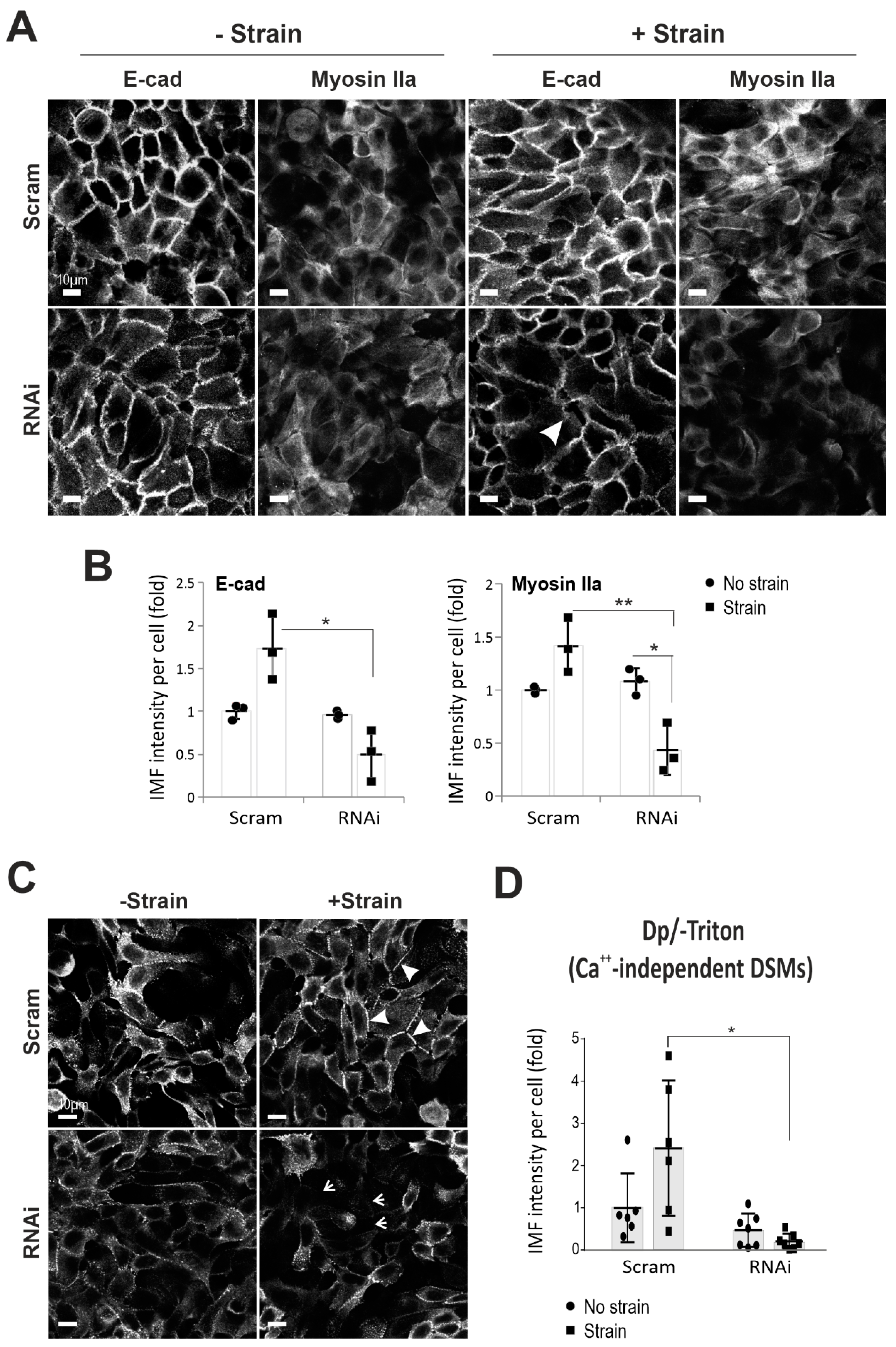
2.2. Dsg3 Is Required for E-cadherin and Actomyosin Junction Assembly in Response to Mechanical Strain
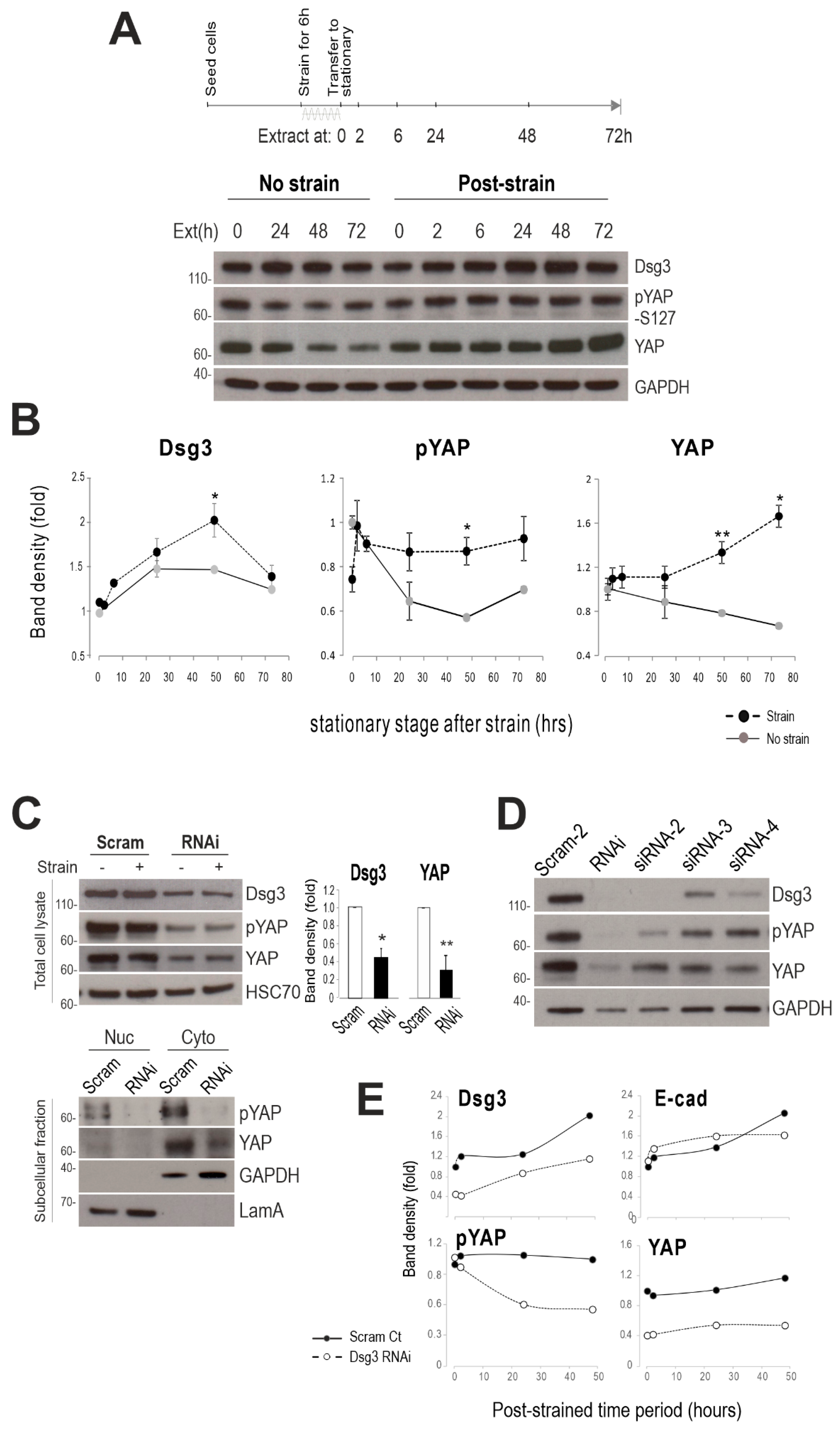
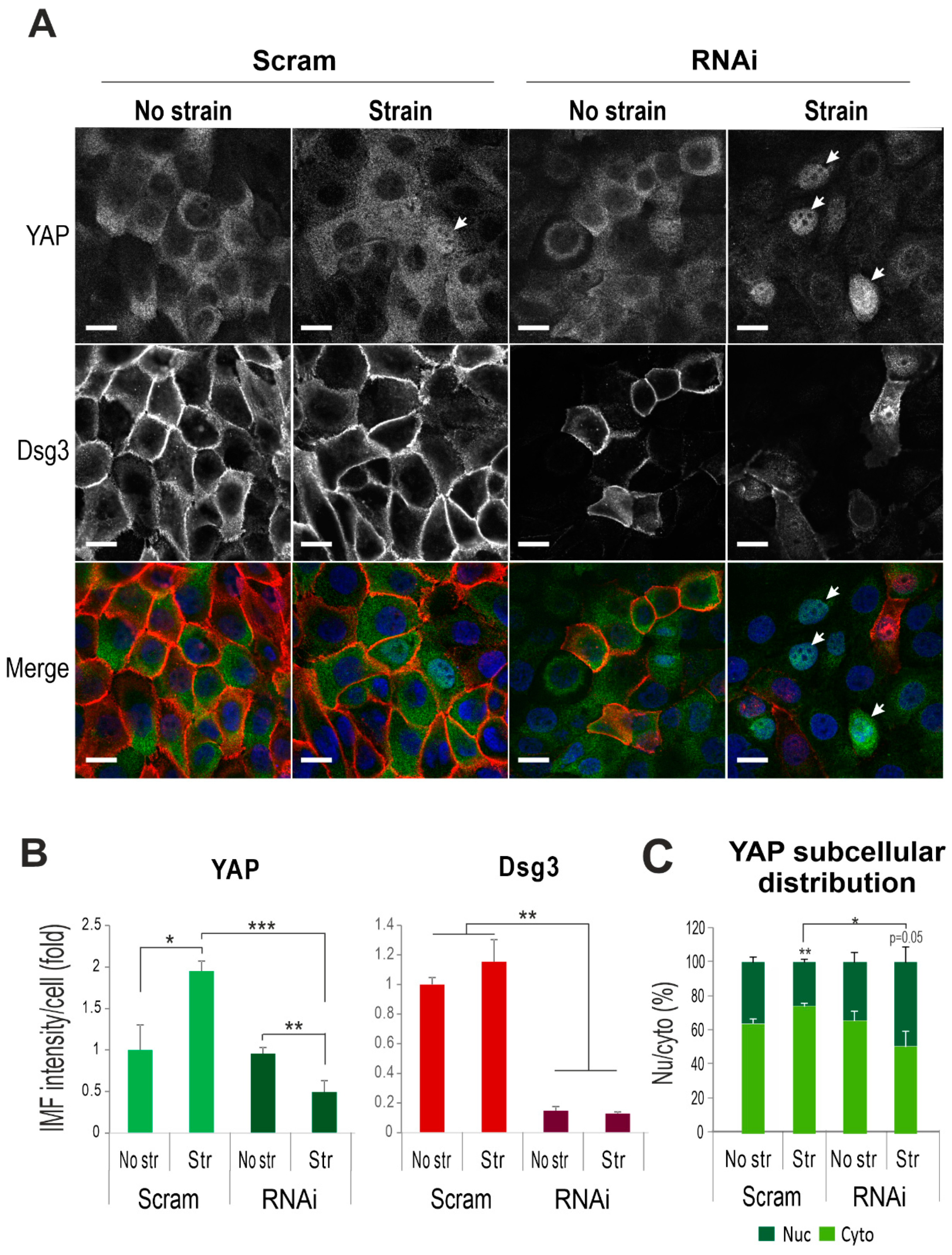
2.3. Dsg3 Regulates the Expression and Localization of YAP in Response to Mechanical Loading
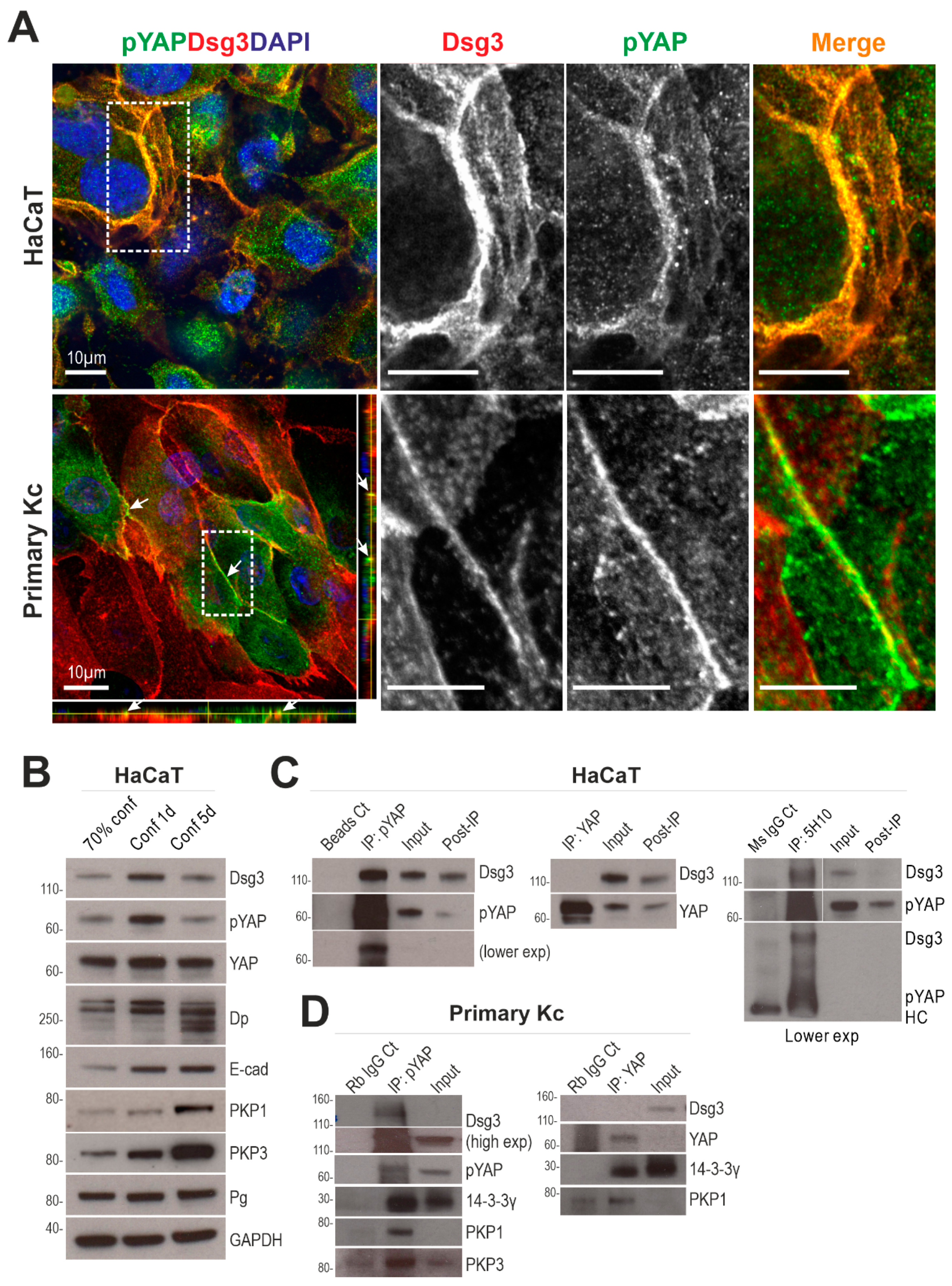
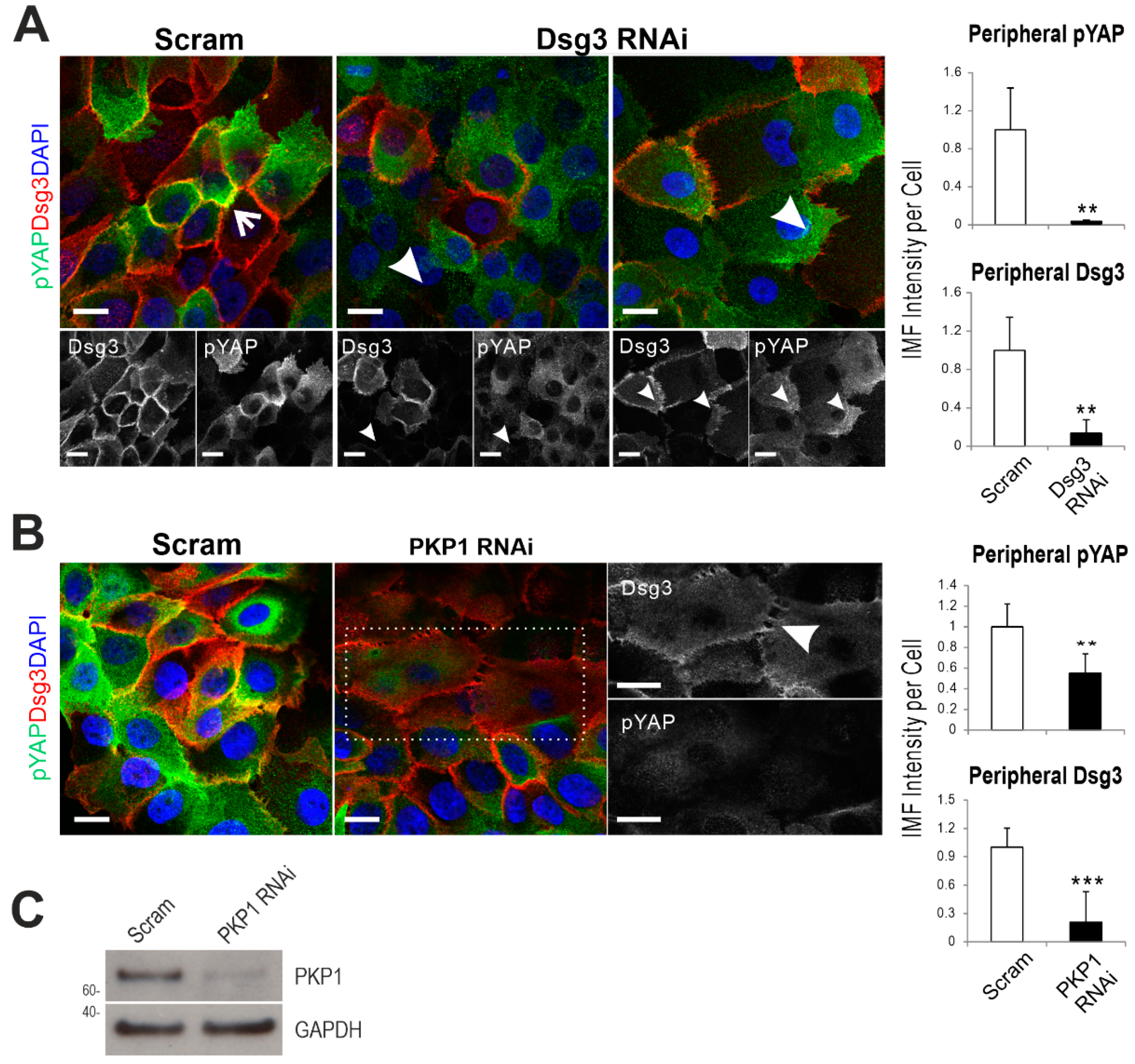
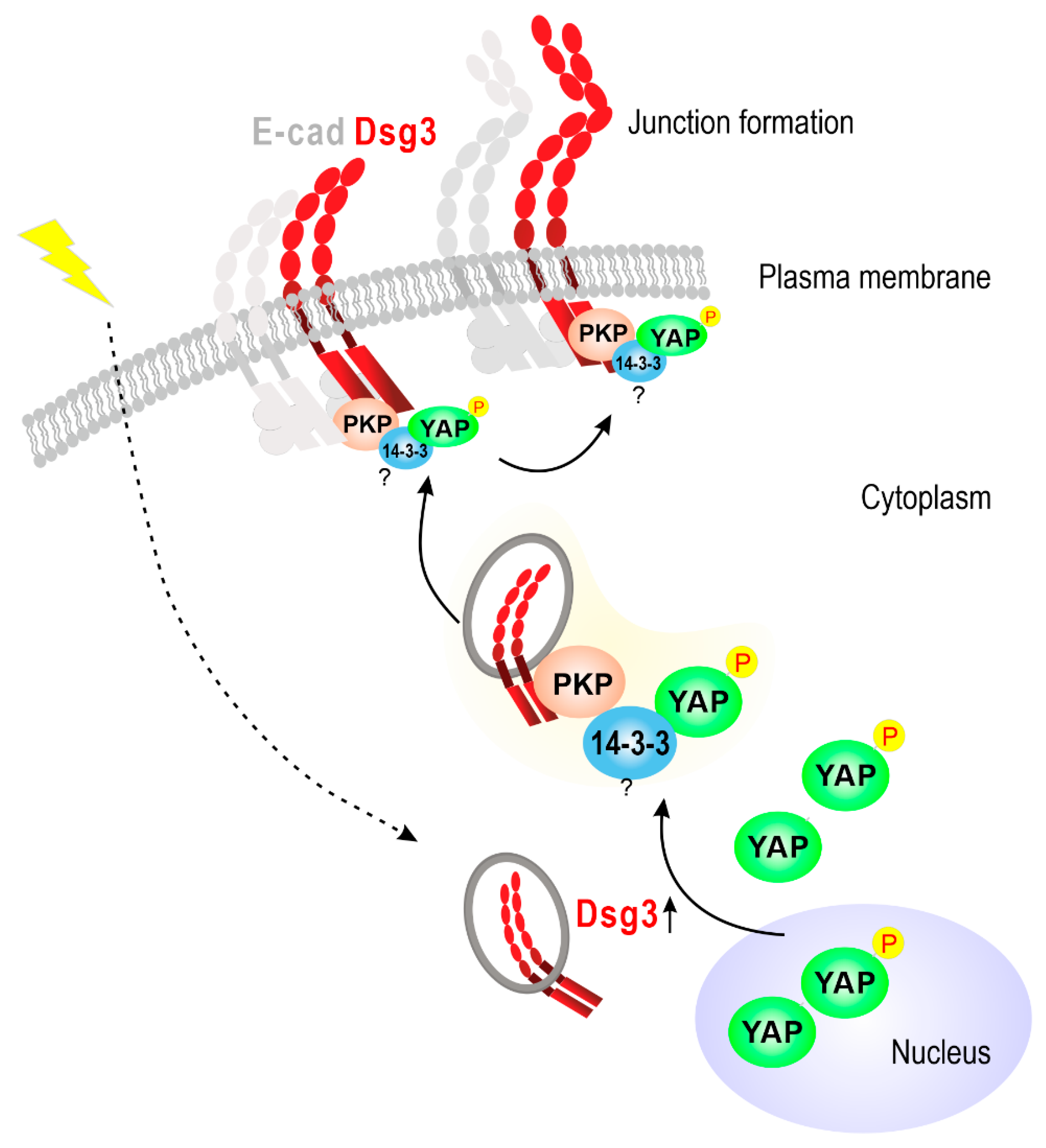
2.4. Dsg3 Colocalizes and Forms a Complex with pYAP, Which Is Sequestered to the Plasma Membrane
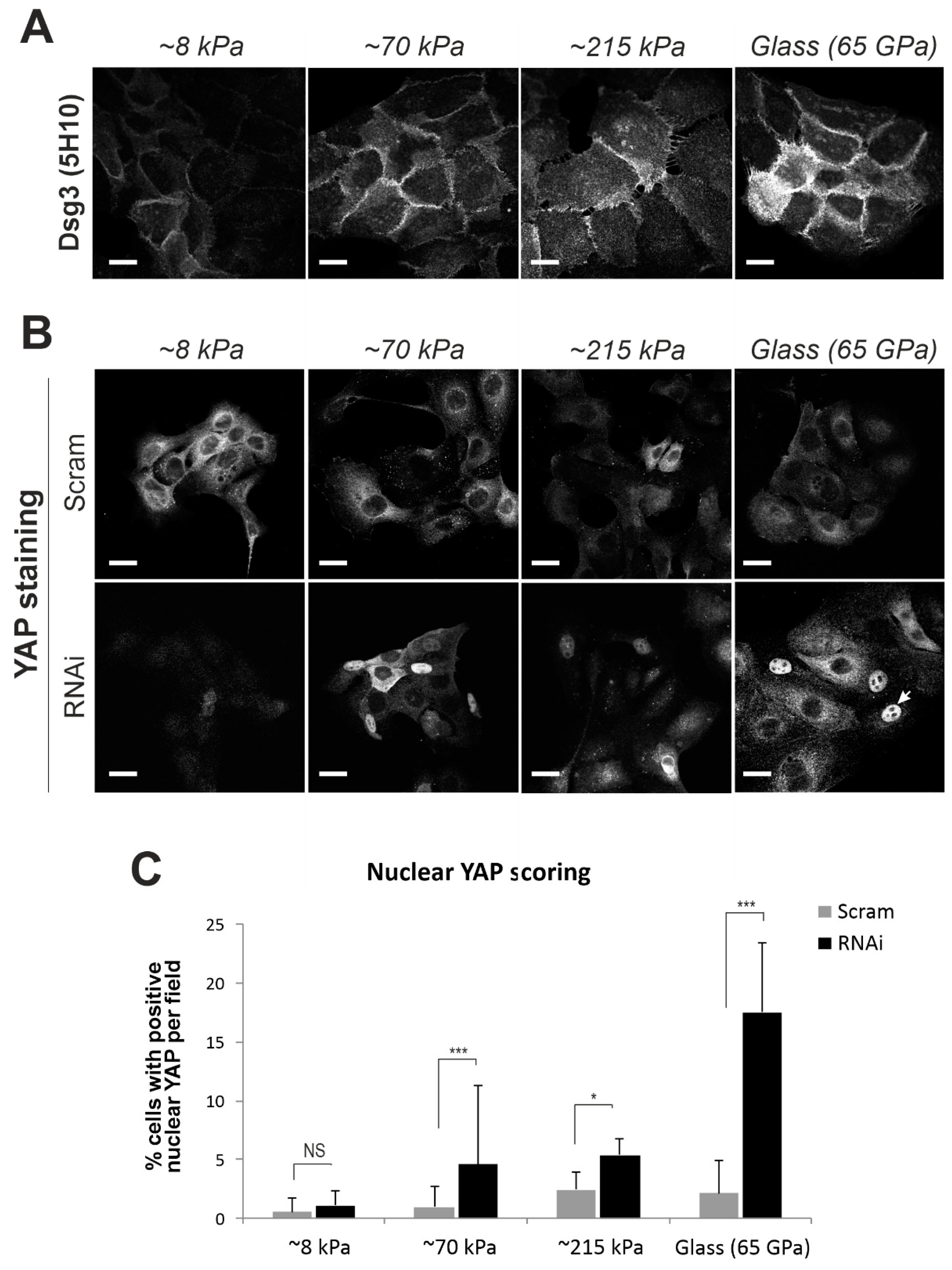
2.5. Elevated Expression of Dsg3 in Response to Substrate Stiffness, Similar to other Force Sensors
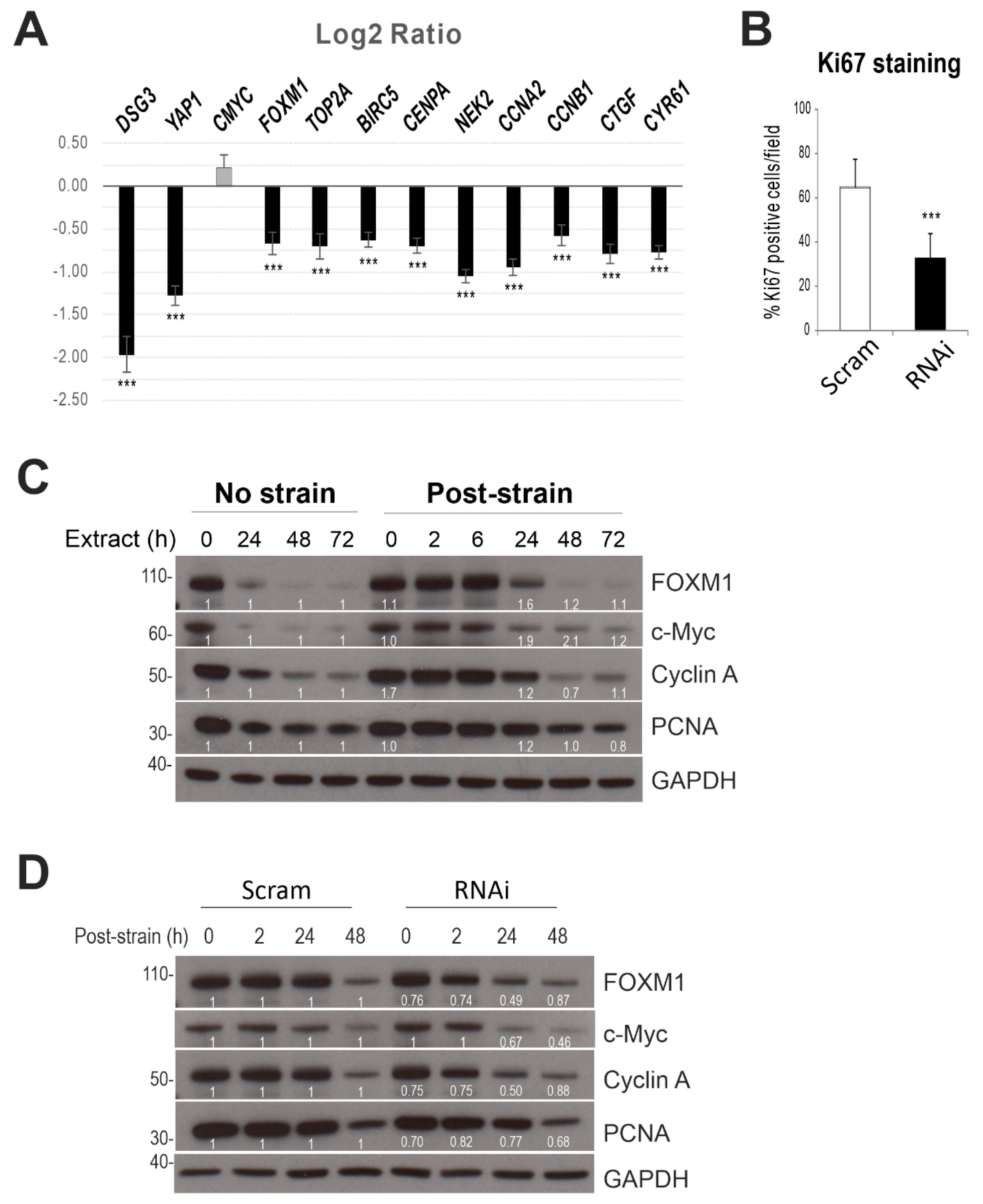
2.6. Dsg3 Knockdown Has an Impact on YAP Target Genes
2.7. Dsg3 Knockdown Has an Impact on Cell Proliferation
3. Discussion
4. Materials and Methods
4.1. Antibodies
4.2. Cell culture and siRNA Transfection
4.3. Application of Cyclic Strain
4.4. Calcium-Independent Desmosome Analysis
4.5. Immunofluorescence, Microscopy and Image Analysis
4.6. Preparation and Functionalization of Polyacrylamide (PA) Gels
4.7. Western Blotting, Co-immunoprecipitation, and Biotinylation Assay
4.8. Reverse Transcription Absolute Quantitative RT-PCR (qPCR)
4.9. Ki67 Staining
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aqeilan, R.I. Hippo signaling: To die or not to die. Cell Death Differ. 2013, 20, 1287–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.G.; Koh, E.; Chen, X.; Gumbiner, B.M. E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc. Natl. Acad. Sci. USA 2011, 108, 11930–11935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le, D.J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Low, B.C.; Pan, C.Q.; Shivashankar, G.V.; Bershadsky, A.; Sudol, M.; Sheetz, M. YAP/TAZ as mechanosensors and mechanotransducers in regulating organ size and tumor growth. FEBS Lett. 2014, 588, 2663–2670. [Google Scholar] [CrossRef] [Green Version]
- Panciera, T.; Azzolin, L.; Cordenonsi, M.; Piccolo, S. Mechanobiology of YAP and TAZ in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 758–770. [Google Scholar] [CrossRef]
- Ladoux, B.; Nelson, W.J.; Yan, J.; Mege, R.M. The mechanotransduction machinery at work at adherens junctions. Integr. Biol. 2015, 7, 1109–1119. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, M.A.; DeSimone, D.W. Cell adhesion receptors in mechanotransduction. Curr. Opin. Cell Biol. 2008, 20, 551–556. [Google Scholar] [CrossRef] [Green Version]
- De Rooij, J. Cadherin adhesion controlled by cortical actin dynamics. Nat. Cell Biol. 2014, 16, 508–510. [Google Scholar] [CrossRef]
- Jasaitis, A.; Estevez, M.; Heysch, J.; Ladoux, B.; Dufour, S. E-cadherin-dependent stimulation of traction force at focal adhesions via the Src and PI3K signaling pathways. Biophys. J. 2012, 103, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Leckband, D.E.; le, D.Q.; Wang, N.; de Rooij, J. Mechanotransduction at cadherin-mediated adhesions. Curr. Opin. Cell Biol. 2011, 23, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Leckband, D.E.; de Rooij, J. Cadherin adhesion and mechanotransduction. Annu Rev. Cell Dev. Biol. 2014, 30, 291–315. [Google Scholar] [CrossRef] [PubMed]
- Tzima, E.; Irani-Tehrani, M.; Kiosses, W.B.; Dejana, E.; Schultz, D.A.; Engelhardt, B.; Cao, G.; DeLisser, H.; Schwartz, M.A. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 2005, 437, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Thomason, H.A.; Scothern, A.; McHarg, S.; Garrod, D.R. Desmosomes: Adhesive strength and signalling in health and disease. Biochem. J. 2010, 429, 419–433. [Google Scholar] [CrossRef]
- Chen, S.N.; Gurha, P.; Lombardi, R.; Ruggiero, A.; Willerson, J.T.; Marian, A.J. The hippo pathway is activated and is a causal mechanism for adipogenesis in arrhythmogenic cardiomyopathy. Circ. Res. 2014, 114, 454–468. [Google Scholar] [CrossRef]
- Baddam, S.R.; Arsenovic, P.T.; Narayanan, V.; Duggan, N.R.; Mayer, C.R.; Newman, S.T.; Abutaleb, D.A.; Mohan, A.; Kowalczyk, A.P.; Conway, D.E. The Desmosomal Cadherin Desmoglein-2 Experiences Mechanical Tension as Demonstrated by a FRET-Based Tension Biosensor Expressed in Living Cells. Cells 2018, 7, 66. [Google Scholar] [CrossRef] [Green Version]
- Price, A.J.; Cost, A.L.; Ungewiss, H.; Waschke, J.; Dunn, A.R.; Grashoff, C. Mechanical loading of desmosomes depends on the magnitude and orientation of external stress. Nat. Commun. 2018, 9, 5284. [Google Scholar] [CrossRef]
- Teh, M.T.; Parkinson, E.K.; Thurlow, J.K.; Liu, F.; Fortune, F.; Wan, H. A molecular study of desmosomes identifies a desmoglein isoform switch in head and neck squamous cell carcinoma. J. Oral Pathol. Med. 2011, 40, 67–76. [Google Scholar] [CrossRef]
- Amagai, M.; Klaus-Kovtun, V.; Stanley, J.R. Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell 1991, 67, 869–877. [Google Scholar] [CrossRef]
- Shimizu, A.; Ishiko, A.; Ota, T.; Tsunoda, K.; Koyasu, S.; Amagai, M.; Nishikawa, T. Ultrastructural changes in mice actively producing antibodies to desmoglein 3 parallel those in patients with pemphigus vulgaris. Arch. Dermatol. Res. 2002, 294, 318–323. [Google Scholar] [CrossRef]
- Brown, L.; Wan, H. Desmoglein 3: A help or a hindrance in cancer progression? Cancers 2015, 7, 266–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotzer, V.; Hartlieb, E.; Vielmuth, F.; Gliem, M.; Spindler, V.; Waschke, J. E-cadherin and Src associate with extradesmosomal Dsg3 and modulate desmosome assembly and adhesion. Cell. Mol. Life Sci. 2015, 72, 4885–4897. [Google Scholar] [CrossRef] [PubMed]
- Tsang, S.M.; Liu, L.; Teh, M.T.; Wheeler, A.; Grose, R.; Hart, I.R.; Garrod, D.R.; Fortune, F.; Wan, H. Desmoglein 3, via an interaction with E-cadherin, is associated with activation of Src. PLoS ONE 2010, 5, e14211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, S.M.; Brown, L.; Lin, K.; Liu, L.; Piper, K.; O’Toole, E.A.; Grose, R.; Hart, I.R.; Garrod, D.R.; Fortune, F.; et al. Non-junctional human desmoglein 3 acts as an upstream regulator of Src in E-cadherin adhesion, a pathway possibly involved in the pathogenesis of pemphigus vulgaris. J. Pathol. 2012, 227, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.; Waseem, A.; Cruz, I.N.; Szary, J.; Gunic, E.; Mannan, T.; Unadkat, M.; Yang, M.; Valderrama, F.; O’Toole, E.A.; et al. Desmoglein 3 promotes cancer cell migration and invasion by regulating activator protein 1 and protein kinase C-dependent-Ezrin activation. Oncogene 2014, 33, 2363–2374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, S.M.; Brown, L.; Gadmor, H.; Gammon, L.; Fortune, F.; Wheeler, A.; Wan, H. Desmoglein 3 acting as an upstream regulator of Rho GTPases, Rac-1/Cdc42 in the regulation of actin organisation and dynamics. Exp. Cell Res. 2012, 318, 2269–2283. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.J.; Chang, J.T.; Lee, L.; Wang, H.M.; Liao, C.T.; Chiu, C.C.; Chen, P.J.; Cheng, A.J. DSG3 is overexpressed in head neck cancer and is a potential molecular target for inhibition of oncogenesis. Oncogene 2007, 26, 467–476. [Google Scholar] [CrossRef] [Green Version]
- Ferris, R.L.; Xi, L.; Raja, S.; Hunt, J.L.; Wang, J.; Gooding, W.E.; Kelly, L.; Ching, J.; Luketich, J.D.; Godfrey, T.E. Molecular staging of cervical lymph nodes in squamous cell carcinoma of the head and neck. Cancer Res. 2005, 65, 2147–2156. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.C.; Lee, T.J.; Chang, P.H.; Lee, Y.S.; Chuang, C.C.; Jhang, Y.J.; Chen, Y.W.; Chen, C.W.; Tsai, C.N. Desmoglein 3 is overexpressed in inverted papilloma and squamous cell carcinoma of sinonasal cavity. Laryngoscope 2010, 120, 26–29. [Google Scholar] [CrossRef]
- Savci-Heijink, C.D.; Kosari, F.; Aubry, M.C.; Caron, B.L.; Sun, Z.; Yang, P.; Vasmatzis, G. The role of desmoglein-3 in the diagnosis of squamous cell carcinoma of the lung. Am. J. Pathol. 2009, 174, 1629–1637. [Google Scholar] [CrossRef] [Green Version]
- Solassol, J.; Burcia, V.; Costes, V.; Lacombe, J.; Mange, A.; Barbotte, E.; De Verbizier, D.; Cartier, C.; Makeieff, M.; Crampette, L.; et al. Pemphigus vulgaris antigen mRNA quantification for the staging of sentinel lymph nodes in head and neck cancer. Br. J. Cancer 2010, 102, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Borghi, N.; Sorokina, M.; Shcherbakova, O.G.; Weis, W.I.; Pruitt, B.L.; Nelson, W.J.; Dunn, A.R. E-cadherin is under constitutive actomyosin-generated tension that is increased at cell-cell contacts upon externally applied stretch. Proc. Natl. Acad. Sci. USA 2012, 109, 12568–12573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dufour, S.; Mege, R.M.; Thiery, J.P. alpha-catenin, vinculin, and F-actin in strengthening E-cadherin cell-cell adhesions and mechanosensing. Cell Adhes. Migr. 2013, 7, 345–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Tan, J.L.; Cohen, D.M.; Yang, M.T.; Sniadecki, N.J.; Ruiz, S.A.; Nelson, C.M.; Chen, C.S. Mechanical tugging force regulates the size of cell-cell junctions. Proc. Natl. Acad. Sci. USA 2010, 107, 9944–9949. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.E.; Merritt, A.J.; Garrod, D.R. Calcium-independent desmosomes of keratinocytes are hyper-adhesive. J. Investig. Dermatol. 2007, 127, 775–781. [Google Scholar] [CrossRef]
- Huelter-Hassler, D.; Tomakidi, P.; Steinberg, T.; Jung, B.A. Orthodontic strain affects the Hippo-pathway effector YAP concomitant with proliferation in human periodontal ligament fibroblasts. Eur. J. Orthod. 2017, 39, 251–257. [Google Scholar] [CrossRef]
- Price, A.J.; Huang, E.Y.; Sebastiano, V.; Dunn, A.R. A semi-interpenetrating network of polyacrylamide and recombinant basement membrane allows pluripotent cell culture in a soft, ligand-rich microenvironment. Biomaterials 2017, 121, 179–192. [Google Scholar] [CrossRef] [Green Version]
- Moftah, H.; Dias, K.; Apu, E.H.; Liu, L.; Uttagomol, J.; Bergmeier, L.; Kermorgant, S.; Wan, H. Desmoglein 3 regulates membrane trafficking of cadherins, an implication in cell-cell adhesion. Cell Adhes. Migr. 2017, 11, 211–232. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Wang, K.; Gu, X.; Leong, K.W. Biophysical Regulation of Cell Behavior—Cross Talk between Substrate Stiffness and Nanotopography. Engineering 2017, 3, 36–54. [Google Scholar] [CrossRef]
- Collins, C.; Denisin, A.K.; Pruitt, B.L.; Nelson, W.J. Changes in E-cadherin rigidity sensing regulate cell adhesion. Proc. Natl. Acad. Sci. USA 2017, 114, E5835–E5844. [Google Scholar] [CrossRef] [Green Version]
- Iivarinen, J.T.; Korhonen, R.K.; Jurvelin, J.S. Experimental and numerical analysis of soft tissue stiffness measurement using manual indentation device—Significance of indentation geometry and soft tissue thickness. Skin Res. Technol. 2014, 20, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; Cai, Q.; Xu, Y. FOXM1 is a downstream target of LPA and YAP oncogenic signaling pathways in high grade serous ovarian cancer. Oncotarget 2015, 6, 27688–27699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, A.W.; Meng, Z.; Yuan, H.X.; Plouffe, S.W.; Moon, S.; Kim, W.; Jho, E.H.; Guan, K.L. Osmotic stress-induced phosphorylation by NLK at Ser128 activates YAP. EMBO Rep. 2017, 18, 72–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Rietscher, K.; Keil, R.; Jordan, A.; Hatzfeld, M. 14-3-3 proteins regulate desmosomal adhesion via plakophilins. J. Cell Sci. 2018, 131, jcs212191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonne, S.; Gilbert, B.; Hatzfeld, M.; Chen, X.; Green, K.J.; van Roy, F. Defining desmosomal plakophilin-3 interactions. J. Cell Biol. 2003, 161, 403–416. [Google Scholar] [CrossRef] [PubMed]
- De Beco, S.; Perney, J.B.; Coscoy, S.; Amblard, F. Mechanosensitive Adaptation of E-Cadherin Turnover across adherens Junctions. PLoS ONE 2015, 10, e0128281. [Google Scholar] [CrossRef]
- Samak, G.; Gangwar, R.; Crosby, L.M.; Desai, L.P.; Wilhelm, K.; Waters, C.M.; Rao, R. Cyclic stretch disrupts apical junctional complexes in Caco-2 cell monolayers by a JNK-2-, c-Src-, and MLCK-dependent mechanism. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G947–G958. [Google Scholar] [CrossRef] [Green Version]
- Nasrollahi, S.; Walter, C.; Loza, A.J.; Schimizzi, G.V.; Longmore, G.D.; Pathak, A. Past matrix stiffness primes epithelial cells and regulates their future collective migration through a mechanical memory. Biomaterials 2017, 146, 146–155. [Google Scholar] [CrossRef]
- Yang, C.; Tibbitt, M.W.; Basta, L.; Anseth, K.S. Mechanical memory and dosing influence stem cell fate. Nat. Mater. 2014, 13, 645–652. [Google Scholar] [CrossRef]
- Chen, L.; Arbieva, Z.H.; Guo, S.; Marucha, P.T.; Mustoe, T.A.; DiPietro, L.A. Positional differences in the wound transcriptome of skin and oral mucosa. BMC Genom. 2010, 11, 471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szpaderska, A.M.; Walsh, C.G.; Steinberg, M.J.; DiPietro, L.A. Distinct patterns of angiogenesis in oral and skin wounds. J. Dent. Res. 2005, 84, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Turabelidze, A.; Guo, S.; Chung, A.Y.; Chen, L.; Dai, Y.; Marucha, P.T.; DiPietro, L.A. Intrinsic differences between oral and skin keratinocytes. PLoS ONE 2014, 9, e101480. [Google Scholar] [CrossRef] [PubMed]
- Elosegui-Artola, A.; Andreu, I.; Beedle, A.E.M.; Lezamiz, A.; Uroz, M.; Kosmalska, A.J.; Oria, R.; Kechagia, J.Z.; Rico-Lastres, P.; Le Roux, A.L.; et al. Force Triggers YAP Nuclear Entry by Regulating Transport across Nuclear Pores. Cell 2017, 171, 1397–1410. [Google Scholar] [CrossRef]
- Mannan, T.; Jing, S.; Foroushania, S.H.; Fortune, F.; Wan, H. RNAi-mediated inhibition of the desmosomal cadherin (desmoglein 3) impairs epithelial cell proliferation. Cell Prolif. 2011, 44, 301–310. [Google Scholar] [CrossRef]
- Yang, C.C.; Graves, H.K.; Moya, I.M.; Tao, C.; Hamaratoglu, F.; Gladden, A.B.; Halder, G. Differential regulation of the Hippo pathway by adherens junctions and apical-basal cell polarity modules. Proc. Natl. Acad. Sci. USA 2015, 112, 1785–1790. [Google Scholar] [CrossRef] [Green Version]
- Wan, H.; South, A.P.; Hart, I.R. Increased keratinocyte proliferation initiated through downregulation of desmoplakin by RNA interference. Exp. Cell Res. 2007, 313, 2336–2344. [Google Scholar] [CrossRef]
- Russell, D.; Andrews, P.D.; James, J.; Lane, E.B. Mechanical stress induces profound remodelling of keratin filaments and cell junctions in epidermolysis bullosa simplex keratinocytes. J. Cell Sci. 2004, 117, 5233–5243. [Google Scholar] [CrossRef] [Green Version]
- Fischer, R.S.; Myers, K.A.; Gardel, M.L.; Waterman, C.M. Stiffness-controlled three-dimensional extracellular matrices for high-resolution imaging of cell behavior. Nat. Protoc. 2012, 7, 2056–2066. [Google Scholar] [CrossRef] [Green Version]
- Peyton, S.R.; Putnam, A.J. Extracellular matrix rigidity governs smooth muscle cell motility in a biphasic fashion. J. Cell Physiol. 2005, 204, 198–209. [Google Scholar] [CrossRef]
- Gemenetzidis, E.; Bose, A.; Riaz, A.M.; Chaplin, T.; Young, B.D.; Ali, M.; Sugden, D.; Thurlow, J.K.; Cheong, S.C.; Teo, S.H.; et al. FOXM1 upregulation is an early event in human squamous cell carcinoma and it is enhanced by nicotine during malignant transformation. PLoS ONE 2009, 4, e4849. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uttagomol, J.; Ahmad, U.S.; Rehman, A.; Huang, Y.; Laly, A.C.; Kang, A.; Soetaert, J.; Chance, R.; Teh, M.-T.; Connelly, J.T.; et al. Evidence for the Desmosomal Cadherin Desmoglein-3 in Regulating YAP and Phospho-YAP in Keratinocyte Responses to Mechanical Forces. Int. J. Mol. Sci. 2019, 20, 6221. https://doi.org/10.3390/ijms20246221
Uttagomol J, Ahmad US, Rehman A, Huang Y, Laly AC, Kang A, Soetaert J, Chance R, Teh M-T, Connelly JT, et al. Evidence for the Desmosomal Cadherin Desmoglein-3 in Regulating YAP and Phospho-YAP in Keratinocyte Responses to Mechanical Forces. International Journal of Molecular Sciences. 2019; 20(24):6221. https://doi.org/10.3390/ijms20246221
Chicago/Turabian StyleUttagomol, Jutamas, Usama Sharif Ahmad, Ambreen Rehman, Yunying Huang, Ana C. Laly, Angray Kang, Jan Soetaert, Randy Chance, Muy-Teck Teh, John T. Connelly, and et al. 2019. "Evidence for the Desmosomal Cadherin Desmoglein-3 in Regulating YAP and Phospho-YAP in Keratinocyte Responses to Mechanical Forces" International Journal of Molecular Sciences 20, no. 24: 6221. https://doi.org/10.3390/ijms20246221