Phenotypic Plasticity of Fibroblasts during Mammary Carcinoma Development

 , ,
, ,  , , and
, , and
Abstract
:1. Introduction
2. Results
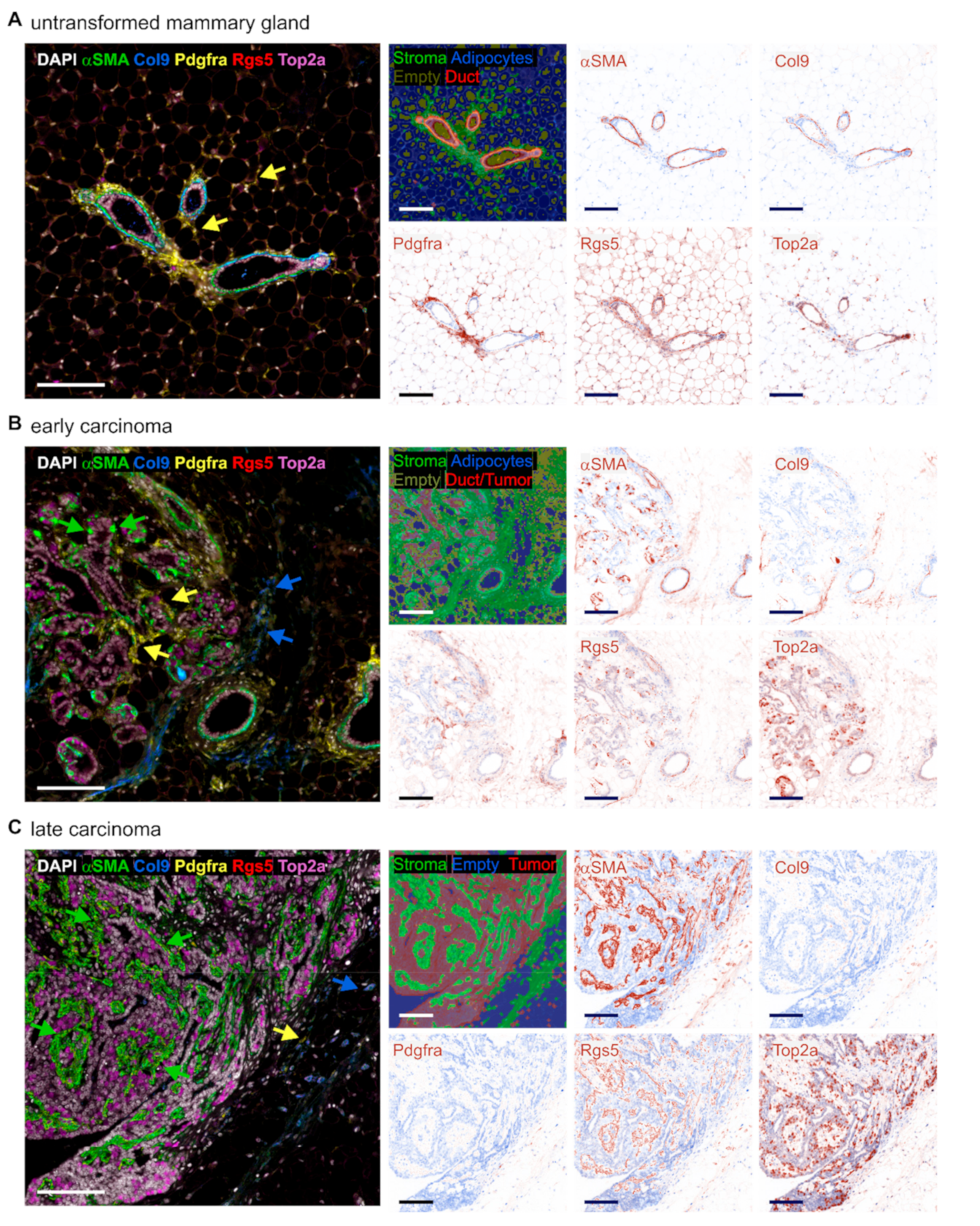
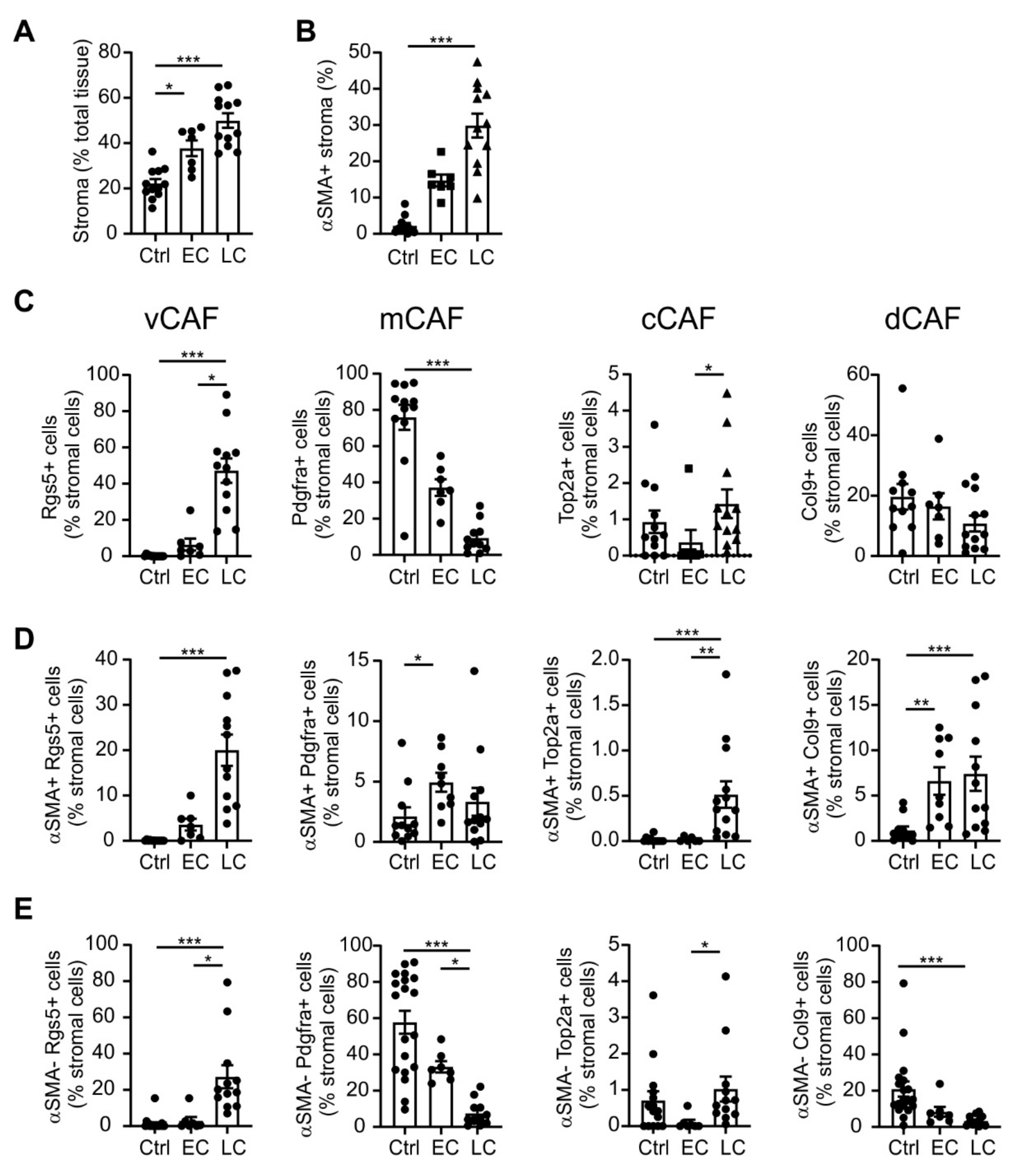
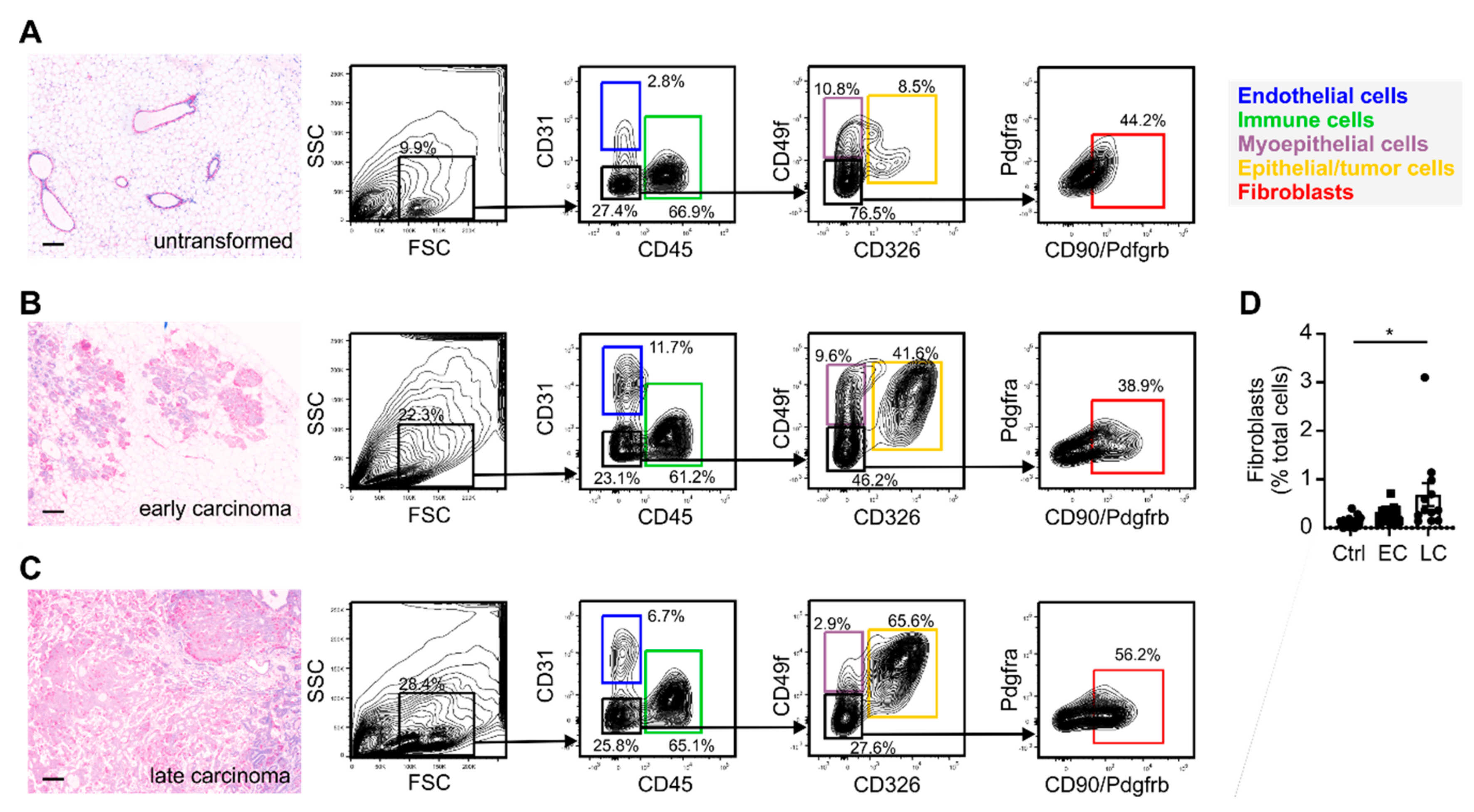
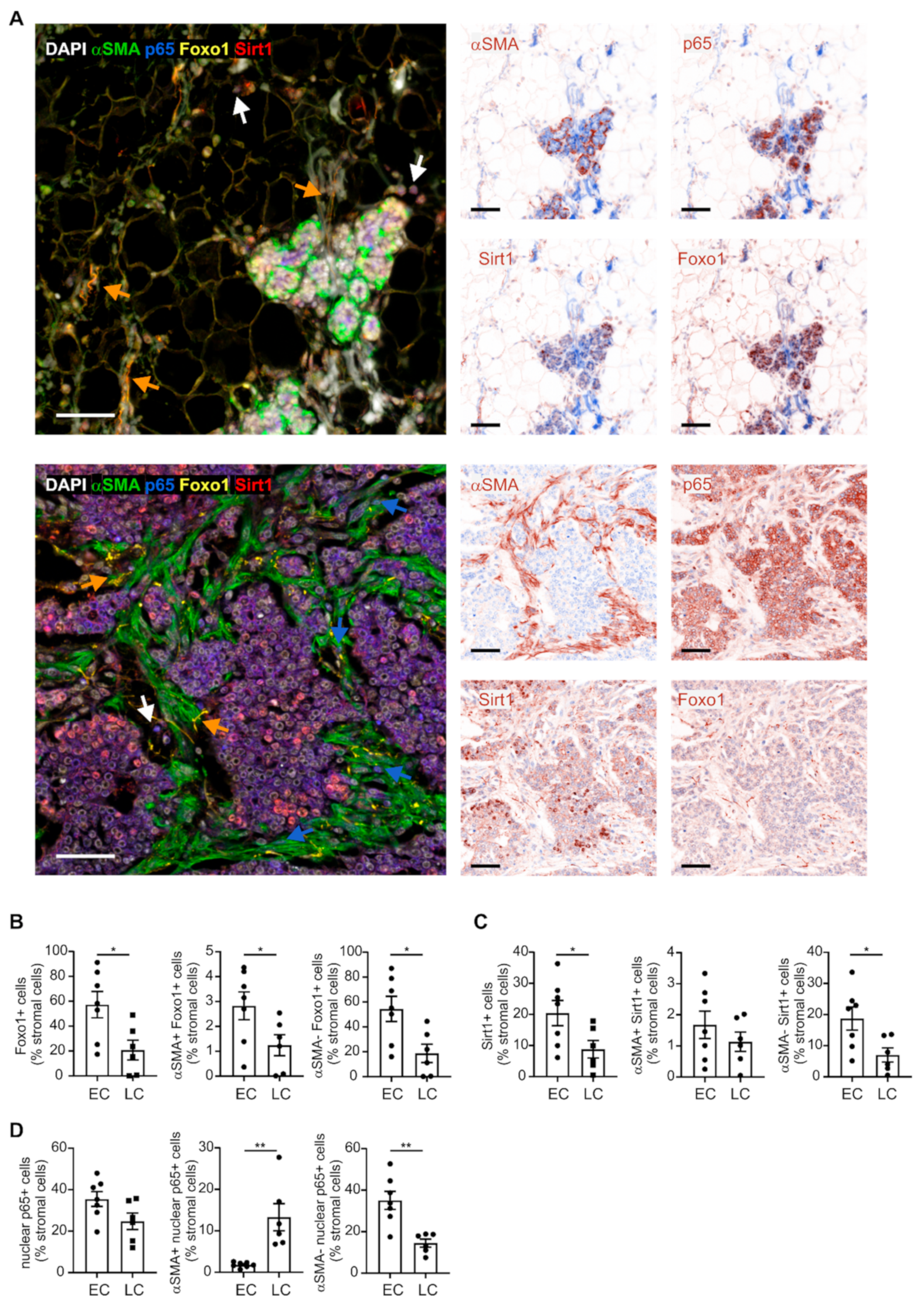
2.1. Varying Fibroblast Subtypes during Mammary Gland Transformation and Tumor Development
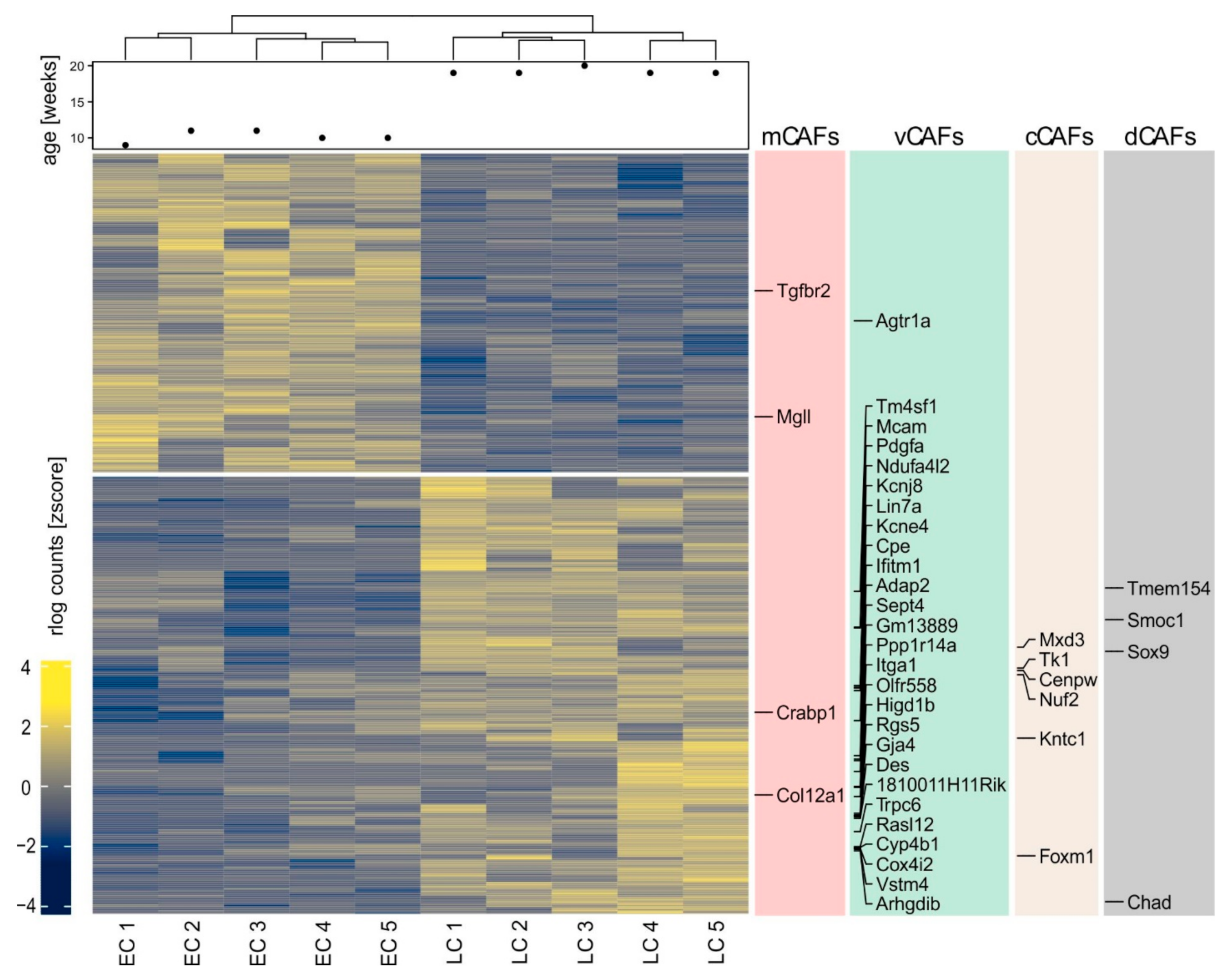
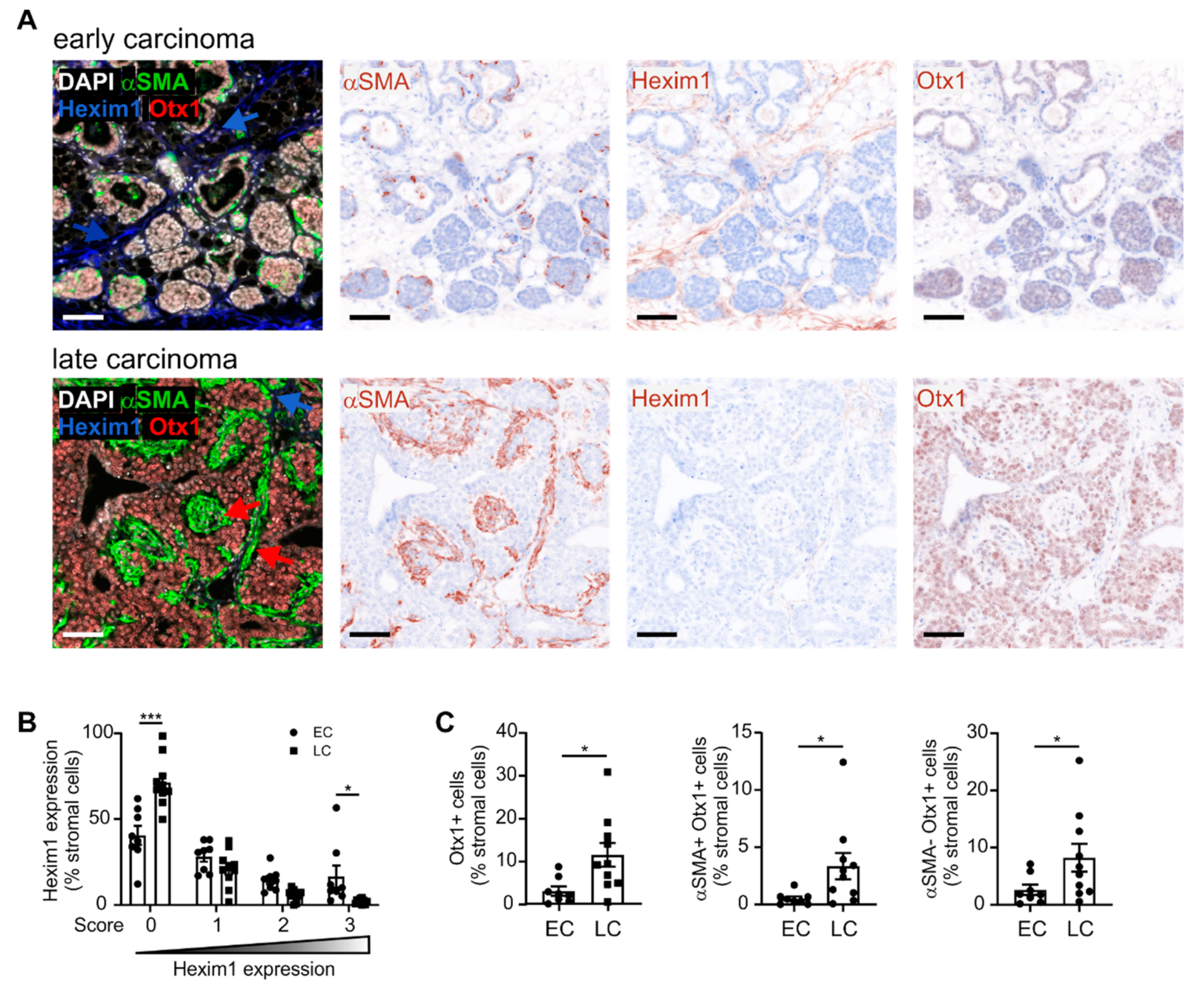
2.2. A Gene Signature Separates Early versus Late-stage CAFs
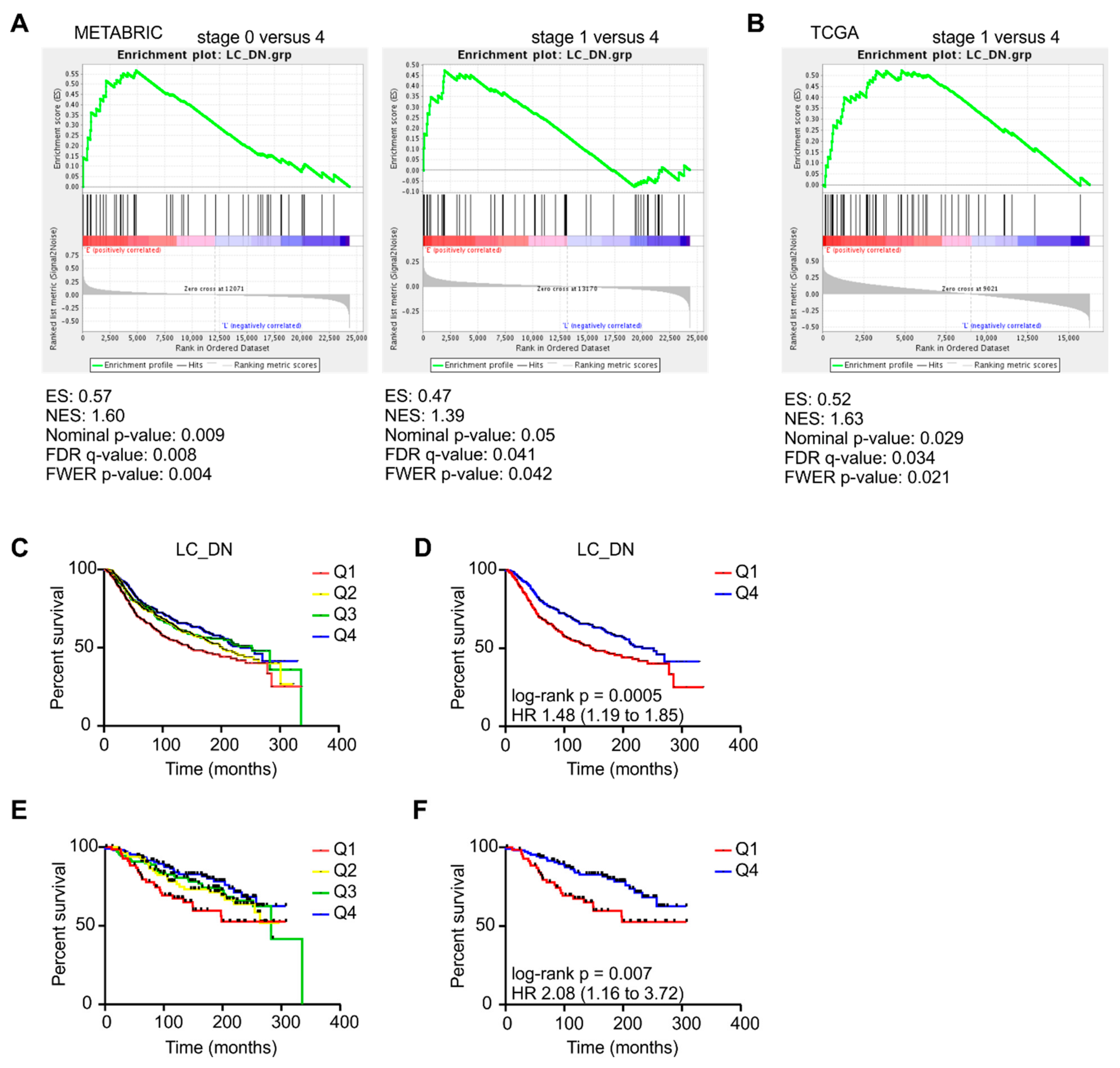
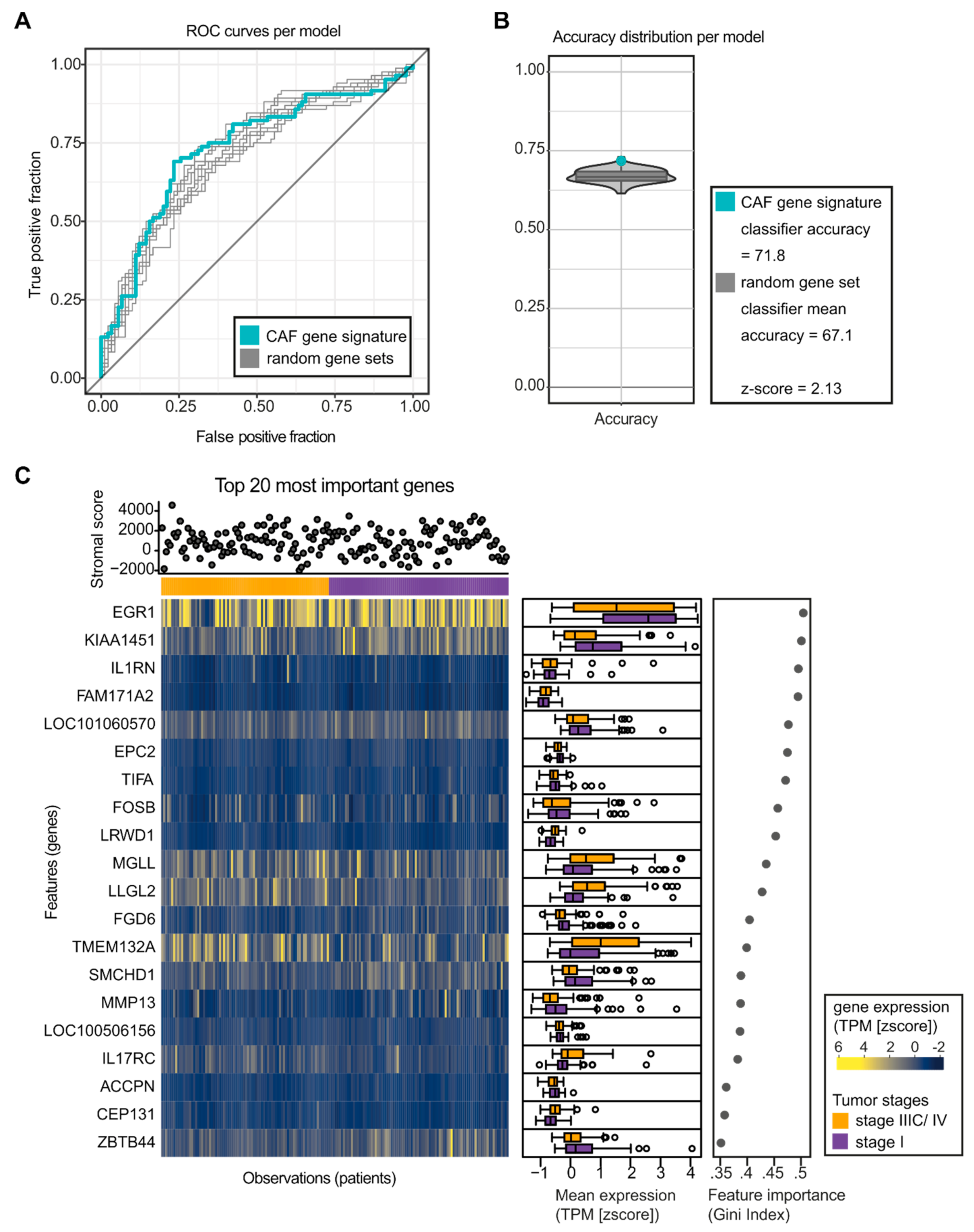
2.3. Changes in CAF Development in PyMT Tumors are reflected in Mammary Carcinoma Patients
3. Discussion
4. Materials and Methods
4.1. Animal Experiments
4.2. Flow Cytometry
4.3. cDNA Synthesis, Library Generation and Whole Transcriptome RNA Sequencing
4.4. RNA-seq Data Processing
4.5. Differential Expression Analysis
4.6. Analysis of Publicly Available Human Mammary Carcinoma Datasets
4.7. Assignment of Human-mouse Orthologs
4.8. GSEA, Reactome Pathway and GO Term Analysis
4.9. PhenOptics Immunofluorescence Analysis
4.10. Random Forest Analysis
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sorrell, J.M.; Caplan, A.I. Fibroblasts-a diverse population at the center of it all. Int. Rev. Cell Mol. Biol. 2009, 276, 161–214. [Google Scholar] [CrossRef] [PubMed]
- Driskell, R.R.; Watt, F.M. Understanding fibroblast heterogeneity in the skin. Trends Cell Biol. 2015, 25, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.D.; Watt, F.M. Fibroblast heterogeneity: Implications for human disease. J. Clin. Invest. 2018, 128, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B. The role of myofibroblasts in wound healing. Curr. Res. Transl. Med. 2016, 64, 171–177. [Google Scholar] [CrossRef]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef]
- Weiskirchen, R.; Weiskirchen, S.; Tacke, F. Organ and tissue fibrosis: Molecular signals, cellular mechanisms and translational implications. Mol. Asp. Med. 2019, 65, 2–15. [Google Scholar] [CrossRef]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349. [Google Scholar] [CrossRef]
- Jun, J.I.; Lau, L.F. Resolution of organ fibrosis. J. Clin. Investig. 2018, 128, 97–107. [Google Scholar] [CrossRef] [Green Version]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar]
- Zeisberg, E.M.; Zeisberg, M. The role of promoter hypermethylation in fibroblast activation and fibrogenesis. J. Pathol. 2013, 229, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Albrengues, J.; Bertero, T.; Grasset, E.; Bonan, S.; Maiel, M.; Bourget, I.; Philippe, C.; Herraiz Serrano, C.; Benamar, S.; Croce, O.; et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat. Commun. 2015, 6, 10204. [Google Scholar] [CrossRef] [PubMed]
- Grugan, K.D.; Miller, C.G.; Yao, Y.; Michaylira, C.Z.; Ohashi, S.; Klein-Szanto, A.J.; Diehl, J.A.; Herlyn, M.; Han, M.; Nakagawa, H.; et al. Fibroblast-secreted hepatocyte growth factor plays a functional role in esophageal squamous cell carcinoma invasion. Proc. Natl. Acad. Sci. USA 2010, 107, 11026–11031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Wever, O.; Pauwels, P.; De Craene, B.; Sabbah, M.; Emami, S.; Redeuilh, G.; Gespach, C.; Bracke, M.; Berx, G. Molecular and pathological signatures of epithelial-mesenchymal transitions at the cancer invasion front. Histochem. Cell Biol. 2008, 130, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Harper, J.; Sainson, R.C. Regulation of the anti-tumour immune response by cancer-associated fibroblasts. Semin. Cancer Biol. 2014, 25, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Augsten, M. Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front. Oncol. 2014, 4, 62. [Google Scholar] [CrossRef] [PubMed]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Weaver, V.M. Mechanics, malignancy, and metastasis: The force journey of a tumor cell. Cancer Metastasis Rev. 2009, 28, 113–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankova, D.; Chen, Y.; Terajima, M.; Schliekelman, M.J.; Baird, B.N.; Fahrenholtz, M.; Sun, L.; Gill, B.J.; Vadakkan, T.J.; Kim, M.P.; et al. Cancer-Associated Fibroblasts Induce a Collagen Cross-link Switch in Tumor Stroma. Mol. Cancer Res. 2016, 14, 287–295. [Google Scholar] [CrossRef]
- Yamauchi, M.; Barker, T.H.; Gibbons, D.L.; Kurie, J.M. The fibrotic tumor stroma. J. Clin. Investig. 2018, 128, 16–25. [Google Scholar] [CrossRef]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Madar, S.; Goldstein, I.; Rotter, V. ‘Cancer associated fibroblasts’--more than meets the eye. Trends Mol. Med. 2013, 19, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Costea, D.E.; Hills, A.; Osman, A.H.; Thurlow, J.; Kalna, G.; Huang, X.; Pena Murillo, C.; Parajuli, H.; Suliman, S.; Kulasekara, K.K.; et al. Identification of two distinct carcinoma-associated fibroblast subtypes with differential tumor-promoting abilities in oral squamous cell carcinoma. Cancer Res. 2013, 73, 3888–3901. [Google Scholar] [CrossRef] [PubMed]
- Bartoschek, M.; Oskolkov, N.; Bocci, M.; Lovrot, J.; Larsson, C.; Sommarin, M.; Madsen, C.D.; Lindgren, D.; Pekar, G.; Karlsson, G.; et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 2018, 9, 5150. [Google Scholar] [CrossRef]
- Lin, E.Y.; Jones, J.G.; Li, P.; Zhu, L.; Whitney, K.D.; Muller, W.J.; Pollard, J.W. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am. J. Pathol. 2003, 163, 2113–2126. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Sesto, A.; Navarro, M.; Burslem, F.; Jorcano, J.L. Analysis of the ultraviolet B response in primary human keratinocytes using oligonucleotide microarrays. Proc. Natl. Acad. Sci. USA 2002, 99, 2965–2970. [Google Scholar] [CrossRef] [Green Version]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef]
- Zerr, P.; Palumbo-Zerr, K.; Huang, J.; Tomcik, M.; Sumova, B.; Distler, O.; Schett, G.; Distler, J.H. Sirt1 regulates canonical TGF-beta signalling to control fibroblast activation and tissue fibrosis. Ann. Rheum. Dis. 2016, 75, 226–233. [Google Scholar] [CrossRef]
- Essaghir, A.; Dif, N.; Marbehant, C.Y.; Coffer, P.J.; Demoulin, J.B. The transcription of FOXO genes is stimulated by FOXO3 and repressed by growth factors. J. Biol. Chem. 2009, 284, 10334–10342. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhou, Y.; Graves, D.T. FOXO transcription factors: Their clinical significance and regulation. Biomed Res. Int. 2014, 2014, 925350. [Google Scholar] [CrossRef] [PubMed]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, K.; Shahmoradgoli, M.; Martinez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, D.; Wang, L.; Wang, S.; Roden, A.C.; Zhao, H.; Li, X.; Prakash, Y.S.; Matteson, E.L.; Tschumperlin, D.J.; et al. Profibrotic effect of IL-17A and elevated IL-17RA in idiopathic pulmonary fibrosis and rheumatoid arthritis-associated lung disease support a direct role for IL-17A/IL-17RA in human fibrotic interstitial lung disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 316, L487–L497. [Google Scholar] [CrossRef]
- Mengshol, J.A.; Vincenti, M.P.; Brinckerhoff, C.E. IL-1 induces collagenase-3 (MMP-13) promoter activity in stably transfected chondrocytic cells: requirement for Runx-2 and activation by p38 MAPK and JNK pathways. Nucleic Acids Res. 2001, 29, 4361–4372. [Google Scholar] [CrossRef] [Green Version]
- Takatsuna, H.; Kato, H.; Gohda, J.; Akiyama, T.; Moriya, A.; Okamoto, Y.; Yamagata, Y.; Otsuka, M.; Umezawa, K.; Semba, K.; et al. Identification of TIFA as an adapter protein that links tumor necrosis factor receptor-associated factor 6 (TRAF6) to interleukin-1 (IL-1) receptor-associated kinase-1 (IRAK-1) in IL-1 receptor signaling. J. Biol. Chem. 2003, 278, 12144–12150. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Fang, F.; Tourtellotte, W.; Varga, J. Egr-1: New conductor for the tissue repair orchestra directs harmony (regeneration) or cacophony (fibrosis). J. Pathol. 2013, 229, 286–297. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, G.; Wang, Z.; Qin, H.; Mo, B.; Wang, C. Elongation factor-2 kinase acts downstream of p38 MAPK to regulate proliferation, apoptosis and autophagy in human lung fibroblasts. Exp. Cell Res. 2018, 363, 291–298. [Google Scholar] [CrossRef]
- Ponten, F.; Jirstrom, K.; Uhlen, M. The Human Protein Atlas—A tool for pathology. J. Pathol. 2008, 216, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, T.W.; Byrne, C.; Colditz, G.; Connolly, J.L.; Schnitt, S.J. Radial scars in benign breast-biopsy specimens and the risk of breast cancer. N. Engl. J. Med. 1999, 340, 430–436. [Google Scholar] [CrossRef] [PubMed]
- DeFilippis, R.A.; Chang, H.; Dumont, N.; Rabban, J.T.; Chen, Y.Y.; Fontenay, G.V.; Berman, H.K.; Gauthier, M.L.; Zhao, J.; Hu, D.; et al. CD36 repression activates a multicellular stromal program shared by high mammographic density and tumor tissues. Cancer Discov. 2012, 2, 826–839. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, K.; Vierkant, R.A.; Frank, R.D.; Winham, S.; Visscher, D.W.; Pankratz, V.S.; Scott, C.G.; Brandt, K.; Sherman, M.E.; Radisky, D.C.; et al. Association between mammographic breast density and histologic features of benign breast disease. Breast Cancer Res. 2017, 19, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brucher, B.L.; Lyman, G.; van Hillegersberg, R.; Pollock, R.E.; Lordick, F.; Yang, H.K.; Ushijima, T.; Yeoh, K.G.; Skricka, T.; Polkowski, W.; et al. Imagine a world without cancer. BMC Cancer 2014, 14, 186. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Boyd, N.F. Mammographic density. Potential mechanisms of breast cancer risk associated with mammographic density: hypotheses based on epidemiological evidence. Breast Cancer Res. 2008, 10, 201. [Google Scholar] [CrossRef] [PubMed]
- Etzold, A.; Galetzka, D.; Weis, E.; Bartsch, O.; Haaf, T.; Spix, C.; Itzel, T.; Schweiger, S.; Strand, D.; Strand, S.; et al. CAF-like state in primary skin fibroblasts with constitutional BRCA1 epimutation sheds new light on tumor suppressor deficiency-related changes in healthy tissue. Epigenetics 2016, 11, 120–131. [Google Scholar] [CrossRef]
- Terrinoni, A.; Pagani, I.S.; Zucchi, I.; Chiaravalli, A.M.; Serra, V.; Rovera, F.; Sirchia, S.; Dionigi, G.; Miozzo, M.; Frattini, A.; et al. OTX1 expression in breast cancer is regulated by p53. Oncogene 2011, 30, 3096–3103. [Google Scholar] [CrossRef]
- Wagenblast, E.; Soto, M.; Gutierrez-Angel, S.; Hartl, C.A.; Gable, A.L.; Maceli, A.R.; Erard, N.; Williams, A.M.; Kim, S.Y.; Dickopf, S.; et al. A model of breast cancer heterogeneity reveals vascular mimicry as a driver of metastasis. Nature 2015, 520, 358–362. [Google Scholar] [CrossRef] [Green Version]
- Raz, Y.; Cohen, N.; Shani, O.; Bell, R.E.; Novitskiy, S.V.; Abramovitz, L.; Levy, C.; Milyavsky, M.; Leider-Trejo, L.; Moses, H.L.; et al. Bone marrow-derived fibroblasts are a functionally distinct stromal cell population in breast cancer. J. Exp. Med. 2018, 215, 3075–3093. [Google Scholar] [CrossRef]
- Erez, N.; Truitt, M.; Olson, P.; Arron, S.T.; Hanahan, D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Pires, B.R.B.; Silva, R.; Ferreira, G.M.; Abdelhay, E. NF-kappaB: Two Sides of the Same Coin. Genes 2018, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.Y.; Chinnaiyan, A.M.; et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiquet, M.; Birk, D.E.; Bonnemann, C.G.; Koch, M. Collagen XII: Protecting bone and muscle integrity by organizing collagen fibrils. Int. J. Biochem. Cell Biol. 2014, 53, 51–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manon-Jensen, T.; Karsdal, M.A. Type XII Collagen. In Biochemistry of Collagens, Laminins and Elastin: Structure, Function and Biomarkers, 1st ed.; Academic Press: Cambridge, MA, USA, 2016; pp. 81–85. [Google Scholar] [CrossRef]
- Datar, I.; Feng, J.; Qiu, X.; Lewandowski, J.; Yeung, M.; Ren, G.; Aras, S.; Al-Mulla, F.; Cui, H.; Trumbly, R.; et al. RKIP Inhibits Local Breast Cancer Invasion by Antagonizing the Transcriptional Activation of MMP13. PLoS ONE 2015, 10, e0134494. [Google Scholar] [CrossRef] [PubMed]
- Decock, J.; Thirkettle, S.; Wagstaff, L.; Edwards, D.R. Matrix metalloproteinases: Protective roles in cancer. J. Cell Mol. Med. 2011, 15, 1254–1265. [Google Scholar] [CrossRef]
- Kloudova, A.; Guengerich, F.P.; Soucek, P. The Role of Oxysterols in Human Cancer. Trends Endocrinol. Metab. 2017, 28, 485–496. [Google Scholar] [CrossRef]
- Sato, T.; Tran, T.H.; Peck, A.R.; Liu, C.; Ertel, A.; Lin, J.; Neilson, L.M.; Rui, H. Global profiling of prolactin-modulated transcripts in breast cancer in vivo. Mol. Cancer 2013, 12, 59. [Google Scholar] [CrossRef]
- Moon, H.G.; Kim, N.; Jeong, S.; Lee, M.; Moon, H.; Kim, J.; Yoo, T.K.; Lee, H.B.; Kim, J.; Noh, D.Y.; et al. The Clinical Significance and Molecular Features of the Spatial Tumor Shapes in Breast Cancers. PLoS ONE 2015, 10, e0143811. [Google Scholar] [CrossRef]
- Horibata, S.; Rice, E.J.; Zheng, H.; Mukai, C.; Chu, T.Y.; Marks, B.A.; Coonrod, S.A.; Danko, C.G. A bi-stable feedback loop between GDNF, EGR1, and ER alpha contribute to endocrine resistant breast cancer. PLoS ONE 2018, 13, e0194522. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Song, N.; Liu, L.; Liu, X.H.; Ding, X.; Song, X.; Yang, S.D.; Shan, L.; Zhou, X.; Su, D.X.; et al. USP9X regulates centrosome duplication and promotes breast carcinogenesis. Nat. Commun. 2017, 8, 14866. [Google Scholar] [CrossRef] [PubMed]
- Weichand, B.; Popp, R.; Dziumbla, S.; Mora, J.; Strack, E.; Elwakeel, E.; Frank, A.C.; Scholich, K.; Pierre, S.; Syed, S.N.; et al. S1PR1 on tumor-associated macrophages promotes lymphangiogenesis and metastasis via NLRP3/IL-1beta. J. Exp. Med. 2017, 214, 2695–2713. [Google Scholar] [CrossRef] [PubMed]
- Roehr, J.T.; Dieterich, C.; Reinert, K. Flexbar 3.0-SIMD and multicore parallelization. Bioinformatics 2017, 33, 2941–2942. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.; Achuthan, P.; Akanni, W.; Allen, J.; Amode, M.R.; Armean, I.M.; Bennett, R.; Bhai, J.; Billis, K.; Boddu, S.; et al. Ensembl 2019. Nucleic Acids Res. 2019, 47, D745–D751. [Google Scholar] [CrossRef] [PubMed]
- Train, C.M.; Glover, N.M.; Gonnet, G.H.; Altenhoff, A.M.; Dessimoz, C. Orthologous Matrix (OMA) algorithm 2.0: More robust to asymmetric evolutionary rates and more scalable hierarchical orthologous group inference. Bioinformatics 2017, 33, i75–i82. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019, 47, D419–D426. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Statnikov, A.; Wang, L.; Aliferis, C.F. A comprehensive comparison of random forests and support vector machines for microarray-based cancer classification. BMC Bioinform. 2008, 9, 319. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Upregulated in LC | ||||
|---|---|---|---|---|
| GSEA | ||||
| GENE SET NAME | ES | NES | NOMp-value | FDR q-val |
| HINATA_NFKB_TARGETS_KERATINOCYTE_UP | 0.79 | 1.81 | <0.001 | 0.15 |
| SESTO_RESPONSE_TO_UV_C0 | 0.78 | 1.78 | <0.001 | 0.15 |
| Reactome | ||||
| Reactome pathways | Fold enrichment | +/− | Raw p-value | FDR |
| Chemokine receptors bind chemokines | 12.08 | + | 1.05E-04 | 2.41E-02 |
| G alpha (i) signaling events | 5.00 | + | 6.87E-07 | 1.10E-03 |
| GO | ||||
| PANTHER GO-Slim Biological Process | Fold enrichment | +/− | Raw p-value | FDR |
| Granulocyte chemotaxis | 14.49 | + | 8.35E-06 | 2.46E-03 |
| Cytokine-mediated signaling pathway | 8.45 | + | 3.45E-05 | 7.62E-03 |
| Regulation of MAPK cascade | 7.51 | + | 2.31E-04 | 2.55E-02 |
| Inflammatory response | 6.63 | + | 1.42E-04 | 1.80E-02 |
| PANTHER GO-Slim Molecular Function | Fold enrichment | +/− | Raw p-value | FDR |
| Potassium channel regulator activity | 19.10 | + | 8.92E-04 | 2.63E-02 |
| Endopeptidase inhibitor activity | 10.66 | + | 8.80E-06 | 4.90E-04 |
| Cytokine receptor binding | 9.87 | + | 1.93E-07 | 2.42E-05 |
| Protease binding | 9.25 | + | 2.03E-05 | 9.24E-04 |
| Cytokine activity | 8.76 | + | 5.27E-07 | 4.40E-05 |
| G-protein coupled receptor binding | 6.29 | + | 1.93E-04 | 6.90E-03 |
| G-protein coupled receptor activity | 3.45 | + | 2.78E-04 | 8.71E-03 |
| Upregulated in EC | ||||
| GO | ||||
| PANTHER GO-Slim Biological Process | Fold enrichment | +/− | Raw p-value | FDR |
| Regulation of transcription by RNA polymerase II | 3.97 | + | 9.12E-07 | 8.06E-04 |
| Transcription by RNA polymerase II | 3.10 | + | 2.84E-06 | 8.37E-04 |
| PANTHER GO-Slim Molecular Function | Fold enrichment | +/− | Raw p-value | FDR |
| Transcription regulatory region DNA binding | 4.55 | + | 1.88E-05 | 4.71E-03 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elwakeel, E.; Brüggemann, M.; Fink, A.F.; Schulz, M.H.; Schmid, T.; Savai, R.; Brüne, B.; Zarnack, K.; Weigert, A. Phenotypic Plasticity of Fibroblasts during Mammary Carcinoma Development. Int. J. Mol. Sci. 2019, 20, 4438. https://doi.org/10.3390/ijms20184438
Elwakeel E, Brüggemann M, Fink AF, Schulz MH, Schmid T, Savai R, Brüne B, Zarnack K, Weigert A. Phenotypic Plasticity of Fibroblasts during Mammary Carcinoma Development. International Journal of Molecular Sciences. 2019; 20(18):4438. https://doi.org/10.3390/ijms20184438
Chicago/Turabian StyleElwakeel, Eiman, Mirko Brüggemann, Annika F. Fink, Marcel H. Schulz, Tobias Schmid, Rajkumar Savai, Bernhard Brüne, Kathi Zarnack, and Andreas Weigert. 2019. "Phenotypic Plasticity of Fibroblasts during Mammary Carcinoma Development" International Journal of Molecular Sciences 20, no. 18: 4438. https://doi.org/10.3390/ijms20184438