p53 at the Crossroads between Different Types of HDAC Inhibitor-Mediated Cancer Cell Death


1
Department of Cranio-Maxillofacial Surgery, University of Münster, Albert-Schweitzer-Campus 1, 48149 Münster, Germany
2
Department of Medical Microbiology, University of Münster, Albert-Schweitzer-Campus 1, 48149 Münster, Germany
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(10), 2415; https://doi.org/10.3390/ijms20102415
Submission received: 29 March 2019
/
Revised: 6 May 2019
/
Accepted: 13 May 2019
/
Published: 15 May 2019
(This article belongs to the Special Issue Apoptosis and Autophagy: The Double Edge in Cancer Development and Progression and in Other Human Diseases)
Abstract
:Cancer is a complex genetic and epigenetic-based disease that has developed an armada of mechanisms to escape cell death. The deregulation of apoptosis and autophagy, which are basic processes essential for normal cellular activity, are commonly encountered during the development of human tumors. In order to assist the cancer cell in defeating the imbalance between cell growth and cell death, histone deacetylase inhibitors (HDACi) have been employed to reverse epigenetically deregulated gene expression caused by aberrant post-translational protein modifications. These interfere with histone acetyltransferase- and deacetylase-mediated acetylation of both histone and non-histone proteins, and thereby exert a wide array of HDACi-stimulated cytotoxic effects. Key determinants of HDACi lethality that interfere with cellular growth in a multitude of tumor cells are apoptosis and autophagy, which are either mutually exclusive or activated in combination. Here, we compile known molecular signals and pathways involved in the HDACi-triggered induction of apoptosis and autophagy. Currently, the factors that determine the mode of HDACi-elicited cell death are mostly unclear. Correspondingly, we also summarized as yet established intertwined mechanisms, in particular with respect to the oncogenic tumor suppressor protein p53, that drive the interplay between apoptosis and autophagy in response to HDACi. In this context, we also note the significance to determine the presence of functional p53 protein levels in the cancer cell. The confirmation of the context-dependent function of autophagy will pave the way to improve the benefit from HDACi-mediated cancer treatment.
{kind=link}
{kind=link}
{kind=link}
1. Introduction: Cell Death and Cancer
Resistance to programmed cell death and uncontrolled cell proliferation are pivotal landmarks supporting the initiation and progression of the complex and widespread disease known as cancer. The concept of programmed cell death as a multi-step, non-accidental, tightly controlled and defined self-destruction process was uncovered in 1964 [1]. The main forms of programmed cell death that were initially identified based on morphology are apoptosis, autophagy, and necroptosis; which, following their disruption, facilitate the development and growth of cancer [2,3,4,5]. An imbalance between cell division and cell death is a crucial Janus-faced factor, not only in tissue homeostasis and development, but also in the initiation, progression, and treatment of cancer, as it can be the cause as well as the solution to the dilemma [6]. Comparable to other defined common traits that govern oncobiology, such as limitless replicative potential, insensitivity to anti-growth signals, sustainment of angiogenesis, the ability to form metastases, deregulated metabolism, and the escape from the immune system, the inactivation of cell death mechanisms by cancer cells has been related to genetic defects in oncogenes or tumor suppressor genes [7,8]. In recent years, tumor initiation and progression has been increasingly linked to the disruption of epigenetic modifications, including DNA methylation, posttranslational histone or non-histone modifications, and microRNAs which may be considered as a further distinguishing mark of cancer [9]. In fact, several studies have confirmed that epimutations concerning the structure of chromatin are mutational hotspots in cancer representing about one third of so-called tumor-causing driver mutations [10,11]. Consequently, these aberrant epigenetic determinants have also been identified in the deregulation of different key signaling pathways of the cell, including those for cell death mechanisms. Targets for these epigenetic changes include, oncogenes and tumor suppressor genes, other key regulatory molecules, or even microRNAs that regulate apoptosis and autophagy.
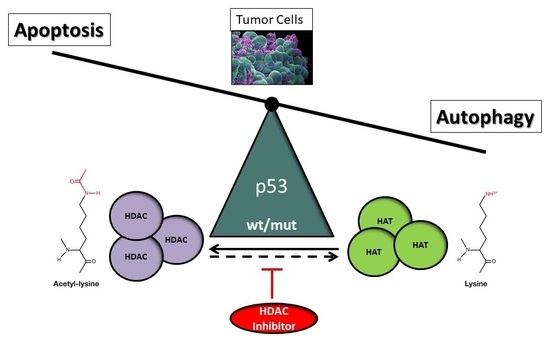
The tumor suppressor and non-histone protein p53 is allocated an important role in this context. The modification of the acetylation of p53 regulates its transcriptional activity, related among many other functions to the regulation of apoptosis and autophagy, which is pivotal for the elimination of malignant cells [12]. The present review aims to address the ambiguous role of p53 as a modulator of the nature of histone deacetylase inhibitor (HDACi)-mediated cell death response in tumor cells, in particular for the induction of autophagy in cancer therapy. In view of our recent finding, we highlight the supposed function of p53 as a molecular switch that directs HDACi-mediated tumor response towards either forms of programmed cell death depending on its presence in the cell [13,14,15].
2. Post-Translational Acetylation Defects in Cancer
Histones are the core proteins that enable the higher organization of eukaryotic genomic DNA into nucleosomes [16]. Among the increasing number of possible post-translational histone modifications, the acetylation of histones, as well as non-histone proteins, have been recognized as a major epigenetic regulatory mechanism of gene transcription [17]. Thereby, the chromatin structure and regulation of gene expression of numerous key cellular processes, such as cell proliferation, differentiation, adhesion, and cell death is strongly affected [18,19]. Conclusively, histone acetylation has become apparent as a mechanism that enables the cell to deal with a variety of stressful conditions. Histone and non-histone protein acetylation is accomplished by different enzyme classes of histone acetyltransferases (HAT) and histone deacetylases (HDAC) that regulate the structure of chromatin and thereby modulate the accessibility of transcription factors to their target DNA sequences [20]. They belong to the so-called bromodomain proteins which are able to identify acetylated lysine residues on the N-terminal tails of histones and translate this signal [21]. Histone acetylation, particularly as proximal promoters and enhancers of transcriptionally active regions, untighten the connection between core nucleosome proteins and DNA, and thus facilitate the binding of transcription factors. One of the first recognized and decisive non-histone proteins targeted by HDACs and HATs, is the transcription factor p53 that regulates cell apoptosis and autophagy among its multitude of other functions as the “gatekeeper” of the cell [22].
Aberrant histone and non-histone deacetylation were also identified as the cause of transcriptional repression of tumor-related genes that interfere with important physiological cellular processes such as cell death regulation, and thereby promote malignant transformation [23,24]. Dysregulation, caused by the overexpression, abnormal HDAC recruitment, or loss-of-function mutations of HDACs in human cancer have been detected in solid (neuroblastoma, medulloblastoma, lung, gastric, liver, pancreatic, colorectal, breast, ovarian, prostate, renal, bladder, melanoma, oral, endometrial, pancreatic, thyroid, esophageal) as well as hematological tumors (ALL, CLL, AML, DLBCL, CTCL, HL, Myeloma) [25,26,27]. In particular, the loss of acetylation of histone H4 at lysine 16 (H4K16ac) was found to be decisive in tumorigenesis [28,29]. This finding is underlined by the fact that global histone modifications, such as acetylation and dimethylation, or even trimethylation levels of histone H3 and H4 (H3K9ac, K18ac, H4 K12ac; H3 K4diMe, H4R3diMe; H4K20triMe) correlate with the risk of cancer recurrence and allow a prognosis of poor survival in distinct types of cancer [30,31]. In addition, also HATs have been shown to be involved in cancer among other malignancies caused by the altered acetylation of histone proteins and non-histone proteins [26,32]. However, their effects could be either tumor-promoting or tumor-suppressive due to the hypo- or hyperacetylation effects of oncogenes, respectively. Hyperacetylation via HAT p300, for example, was correlated with the occurrence of hepatocellular carcinoma and the specific acetylation of H3K18 with prostate cancer [33,34]. Furthermore, p300 as an important trascriptional co-activator of androgen receptor activity, was shown to be upregulated during prostate or colorectal cancer progression [35,36,37,38]. HATs have also been reported to acetylate non-histone targets, such as the oncoprotein c-Myc promoting increased stability and cancer progression, or the tumor suppressor protein p53, promoting its transcriptional activity and cytoprotective function [39,40,41].
3. HATs and HDACs, the Effectors of Post-Translational Acetylation and Deacetylation
The dynamic and reversible acetylation of proteins is catalyzed by the post-translational actions of the families of HAT and HDAC enzymes which represent lysine acetyltransferases or deacetyltransferases, respectively [42]. Acetylation belongs to a myriad of possible histone modifications such as methylation (mono-, di-, and tri-methylation), phosphorylation, ubiquitination, sumoylation, or unfamiliar variants, such as succinylation, butyrylation, and neddylation, that have been intensively investigated because of their fundamental role in regulating gene expression by altered chromatin structure [19,43,44,45,46,47]. While HATs assist in the transfer of an acetyl group from acetyl-CoA to form ε-N-acetyllysine residues of histone and non-histone proteins and activate gene transcription by forming an open chromatin configuration associated with euchromatin, HDACs remove it, and consequently suppress transcription by supporting a condensed configuration [17,48,49]. Particularly, DNA elements that have regulatory function, such as promoters, or the histones H3 and H4, that are part of the tetrameric nucleosome structure and regulate transcriptional access, undergo specific acetylation [19,50]. In addition to transcriptional regulation, many essential cellular basic activities are governed by specific HAT/HDAC activity, which is primarily due to the vast number of non-histone proteins that have been identified as their targets, and seem to be their evolutionary primary targets [18,51]. These include, in addition to the regulation of cell death, replication, DNA repair, cell cycle regulation, DNA stress response, and senescence among others. Via modifying the acetylation of non-histone proteins, many molecular functions can be influenced, such as mRNA splicing, mRNA transport and integrity, protein translation, protein activity, localization, stability and interactions [18]. As non-histone substrates, tumor suppressor proteins (e.g., p53, RUNX3), signaling mediators (e.g., STAT3, β-catenin, SMAD7), steroid receptors (e.g., androgen, estrogen, SHP), transcriptional factors, and co-regulators (e.g., c-MYC, HMG, YY1, EKLF, E2F1, GATA factors, HIF-1α, MyoD, NF-κB, FoxB3), as well as structural (e.g., cell motility proteins), chaperone proteins, and nuclear import proteins (e.g., α-tubulin, importin-α, Ku70, HSP90) have been identified [52,53,54].
Depending on their subcellular localization, in either the nucleus or cytoplasm, HATs are categorized into Type A or B, that either contain or lack a bromodomain, respectively [55,56]. Based on sequence homology as well as shared structural features and functional roles, they have been grouped into the three major categories of GNAT (GCN5-related N-acetyltransferases), EP300/CREBBP (E1A binding protein p300 and CREB-binding protein), and MYST family [48]. PCAF (p300/CBP-associated factor), belonging to the GNAT family, and the paralogues p300 and CBP, which are key transcriptional co-activators of the cell and the only members of the EP300/CREBBP family, are the most important non-histone acetylating HATs. Although HATs are implicated in tumor progression, and in vitro designed inhibitors might be potentially useful, these are not clinically applicable thus far. Due to their function in protein complexes, current HAT inhibitors experience disadvantageous properties such as instability, low potency, or a lack of selectivity [57]; nevertheless, although progress on the advancement of clinically useful HAT inhibitors is delayed, they will be of interest for future cancer therapy [58].
According to their structure and homology to yeast proteins, the 18 known HDACs of humans have been divided into four classes. These comprise class I HDACs (with members HDAC1, 2, 3, 8), class II HDACs (with members HDAC4, 5, 6, 7, 9, 10), class III HDACs which were termed sirtuins (with members SIRT 1-7), and class IV HDACs (with member HDAC11). Only class III HDACs, containing the sirtuins, require NAD+ for their activity while all other classes contain a zinc-binding site. Also, differing subcellular localization can be applied for HDAC classification [49]. In contrast to the more restricted tissue-specific expression pattern of the HDAC classes II to IV with subcellular localization in nucleus, cytoplasm, or mitochondria, class I exhibits a ubiquitous distribution pattern due to the exclusive presence in the cell nucleus explaining the high enzymatic activity of its members. Thus, for example, sirtuins have been found to reside in all three compartments including SIRT1 and 2 in the cytoplasm, SIRT3, 4, and 5 in mitochondria, and SIRT1, 2, 6, and 7 in the nucleus. The subcellular presence of HDAC6 and 10 mostly in the cytoplasm is another reason for the separation of these members of HDAC class II A from B, as they can be localized in the nucleus, cytoplasm (HDAC4, 5, 9), as well as in mitochondria (HDAC7).
4. Inhibition of Post-Translational Deacetylation in Cancer Cells
Related to these findings of post-translational acetylation defects in cancer, increasing interest has been attracted by the development of HDAC inhibitors (HDACi) as encouraging candidates for cancer therapy that are able to reverse the actions of deregulated HDAC expression [59,60]. Comparable to other epigenetic drugs, such as to DNA methyltransferase (DNMT) inhibitors, HDACi are able to lift the silenced expression of tumor suppressor genes and reactivate associated signaling pathways such as programmed cell death. As opposed to unalterable genetic mutations, these compounds that modify acetylation or methylation attachments of protein and DNA molecules make use of the partial reversibility of the epigenome. Although several drawbacks for solid tumors exist, many in vitro data and clinical studies have already attested positive effects for the use of HDAC targeting; however, their detailed mechanisms of action are still are under investigation. Moreover, further inhibitors are in the process of development. Nevertheless, the prediction of the cellular or molecular response following HDACi treatment remains elusive as they may exert pleiotropic effects that vary, depending on the class of HDACi and the type of tumor. Therefore, a clear-cut strategy or definition of predictors for the use of HDACi in cancer therapy would be required. Improved molecular mechanistic insights related to cell death and resistance will therefore be useful for enabling a more targeted use of HDACi and the prediction of beneficiaries in cancer therapy [61].
HDACi categories encompass hydroxamic acids (hydroxamates), cyclic tetrapeptides, benzamides, electrophilic ketones, and aliphatic acids that include natural but also synthetic derivatives [62]. This classification mainly relies on the chemical structure of their zinc-binding group; in addition, HDACi can also be subdivided into zinc-dependent pan- or broad-spectrum inhibitors that inhibit class I, II and IV HDACs in contrast to primarily class I-specific HDACi [23,63,64]. Favored representatives of the hydroxamates are SAHA (suberoylanilide hydroxamic acid, vorinostat, Zolinza), which is a preferred derivative of naturally occurring TSA (trichostatin A), as well as the CBHA (m-carboxycinnamic acid bishydroxamate)-derived tubacin, LAQ-824 (dacinostat), LBH-589 (panobinostat), or PXD-101 (belinostat) [64,65,66,67]. The class I-selective FK-228 (romidepsin, FR901228, istodax) belongs to the group of cyclic tetrapeptides [68,69,70]. MS-275 (entinostat) and MGCD0103 (mocetinostat), exhibiting enhanced HDAC class I selectivity, are members of benzamide-based HDACi [71,72,73]. The minor effective class I- and IIa-specific HDACi, VPA (valproic acid), PBA (phenylbutyrate), NaB (sodium butyrate), or AN-9 (pivaloyloxymethyl butyrate) belong to the category of aliphatic acids [74,75].
The unexpected, tumor-specific effects of HDACi in contrast to the ubiquitous functions of HDACs in gene regulation remain puzzling; however, they commonly induce chromatin relaxation and transcriptional de-repression in cancer cells. By the reactivation of a small percentage of silenced key regulatory genes, e.g., tumor suppressor or oncoproteins, due to the restored or increased acetylation, re-expression of genes associated with multiple detrimental tumor-defensive mechanisms, such as cell cycle arrest, differentiation, and cell death, but also the inhibition of metastasis and angiogenesis, is implemented [76,77,78,79,80,81]. Accordingly, major efforts have been undertaken to employ HDACi for the development of promising novel anticancer strategies which have ended in the establishment of numerous clinical trials that involve HDACi (www.clinicaltrials.gov). Either monotherapeutic or combinatorial treatments of several HDACi in hematological and solid malignancies with varying outcomes, have been, or are in the process of being tested [82,83,84]. Up to now, these evaluations have led to the admittance of four licensed HDACi (SAHA, panobinostat (LBH589), belinostat (PXD-101), and romidepsin (FK228)) for the treatment of cutaneous T cell lymphoma, multiple myeloma, or peripheral T cell lymphoma representing exclusively pan-inhibitors [85,86,87,88,89]. Although preclinical studies involving single treatment regimen of many HDACi were promising, almost all types of solid tumor (e.g., ovarian, breast, renal, prostate, and head and neck cancer) failed to demonstrate benefits in phase II clinical trials [90,91,92,93,94,95,96]. Moreover, patients experienced considerable non-selective off-target effects ranging from negligible (such as diarrhea, anorexia, dehydration) to serious (myelosuppression, thrombocytopenia, and cardiotoxicity) outcomes [75,84,97,98]. The reasons therefore are currently unclear and were discussed to be due to a combination of divergent blood vessel supply, intrinsic molecular heterogeneity involving epigenetic modifications, and the progression of treatment resistance. Consequently, nanoparticle-supported targeting has been proposed as a future drug-delivery method to avoid potential stability issues with HDACi drugs; however, also in this case negative long-term treatment effects might be predicted [99,100]. Therefore, to bypass clinical limitations and reduce adverse effects, the development of isoform-specific and/or tissue-specific HDACi with improved selective efficacy, in place of pan-inhibitors, is enforced [63,101]. Collectively, the improved understanding of the context-dependent effects in epigenetic interference mediated by individual HDAC enzymes will provide a synergistic advantage for designing specifically targeted cancer treatment strategies.
5. Effector Mechanisms of HDACi-Induced Cell Death
Due to a range of histone and non-histone targets and multitude of effects exerted by the posttranslational modification of acetylation, HDACi can arrange an array of cellular effects with respect to growth, differentiation, migration, senescence, and death that contribute to their eminent anti-tumor activities [102]. These may differ, depending on the type of tumor and applied dose; nevertheless, specific mechanisms are crystallizing for many HDACi [98,102,103,104]. It is very likely that future research will shed light on additional, non-elucidated mechanisms contributing to HDACi-mediated anticancer activity.
As a prerequisite for the induction of programmed cell death, a fundamental effect of HDACi was determined by their ability to re-induce cell cycle arrest and induce cell differentiation in transformed cells by downregulating positive regulators of cell proliferation, such as c-MYC and c-SRC [65,105,106,107,108,109,110,111]. Cell cycle arrest, in the G1 or G2 phase of the cell, is induced by p53-dependent or -independent induction of p21 (encoded by the gene CDKN1A) expression [112,113,114,115]. p21 is a cyclin-dependent kinase inhibitor that interferes with cyclin D1 and D2 activity [116,117]. Presumably, due to accumulated DNA damage (e.g., by DNA double-strand breaks caused by oxidative stress), this failure to exit the cell cycle from an incomplete mitosis might result in the induction of apoptosis [118,119,120,121]. Consistently, upon inhibiting the SIRT1-mediated deacetylation activity of p53, DNA damage-induced apoptosis was activated by p53-mediated target gene expression in a representative report [122]. The induction of DNA damage and cell death goes along with another very important characteristic mechanism of HDACi, which is the generation of reactive oxygen species (ROS). Accordingly, the accumulation of thioredoxin (TXN), a natural scavenger of ROS and intracellular antioxidant, was detected in normal, but not in malignant, human fibroblasts [123]. Interference with multiple DNA repair processes in response to various genotoxic insults, or the induction of autophagy, is a novel discovered effector mechanism important for the maintenance of genomic integrity. The inhibition of HDAC6, for example, was documented to provoke cell death as the MSH2-regulated DNA mismatch repair capability of the cell was deregulated [124]. This is a very obvious finding, as HDACs have been reported to modulate deacetylation of histones at DNA damage sites, and are fundamentally involved in regulating the expression of DNA damage-related response proteins [25,125,126,127,128,129,130,131,132,133]. In support of this, increased expression of a marker of DNA double strand breaks, H2AX, was disclosed in transformed but not in normal cells in response to SAHA treatment, explaining its tumor cell-specific activity [120]. Furthermore, by directly altering the acetylation pattern of non-histone proteins such as transcription factors, the signaling pathways that are involved in cell death (e.g., NF-κB, p53, and STATs), can be re-activated by HDACi in cancer cells [18]. For instance, the half-life of the transcriptional activity of p53 is determined by its acetylation status which regulates its degradation through the directly interacting MDM2 E3-ligase in H1299 carcinoma cells [134]. Further detrimental effects, resulting from HDACi treatment, are the interference with chaperone protein function and the inhibition of stress response pathways in the endoplasmic reticulum [107,135]. Thereby, the physiological elimination of misfolded proteins, as well as the stability and expression of oncoproteins, can be affected. Moreover, HDACi were found to interfere with the migration and invasion of human tumor cell lines by de-repression of metastasis-related genes [136,137]. Additionally, the inhibition of angiogenesis by changing the expression of many pro- and anti-angiogenic genes was attested for HDACi [138,139,140].
Overall, key determinants of HDACi lethality that interfere with cellular growth in a multitude of tumor cells, are apoptosis and autophagy [141,142,143,144]. These morphologically distinct two major forms of programmed cell death which promoted the use of HDACi as promising therapeutic agents will be discussed in the next two sections.
5.1. HDAC Inhibitor-Induced Apoptosis
For many years, apoptosis has been considered as the prevailing mechanism of programmed cell death in eukaryotic cells which is traditionally targeted in cancer therapy. Also, cells undergoing HDACi treatment exhibit caspase-mediated induction of apoptosis, as the most commonly encountered form of cytotoxic response [145,146,147]. Two different modes—either the intrinsic (mitochondrial) pathway, or the extrinsic (death-receptor) pathway—can be employed, alone or in combination, which finally both discharge executioner caspases that degrade and eliminate the cell [145,148]. As already mentioned above, this programmed type-I cell death is activated by HDACi exclusively in cancer cells, presumably by rendering them sensitive due to the imposition of cell cycle arrest [149].
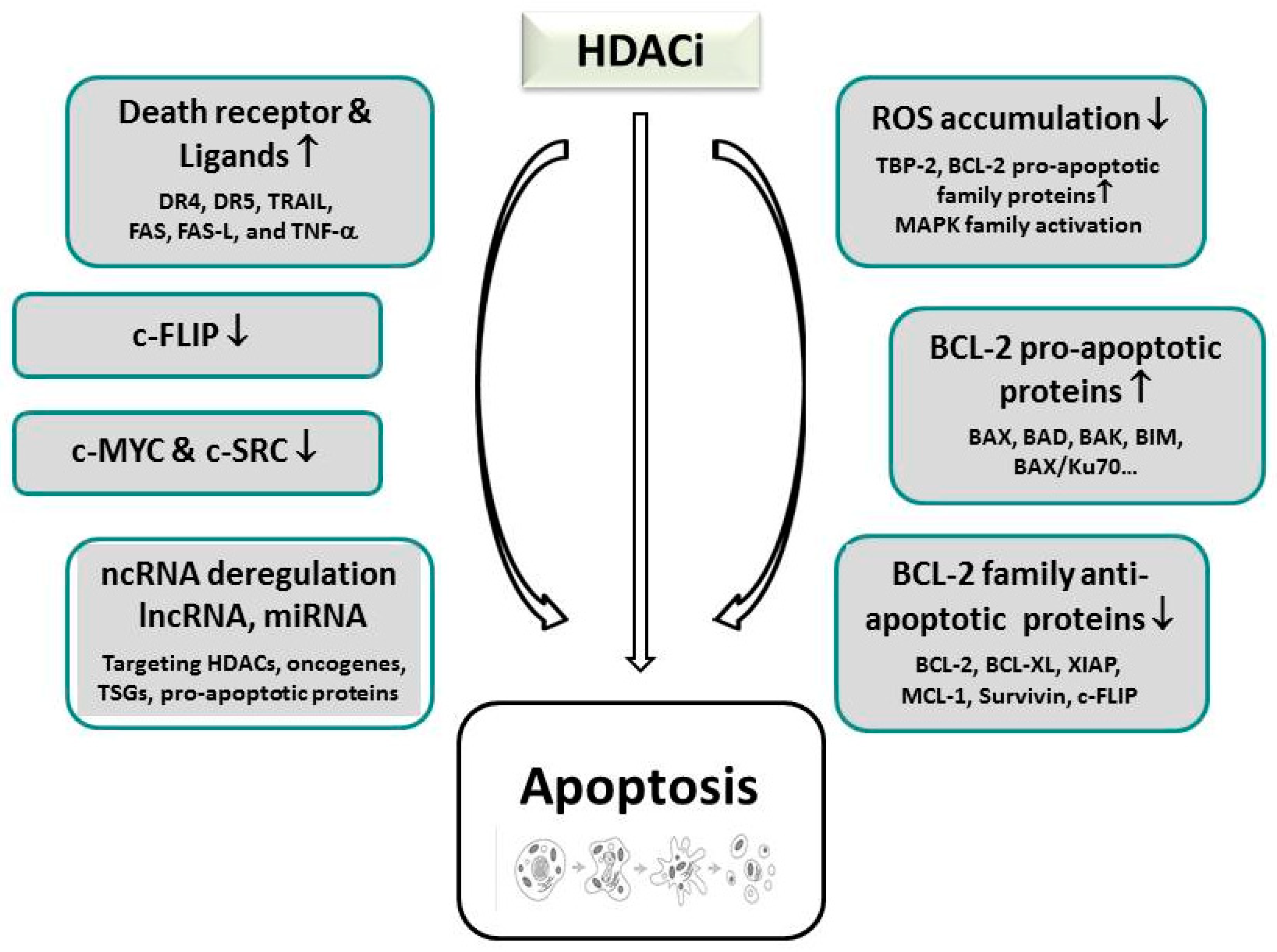
Upregulation of pro-apoptotic proteins belonging to the B-cell Lymphoma-2 (BCL-2) family (e.g., BAX and BAK) that comprise the modifying members of the BH3 (BCL-2 homology 3)-only protein family, including sensitizers (NOXA, BAD) and activators (BID, BIM, BMF, PUMA), lead to the disruption of the mitochondrial membrane, the release of cytochrome C, apoptosome formation, and the final activation of executioner caspases in the intrinsic pathway of apoptosis [112,119,150,151,152,153]. Mechanistically, to elicit apoptosis in the intrinsic pathway, HDACi predominantly seem to interfere with the balanced expression between pro- and anti-apoptotic proteins, whereas a single case documented SAHA-induced cleavage of BID (see Figure 1) [119,149,154]. In addition to p53-dependent or -independent pathways, that will be discussed in Section 6.2, HDACi treatment can additionally activate ROS-dependent apoptosis by upregulation of pro-apoptotic proteins, and the induction of DNA damage [107,119,123,155,156]. As an underlying mechanism, elevated expression of the thioredoxin (TRX)-binding protein-2 (TBP-2) are assumed that are present in malignant cells; these inhibit the ROS scavenger TRX, which is able to compensate oxidative stress, [107,123]. For the enhancement of apoptosis, HDACi can also promote the down-regulation of anti-apoptotic genes (e.g., BCL-2, BCL-XL, XIAP, MCL-1, Survivin) [112,113,157,158]. As a consequence, HDACi-mediated apoptosis can be suppressed by the overexpression of BCL-2 and BCL-XL [119,145]. Similarly, interference of p53 with the interaction of BAX and Ku70, an acetylation-sensitive DNA repair protein, was shown to promote mitochondria-mediated apoptosis in neuroblastoma cells [159]; the involvement of p53 could be demonstrated by transactivation-deficient p53 mutants that were still able to mediate BAX-dependent apoptosis in HDACi-treated p53-null cells due to the transcription-independent activation of BAX.
In the extrinsic pathway, HDACi primarily restore the expression of death receptors such as DR4 and DR5, or their corresponding ligands, such as TRAIL, FAS, FAS-L, and TNF-α [160,161,162,163,164,165,166,167]; this reactivation very often triggers a bottleneck of apoptosis resistance along this pathway in untreated tumor cells and link the death receptor with the intrinsic pathway [145,168]. The interaction of these members of the TNF-superfamily then further support the recruitment of the death-inducing signaling complex (DISC), which contains the FADD adapter protein and the initiator caspases-8 and -10 [169,170]. Also for the death receptor pathway, HDACi-stimulated expression of anti-apoptotic proteins such as of cytoplasmic FLICE-like inhibitory protein (c-FLIP) were noticed [171].
Recently, also non-coding RNA (ncRNA) expression, mostly in the form of microRNAs, has been implicated in HDACi-reactivated induction of apoptosis in cancer cells. Importantly, these have been demonstrated to take over the functions of tumor suppressor proteins and underlie epigenetic regulation themselves; accordingly, microRNAs such as microRNA (miR)-127, miR-124a, and miR-34b/c, have been methylated in cancer cells, thus generating many potential targets of cancer therapy [172]. Apoptosis was mediated in these reports either by deregulating miRs that target HDACs, thereby mimicking HDACi-inducible effects, or by directly interfering with the expression of miRs. The first mechanism was documented for instance for HDAC1 that was repressed by miR-449a in prostate cancer [173]. Examples for direct regulation of miR-supported apoptosis were documented by TSA and SAHA-induced overexpression of miR-129-5p in thyroid cancer cells, or c-MYC-mediated silencing of miR-15 and let-7 miRs that cause transcriptional suppression of BCL-2 and BCL-XL in B-cell lymphomas [174,175]; furthermore, sodium butyrate and panobinostat-induced expression of miR-31 caused apoptosis and cellular senescence via downregulation of BIM1 in breast cancer cells and fibroblasts [176]. A sophisticated role could be unraveled for miR-34a in pancreatic cancer by engaging as a tumor suppressor miR and even regulating acetylation in a feedback loop [177]. SAHA treatment re-established miR-34a expression that led to p53 acetylation and consequent upregulation of CDKN1A as well as PUMA (p53 upregulated modulator of apoptosis) target genes, thereby initiating the induction of apoptosis.
Direct assessment of the role of HDACs has also yielded several candidates that were implicated in the regulation of intrinsic apoptosis by interfering with the subtle balance of pro-apoptotic and anti-apoptotic factors. The deletion of HDAC2 in gastric cancer cells promoted the upregulation of the proapoptotic proteins BAX, AIF, and APAF-1, while it silenced the expression of BCL-2 [178]. The BCL-2 modifying factor, BMF which is a pro-apoptotic activator, was reported to be conjointly downregulated by HDAC1 and 8; inhibition of HDAC8 by methylselenopyruvate in colon cancer cells restored BMF downregulation and thereby activated apoptosis [150,179]. HDAC3 was found to suppress the pro-apoptotic protein PUMA in gastric cancer cells which can be restored by HDACi (TSA) treatment [180].
5.2. HDAC Inhibitor-Induced Autophagy
Autophagy is a conserved catabolic cellular mechanism of self-degradation of cytoplasmic constituents. Autophagy has been categorized into macroautophagy, microautophagy, and chaperone-mediated autophagy of which we further discuss macroautophagy in here, if not stated otherwise [181,182,183,184]. Known signals triggering autophagy are quite diverse and include mostly shortage of nutrients but also the presence of protein aggregates, damaged organelles, hypoxia, and ROS. Aged or damaged molecules and organelles are recycled in specifically for this process formed autophagosomes which is governed by a complex genetic program required in cellular homeostasis or cell death [185,186]. Unlike apoptosis or necrosis, autophagy has been attributed with a dual role in cancer which can result either in a survival- or a death-promoting response to encounter adverse genotoxic or pharmacological stress. This form of HDACi-incurred lethality has only recently been brought into evidence as an effector mechanism that interferes with cellular growth [187]. Epigenetic interference in the regulation of autophagy can either inhibit, or support, the formation of a malignant phenotype. The complex cytoprotective or cytotoxic response of autophagy in tumor cells thereby seems to depend on the type and stage of cancer, its genetic predisposition, as well as the duration and dose of HDACi treatment [188,189,190,191]. The cellular response might also reflect the diverse mutational status of cancer cells, in particular with regard to the highly altered oncoproteins or oncosuppressor genes, such as AKT-1, PTEN, BECLIN-1, or p53 that promote tumorigenesis and are crucial regulators of autophagy [192]. A further issue, why this type of mostly pathological or drug-induced cell death is controversially discussed, might be found in the largely unknown mechanisms that determine how autophagy eliminates cells. One explanation could be the selective accumulation and degradation of cell survival factors in autophagosomes; thus, accumulation of ROS and cell death could be induced by the recruitment of catalase in autophagososmes [193,194]. In general, it has been elaborated that autophagy prevailingly exerts a protective and tumor-suppressive role during the initial phases of tumor development, but also in normal cells. This kind of surveillance mechanism helps to reduce the effects of ROS accumulation by removing damaged organelles and cellular components, and by decelerating the transformation potential of healthy towards malignant cells [195]. For instance, it was evident in mice with a hemizygous Beclin-1 deletion that predisposed for increased tumor formation, or in autophagy-mediated clearance of SAHA-treated apoptosis-resistant uterine sarcoma cells [196]. The tumor-promoting effects of autophagy, however, seem to outweigh in the later progression and metastasis stages of established tumors. Here, autophagy eradicates ROS-induced metabolic stress products, provides nutrients required for cancer cell survival, and mediates resistance during chemotherapy to prevent tumor cell elimination [197]. Under these pathological stressful conditions, autophagy can be stimulated above the usual basal levels that are present in normal cells, and cell death is presumably favored by enhancing the “self-eating” program by not yet elucidated mechanisms [198]. The disruption of autophagy, either by endogenous defects, or by pharmacological interference, as for instance by HDACi, will therefore lead to prolonged tumor survival as it potentiates cell death. This insight is particularly relevant, since HDACi-mediated inhibition of autophagy in combination with chemotherapeutic treatment has been favored as a novel approach in cancer therapy. Consequently, the confirmation of the context-dependent function of autophagy and investigation of limiting molecular determinants gain tremendous importance for a positive therapeutic outcome [191,199,200].
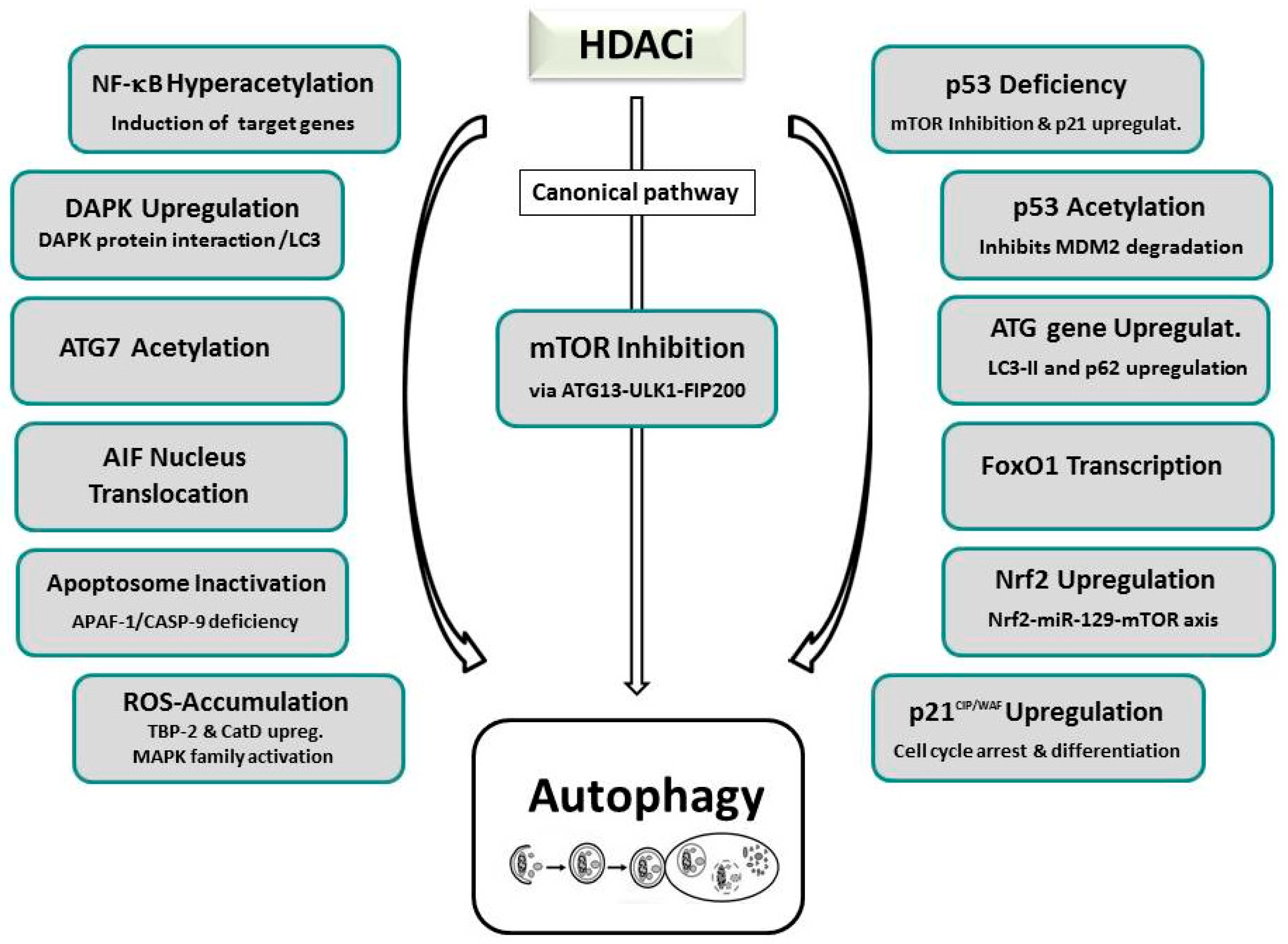
Research thus far has unveiled a multitude of HDACi-induced suppressive or activating autophagic signaling pathways for autophagy involving primarily the modification of HAT/HDAC-mediated acetylation of many “AuTophaGy-related” (ATG) proteins, including ULK1 (UNC-51-like kinase 1), ATG3, or ATG7 (see Figure 2) [14,98,201]. A major frequented pathway of HDACi-induced autophagy is represented by suppression of mTOR, the nutrient-sensing kinase mammalian target of rapamycin, that is often accompanied by increased expression of ATG proteins or regulators, including LC3 (microtubule-associated protein 1A/1B-light chain 3) and BECLIN-1 [13,187,202,203,204,205,206,207,208]. mTOR is a known major molecular hub of autophagy, that activates autophagosome formation by inactivating another major downstream autophagic switch, the ULK1 complex. The fundamental role of mTOR by SAHA-induced inactivation leading to re-establishment of ULK1 function could initially be documented by studies of our own group and by Gammoh et al. on endometrial sarcoma cells, and were confirmed in several subsequent cases [13,202,203,204,205,206]. Many research groups also published an additional occurrence of apoptosis in cells that underwent HDACi-induced autophagy. Alone or in combination with mTOR deactivation, massive intracellular ROS generation impairing mitochondrial respiration and energy metabolism has been reported to mediate SAHA-facilitated autophagy in tumor cells. In several tumors undergoing HDACi-dependent ROS accumulation, additional increased expression of the lysosomal protease cathepsin D, decreased expression of its substrate, TRX, or activation of ERK1/2 and JNK, that belong to the MAPK (mitogen activated protein kinase) family, has been resolved [203,209]. Mechanistically, enzymes related to energy metabolism, anti-oxidative stress, and cellular redox control are presumed to be involved in ROS-activated autophagy as evidenced by proteomic analysis of SAHA-induced Jurkat T-leukemia cells [203,210].
Either induction of NF-κB associated target genes involving hyperacetylation of NF-κB RELA/p65 in SAHA/MS-275 treated PC3 cells, or attenuation of pERK/NF-κB signaling, accompanied by increased transcriptional activity of CDKN1A, were also determined as the cause of HDACi-induced autophagy [211]. HDACi-stimulated p21 expression was documented in H40 and SAHA-treated PC-3M and HL-60 cells resulting in cell cycle arrest, cell differentiation, and autophagy [212]. Furthermore, treatment of prostate cancer cells with the novel HDACi, MRJF4, promoted p21-elicited autophagy [213]. Further single reports, supporting other regulatory mechanisms of HDACi-promoted autophagy, include nuclear translocation of the apoptosis inducing factor (AIF), apoptosome inactivation, transcriptional activity of FoxO1, and the upregulation of death-associated protein kinase (DAPK) [13,211,214,215,216,217]. A very recent study even implicated microRNA-mediated regulation of mTOR by the transcription factor Nrf2 (nuclear factor erythroid 2 like-2) as a mechanism of HDACi-induced autophagy [218]. In response to HDACi treatment, increased Nrf2 mRNA and protein levels accumulated that led to the increased transcription of miR-129-3p which in turn targeted mTOR.
Nevertheless, the HDACi-mediated suppression of autophagy could be verified in two reports. In the first case, treatment of myeloid-leukemic cells with valproic acid, SAHA, TSA, panobinostat, or JQ2, decreased the expression of ATG7, an autophagic regulatory protein required for fusion of peroxisomal and vacuolar membranes, and thereby downregulated the autophagic flux but promoted autophagy [219]. Moreover, the sirtuin inhibitor tenovin-6 was demonstrated to suppress autophagy in chronic lymphocytic leukemia (CLL) cells as evidenced by increased expression of autophagy-lysosomal pathway genes and the autophagic markers LC3-II and p62 [220]. HDACi-dependent autophagic induction entailing p53 acetylation and p53-deficiency, based on our own findings, will be discussed in Section 6.2.
Involvement of members of different HAT families in the epigenetic control of autophagy via acetylation of different ATG proteins and effectors has also been observed. Thus, ATG7 has been reported to co-localize and interact with the EP300 acetyltransferase [221]. Silencing of EP300 by RNAi promoted the induction of autophagy while starvation-induced autophagy, a process controlled by BAG6/BAT3 (BCL-2-associated athanogene 6)-mediated regulation of EP300, was suppressed upon overexpression of EP300 [221,222,223]. Furthermore, beside the EP300 family, also the MYST family of HATs was associated with the induction of autophagy. TIP60-dependent acetylation of ULK-1 via glycogen synthase kinase-3 (GSK3) serves as a fundamental cellular mechanism that is initiated during starvation-induced autophagy [224]. The interaction of ATG3 with ATG8, that is required for autophagic pathway signaling, was found to depend on Esa1-mediated acetylation [49]. Histone H4 (H4K16ac)-mediated activation of autophagy was induced by the downregulation of hMOF (KAT8/MYST1) [50]. Also, histone H3 acetylation seems to induce autophagy as it was demonstrated to limit the development of resistance due to chronic mTOR inhibition in renal cell carcinoma cells that were targeted by valproic acid [225].
In contrast to the stimulatory autophagy-triggering signals caused by the majority of HDACi, HDACs, that initially trigger deacetylation activity, have been established as inhibitors of autophagy [226]. Either depletion of HDAC1, or HDACi (FK228)-mediated inhibition of HDAC1 in HeLa cells, induces autophagy as evidenced by accumulation of the autophagosomal marker LC3-II [227,228,229]. Combined inhibition of HDAC1 and 2 by TSA triggered phenylephrine-induced autophagy in cardiomyocytes by entangling the autophagic regulators ATG5 or BECLIN-1 [227,228]. Skeletal muscle-specific deletion of both HDACs therefore resulted in perinatal lethality in a subset of mice as they lacked skeletal muscle homeostasis and autophagic flux [230]. HDAC6 was established initially as a microtubule-associated deacetylase that, as a compensatory degradation mechanism, activates autophagy when the ubiquitin-proteasome-system is blocked [231]. In mitophagy—the autophagic degradation of mitochondria—a similar mechanism involving HDAC6 and SQSTM1/p62 recruitment by parkin-mediated ubiquitination was observed which assists in mitochondrial clearance [232]. The key role of HDAC6, that is able to bind to polyubiquitinated proteins and can be inhibited by tubacin, is the mediation of autophagosome-lysosome fusion by controlling the acetylation of SIK2 (salt-inducible kinase 2) [64,233]. HDAC6 seems to be a primary object regulating the anti-cancer response of HDACi by deacetylation of non-nuclear proteins; it is involved in the clearance of toxic misfolded protein aggregates through aggresome-mediated autophagy whose deficiency impairs tumor growth [234,235]. The induction of apoptosis as well as autophagy, in combination with cell cycle arrest and diminished ERK activation, were activated by Apicidin-elicited suppression of HDAC7 in salivary mucoepidermoid carcinoma cells [236]. HDAC10, presumably by modifying the acetylation of Hsp70 protein families, adds to autophagy-mediated cell survival as its disabling, either by ablation, or by treatment with class II-B inhibitors bufexamac and tubastatin, blocked the autophagic flux and prevented efficient autophagosome-lysosome fusion; hence, neuroblastoma cells were rendered more sensitive to cytotoxic drugs [237].
Among class III HDACs, SIRT1, 2, 3, 5, and 6 have been implicated in autophagic signaling. SIRT1 activity directly deacetylates ATG 5, 7, and 8 which are required for induction of starvation-induced autophagy [238]. Furthermore, participation in oxidative stress-induced autophagy in embryonic stem cells and PLB (Plumbagin)-induced autophagy in prostate cancer cells, involving the mTOR pathway, has been attested [239,240]. SIRT2 drives acetylation of FoxO1, a transcription factor that is regulated by ATG7 interaction and stimulates autophagy [241]. The mitochondrial deacetylase SIRT3 activates cytoprotective mitophagy in breast cancer and other cells during various conditions, such as ROS induction, starvation, or proteotoxic stress [242,243,244]. SIRT5 has been established in the downregulation of ammonia-induced autophagy and mitophagy by controlling glutamine metabolism [245]. By engaging the IGF-AKT-mTOR pathway, or by ROS-induced activation of mTOR, SIRT6 stimulated autophagy in human bronchial epithelial cells or neuronal cells, respectively [246,247].
6. The Role of p53 in HDACi-Mediated Cancer Cell Death
6.1. Post-Translational Regulation of Wild-Type and Mutant p53 by Acetylation
The transcription factor and tumor suppressor protein p53, considered as a “custodian” of the cell, has a detrimental role in the regulation of cell integrity and homeostasis and thus in tumor defense. This protein, p53, coordinates cellular responses such as cell cycle arrest, apoptosis, senescence, metabolism, differentiation, angiogenesis, and even modulates autophagy. Among many other post-translational modifications, acetylation assists this master regulator to sense and integrate a variety of internal and external cellular stress signals, such as DNA damage and changes in DNA methylation, genotoxicity, hypoxia, oxidative stress, or oncogene activation [22,44]. In response, p53, as a central transcription factor that translocates to the nucleus by detaching from the E3 ubiquitin ligase MDM2 (mouse double minute 2 homolog), modulates multiple downstream target genes that regulate processes such as cell cycle progression and cell death [134,248,249]. In appropriate conditions, p53 induces apoptosis by transactivating, i.e., transcriptional activation of pro-apoptotic genes, or by direct interaction with anti-apoptotic proteins located in the mitochondrial membrane [250,251].
p53 is a well-investigated representative of crosstalk with respect to acetylation, and other post-translational modifications [22,44,252]. Currently, 13 lysine residues of p53, that are located mostly at the C-terminal end, have been reported to undergo acetylation [253,254]. Acetylation and deacetylation of p53 are managed by the CBP/p300 or the MYST family (TIP60, hMOF) of HATs and by the HDAC1/mSin3 complex or Sirt1, respectively [22,255,256,257,258,259]. HDAC2 that is overexpressed in numerous cancer types has also been linked to the regulation of deacetylation of C-terminal lysine side-chains in p53, and functions as a corepressor that is involved in regulating to the interaction with target genes [260,261]. Furthermore, in human hepatocellular carcinoma tissue p53 protein levels were found overexpressed through downregulation of MDM2-mediated p53 degradation by SIRT3 [262,263]. Disruption of acetylation by HDACi at distinct sites could either affect the sequence-specific DNA binding activity which transactivate target genes or modify nuclear export, co-activator recruitment, or stability properties of p53 [12,22,252,256,257,264]. Examples are the failure of p53 to activate p21 (p21waf-1/cip1) transcription, the ability to induce cell growth arrest upon deleting a C-terminal acetylation site, or the interference with ubiquitin–proteasome-dependent degradation of p53 by MDM2 in a feedback loop [265,266]. Reciprocally, by the HDACi-mediated hyperacetylation of key residues of p53, enhanced stabilization, cell cycle arrest, and the expression of pro-apoptotic genes can be achieved [267,268]. This finding gains particular importance as variants of p53, provoking a loss of the sequence-specific DNA binding ability of transcriptional targets, are among the most common mutations encountered in human cancer [248,269]. Hyperstable mutant gain-of-function variants which elicit overexpression of chaperone or MDM2 short isoforms, and accumulate in the cell due to increased half-life, can even have a dominant-negative effect on the remaining wild-type allele (e.g., by blocking access to the promoter, or by assuming pro-oncogenic functions) [270,271,272,273,274,275]. Consistently, HDACi treatment exerts a destabilizing effect by enabling the cell to degrade mutant p53, either by reactivating, or by mimicking its transactivation capability, resulting in increased p21 and MDM-2 expression [276,277]. Accordingly, a study using selective HDACi, exposed a regulatory mechanism of mutant p53 transcription which presumably involves direct interaction of HDAC8 via HoxA5, as histone acetylation is not altered [278]. Alternatively, HDACi (SAHA)-induced inhibition of HDAC6 was uncovered to facilitate the degradation of mutant p53 by MDM2 and CHIP ligases due to its release from the chaperone HSP90 [279]. Moreover, HDACi-induced autophagy was suggested as a further mechanism in eliminating mutant p53 [280,281].
6.2. HDACi-Mediated Apoptosis and Autophagy by p53
As mentioned earlier, the induction of mitochondrial and death receptor-mediated apoptosis that is often accompanied by ROS generation and p21-induced cell cycle arrest, has been denoted as the most common form of HDACi-mediated cell death [145,147,148]. Nevertheless, the role of p53 in this context is controversial as by HDACi caused post-translational acetylation, the capability of p53 to transcriptionally transactivate several pro-apoptotic genes, such as BAX, PUMA, and NOXA, is hindered. Thus, many studies demonstrate p53-independent p21 induction and apoptosis upon HDACi administration and the anticancer effect of HDACi is not influenced by the mutational status of p53 in the tumor [104,105,106,217,282]. Nevertheless, several reports demonstrate acetylation and stabilization of p53 in various tumor models associated with the induction of cell cycle arrest and apoptosis [107,108,112]. Consistently, the combined inhibition of HDACs by TSA, and SIRT1 by nicotineamide, reversed the inhibitory effects of cAMP on DNA damage-induced p53 stabilization and apoptosis, providing evidence that p53 acetylation is involved in anti-apoptotic activities [283]. Also, TSA-mediated induction of apoptosis by PUMA upregulation in gastric cancer cells could be achieved, either by direct p53 modulation, or by HDAC3 inhibition, promoting the interaction of the PUMA promoter with p53 [113]. Furthermore, experiments of isogenic HCT-116 colon cancer cell lines with different mutant or wild-type p53 variants demonstrated either p53-dependent or independent antitumor responses depending on the type of HDACi treatment [284,285]. In addition, a backup mechanism of HDACi-induced stabilization of the transcription factor RUNX3 could be demonstrated that induces cell cycle arrest and apoptosis, due to concomitant upregulation of p21 and BID in tumor cells [286,287,288]. Consequently, HDACi may activate p53, but do not unconditionally need p53 for exerting anticancer effects, and p53-dependent as well as independent pathways may contribute to HDACi-mediated apoptotic processes.
In addition to canonical cell death, programmed activation of autophagy can occur alone or in combination in tumor cells expanding the repertoire of HDACi-elicited anti-cancer mechanisms [13,187,202,205,206,207,216,226,289,290]. Recent evidence implies that also p53 is involved in HDACi-inflicted autophagic cell death reflecting the importance of acetylation in autophagy control. Particularly, forapoptosis resistance in p53-deficient malignant cells, HDACi-elicited autophagy could provide an alternative pathway to sustain programmed cell death. The results of our own research studies performed in endometrial sarcoma (ESS) cells suggest that p53 is a major regulatory switch between HDACi-triggered autophagic and apoptotic cell death [13]. In this model, we strived to unearth the molecular cause for the previously published prevalent cell death due to dose-dependent induction of autophagy in ESS-1 cells, in contrast to predominant activation of apoptosis in MES-SA cells, following the treatment with the potent small-molecule pan-inhibitor SAHA [291]. Caspase-dependent and independent cell death resulted thereby in the elimination of 48% MES-SA cells and 80% ESS-1 cells after 24 hours of HDACi administration, respectively, and included prominent p21 upregulation and cell cycle arrest at the G1/S transition phase in both cell types. Consistent with the induction of the autophagic pathway, we were also able to detect a decrease in mTOR expression in the ESS-1 cell line that was not detectable in MES-SA cells [13]. Therefore, we were keen to identify differences in upstream signaling pathways of both cell lines that link mTOR with SAHA-induced autophagy. Upon investigating major autophagic and apoptotic regulators upstream of mTOR, we determined a complete absence of p53 protein coupled with decreased levels of PUMA expression in ESS-1 cells. p53-deficient expression, promoted by a presumable degradation of the p53 transcript, was obviously caused by a novel nonsense mutation (p53R213X) that we uncovered in the transactivating domain of p53 and was not present in MES-SA cells. Transfection of ESS-1 cells with wild-type p53 mRNA therefore re-induced apoptosis, as documented by upregulated PUMA, caspases-9, as well as effector caspase-3 and -7 expression, in addition to PARP-1 cleavage. At the same time, expression levels of mTOR were re-elevated accompanied by the return to basic autophagy, as supported by LC3 staining. This HDACi-mediated shift between predominant induction of autophagy or apoptosis was therefore depending on the presence of functional p53 protein. This finding could be further confirmed by repeating the p53-mediated rescue in several other p53-deficient tumor cell lines (such as PANC-1, Jurkat, HL-60, and U937) that previously have been reported to undergo SAHA-induced apoptosis or autophagy.
Similar to our study, HDAC-induced autophagy involving a major regulatory role of the transcription factor p53 was noted in several other reports [281,292,293,294]. Concomitant induction of apoptosis and autophagy occurred in the valproic acid (VPA) and TSA treated p53-deficient pancreatic cancer cells [281]. Intriguingly, the identified mechanism included the ability to reactivate wild-type p53 expression from the non-mutant allele and ERK-mediated stabilization of the oncogenic protein c-Myc; the second previously published effect, which activates cell proliferation in cancer cells, is based on the inhibition of ERK phosphorylation and associated c-Myc expression potentially involving acetylation. Attenuated activation of autophagy evoked by re-established p53 wild-type expression correlated with increased expression of the p53 transcriptionally regulated proteins, p21 and PUMA. Additionally, by screening TSA-treated pancreatic cells comparatively low down-regulation of the anti-apoptotic protein Mcl-1 and reduced ROS production occurred in Panc-1 cells. The Sirt1 and 2-specific deacetylase inhibitor sirtinol has also been attested to effect HDACi-induced cell fate in several studies as it interferes with p53-specific deacetylation. Thus, p53 was implicated in regulating the counterbalance between sirtinol-induced apoptosis and autophagy in MCF-7 breast cancer cells [292]. While sirtinol administration favored increased autophagy as demonstrated by LC3-II expression, increased cytochrome C-mediated apoptotic cell death and cell cycle arrest were provoked by application of the autophagy inhibitor 3-methyladenine resulting in elevated BAX levels, but decreased BCL-2 protein expression. A second study also included the novel Sirt1, -2, and -3 protein inhibitor, MHY2256, in addition to sirtinol inhibition in MCF-7 cells, which resulted similarly in cell death type-I and -II including cell cycle arrest [293]. Here, specifically SIRT1/2-stimulated acetylation of p53 at lysine 382 suppressing MDM2-mediated ubiquitination of p53 was found to result in stabilization and increased functional activity of p53. Similarly, MHY2256 treatment caused MDM2-mediated degradation of p53 in endometrial cancer-derived Ishikawa cells and thereby triggered induction of autophagy as well as apoptosis [294]. MHY2256-elicited apoptosis was associated with elevated BAX and slightly increased BCL-2 expression, cytochrome C release, and elevated PARP-1 cleavage, as well as p21 upregulation that was linked to heightened autophagy in Ishikawa cells. The specific involvement of SIRT1 in the regulation of autophagy has also been confirmed by rescue experiments of homozygous Sirt1-deficient mouse embryonic fibroblasts [238]. Only cells that possessed the Sirt1 wild-type gene allowed re-induction of autophagy under starvation conditions and was potentially promoted by the Sirt1 target proteins ATG5, ATG7, and ATG8 whose acetylation was significantly increased in Sirt1-deficient fibroblasts.
The role of p53 as a central factor in coordinating the mode of HDACi-induced cell death is in agreement with a previously determined dual role of p53 in autophagy regulation, depending on its subcellular location in non-transformed cells [295]. In contrast to the nuclear function of p53 in the activation of autophagy, in response to stress conditions, a kind of steady-state function of p53 in arresting the induction of autophagy in the cytoplasm under normal physiological settings was unraveled. Although this novel discovered function of p53 does evidently not involve transactivation-dependent activity but rather direct interaction of the protein with target proteins, it employs also the canonical AMPK–mTOR signaling pathway. In contrast to the nuclear pathway however, inhibition of the AMP-dependent kinase—resulting in activation of mTOR signaling and associated hyperphosphorylation of AMPK, TSC2, acetyl CoA carboxylase, and the mTOR substrate p70S6K—is enforced by cytoplasmic p53 protein. Presently, the involved mechanism is still unclear. Experimental evidence involving mutational analyses indicate that p53 directly interacts with the autophagy-related protein FIP200 (focal adhesion kinase interacting protein of 200 kDa; ATG17) [296]. This interaction inhibits the activation of the ATG13–ULK–FIP200 complex that is responsible for phagopore/autophagosome formation and is indispensable for nutrient starvation-induced autophagy. Interestingly, p53 variants with a loss-of-function promoted the activation of autophagy, while several variants with gain-of-function mutations could still fulfill cytoplasm-controlled inhibition of autophagy. As a proof-of-concept, the drug-induced inhibition of basal cytoplasmic p53, or conversely, a nuclear export domain-deficient form of p53 either exerted an activating or inhibiting influence on autophagy, respectively. Confirmation of this mechanism comes from embryonal carcinoma cells where genetic deletion of p53 activated autophagy involving Beclin-1-mediated degradation of p53 via USP10 and USP13 ubiquitin-specific peptidases in a feedback loop [297]. Evolutionary, cytoplasm-induced transcription-independent autophagy seems to represent a backup mechanism, in case p53 is mutated, to provide increased resilience towards metabolic stress and ensure cell survival.
6.3. Balanced Control of HDACi-Mediated Apoptosis and Autophagy by p53
Programmed cell death by apoptosis and autophagy are in general two different stress-induced biological responses that are mutually exclusive and commonly block each other as they are governed by two distinct genetic programs. Nonetheless, in order to avoid concurrent activation these processes have to undergo coordinated regulation. Thus, initiation of the apoptotic program, as one of the most frequently ensued cell death modalities, also involves the activation of a series of caspases. By proteolytic cleavage of several key regulatory proteins of the autophagic pathway, caspases are likewise able to contribute to the shutdown of autophagy. Thereby, the cytoprotective response of autophagy might be suppressed whereas apoptotic cell death is enhanced. Anti-apoptotic mechanisms, conducted by the autophagic program include the selective degradation of ubiquitinylated cytoplasmic pro-apoptotic proteins. Alternate effects involve targeting of specific molecules such as SQSTM1/p62 that stimulate ROS generation and ROS-induced apoptosis when present in excess amounts in the cell [298,299]. The ubiquitin-binding scaffold protein SQSTM1/p62, is an autophagosome cargo protein that targets other interacting proteins for selective autophagy. It binds to LC3 and thereby acts as a signaling hub that controls cell survival, apoptosis, and inhibition of tumorigenesis by autophagy [300,301,302,303]. Furthermore, the removal of mitochondria by mitophagy can lower the propensity of the cell for intrinsic apoptosis [184]. Under certain conditions which are not well elaborated however, combined induction of autophagic and apoptotic cell death has been observed which is also relevant for many studies involving HDACi-treated cancer cells.
On the molecular level, this dual activation of autophagy and apoptosis involves modifying pro-apoptotic members of the BH3-only family that compensate anti-apoptotic BCL-2 family proteins. These proteins directly interact with the BH3 domain of BECLIN-1 and block BECLIN-1–dependent autophagic activation [304,305]. Either transcriptional downregulation of BCL-2, BCL-XL, and MCL-1, or transcriptional upregulation of BAX, BAD, BNIP3, or PUMA, releases BECLIN-1 from its protein complex that initiates autophagy [306]. Consistently, p53-regulated pro-apoptotic proteins have also been identified as autophagy-promoting proteins [307,308]. As a debated mechanism of autophagy activation, p53 was described to stimulate the phosphorylation of BCL-2 [309]. This causes dissociation of the BECLIN-1/BCL-2 complex since BCL-2 functions as an anti-apoptotic as well as an anti-autophagic protein via its inhibitory interaction with BECLIN-1. The released BECLIN-1 (ATG6) then stimulates phagophore, and finally autophagosome formation, by recruiting and interacting with many proteins [310]. Contrastingly, there is evidence suggesting that the effect of autophagy-promoting BCL-2 family members is owned to their antagonizing inhibitory activity on the apoptosis mediators BAX and BAK. Thus, when BAX and BAK are absent in the cell, BCL-2 family members have no measurable impact on autophagy [311]. A second path is mediated by the upregulation of DRAM (damage-regulated autophagy modulator) through stress-activated p53 [312]. As a lysosomal protein regulating autophagosome-lysosome fusion and direct mediator of autophagy and apoptosis, DRAM can interfere with several different stages of autolysosome formation [313].
Recent reports indicated that also the frequently cancer-deregulated MAPK members, JNK and p38, have a master regulatory role in these processes [309]. MAPK members regulate a variety of cellular activities and can modulate processes, such as proliferation, differentiation, apoptosis and autophagy in response to extracellular stimuli that include UV irradiation, inflammatory cytokines, protein synthesis inhibitors, DNA damaging agents, or chemotherapeutic agents. Interestingly, both of the above specified cell death crosstalk mechanisms were promoted through the activation of p53 and JNK pathways in 2-methoxyestradiol-treated Ewing sarcoma cells [314]. Thus, p53 was assumed to regulate JNK activation which itself regulates DRAMin its function as a pro-apoptotic and pro-autophagic protein. The eminent role of DRAM in 2-methoxyestradiol -mediated autophagy and apoptosis could be demonstrated by silencing of DRAM which suppressed the induction of both processes. Additionally, BCL-2 phosphorylation resulting in the dissociation of the BECLIN-1-BCL-2 complex could be confirmed in this study. As a further rare p53-independent mechanism, calpain-mediated ATG5 fragmentation was inducing mitochondria- and caspase-dependent apoptotic cell death, thereby representing a molecular link between autophagy and apoptosis [315]. Interacting apoptosis and autophagy, including involvement of ATG proteins through caspase- and calpain-1-induced cleavage, were reported, for example, in a case of MGCD0103-treated primary chronic leukocytic leukemia cells, where the PI3K-AKT-mTOR pathway was also activated [208]. The mocetinostat-mediated inhibition of autophagy also involved direct transcriptional modulation of the expression of critical autophagy genes.
As already specified in Section 5.1, for the enhancement of concurrent stimulation of apoptosis and autophagy, also HDACi prevalently impede the equilibrium between pro- and anti-apoptotic proteins, and have been documented to facilitate the down-regulation of anti-apoptotic genes (such as BCL-2, BCL-XL, XIAP, MCL-1, Survivin) [112,113,119,135,157,158]. BCL-2/BECLIN-1-dependent autophagic cell death has not been thoroughly investigated in HDACi-treated cancer cells but is nevertheless presumed to mediate combined activation of both cell types in most cases. Thus, as we previously reviewed, Bcl-2 downregulation was indicated in several reports involving HDACi-mediated dual activation of apoptosis and autophagy involving either p53-specific acetylation or other/unknown mechanisms [15,292,293,316]; On a similar note, also Beclin-1 upregulation has been determined in a few cases among these summarized studies [15,203,204,317]. A direct support for this mechanism comes from a study applying a knockdown of anti-apoptotic mRNAs for BCL-2, BCL-XL, and MCL-1 or using a BCL-2 family inhibitor (GX15-070) in pancreatic cancer cells with combined administration of SAHA or sodium valproate and sorafenib; these inhibitory agents induced autophagy and intrinsic apoptosis, thus bypassing extrinsic apoptosis resistance [318]. In addition, the coupling of apoptosis to suppressive autophagy has been proven by combined with the HDACi-mediated upregulation of BIM and BH3 mimetic-induced liberation of BIM from anti-apoptotic proteins, a process that is normally reliant on p53 [319,320].
Further studies support ROS-induced dual activation of apoptosis and autophagy including a report where autophagy was downregulated [203,219,321]. With respect to MAPK-activated cell death by HDACi, a key regulatory role for ROS-induced p38 MAPK in the balance between apoptosis and autophagy in MS-275 treated HCT116 colon cancer cells could be clearly demonstrated [322]. The type of induced cell death correlated with the duration of HDACi treatment and the resulting expression levels of p38 in this study. While short-term treatment and low ROS-induced ERK activation via p38 resulted in ATG7-induced autophagy, the apoptotic pathway could only be induced by high levels of phosphorylated ERK, JNK, and AKT proteins, provoked by long-term treatment of more than 48 hours or by a knockdown of ATG7. As already detailed above in Section 5.2, the Nrf2-miR-129-3p-mTOR Axis was described to regulate HDACi-induced autophagy [218]. As generally the Nrf2 signaling pathway is activated according to to the MAPK, JNK and p38, a link to the canonical mTOR-regulated pathway of autophagy was established by this report and might also connect to p53. This may be of high relevance as the regulation of Nrf2-mediated autophagy is one mechanism of HDACi-caused chemoresistance.
Nevertheless, beside p53 and MAPK other central coordinators that balance HDACi-induced cell death, favoring either one of both types of cell death, or concurrent activation of apoptosis and autophagy, are very likely to exist. Accordingly, p53-independent induction of p21 expression in combination with dual activation of apoptosis and autophagy have been exemplified in apicidin-treated YD-8 and YD-10B human oral squamous carcinoma cells (OSCC) without defining its molecular causes [282]. Many studies that report simultaneous activation of both types of HDACi-induced cell death also evidenced increased acetylation of the core histones H3 and H4 (for review see [15]). However, as these have been associated with increased transcription of distinct genes implicated in tumor growth suppression it is not clear whether these have a role as HDACi-mediating cell death regulators [81,323]. Therefore, the question of which factors and which exact molecular mechanisms influence the cell’s decision to modulate the interplay and balance between the different modes of HDACi-elicited cell death awaits further detailed research.
7. Perspectives and Future Directions
Identifying the mechanisms of programmed cell death is one of the key requirements to achieve progress, not only in unraveling pathogenetic insights, but also in the development of novel therapeutic strategies for diseases such as cancer. Traditionally, in the last few decades cancer therapy has mainly focused on the enhancement of cellular apoptosis. More recently, clinical strategies for cancer therapy have enforced HDACi administration combined with inhibitors of autophagy. The ability to sensitize apoptosis-resistant tumor cells by the disruption of autophagy was considered a promising route for cancer therapy as this process augments the pro-apoptotic effects of HDACi. In addition, to restrain the extents of tumor necrosis and inflammation, autophagy might be required for the cancer cell to deal with metabolic stress and cytotoxicity during chemotherapy. Furthermore, by expediting the autophagic pathway in advanced stages of the cancer cell, autophagy may promote cell death by mostly non-elucidated mechanisms. This might be also a significant preference for apoptosis resistant cancer cells due to the inactivated caspase-mediated pathways or specifically acquired HDACi-resistance. In this context, the disruption of the autophagic program will facilitate tumor survival. In line with this, it is of crucial importance to define the factors and mechanisms that influence the balance between HDACi-elicited apoptosis, autophagy or even necrosis in the cancer cell. Studying the detailed mechanisms of the modulation of autophagy and apoptosis may lead to a better understanding of the discriminatory role of different p53 variants in controlling cell death and provide the potential to bypass the resistance of cancer cells.
Author Contributions
All authors, M.M., J.K., and L.F.F. were responsible for collecting literature, drafting, writing, reviewing, editing, and illustration of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lockshin, R.; Williams, C. Programmed cell death—II. Endocrine potentiation of the breakdown of the intersegmental muscles of silkmoths. J. Insect Physiol. 1964, 10, 643–649. [Google Scholar] [CrossRef]
- Lockshin, R.A.; Zakeri, Z. Apoptosis, autophagy, and more. Int. J. Biochem. Cell Biol. 2004, 36, 2405–2419. [Google Scholar] [CrossRef]
- Tan, M.; Ooi, J.; Ismail, N.; Moad, A.; Muhammad, T. Programmed cell death pathways and current antitumor targets. Pharmacol. Res. 2009, 26, 1547–1560. [Google Scholar] [CrossRef] [PubMed]
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Schweichel, J.-U.; Merker, H.-J. The morphology of various types of cell death in prenatal tissues. Teratology 1973, 7, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Salehi, B.; Sharifi-Rad, M.; Pezzani, R.; Kobarfard, F.; Sharifi-Rad, J.; Nigam, M. Programmed Cell Death from a Cancer Perspective: An Overview. Mol. Diagn. Ther. 2018, 22, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Sarkar, S.; Horn, G.; Moulton, K.; Oza, A.; Byler, S.; Kokolus, S.; Longacre, M. Cancer development, progression, and therapy: An epigenetic overview. Int. J. Mol. Sci. 2013, 14, 21087–21113. [Google Scholar] [CrossRef]
- Garraway, L.A.; Lander, E.S. Lessons from the Cancer Genome. Cell 2013, 153, 17–37. [Google Scholar] [CrossRef] [Green Version]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A.J.; Kinzler, K.W. Cancer Genome Landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.G.; Piluso, L.G.; Cai, X.; Gadd, B.J.; Ladurner, A.G.; Liu, X. An Acetylation Switch in p53 Mediates Holo-TFIID Recruitment. Mol. Cell 2007, 28, 408–421. [Google Scholar] [CrossRef]
- Fröhlich, L.F.; Mrakovcic, M.; Smole, C.; Zatloukal, K. Molecular mechanism leading to SAHA-induced autophagy in tumor cells: Evidence for a p53-dependent pathway. Cancer Cell Int. 2016, 16, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Mrakovcic, M.; Kleinheinz, J.; Fröhlich, L.F. Histone deacetylase inhibitor-induced autophagy in tumor cells: Implications for p53. Int. J. Mol. Sci. 2017, 18, 1883. [Google Scholar] [CrossRef] [PubMed]
- Mrakovcic, M.; Bohner, L.; Hanisch, M.; Fröhlich, L.F. Epigenetic Targeting of Autophagy via HDAC Inhibition in Tumor Cells: Role of p53. Int. J. Mol. Sci. 2018, 19, 3952. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, R.D. Chromatin structure: A repeating unit of histones and DNA. Science 1974, 184, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Grunstein, M. Histone acetylation in chromatin structure and transcription. Nature 1997, 389, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Spange, S.; Wagner, T.; Heinzel, T.; Krämer, O.H. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int. J. Biochem. Cell Biol. 2009, 41, 185–198. [Google Scholar] [CrossRef]
- Lee, D.Y.; Hayes, J.J.; Pruss, D.D.; Wolffe, A.P. A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell 1993, 72, 73–84. [Google Scholar] [CrossRef]
- Roth, S.Y.; Allis, C.D. Histone acetylation and chromatin assembly: A single escort, multiple dances? Cell 1996, 87, 5–8. [Google Scholar] [CrossRef]
- Kazanets, A.; Shorstova, T.; Hilm, I.K.; Marques, M.; Witcher, M. Epigenetic silencing of tumor suppressor genes: Paradigms, puzzles, and potential. Biochim. Biophys. Acta Rev. Cancer. 2016, 1865, 275–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, W.; Roeder, R.G. Activation of p53 Sequence-Specific DNA Binding by Acetylation of the p53 C-Terminal Domain. Cell 1997, 90, 595–606. [Google Scholar] [CrossRef] [Green Version]
- Glozak, M.A.; Seto, E. Histone deacetylases and cancer. Oncogene 2007, 26, 5420–5432. [Google Scholar] [CrossRef] [Green Version]
- Ropero, S.; Esteller, M. The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 2007, 1, 19–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chrun, E.; Modolo, F.; Ivan, F.D. Histone modifications: A review about the presence of this epigenetic phenomenon in carcinogenesis. Pathol. Pract. 2017, 213, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Barneda-Zahonero, B.; Parra, M. Histone deacetylases and cancer. Mol. Oncol. 2012, 6, 579–589. [Google Scholar] [CrossRef] [Green Version]
- Fraga, M.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef]
- Toh, Y.; Ohga, T.; Endo, K.; Adachi, E.; Kusumoto, H.; Haraguchi, M.; Okamura, T.; Nicolson, G.L. Expression of the metastasis-associated MTA1 protein and its relationship to deacetylation of the histone H4 in esophageal squamous cell carcinomas. Int. J. Cancer 2004, 110, 362–367. [Google Scholar] [CrossRef] [Green Version]
- Seligson, D.; Horvath, S.; Shi, T.; Yu, H.; Tze, S.; Grunstein, M.; Kurdistani, S. Global histone modification patterns predict risk of prostate cancer recurrence. Nature 2005, 435, 1262–1266. [Google Scholar] [CrossRef]
- Seligson, D.; Horvath, S.; McBrian, M.; Mah, V.; Yu, H.; Tze, S.; Wang, Q.; Chia, D.; Goodglick, L.; Kurdistani, S. Global levels of histone modifications predict prognosis in different cancers. Am. J. Pathol. 2009, 174, 1619–1628. [Google Scholar] [CrossRef] [PubMed]
- Gayther, S.; Batley, S.; Linger, L.; Bannister, A.; Thorpe, K.; Chin, S.; Daigo, Y.; Russell, P.; Wilson, A.; Sowter, H.; et al. Mutations truncating the EP300 acetylase in human cancers. Nat. Genet. 2000, 24, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Gural, A.; Sun, X.; Zhao, X.; Perna, F.; Huang, G.; Hatlen, M.; Vu, L.; Liu, F.; Xu, H.; et al. The leukemogenicity of AML1-ETO is dependent on site-specific lysine acetylation. Science 2011, 333, 765–769. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Wu, L.; Liang, T.; Liu, Z.; Li, J.; Li, D.; Xie, H.; Yin, S.; Yu, J.; Lin, Q.; et al. Overexpression of myocyte enhancer factor 2 and histone hyperacetylation in hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2008, 134, 83–91. [Google Scholar] [CrossRef]
- Zhong, J.; Ding, L.; Bohrer, L.; Pan, Y.; Liu, P.; Zhang, J.; Sebo, T.; Karnes, R.; Tindall, D.; van Deursen, J.; et al. p300 acetyltransferase regulates androgen receptor degradation and PTEN-deficient prostate tumorigenesis. Cancer Res. 2014, 74, 1870–1880. [Google Scholar] [CrossRef] [PubMed]
- Debes, J.; Sebo, T.; Lohse, C.; Murphy, L.; Haugen, D.; Tindall, D. p300 in prostate cancer proliferation and progression. Cancer Res. 2003, 63, 7638–7640. [Google Scholar]
- Iyer, N.G.; Ozdag, H.; Caldas, C. p300/CBP and cancer. Oncogene 2004, 23, 4225–4231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishihama, K.; Yamakawa, M.; Semba, S.; Takeda, H.; Kawata, S.; Kimura, S.; Kimura, W. Expression of HDAC1 and CBP/p300 in human colorectal carcinomas. J. Clin. Pathol. 2007, 60, 1205–1210. [Google Scholar] [CrossRef]
- Patel, H.; Du, Y.; Ard, P.; Phillips, C.; Carella, B.; Chen, C.; Rakowski, C.; Chatterjee, C.; Liebermann, P.; Lane, W.; et al. The c-MYC oncoprotein is a substrate of the acetyltransferases hGCN5/PCAF and TIP60. Mol. Cell. Biol. 2004, 24, 10826–10834. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, S. p300/CBP/p53 interaction and regulation of the p53 response. Eur. J. Biochem. 2001, 268, 2773–2778. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Scolnick, D.; Trievel, R.; Zhang, H.; Marmorstein, R.; Halazonetis, T. p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol. Cell. Biol. 1999, 19, 1202–1209. [Google Scholar] [CrossRef] [PubMed]
- Tessarz, P.; Kouzarides, T. Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 2014, 15, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Shilatifard, A. Chromatin modifications by methylation and ubiquitination: Implications in the regulation of gene expression. Annu. Rev. Biochem. 2006, 75, 243–269. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Sarma, K.; Reinberg, D. Histone variants meet their match. Nat. Rev. Mol. Cell Biol. 2005, 6, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.; Allis, C. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Huang, H.; Sabari, B.R.; Garcia, B.A.; Allis, C.D.; Zhao, Y. SnapShot: Histone modifications. Cell 2014, 159, 458. [Google Scholar] [CrossRef] [PubMed]
- Roth, S.Y.; Denu, J.M.; Allis, C.D. Histone acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81–120. [Google Scholar] [CrossRef] [PubMed]
- de Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Morales, V.; Richard-Foy, H. Role of histone N-terminal tails and their acetylation in nucleosome dynamics. Mol. Cell. Biol. 2000, 20, 7230–7237. [Google Scholar] [CrossRef] [PubMed]
- Gregoretti, I.V.; Lee, Y.-M.; Goodson, H.V. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Buchwald, M.; Krämer, O.H.; Heinzel, T. HDACi - Targets beyond chromatin. Cancer Lett. 2009, 280, 160–167. [Google Scholar] [CrossRef]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2011, 10, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Workman, J. Histone acetyltransferase complexes: One size doesn’t fit all. Nat. Rev. Mol. Cell Biol. 2007, 8, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Marmorstein, R.; Roth, S.Y. Histone acetyltransferases: Function, structure, and catalysis. Curr. Opin. Genet. Dev. 2001, 11, 155–161. [Google Scholar] [CrossRef]
- Wapenaar, H.; Dekker, F. Histone acetyltransferases: Challenges in targeting bi-substrate enzymes. Clin. Epigenetics 2016, 8, 59. [Google Scholar] [CrossRef]
- Santer, F.; Höschele, P.; Oh, S.; Erb, H.; Bouchal, J.; Cavaretta, I.; Parson, W.; Meyers, D.; Cole, P.; Culig, Z. Inhibition of the acetyltransferases p300 and CBP reveals a targetable function for p300 in the survival and invasion pathways of prostate cancer cell lines. Mol. Cancer Ther. 2011, 10, 1644–1655. [Google Scholar] [CrossRef]
- Weidle, U.H.; Grossmann, A. Inhibition of histone deacetylases: A new strategy to target epigenetic modifications for anticancer treatment. Anticancer Res. 2000, 20, 1471–1485. [Google Scholar]
- Bolden, J.; Peart, M.; Johnstone, R. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Discov. 2006, 5, 769–784. [Google Scholar] [CrossRef]
- Lee, J.-H.; Choy, M.L.; Marks, P.A. Mechanism of resistance to histone deacetylase inhibitors. Adv. Cancer Res. 2012, 116, 39–86. [Google Scholar] [CrossRef] [PubMed]
- Dokmanovic, M.; Clarke, C.; Marks, P.A. Histone Deacetylase Inhibitors: Overview and Perspectives. Mol. Cancer Res. 2007, 5, 981–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balasubramanian, S.; Verner, E.; Buggy, J.J. Isoform-specific histone deacetylase inhibitors: The next step? Cancer Lett. 2009, 280, 211–221. [Google Scholar] [CrossRef]
- Haggarty, S.J.; Koeller, K.M.; Wong, J.C.; Grozinger, C.M.; Schreiber, S.L. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. USA 2003, 100, 4389–4394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richon, V.; Emiliani, S.; Verdin, E.; Webb, Y.; Breslow, R.; Rifkind, R.; Marks, P. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc. Natl. Acad. Sci. USA 1998, 95, 3003–3007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coffey, D.C.; Kutko, M.C.; Glick, R.D.; Butler, L.M.; Heller, G.; Rifkind, R.A.; Marks, P.A.; Richon, V.M.; La Quaglia, M.P. The Histone Deacetylase Inhibitor, CBHA, Inhibits Growth of Human Neuroblastoma Xenografts in Vivo, Alone and Synergistically with All-Trans Retinoic Acid. Cancer Res. 2001, 61, 3591–3594. [Google Scholar]
- Plumb, J.A.; Finn, P.W.; Williams, R.J.; Bandara, M.J.; Romero, M.R.; Watkins, C.J.; La Thangue, N.B.; Brown, R. Pharmacodynamic Response and Inhibition of Growth of Human Tumor Xenografts by the Novel Histone Deacetylase Inhibitor PXD101. Mol. Cancer Ther. 2003, 2, 721–728. [Google Scholar] [PubMed]
- Furumai, R.; Matsuyama, A.; Kobashi, N.; Lee, K.; Nishiyama, M.; Nakajima, H.; Tanaka, A.; Komatsu, Y.; Nishino, N.; Yoshida, M.; et al. FK228 (Depsipeptide) as a Natural Prodrug That Inhibits Class I Histone Deacetylases. Cancer Res. 2002, 62, 4916–4921. [Google Scholar] [PubMed]
- Liu, T.; Kapustin, G.; Etzkorn, F.A. Design and Synthesis of a Potent Histone Deacetylase Inhibitor. J. Med. Chem. 2007, 50, 2003–2006. [Google Scholar] [CrossRef]
- Jose, B.; Oniki, Y.; Kato, T.; Nishino, N.; Sumida, Y. Novel histone deacetylase inhibitors: Cyclic tetrapeptide with trifluoromethyl and pentafluoroethyl ketones. Bioorg. Med. Chem. Lett. 2004, 14, 5343–5346. [Google Scholar] [CrossRef]
- Maggio, S.C.; Rosato, R.R.; Kramer, L.B.; Dai, Y.; Rahmani, M.; Paik, D.S.; Czarnik, A.C.; Payne, S.G.; Spiegel, S.; Grant, S. The Histone Deacetylase Inhibitor MS-275 Interacts Synergistically with Fludarabine to Induce Apoptosis in Human Leukemia Cells. Cancer Res. 2004, 64, 2590–2600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blum, K.A.; Advani, A.; Fernandez, L.; Van Der Jagt, R.; Kambhampati, S.; Kassis, J.; Davis, M.; Bonfils, C.; Dubay, M.; Dumouchel, J.; et al. Phase II study of the histone deacetylase inhibitor MGCD0103 in patients with previously treated chronic lymphocytic leukaemia. Br. J. Haematol. 2010, 147, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Boumber, Y.; Younes, A.; Garcia-Manero, G. Mocetinostat (MGCD0103): A review of an isotype-specific histone deacetylase inhibitor. Expert. Opin. Investig. Drugs 2011, 20, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Minucci, S.; Zhu, P.; Kra, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Coco, F.L.; Nervi, C.; Pelicci, P.G.; Heinzel, T. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001, 20, 6969–6978. [Google Scholar] [Green Version]
- Rasheed, W.K.; Johnstone, R.W.; Prince, H.M. Histone deacetylase inhibitors in cancer therapy. Expert. Opin. Investig. Drugs 2017, 16, 659–678. [Google Scholar] [CrossRef] [PubMed]
- Krämer, O.H.; Göttlicher, M.; Heinzel, T. Histone deacetylase as a therapeutic target. Trends Endocrinol. Metab. 2001, 12, 294–300. [Google Scholar] [CrossRef]
- Marks, P.A.; Richon, V.M.; Rifkind, R.A. Histone deacetylase inhibitors: Inducers of differentiation or apoptosis of transformed cells. J. Natl. Cancer Inst. 2000, 92, 1210–1216. [Google Scholar] [CrossRef]
- Kim, Y.B.; Ki, S.W.; Yoshida, M.; Horinouchi, S. Mechanism of cell cycle arrest caused by histone deacetylase inhibitors in human carcinoma cells. J. Antibiot. (Tokyo) 2000, 53, 1191–1200. [Google Scholar] [CrossRef]
- Bali, P.; Pranpat, M.; Bradner, J.; Balasis, M.; Fiskus, W.; Guo, F.; Rocha, K.; Kumaraswamy, S.; Boyapalle, S.; Atadja, P.; et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: A novel basis for antileukemia activity of histone deacetylase inhibitors. J. Biol. Chem. 2005, 280, 26729–26734. [Google Scholar] [CrossRef]
- Newbold, A.; Falkenberg, K.J.; Prince, H.M.; Johnstone, R.W. How do tumor cells respond to HDAC inhibition? FEBS J. 2016, 283, 4032–4046. [Google Scholar] [CrossRef] [PubMed]
- Gui, C.Y.; Ngo, L.; Xu, W.S.; Richon, V.M.; Marks, P.A. Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter associated proteins, including HDAC1. Proc. Natl. Acad. Sci. USA 2004, 101, 1241–1246. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.M.; Hackanson, B.; Lübbert, M.; Jung, M. Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin. Epigenetics 2010, 1, 117–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, J.; Cang, S.; Ma, Y.; Petrillo, R.L.; Liu, D. Novel histone deacetylase inhibitors in clinical trials as anti-cancer agents. J. Hematol. Oncol. 2010, 3, 1–13. [Google Scholar] [CrossRef] [PubMed]
- O´Connor, O.A.; Heaney, M.L.; Schwartz, L.; Richardson, S.; Willim, R.; MacGregor-Cortelli, B.; Curly, T.; Moskowitz, C.; Portlock, C.; Horwitz, S.; et al. Clinical Experience with Intravenous and Oral Formulations of the Novel Histone Deacetylase Inhibitor Suberoylanilide Hydroxamic Acid in Patients with Advanced Hematologic Malignancies. J. Clin. Oncol. 2017, 24, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Duvic, M.; Vu, J. Update on the treatment of cutaneous T-cell lymphoma (CTCL): Focus on vorinostat. Biologics 2007, 1, 377. [Google Scholar] [PubMed]
- Duvic, M.; Talpur, R.; Ni, X.; Zhang, C.; Hazarika, P.; Kelly, C.; Chiao, J.H.; Reilly, J.F.; Ricker, J.L.; Richon, V.M.; et al. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 2007, 109, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Bertino, E.M.; Otterson, G.A. Romidepsin: A novel histone deacetylase inhibitor for cancer. Expert. Opin. Investig. Drugs 2011, 20, 1151–1158. [Google Scholar] [CrossRef]
- Rashidi, A.; Cashen, A.F. Belinostat for the treatment of relapsed or refractory peripheral T-cell lymphoma. Futur. Oncol. 2015, 11, 1659–1664. [Google Scholar] [CrossRef] [PubMed]
- Greig, S.L. Panobinostat: A review in relapsed or refractory multiple myeloma. Target. Oncol. 2016, 11, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Slingerland, M.; Guchelaar, H.J.; Gelderblom, H. Histone deacetylase inhibitors: An overview of the clinical studies in solid tumors. Anticancer. Drugs 2014, 25, 140–149. [Google Scholar] [CrossRef]
- Rasheed, W.; Bishton, M.; Johnstone, R.W.; Prince, H.M. Histone deacetylase inhibitors in lymphoma and solid malignancies. Expert Rev. Anticancer. Ther. 2008, 8, 413–432. [Google Scholar] [CrossRef] [PubMed]
- Modesitt, S.; Sill, M.; Hoffman, J.; Bender, D.; Gynecologic Oncology Group. A phase II study of vorinostat in the treatment of persistent or recurrent epithelial ovarian or primary peritoneal carcinoma: A Gynecologic Oncology Group study. Gynecol. Oncol. 2008, 109, 182–186. [Google Scholar] [CrossRef]
- Molife, L.R.; Attard, G.; Fong, P.C.; Karavasilis, V.; Reid, A.H.M.; Patterson, S.; Riggs, C.E.; Higano, C.; Stadler, W.M.; McCulloch, W.; et al. Phase II, two-stage, single-arm trial of the histone deacetylase inhibitor (HDACi) romidepsin in metastatic castration-resistant prostate cancer (CRPC). Ann. Oncol. 2010, 21, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Stadler, W.M.; Margolin, K.; Ferber, S.; McCulloch, W.; Thompson, J.A. A phase II study of depsipeptide in refractory metastatic renal cell cancer. Clin. Genitourin. Cancer. 2006, 5, 57–60. [Google Scholar] [CrossRef]
- Haigentz, M.; Kim, M.; Sarta, C.; Lin, J.; Keresztes, R.S.; Culliney, B.; Gaba, A.G.; Smith, R.V.; Shapiro, G.I.; Chirieac, L.R.; et al. Phase II trial of the histone deacetylase inhibitor romidepsin in patients with recurrent/metastatic head and neck cancer. Oral Oncol. 2012, 48, 1281–1288. [Google Scholar] [CrossRef] [Green Version]
- Luu, T.H.; Morgan, R.J.; Leong, L.; Lim, D.; McNamara, M.; Portnow, J.; Frankel, P.; Smith, D.D.; Doroshow, J.H.; Wong, C.; et al. A phase II trial of vorinostat (suberoylanilide hydroxamic acid) in metastatic breast cancer: A California Cancer Consortium study. Clin. Cancer Res. 2008, 14, 7138–7142. [Google Scholar] [CrossRef] [PubMed]
- Gryder, B.E.; Sodji, Q.H.; Oyelere, A.K. Targeted cancer therapy: Giving histone deacetylase inhibitors all they need to succeed. Futur. Med. Chem. 2012, 4, 505–524. [Google Scholar] [CrossRef] [PubMed]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.; Chen, Z.; Shin, D. Nanoparticle therapeutics: An emerging treatment modality for cancer. Nat. Rev. Drug. Discov. 2008, 7, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Mrakovcic, M.; Absenger, M.; Riedl, R.; Smole, C.; Roblegg, E.; Fröhlich, L.F.; Fröhlich, E. Assessment of long-term effects of nanoparticles in a microcarrier cell culture system. PLoS ONE 2013, 8, e56791. [Google Scholar] [CrossRef]
- Khan, N.; Jeffers, M.; Kumar, S.; Hackett, C.; Boldog, F.; Khramtsov, N.; Qian, X.; Mills, E.; Berghs, S.C.; Carey, N.; et al. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem. J. 2008, 409, 581–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone deacetylase inhibitors: Molecular mechanisms of action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A. The mechanism of the anti-tumor activity of the histone deacetylase inhibitor, suberoylanilide hydroxamic acid (SAHA). Cell Cycle 2004, 3, 534–535. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.; Dai, Y.; Grant, S. Histone deacetylase inhibitor (HDACI) mechanisms of action: Emerging insights. Pharmacol. Ther. 2014, 143, 323–336. [Google Scholar] [CrossRef] [Green Version]
- Richon, V.M.; Sandhoff, T.W.; Rifkind, R.A.; Marks, P.A. Histone deacetylase inhibitor selectively induces p21 WAF1 expression and gene-associated histone acetylation. Proc. Natl. Acad. Sci. USA 2000, 97, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Sandor, V.; Senderowicz, A.; Mertins, S.; Sackett, D.; Sausville, E.; Blagoskonny, M.V.; Bates, S.E. P21-dependent G1 arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. Br. J. Cancer 2000, 83, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.M.; Zhou, X.; Xu, W.-S.; Scher, H.I.; Rifkind, R.A.; Marks, P.A.; Richon, V.M. The histone deacetylase inhibitor SAHA arrests cancer cell growth, up-regulates thioredoxin-binding protein-2, and down-regulates thioredoxin. Proc. Natl. Acad. Sci. USA 2002, 99, 11700–11705. [Google Scholar] [CrossRef] [Green Version]
- Seo, S.-K.; Jin, H.-O.; Woo, S.-H.; Kim, Y.-S.; An, S.; Lee, J.-H.; Hong, S.-I.; Lee, K.-H.; Choe, T.-B.; Park, I.-C. Histone Deacetylase Inhibitors Sensitize Human Non-small Cell Lung Cancer Cells to Ionizing Radiation Through Acetyl p53-Mediated c-myc Down-Regulation. J. Thorac. Oncol. 2011, 6, 1313–1319. [Google Scholar] [CrossRef] [Green Version]
- Takai, N.; Desmond, J.; Kumagai, T.; Gui, D.; Said, J.; Whittaker, S.; Miyakawa, I.; Koeffler, H. Histone deacetylase inhibitors have a profound antigrowth activity in endometrial cancer cells. Clin. Cancer Res. 2004, 10, 1141–1149. [Google Scholar] [CrossRef]
- Souleimani, A.; Asselin, C. Regulation of c-myc expression by sodium butyrate in the colon carcinoma cell line Caco-2. FEBS Lett. 1993, 326, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, C.L.; Smith-Windsor, E.L.; Bonham, K. Src family kinase members have a common response to histone deacetylase inhibitors in human colon cancer cells. Int. J. Cancer 2006, 118, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Vrana, J.; Decker, R.; Johnson, C.; Wang, Z.; Jarvis, W.; Richon, V.; Ehinger, M.; Fisher, P.; Grant, S. Induction of apoptosis in U937 human leukemia cells by suberoylanilide hydroxamic acid (SAHA) proceeds through pathways that are regulated by Bcl-2/Bcl-XL, c-Jun, and p21CIP1, but independent of p53. Oncogene 1999, 18, 7016–7025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawrocki, S.T.; Carew, J.S.; Douglas, L.; Cleveland, J.L.; Humphreys, R.; Houghton, J.A. Histone Deacetylase Inhibitors Enhance Lexatumumab-Induced Apoptosis via a p21 Cip1-Dependent Decrease in Survivin Levels. Cancer Res. 2007, 67, 6987–6995. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, M.; Yu, C.; Reese, E.; Ahmed, W.; Hirsch, K.; Dent, P.; Grant, S. Inhibition of PI-3 kinase sensitizes human leukemic cells to histone deacetylase inhibitor-mediated apoptosis through p44/42 MAP kinase inactivation and abrogation of p21 CIP1/WAF1 induction rather than AKT inhibition. Oncogene 2003, 22, 6231–6242. [Google Scholar] [CrossRef]
- Burgess, A.J.; Pavey, S.; Warrener, R.; Hunter, L.K.; Piva, T.J.; Musgrove, E.A.; Saunders, N.; Parsons, P.G.; Gabrielli, B.G. Up-Regulation of p21 WAF1/CIP1 by Histone Deacetylase Inhibitors Reduces Their Cytotoxicity. Mol. Pharmacol. 2001, 60, 828–837. [Google Scholar]
- Steinmann, R.; Hoffmann, B.; Iro, A.; Guillouf, C.; Liebermann, D.A.; El-Houseini, M.E. Induction of p21 (WAF-1/CIP1) during differentiation. Oncogene 1994, 9, 3389–3396. [Google Scholar]
- Harper, J.W.; Adami, G.R.; Wei, N.; Keyomarsi, K.; Elledge, S.J. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 1993, 75, 805–816. [Google Scholar] [CrossRef]
- Qiu, L.; Burgess, A.; Fairlie, D.; Leonard, H.; Parsons, P.; Gabrielli, B. Histone deacetylase inhibitors trigger a G2 checkpoint in normal cells that is defective in tumor cells. Mol. Biol. Cell 2000, 11, 2069–2083. [Google Scholar] [CrossRef] [PubMed]
- Ruefli, A.A.; Ausserlechner, M.J.; Bernhard, D.; Sutton, V.R.; Tainton, K.M.; Kofler, R.; Smyth, M.J.; Johnstone, R.W. The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway characterized by cleavage of Bid and production of reactive oxygen species. Proc. Natl. Acad. Sci. USA 2001, 98, 10833–10838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Choy, M.L.; Ngo, L.; Foster, S.S.; Marks, P.A. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc. Natl. Acad. Sci. USA 2010, 107, 14639–14644. [Google Scholar] [CrossRef] [Green Version]
- Warrener, R.; Beamish, H.; Burgess, A.; Waterhouse, N.J.; Giles, N.; Fairlie, D.; Gabrielli, B. Tumor cell-selective cytotoxicity by targeting cell cycle checkpoints. FASEB J. 2003, 17, 1550–1552. [Google Scholar] [CrossRef]
- Conrad, E.; Polonio-Vallon, T.; Meister, M.; Matt, S.; Bitomsky, N.; Herbel, C.; Liebl, M.; Greiner, V.; Kriznik, B.; Schumacher, S.; et al. HIPK2 restricts SIRT1 activity upon severe DNA damage by a phosphorylation-controlled mechanism. Cell Death Differ. 2016, 23, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Ungerstedt, J.S.; Sowa, Y.; Xu, W.; Shao, Y.; Dokmanovic, M.; Perez, G.; Ngo, L.; Holmgren, A.; Jiang, X.; Marks, P.A. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 673–678. [Google Scholar] [CrossRef] [Green Version]
- Namdar, M.; Perez, G.; Ngo, L.; Marks, P.A. Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA damage and sensitizes transformed cells to anticancer agents. Proc. Natl. Acad. Sci. USA 2010, 107, 20003–20008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Zhu, W.-G. Targeting Histone Deacetylases for Cancer Therapy: From Molecular Mechanisms to Clinical Implications. Int. J. Biol. Sci. 2014, 10, 757–770. [Google Scholar] [CrossRef] [Green Version]
- Miller, K.M.; Tjeertes, J.V.; Coates, J.; Legube, G.; Polo, S.E.; Britton, S.; Jackson, S.P. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat. Struct. Mol. Biol. 2010, 17, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Bhaskara, S.; Knutson, S.K.; Jiang, G.; Chandrasekharan, M.B.; Wilson, A.J.; Zheng, S.; Yenamandra, A.; Locke, K.; Yuan, J.L.; Bonine-Summers, A.R.; et al. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell 2010, 18, 436–447. [Google Scholar] [CrossRef] [Green Version]
- Thurn, K.T.; Thomas, S.; Raha, P.; Qureshi, I.; Munster, P.N. Histone deacetylase regulation of ATM-mediated DNA damage signaling. Mol. Cancer Ther. 2013, 12, 2078–2087. [Google Scholar] [CrossRef] [PubMed]
- Kao, G.D.; McKenna, W.G.; Guenther, M.G.; Muschel, R.J.; Lazar, M.A.; Yen, T.J. Histone deacetylase 4 interacts with 53BP1 to mediate the DNA damage response. J. Cell Biol. 2003, 160, 1017–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotian, S.; Liyanarachchi, S.; Zelent, A.; Parvin, J.D. Histone deacetylase 9 and 10 are required for homologous recombination. J. Biol. Chem. 2011, 286, 7722–7726. [Google Scholar] [CrossRef] [PubMed]
- Gorospe, M.; de Cabo, R. AsSIRTing the DNA damage response. Trends Cell Biol. 2008, 18, 77–83. [Google Scholar] [CrossRef]
- Kaidi, A.; Weinert, B.T.; Choudhary, C.; Jackson, S.P. Human SIRT6 promotes DNA end resection through CtlP deacetylation. Science 2010, 329, 1348–1353. [Google Scholar] [CrossRef] [PubMed]
- Motoslavsky, R.; Chua, K.F.; Lombard, D.B.; Pang, W.W.; Fischer, M.R.; Gellon, L.; Liu, P.; Motoslavsky, G.; Franco, S.; Murphy, M.M.; et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 2006, 124, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zhao, W.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation Is Indispensable for p53 Activation. Cell 2008, 133, 612–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosato, R.R.; Almenara, J.A.; Grant, S. The Histone Deacetylase Inhibitor MS-275 Promotes Differentiation or Apoptosis in Human Leukemia Cells through a Process Regulated by Generation of Reactive Oxygen Species and Induction of p21 CIP1/WAF1. Cancer Res. 2003, 63, 3637–3645. [Google Scholar] [PubMed]
- Lin, K.; Wang, Y.; Chen, C.; Ho, C.; Su, W.; Jou, Y. HDAC inhibitors augmented cell migration and metastasis through induction of PKCs leading to identification of low toxicity modalities for combination cancer therapy. Clin. Cancer Res. 2012, 18, 4691–4701. [Google Scholar] [CrossRef] [PubMed]
- Pulukuri, S.; Gorantla, B.; Rao, J. Inhibition of histone deacetylase activity promotes invasion of human cancer cells through activation of urokinase plasminogen activator. J. Biol Chem. 2007, 282, 35594–35603. [Google Scholar] [CrossRef] [PubMed]
- Schoepflin, Z.R.; Shapiro, I.M.; Risbud, M.V. Class I and IIa HDACs mediate HIF-1a stability through PHD2-dependent mechanism, while HDAC6, a classIIb member, promotes HIF1a transcriptional activity in nucleus pulposus cells of the intervertebral disc. J. Bone Miner. Res. 2016, 31, 1287–1299. [Google Scholar] [CrossRef]
- Deroanne, C.F.; Bonjean, K.; Servotte, S.; Devy, L.; Colige, A.; Clausse, N.; Blacher, S.; Verdin, E.; Foidart, J.M.; Nusgens, B.V.; et al. Histone deacetylases inhibitors as anti-angiogenic agents altering vascular endothelial growth factor signaling. Oncogene 2002, 21, 427–436. [Google Scholar] [CrossRef] [Green Version]
- Jeong, J.W.; Bae, M.K.; Ahn, M.Y.; Kim, S.H.; Sohn, T.K.; Bae, M.H.; Yoo, M.A.; Song, E.J.; Lee, K.J.; Kim, K.W. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell 2002, 111, 709–720. [Google Scholar] [CrossRef]
- Fournel, M.; Bonfils, C.; Hou, Y.; Yan, P.; Trachy-Bourget, M.C.; Kalita, A.; Liu, J.; Lu, A.H.; Zhou, N.Z.; Robert, M.F.; et al. MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Mol. Cancer Ther. 2008, 7, 759–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigushin, D.M.; Ali, S.; Pace, P.E.; Mirsaidi, N.; Ito, K.; Adcock, I.; Coombes, R.C. Trichostatin A is a histone deacetylase inhibitor with potent antitumor activity against breast cancer in vivo. Clin. Cancer Res. 2001, 7, 971–976. [Google Scholar] [PubMed]
- Sato, N.; Ohta, T.; Kitagawa, H.; Kayahara, M.; Ninomiya, I.; Fushida, S.; Fujimura, T.; Nishimura, G.; Shimizu, K.; Miwa, K. FR901228, a novel histone deacetylase inhibitor, induces cell cycle arrest and subsequent apoptosis in refractory human pancreatic cancer cells. Int. J. Oncol. 2004, 24, 679–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, H.J.; Owa, T.; Hassig, C.A.; Shimada, J.; Schreiber, S.L. Depudecin induces morphological reversion of transformed fibroblasts via the inhibition of histone deacetylase. Proc. Natl. Acad. Sci. USA 1998, 95, 3356–3361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosato, R.; Almenara, J.; Dai, Y.; Grant, S. Simultaneous activation of the intrinsic and extrinsic pathways by histone deacetylase (HDAC) inhibitors and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) synergistically induces mitochondrial damage and apoptosis in human leukemia cells. Mol. Cancer Ther. 2003, 2, 1273–1284. [Google Scholar] [PubMed]
- Emanuele, S.; Lauricella, M.; Tesoriere, G. Histone deacetylase inhibitors: Apoptotic effects and clinical implications. Int. J. Oncol. 2008, 33, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Frew, A.J.; Johnstone, R.W.; Bolden, J.E. Enhancing the apoptotic and therapeutic effects of HDAC inhibitors. Cancer Lett. 2009, 280, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhong, Q. Histone deacetylase inhibitors and cell death. Cell. Mol. Life Sci. 2014, 71, 3885–3901. [Google Scholar] [CrossRef] [Green Version]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Adachi, M.; Kawamura, R.; Imai, K. Bmf is a possible mediator in histone deacetylase inhibitors FK228 and CBHA-induced apoptosis. Cell Death Differ. 2006, 13, 129–140. [Google Scholar] [CrossRef]
- Zhao, Y.; Tan, J.; Zhuang, L.; Jiang, X.; Liu, E.T.; Yu, Q. Inhibitors of histone deacetylases target the Rb-E2F1 pathway for apoptosis induction through activation of proapoptotic protein Bim. Proc. Natl. Acad. Sci. USA 2005, 102, 16090–16095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Wang, X. Cytochrome C-mediated Apoptosis. Annu. Rev. Biochem. 2004, 73, 87–106. [Google Scholar] [CrossRef]
- Elkholi, R.; Floros, K.; Chipuk, J. The Role of BH3-Only Proteins in Tumor Cell Development, Signaling, and Treatment. Genes Cancer. Genes Cancer 2011, 2, 523–537. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Bae, S.C. Histone deacetylase inhibitors: Molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 2011, 3, 166–179. [Google Scholar] [PubMed]
- Robert, C.; Rassool, F. V Chapter Three—HDAC Inhibitors: Roles of DNA Damage and Repair. Adv. Cancer Res. 2012, 116, 87–129. [Google Scholar]
- Sade, H.; Sarin, A. Reactive oxygen species regulate quiescent T-cell apoptosis via the BH3-only proapoptotic protein BIM. Cell Death Differ. 2004, 11, 416–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosato, R.R.; Maggio, S.C.; Almenara, J.A.; Payne, S.G.; Atajda, P.; Spiegel, S.; Dent, P.; Grant, S. The histone deacetylase inhibitor LAQ824 induces human leukemia cell death through a process involving XIAP down-regulation, oxidative injury, and the acid shingomyelinase-dependent generation of ceramide. Mol. Pharmacol. 2006, 69, 216–225. [Google Scholar] [CrossRef]
- Fandy, T.E.; Srivastava, R.K. Trichostatin A sensitizes TRAIL-resistant myeloma cells by downregulation of the antiapoptotic Bcl-2 proteins. Cancer Chemother. Pharmacol. 2006, 58, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, C.; Opipari, A.W.; Bian, X.; Castle, V.P.; Kwok, R.P. Ku70 acetylation mediates neuroblastoma cell death induced by histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 4842–4847. [Google Scholar] [CrossRef] [Green Version]
- Fandy, T.E.; Shankar, S.; Ross, D.D.; Sausville, E.; Srivastava, R.K. Interactive Effects of HDAC Inhibitors and TRAIL on Apoptosis Are Associated with Changes in Mitochondrial Functions and Expressions of Cell Cycle Regulatory Genes in Multiple Myeloma. Neoplasia 2005, 7, 646–657. [Google Scholar] [CrossRef] [Green Version]
- Sonnemann, J.; Dreyer, L.; Hartwig, M.; Palani, C.D.; Hong, L.T.T.; Klier, U.; Bröker, B.; Völker, U.; Beck, J.F. Histone deacetylase inhibitors induce cell death and enhance the apoptosis-inducing activity of TRAIL in Ewing’s sarcoma cells. J. Cancer Res. Clin. Oncol. 2007, 133, 847–858. [Google Scholar] [CrossRef]
- Carlisi, D.; Lauricella, M.; D’Anneo, A.; Emanuele, S.; Angileri, L.; Di Fazio, P.; Santulli, A.; Vento, R.; Tesoriere, G. The histone deacetylase inhibitor suberoylanilide hydroxamic acid sensitises human hepatocellular carcinoma cells to TRAIL-induced apoptosis by TRAIL-DISC activation. Eur. J. Cancer 2009, 45, 2425–2438. [Google Scholar] [CrossRef] [PubMed]
- Nakata, S.; Yoshida, T.; Horinaka, M.; Shiraishi, T.; Wakada, M.; Sakai, T. Histone deacetylase inhibitors upregulate death receptor 5/TRAIL-R2 and sensitize apoptosis induced by TRAIL/APO2-L in human malignant tumor cells. Oncogene 2004, 23, 6261–6271. [Google Scholar] [CrossRef] [Green Version]
- Aquilera, D.G.; Das, C.M.; Sinnappah-Kang, N.D.; Joyce, C.; Taylor, P.H.; Wen, S.; Hasselblatt, M.; Paulus, W.; Fuller, G.; Wolff, J.E.; et al. Reactivation of death receptor 4 (DR4) expression sensitizes medulloblastoma cell lines to TRAIL. J. Neurooncol. 2009, 93, 303–318. [Google Scholar] [CrossRef] [Green Version]
- Fulda, S.; Küfer, M.U.; Meyer, E.; van Valen, F.; Dockhorn-Dworniczak, B.; Debatin, K.M. Sensitization for death receptor- or drug-induced apoptosis by re-expression of caspase-8 through demethylation or gene transfer. Oncogene 2001, 20, 5865–5877. [Google Scholar] [CrossRef]
- Fulda, S. Modulation of TRAIL-induced apoptosis by HDAC inhibitors. Curr. Cancer Drug Targets 2008, 8, 132–140. [Google Scholar] [CrossRef]
- Gillenwater, A.M.; Zhong, M.; Lotan, R. Histone deacetylase inhibitor suberoylanilide hydroxamic acid induces apoptosis through both mitochondrial and Fas (Cd95) signaling in head and neck squamous carcinoma cells. Mol. Cancer Ther. 2007, 6, 2967–2975. [Google Scholar] [CrossRef] [Green Version]
- Shankar, S.; Singh, T.; Fandy, T.; Luetrakul, T.; Ross, D.; Srivastava, R. Interactive effects of histone deacetylase inhibitors and TRAIL on apoptosis in human leukemia cells: Involvement of both death receptor and mitochondrial pathways. Int. J. Mol. Med. 2005, 16, 1125–1138. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Dixit, V.M. Death Receptors: Signaling and Modulation. Science 1998, 281, 1305–1309. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, H.; Ashkenzi, A. Apo2L/TRAIL and its death and decoy receptors. Cell Death Differ. 2003, 10, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Natoni, F.; Diolordi, L.; Santoni, C.; Gilardini Montani, M.S. Sodium butyrate sensitises human pancreatic cancer cells to both the intrinsic and extrinsic apoptotic pathways. Biochim. Biophys. Acta 2005, 1745, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Moutinho, C.; Esteller, M. MicroRNAs and Epigenetics. Adv. Cancer Res. 2017, 135, 189–220. [Google Scholar] [CrossRef] [PubMed]
- Noonan, E.J.; Place, R.F.; Pookot, D.; Basak, S.; Whitson, J.M.; Hirata, H.; Giardina, C.; Dahiya, R. miR-449a targets HDAC-1 and induces growth arrest in prostate cancer. Oncogene 2009, 28, 1714–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brest, P.; Lassalle, S.; Hofmann, V.; Bordone, O.; Gavric Tanga, V.; Bonnetaud, C.; Moreilhon, C.; Rios, G.; Santini, J.; Barbry, P.; et al. miR-129-5p is required for histone deacetylase inhibitor-induced cell death in thyroid cancer cells. Endocr. Relat. Cancer 2011, 18, 711–719. [Google Scholar] [CrossRef]
- Adams, C.M.; Hiebert, S.W.; Eischen, C.M. Myc induces miRNA-mediated apoptosis in response to HDAC inhibition in hematologic malignancies. Cancer Res. 2016, 76, 736–748. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Dimri, M.; Dimri, G.P. MicroRNA-31 is a trascriptional target of histone deacetylase inhibitors and a regulator of cellular senescence. J. Biol. Chem. 2015, 290, 10555–10567. [Google Scholar] [CrossRef]
- Nalls, D.; Tang, S.-N.; Rodova, M.; Srivastava, R.K.; Shankar, S. Targeting epigenetic regulation of miR-34a for treatment of pancreatic cancer by inhibition of pancreatic cancer stem cells. PLoS ONE 2011, 6, e24099. [Google Scholar] [CrossRef]
- Kim, J.; Noh, J.; Eun, J.; Jung, K.; Bae, H.; Shen, Q.; Kim, M.; Chan, Y.; Kim, S.; Park, W.; et al. Targeted inactivation of HDAC2 restores p16INK4a activity and exerts antitumor effects on human gastric cancer. Cancer Res. 2013, 11, 62–73. [Google Scholar] [CrossRef]
- Kang, Y.; Nian, H.; Rajendran, P.; Kim, E.; Dashwood, W.; Pinto, J.; Boardman, L.; Thibodeau, S.; Limburg, P.; Löhr, C.; et al. HDAC8 and STAT3 repress BMF gene activity in colon cancer cells. Cell Death Dis. 2014, 5, e1476. [Google Scholar] [CrossRef]
- Feng, L.; Pan, M.; Sun, J.; Lu, H.; Shen, Q.; Zhang, S.; Jiang, T.; Liu, L.; Jin, W.; Chen, Y.; et al. Histone deacetylase 3 inhibits expression of PUMA in gastric cancer cells. J. Mol. Med. 2013, 91, 49–58. [Google Scholar] [CrossRef]
- Mrakovcic, M.; Fröhlich, L.F. P53-mediated molecular control of autophagy in tumor cells. Biomolecules 2018, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- Li, W.W.; Li, J.; Bao, J. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2012, 69, 1125–1136. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Chaperone-mediated autophagy: A unique way to enter the lysosome world. Trends Cell Biol. 2012, 22, 305–309. [Google Scholar] [CrossRef]
- Youle, R.; Narendra, D. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Noda, T.; Yoshimori, T.; Tanaka, Y.; Ishii, T.; George, M.D.; Klionsky, D.J.; Ohsumi, M.; Ohsumi, Y. A protein conjugation system essential for autophagy. Nature 1998, 395, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.; Gao, Z.; Marks, P.A.; Jiang, X. Apoptotic and autophagic cell death induced by histone deacetylase inhibitors. Proc. Natl. Acad. Sci. USA 2004, 101, 18030–18035. [Google Scholar] [CrossRef] [Green Version]
- Shintani, T.; Klionsky, D.J. Autophagy in health and disease: A double-edged sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef]
- Yang, Z.J.; Chee, C.E.; Hunag, S.; Sinicrope, F.A. The role of autophagy in cancer: Therapeutic implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef]
- Rosenfeldt, M.T.; Ryan, K.M. The role of autophagy in tumour development and cancer therapy. Expert Rev. Mol. Med. 2009, 11, e36. [Google Scholar] [CrossRef]
- Matthew, R. The role of autophagy in cancer. Nat. Rev. Cancer 2007, 12, 961–967. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Baehrecke, E.H. Autophagy, cell death, and cancer. Mol. Cell. Oncol. 2015, 2, e985913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Wan, F.; Dutta, S.; Welsh, S.; Liu, Z.; Freundt, E.; Baehrecke, E.L.M. Autophagic programmed cell death by selective catalase degradation. Proc. Natl. Acad. Sci. USA 2006, 103, 4952–4957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poillet-Perez, L.; Despouy, G.; Delage-Mourroux, R.; Boyer-Guittaut, M. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox. Biol. 2015, 4, 184–192. [Google Scholar] [CrossRef]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.; Heintz, N. Beclin-1, an autophagy gene essential for early embryonic development, is a haploinsuffient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef]
- Mowers, E.E.; Sharifi, M.N.; Macleod, K.F. Autophagy in cancer metastasis. Oncogene 2017, 36, 1619–1630. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [PubMed]
- Lopez, G.; Torres, K.; Lev, D. Autophagy blockade enhances HDAC inhibitors’ pro-apoptotic effects: Potential implications for the treatment of a therapeutic-resistant malignancy. Autophagy 2011, 7, 40–41. [Google Scholar] [CrossRef]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef]
- Yi, C.; Ma, M.; Ran, L.; Zheng, J.; Tong, J.; Zhu, J.; Ma, C.; Sun, Y.; Zhang, S.; Feng, W.; et al. Function and Molecular Mechanism of Acetylation in Autophagy Regulation. Science 2012, 336, 474–477. [Google Scholar] [CrossRef]
- Gammoh, N.; Lam, D.; Puente, C.; Ganley, I.; Marks, P.A.; Jiang, X. Role of autophagy in histone deacetylase inhibitor-induced apoptotic and nonapoptotic cell death. Proc. Natl. Acad. Sci. USA 2012, 109, 6561–6565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Liu, R.; Lei, Y.; Wang, K.; Lau, Q.C.; Xie, N.; Zhou, S.; Nie, C.; Chen, L.; Wei, Y.; et al. Proteomic analysis revealed association of aberrant ROS signaling with suberoylanilide hydroxamic acid-induced autophagy in Jurkat T-leukemia cells. Autophagy 2010, 6, 711–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiao, M.; Cheng, W.; Yang, Y.; Shen, C.; Chiao, M.; Cheng, W.; Yang, Y.; Shen, C.; Ko, J. Suberoylanilide hydroxamic acid (SAHA) causes tumor growth slowdown and triggers autophagy in glioblastoma stem cells. Autophagy 2013, 9, 1509–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.-L.; Yang, P.-M.; Shun, C.-T.; Wu, M.-S.; Weng, J.-R.; Chen, C.-C. Autophagy potentiates the anti-cancer effects of the histone deacetylase inhibitors in hepatocellular carcinoma. Autophagy 2010, 6, 1057–1065. [Google Scholar] [CrossRef] [Green Version]
- Hrzenjak, A.; Kremser, M.; Strohmeier, B.; Moinfar, F.; Zatloukal, K.; Denk, H. SAHA induces caspase-independent, autophagic cell death of endometrial stromal sarcoma cells by influencing the mTOR pathway. J. Pathol. 2008, 216, 495–504. [Google Scholar] [CrossRef]
- Cao, Q.; Yu, C.; Xue, R.; Hsueh, W.; Pan, P.; Chen, Z.; Wang, S.; McNutt, M.; Gu, J. Autophagy induced by suberoylanilide hydroxamic acid in Hela S3 cells involves inhibition of protein kinase B and up-regulation of Beclin 1. Int. J. Biochem. Cell Biol. 2008, 40, 272–283. [Google Scholar] [CrossRef] [PubMed]
- El-Khoury, V.; Pierson, S.; Szwarcbart, E.; Brons, N.H.C.; Roland, O.; Cherrier-De Wilde, S.; Plawny, L.; Van Dyck, E.; Berchem, G. Disruption of autophagy by the histone deacetylase inhibitor MGCD0103 and its therapeutic implication in B-cell chronic lymphocytic leukemia. Leukemia 2014, 28, 1636–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carew, J.S.; Nawrocki, S.T.; Kahue, C.N.; Zhang, H.; Yang, C.; Chung, L.; Houghton, J.A.; Huang, P.; Giles, F.J.; Cleveland, J.L. Targeting autophagy augments the anticancer activity of the histone deacetylase inhibitor SAHA to overcome Bcr-Abl-mediated drug resistance. Blood 2007, 110, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Park, M.A.; Reinehr, R.; Haussinger, D.; Voelkel-Johnson, C.; Ogretmen, B.; Yacoub, A.; Grant, S.; Dent, B. Sorafenib activates CD95 and promotes autophagy and cell death via Src family kinases in gastrointestinal tumor cells. Mol. Cancer Ther. 2010, 9, 2220–2231. [Google Scholar] [CrossRef] [PubMed]
- Shulak, L.; Beljanski, V.; Chiang, C.; Dutta, S.M.; Van Grevenynghe, J.; Belgnaoui, S.M.; Nguyen, T.L.A.; Di Lenardo, T.; Semmes, O.J.; Lin, R.T.; et al. Histone Deacetylase Inhibitors Potentiate Vesicular Stomatitis Virus Oncolysis in Prostate Cancer Cells by Modulating NF-kappa B-Dependent Autophagy. J. Virol. 2014, 88, 2927–2940. [Google Scholar] [CrossRef]
- Long, J.; Zhao, J.; Yan, Z.; Liu, Z.; Wang, N. Antitumor effects of a novel sulfur-containing hydroxamate histone deacetylase inhibitor H40. Int. J. Cancer 2009, 124, 1235–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Giacomo, V.; Di Valerio, V.; Rapino, M.; Bosco, D.; Cacciatore, I.; Ciulla, M.; Marrazzo, A.; Fiorito, J.; Di Stefano, A.; Cataldi, A. MRJF4, a novel histone deacetylase inhibitor, induces p21 mediated autophagy in PC3 prostate cancer cells. Cell Mol. Biol 2015, 61, 17–23. [Google Scholar] [PubMed]
- Watanabe, M.; Adachi, S.; Matsubara, H.; Imai, T.; Yui, Y.; Mizushima, Y. Induction of autophagy in malignant rhabdoid tumor cells by the histone deacetylase inhibitor FK228 through AIF translocation. Int. J. Cancer Res. 2009, 67, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ng, S.; Wang, J.; Zhou, J.; Tan, S.; Yang, N.; Lin, Q.; Xia, D.; Shen, H. Histone deacetylase inhibitors induce autophagy through FOXO1-dependent pathways. Autophagy 2015, 11, 629–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandesiri, M.; Chakilam, S.; Ivanovska, J.; Benderska, N.; Ocker, M.; Di Fazio, P.; Feoktistova, M.; Gali-Muhtasib, H.; Rave-Fränk, M.; Prante, O.; et al. DAPK plays an important role in panobinostat-induced autophagy and commits cells to apoptosis under autophagy deficient conditions. Apoptosis 2012, 17, 1300–1315. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.; Bots, M.; Lindemann, R.K.; Bolden, J.E.; Newbold, A.; Cluse, L.A.; Scott, C.L.; Strasser, A.; Atadja, P.; Lowe, S.W.; et al. The histone deacetylase inhibitors LAQ824 and LBH589 do not require death receptor signaling or a functional apoptosome to mediate tumor cell death or therapeutic efficacy. Blood 2009, 114, 380–393. [Google Scholar] [CrossRef] [Green Version]
- Sun, W.; Yi, Y.; Xia, G.; Zhao, Y.; Yu, Y.; Li, L.; Hua, C.; He, B.; Yang, B.; Yu, C.; et al. Nrf2-miR-129-3p-mTOR Axis Controls an miRNA Regulatory Network Involved in HDACi-Induced Autophagy. Mol. Ther. 2019, 27, 1039–1050. [Google Scholar] [CrossRef] [PubMed]
- Stankov, M.V.; El Khatib, M.; Kumar Thakur, B.; Heitmann, K.; Panayotova-Dimitrova, D.; Schoening, J.; Bourquin, J.P.; Schweitzer, N.; Leverkus, M.; Welte, K.; et al. Histone deacetylase inhibitors induce apoptosis in myeloid leukemia by suppressing autophagy. Leukemia 2014, 28, 577–588. [Google Scholar] [CrossRef]
- Maccallum, S.F.; Groves, M.J.; James, J.; Murray, K.; Appleyard, V.; Prescott, A.R.; Drbal, A.A.; Nicolaou, A.; Cunningham, J.; Haydock, S.; et al. Dysregulation of autophagy in chronic lymphocytic leukemia with the small-molecule Sirtuin inhibitor Tenovin-6. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.H.; Finkel, T. Regulation of autophagy by the p300 acetyltransferase. J. Biol. Chem. 2009, 284, 6322–6328. [Google Scholar] [CrossRef] [PubMed]
- Sebti, S.; Prébois, C.; Pérez-Gracia, E.; Bauvy, C.; Desmots, F.; Pirot, N.; Gongora, C.; Bach, A.; Hubberstey, A.; Palissot, V.; et al. BAT3 modulates p300-dependent acetylation of p53 and autophagy-related protein 7 (ATG7) during autophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 4115–4120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebti, S.; Prébois, C.; Pérez-Gracia, E.; Bauvy, C.; Desmots, F.; Pirot, N.; Gongora, C.; Bach, A.; Hubberstey, A.; Palissot, V.; et al. BAG6/BAT3 modulates autophagy by affecting EP300/p300 intracellular localization. Autophagy 2014, 10, 1341–1342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.-Y.; Li, T.Y.; Liu, Q.; Zhang, C.; Li, X.; Chen, Y.; Zhang, S.-M.; Lian, G.; Liu, Q.; Ruan, K.; et al. Protein phosphorylation-acetylation cascade connects growth factor deprivation to autophagy. Autophagy 2012, 9, 1385–1386. [Google Scholar] [CrossRef] [PubMed]
- Juengel, E.; Dauselt, A.; Makarevic, J.; Wiesner, C.; Tsaur, I.; Bartsch, G.; Haferkamp, A.; Blaheta, R. Acetylation of histone H3 prevents resistance development caused by chronic mTOR inhibition in renal carcinoma cells. Cancer Lett. 2012, 324, 83–90. [Google Scholar] [CrossRef]
- Koeneke, E.; Witt, O.; Oehme, I. HDAC Family Members Intertwined in the Regulation of Autophagy: A Druggable Vulnerability in Aggressive Tumor Entities. Cells 2015, 4, 135–168. [Google Scholar] [CrossRef]
- Cao, D.J.; Wang, Z.V.; Battiprolu, P.K.; Jiang, N.; Morales, C.R.; Kong, Y.; Rothermel, B.A.; Gillette, T.G.; Hill, J.A. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc. Natl. Acad. Sci. USA 2011, 108, 4123–4128. [Google Scholar] [CrossRef] [Green Version]
- Oh, M.; Choi, I.-K.; Kwon, H.J. Inhibition of histone deacetylase1 induces autophagy. Biochem. Biophys. Res. Commun. 2008, 369, 1179–1183. [Google Scholar] [CrossRef]
- Xie, H.; Noh, J.; Kim, J.; Jung, K.; Eun, J.; Bae, H.; Kim, M.; Chang, Y.; Lee, J.; Park, H.; et al. HDAC1 inactivation induces mitotic defect and caspase-independent autophagic cell death in liver cancer. PLoS ONE 2012, 7, e34265. [Google Scholar] [CrossRef] [PubMed]
- Moresi, V.; Carrer, M.; Grueter, C.; Rifki, O.; Shelton, J.; Richardson, J.; Bassel-Duby, R.; Olson, E. Histone deacetylases 1 and 2 regulate autophagy flux and skeletal muscle homeostasis in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 1649–1654. [Google Scholar] [CrossRef] [Green Version]
- Pandey, U.; Nie, Z.; Batlevi, Y.; McCray, B.; Ritson, G.; Nedelsky, N.; Schwartz, S.; DiProspero, N.; Knight, M.; Schuldiner, O.; et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007, 447, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Nagano, Y.; Taylor, J.; Lim, K.; Yao, T. Disease causing mutations in parkin impair mitochondrial ubiquitination, aggregation, and HDAC6-dependent mitophagy. J. Cell Biol. 2010, 189, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Tan, B.; Chen, W.; Lin, Y.; Huang, J.; Chang, H.; Sun, H.; Hsu, P.; Liou, G.; Shen, J.; et al. Reversible acetylation regulates salt-inducible kinase (SIK2) and its function in autophagy. J. Biol. Chem. 2013, 288, 6227–6237. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef]
- Iwata, A.; Riley, B.E.; Johnston, J.A.; Kopito, R.R. HDAC6 and microtubules are required for autophagic degradation of aggregated Huntingtin. J. Biol. Chem. 2005, 280, 40282–40292. [Google Scholar] [CrossRef]
- Ahn, M.-Y.; Yoon, J.-H. Histone deacetylase 7 silencing induces apoptosis and autophagy in salivary mucoepidermoid carcinoma cells. J. Oral Pathol. Med. 2017, 46, 276–283. [Google Scholar] [CrossRef]
- Oehme, I.; Linke, J.-P.; Bock, B.C.; Milde, T.; Lodrini, M.; Hartenstein, B.; Wiegand, I.; Eckert, C.; Roth, W.; Kool, M.; et al. Histone deacetylase 10 promotes autophagy-mediated cell survival. Proc. Natl. Acad. Sci. USA 2013, 110, E2592–E2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Li, X.; He, Z.; Pan, S.; Yang, Y.; Zhang, X.; Chow, K.; Yang, T.; Qiu, J.; Zhou, Q.; et al. Induction of apoptosis and autophagy via sirtuin1- and PI3K/Akt/mTOR-mediated pathways by plumbagin in human prostate cancer cells. Drug Des. Dev. Ther. 2015, 9, 1511–1554. [Google Scholar] [CrossRef]
- Ou, X.; Lee, M.; Huang, X.; Messina-Graham, S.; Broxmeyer, H. SIRT1 positively regulates autophagy and mitochondria function in embryonic stem cells under oxidative stress. Stem Cells 2014, 32, 1183–1194. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Yang, J.; Liao, W.; Liu, X.; Zhang, H.; Wang, S.; Wang, D.; Feng, J.; Yu, L.; Zhu, W.-G. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat. Cell Biol. 2010, 12, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Tseng, A.; Shieh, S.; Wang, D. SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic. Biol. Med. 2013, 63, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Webster, B.; Scott, I.; Han, K.; Li, J.; Lu, Z.; Stevens, M.; Malide, D.; Chen, Y.; Samsel, L.; Connelly, P.; et al. Restricted mitochondrial protein acetylation initiates mitochondrial autophagy. J. Cell Sci. 2013, 126, 4843–4849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papa, L.; Germain, D. SirT3 regulates the mitochondrial unfolded protein response. Mol. Cell. Biol. 2014, 34, 699–710. [Google Scholar] [CrossRef] [PubMed]
- Polletta, L.; Vernucci, E.; Carnevale, I.; Arcangeli, T.; Rotili, D.; Palmerio, S.; Steegborn, C.; Nowak, T.; Schutkowski, M.; Pellegrini, L.; et al. SIRT5 regulation of ammonia-induced autophagy and mitophagy. Autophagy 2015, 11, 253–270. [Google Scholar] [CrossRef] [Green Version]
- Takasaka, N.; Araya, J.; Hara, H.; Ito, S.; Kobayashi, K.; Kurita, Y.; Wakui, H.; Tsurushige, C.; Kojima, J.; Numata, T.; et al. Autophagy Induction by SIRT6 through Attenuation of Insulin-like Growth Factor Signaling Is Involved in the Regulation of Human Bronchial Epithelial Cell Senescence. J. Immunol. 2014, 192, 958–968. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Yang, X.; Liu, T.; Zhang, T.; Xie, Q.R.; Xia, W. Autophagy induction by SIRT6 is involved in oxidative stress-induced neuronal damage. Protein Cell 2016, 7, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Vousden, K.H.; Lu, X. Live or let die: The cell´s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef]
- Brooks, C.L.; Gu, W. p53 Ubiquitination: Mdm2 and Beyond Review. Mol. Cell 2006, 21, 307–315. [Google Scholar] [CrossRef]
- Schuler, M.; Bossy-wetzel, E.; Goldstein, J.C.; Fitzgerald, P.; Green, D.R. p53 Induces Apoptosis by Caspase Activation through Mitochondrial Cytochrome c Release. J. Biol. Chem. 2000, 275, 7337–7342. [Google Scholar] [CrossRef] [Green Version]
- Chipuk, J.E.; Green, D.R. Dissecting p53-dependent apoptosis. Cell Death Differ. 2006, 13, 994–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Li, M.; Tang, Y.; Laszkowska, M.; Roeder, R.G.; Gu, W. Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2003, 101, 2259–2264. [Google Scholar] [CrossRef] [PubMed]
- Beckerman, R.; Prives, C. Transcriptional regulation by p53. Cold Spring Harb. Perspect. Biol. 2010, 2, a000935. [Google Scholar] [CrossRef] [PubMed]
- Brooks, C.L.; Gu, W. The impact of acetylation and deacetylation on the p53 pathway. Protein Cell 2011, 2, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, H.; Dessain, S.; Ng Eaton, E.; Imai, S.; Frye, R.; Pandita, T.; Guarente, L.; Weinberg, R. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef]
- Sykes, S.M.; Mellert, H.S.; Holbert, M.A.; Li, K.; Lane, W.S.; Mcmahon, S.B. Acetylation of the p53 DNA binding domain regulates apoptosis induction. Mol. Cell 2006, 24, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Luo, J.; Zhang, W. Tip60-Dependent Acetylation of p53 Modulates the Decision between Cell-Cycle Arrest and Apoptosis. Mol. Cell 2006, 24, 827–839. [Google Scholar] [CrossRef]
- Luo, J.; Su, F.; Chen, D.; Shiloh, A.; Gu, W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 2000, 408, 377–381. [Google Scholar] [CrossRef]
- Luo, J.; Nikolaev, A.Y.; Imai, S.; Chen, D.; Su, F.; Shiloh, A.; Guarente, L.; Gu, W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 2001, 107, 137–148. [Google Scholar] [CrossRef]
- Brandl, A.; Wagner, T.; Uhlig, K.M.; Knauer, S.K.; Stauber, R.H.; Melchior, F.; Schneider, G.; Heinzel, T.; Krämer, O.H. Dynamically regulated sumoylation of HDAC2 controls p53 deacetylation and restricts apoptosis following genotoxic stress. Mol. Cell. Biol. 2012, 4, 284–293. [Google Scholar] [CrossRef] [Green Version]
- Wagner, T.; Brand, P.; Heinzel, T.; Krämer, O.H. Histone deacetylase 2 controls p53 and is a critical factor in tumorigenesis. Biochim. Biophys. Acta 2012, 1846, 524–538. [Google Scholar] [CrossRef] [PubMed]
- Carlisi, D.; Vassallo, B.; Lauricella, M.; Emanuele, S.; D’Anneo, A.; Di Leonardo, E.; Di Fazio, P.; Vento, R.; Tesoriere, G. Histone deacetylase inhibitors induce in human hepatoma HepG2 cells acetylation of p53 and histones in correlation with apoptotic effects. Int. J. Oncol. 2008, 32, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-Y.; Zhou, L.-M. Sirt3 inhibits hepatocellular carcinoma cell growth through reducing Mdm2-mediated p53 degradation. Biochem. Biophys. Res. Commun. 2012, 423, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Barlev, N.A.; Liu, L.; Chehab, N.H.; Mansfield, K.; Harris, K.G.; Halazonetis, T.D.; Berger, S.L. Acetylation of p53 Activates Transcription through Recruitment of Coactivators/Histone Acetyltransferases. Mol. Cell 2001, 8, 1243–1254. [Google Scholar] [CrossRef]
- Wang, X.; Taplick, J.; Geva, N.; Oren, M. Inhibition of p53 degradation by Mdm2 acetylation. FEBS Lett. 2004, 561, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Lu, S.; Wu, L.; Chai, G.; Wang, H.; Chen, Y.; Sun, J.; Yu, Y.; Zhou, W.; Zheng, Q.; et al. Acetylation of p53 at Lysine 373/382 by the Histone Deacetylase Inhibitor Depsipeptide Induces Expression of p21 Waf1/Cip1. Mol. Cell. Biol. 2006, 26, 2782–2790. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y. Regulation of p53 responses by post-translational modifications. Cell Death Differ. 2003, 10, 400–403. [Google Scholar] [CrossRef]
- Roy, S.; Packman, K.; Jeffrey, R.; Tenniswood, M. Histone deacetylase inhibitors differentially stabilize acetylated p53 and induce cell cycle arrest or apoptosis in prostate cancer cells. Cell Death Differ. 2005, 12, 482–491. [Google Scholar] [CrossRef] [Green Version]
- Muller, P.A.J.; Vousden, K.H. P53 Mutations in Cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef]
- Liu, J.; Ma, Q.; Zhang, M.; Wang, X.; Zhang, D.; Li, W.; Wang, F.; Wu, E. Alterations of TP53 are associated with a poor outcome for patients with hepatocellular carcinoma: Evidence from a systematic review and metaanalysis. Eur. J. Cancer 2012, 48, 2328–2338. [Google Scholar] [CrossRef]
- Zheng, T.; Wang, J.; Zhao, Y.; Zhang, C.; Lin, M.; Wang, X.; Yu, H.; Liu, L.; Feng, Z.; Hu, W. Spliced MDM2 isoforms promote mutant p53 accumulation and gain-of-function in tumorigenesis. Nat. Commun. 2013, 4, 2996. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Zhao, Y.; Xu, Y.; Zheng, M. Mutant p53 in Cancer: Accumulation, Gain-of-Function, and Therapy. J. Mol. Biol. 2017, 429, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, D.; Pati, S.; Zambetti, G.; Chu, S.; Teresky, A.K.; Moore, M.; Finlay, C.; Levine, A.J. Gain of function mutations in p53. Nat. Genet. 1993, 4, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Santoro, R.; Strano, S.; Blandino, G. Transcriptional regulation by mutant p53 and oncogenesis. Subcell. Biochem. 2014, 85, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Willis, A.; Jung, E.J.; Wakefield, T.; Chen, X. Mutant p53 exerts a dominant negative effect by preventing wild-type from binding to the promoter of its target genes. Oncogene 2004, 23, 2330–2338. [Google Scholar] [CrossRef]
- Li, D.; Marchenko, N.D.; Schulz, R. Functional Inactivation of Endogenous MDM2 and CHIP by HSP90 Causes Aberrant Stabilization of Mutant p53 in Human Cancer Cells. Mol. Cancer Res. 2011, 9, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V.; Trostel, S.; Kayastha, G.; Demidenko, Z.N.; Vassilev, L.T.; Romanova, L.Y.; Bates, S.; Fojo, T. Depletion of Mutant p53 and Cytotoxicity of Histone Deacetylase Inhibitors. Cancer Res. 2005, 65, 7386–7392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, W.; Liu, S.; Xu, E.; Zhang, J.; Zhang, Y.; Chen, X.; Chen, X. Histone deacetylase inhibitors suppress mutant p53 transcription via histone deacetylase 8. Oncogene 2013, 32, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Marchenko, N.; Moll, U. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Diff. 2011, 18, 1904–1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garufi, A.; Pucci, D.; D’Orazi, V.; Circone, M.; Bossi, G.; Avantaggiati, M.L.; D’Orazi, G. Degradation of mutant p53H175 protein by Zn(II) through autophagy. Cell Death Dis. 2014, 5, e1271. [Google Scholar] [CrossRef]
- Saveria, M.; Montani, G.; Granato, M.; Santoni, C.; Del Porto, P.; Merendino, N.; Orazi, G.D.; Faggioni, A.; Cirone, M. Histone deacetylase inhibitors VPA and TSA induce apoptosis and autophagy in pancreatic cancer cells. Cell Oncol. 2017, 40, 167–180. [Google Scholar] [CrossRef]
- Ahn, M.-Y.; Ahn, S.-G.; Yoon, J.-H. Apicidin, a histone deaceylase inhibitor, induces both apoptosis and autophagy in human oral squamous carcinoma cells. Oral Oncol. 2011, 47, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Kloster, M.; Naderi, E.; Haaland, I.; Gjertsen, B.; Blomhoff, H.; Naderi, S. cAMP signalling inhibits p53 acetylation and apoptosis via HDAC and SIRT deacetylases. Int. J. Oncol. 2013, 42, 1815–1821. [Google Scholar] [CrossRef] [Green Version]
- Sonnemann, J.; Hartwig, M.; Plath, A.; Saravana Kumar, K.; Müller, C.; Beck, J.F. Histone deacetylase inhibitors require caspase activity to induce apoptosis in lung and prostate carcinoma cells. Cancer Lett. 2006, 232, 148–160. [Google Scholar] [CrossRef]
- Sonnemann, J.; Marx, C.; Becker, S.; Wittig, S.; Palani, C.; Kramer, O.; Beck, J. p53–dependent and p53- independent anticancer effects of different histone deacetylase inhibitors. Br. J. Cancer 2014, 110, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Chi, X.Z.; Yang, J.O.; Lee, K.Y.; Ito, K.; Sakakura, C.; Li, Q.L.; Kim, H.R.; Cha, E.J.; Lee, Y.H.; Kaneda, A.; et al. RUNX3 suppresses gastric epithelial cell growth by inducing p21(WAF1/cip1) expression in cooperation with transforming factor (beta)-activated SMAD. Mol. Cell. Biol. 2005, 25, 8097–8107. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Ito, K.; Fukamachi, H.; Chi, X.Z.; Wee, H.J.; Inoue, K.; Ida, H.; Bouillet, P.; Strasser, A.; Bae, S.C.; et al. The RUNX3 tumor suppressor upregulates Bim in gastric epithelial cells undergoing transforming growth factor beta induced apoptosis. Mol. Cell. Biol. 2006, 26, 4474–4488. [Google Scholar] [CrossRef]
- Jin, Y.H.; Jeon, E.J.; Li, Q.L.; Lee, Y.H.; Choi, J.K.; Kim, W.J.; Lee, K.Y.; Bae, S.C. Transforming growth factor-beta stimulates p300-dependent RUNX3 acetylation, which inhibits ubiquitination-mediated degradation. J. Biol. Chem. 2004, 279, 29409–29417. [Google Scholar] [CrossRef]
- Robert, T.; Vanoli, F.; Chiolo, I.; Shubassi, G.; Bernstein, K.; Rothstein, R.; Botrugno, O.; Parazzoli, D.; Oldani, A.; Minucci, S.; et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 2011, 471, 74–79. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S.; Tanaka, K.; Sakimura, R.; Okada, T.; Nakamura, T.; Li, Y.; Takasaki, M.; Nakabeppu, Y.; Iwamoto, Y. Suberoylanilide hydroxamic acid (SAHA) induces apoptosis or autophagy-associated cell death in chondrosarcoma cell lines. Anticancer Res. 2008, 28, 1585–1591. [Google Scholar] [PubMed]
- Fröhlich, L.F.; Mrakovcic, M.; Smole, C.; Lahiri, P.; Zatloukal, K. Epigenetic silencing of apoptosis-inducing gene expression can be efficiently overcome by combined SAHA and TRAIL treatment in uterine sarcoma cells. PLoS ONE 2014, 9, e91558. [Google Scholar] [CrossRef]
- Wang, J.; Kim, T.H.; Ahn, M.Y.; Lee, J.; Jung, J.H.; Choi, W.S.; Lee, B.M.; Yoon, K.S.; Yoon, S.; Kim, H.S. Sirtinol, a class III HDAC inhibitor, induces apoptotic and autophagic cell death in MCF-7 human breast cancer cells. Int. J. Oncol. 2012, 41, 1101–1109. [Google Scholar] [CrossRef]
- Park, E.Y.; Woo, Y.; Kim, S.J.; Kim, D.H.; Lee, E.K.; De, U.; Kim, K.S.; Lee, J.; Jung, J.H.; Ha, K.T.; et al. Anticancer effects of a new SIRT inhibitor, MHY2256, against human breast cancer MCF-7 cells via regulation of MDM2-p53 binding. Int. J. Biol. Sci. 2016, 12, 1555–1567. [Google Scholar] [CrossRef]
- De, U.; Son, J.Y.; Sachan, R.; Park, Y.J.; Kang, D.; Yoon, K.; Lee, B.M.; Kim, I.S.; Moon, H.R.; Kim, H.S. A New Synthetic Histone Deacetylase Inhibitor, MHY2256, Induces Apoptosis and Autophagy Cell Death in Endometrial Cancer Cells via p53 Acetylation. Int. J. Mol. Sci. 2018, 19, 2743. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, E.M.; Maiuri, C.; Galluzzi, L.; Vitale, I.; Djavaheri-Mergny, M.; D’Amelio, M.; Criollo, A.; Morselli, E.; Zhu, C.; Harper, F.; et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell Biol. 2008, 10, 676–687. [Google Scholar] [CrossRef] [Green Version]
- Morselli, E.; Shen, S.; Ruckenstuhl, C.; Bauer, M.; Mariño, G.; Galluzzi, L.; Criollo, A.; Michaud, M.; Maiuri, M.; Chano, T.; et al. p53 inhibits autophagy by interacting with the human ortholog of yeast Atg17, RB1CC1/FIP200. Cell Cycle 2011, 10, 2763–2769. [Google Scholar] [CrossRef]
- Tripathi, R.; Ash, D.; Shaha, C. Beclin-1–p53 interaction is crucial for cell fate determination in embryonal carcinoma cells. J. Cell Mol. Med. 2014, 18, 2275–2286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaid, S.; Brandts, C.; Serve, H.; Dikic, I. Ubiquitination and selective autophagy. Cell Death Differ. 2013, 20, 21–30. [Google Scholar] [CrossRef]
- Shi, C.; Pan, B.-Q.; Shi, F.; Xie, Z.-H.; Jiang, Y.-Y.; Shang, L.; Zhang, Y.; Xu, X.; Cai, Y.; Hao, J.-J. Sequestosome 1 protects esophageal squamous carcinoma cells from apoptosis via stabilizing SKP2 under serum starvation condition. Oncogene 2018, 37, 3260–3274. [Google Scholar] [CrossRef]
- Seibenhener, M.L.; Geetha, T.; Wooten, M.W. Sequestosome 1/p62 - more than just a scaffold. FEBS Lett. 2007, 581, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Seibenhener, M.L.; Calderilla-Barbosa, L.; Diaz-Meco, M.-T.; Moscat, J.; Jiang, J.; Wooten, M.W.; Wooten, M.C. SQSTM1/p62 interacts with HDAC6 and regulates deacetylase activity. PLoS ONE 2013, 8, e76016. [Google Scholar] [CrossRef]
- Lahiri, P.; Schmidt, V.; Smole, C.; Kufferath, I.; Denk, H.; Strnad, P.; Rölicke, T.; Fröhlich, L.F.; Zatloukal, K. P62/Sequestosome-1 Is indispensable for maturation and stabilization of Mallory-Denk bodies. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ni, H.-M.; Guo, F.; Ding, Y.; Shi, Y.-H.; Lahiri, P.; Fröhlich, L.F.; Rülicke, T.; Smole, C.; Schmidt, V.C.; et al. Sequestosome 1/p62 protein is associated with autophagic removal of excess hepatic endoplasmic reticulum in mice. J. Biol. Chem. 2016, 291. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.; Le Toumelin, G.; Criollo, A.; Rain, J.; Gautier, F.; Juin, P.; Tasdemir, E.; Pierron, G.; Troulinaki, K.; Tavernarakis, N.; et al. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007, 26, 2527–2539. [Google Scholar] [CrossRef]
- Zalckvar, E.; Berissi, H.; Mizrachy, L.; Idelchuk, Y.; Koren, I.; Eisenstein, M.; Sabanay, H.; Pinkas-Kramarski, R.; Kimchi, A. DAP-kinase mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009, 10, 285–292. [Google Scholar] [CrossRef]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.; Mizushima, N.; Packer, M.; Schneider, M.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.Y.; Gang, H.; Aviv, Y.; Dhingra, R.; Margulets, V.; Kirshenbaum, L.A. p53 Mediates Autophagy and Cell Death by a Mechanism Contingent on Bnip3. Hypertension 2013, 62, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Yee, K.S.; Wilkinson, S.; James, J.; Ryan, K.M.; Vousden, K.H. PUMA and Bax-induced Autophagy Contributes to Apoptosis. Cell Death Differ. 2010, 16, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Lorrin, S.; Pierron, G.; Ryan, K.; Codogno, P.; Djavaheri-Mergny, M. Evidence for the interplay between JNK and p53-DRAM signaling pathways in the regulation of autophagy. Autophagy 2010, 6, 153. [Google Scholar] [CrossRef]
- Maiuri, M.; Criollo, A.; Kroemer, G. Crosstalk between apoptosis and autophagy within the Beclin 1 interactome. EMBO J. 2010, 29, 515–516. [Google Scholar] [CrossRef] [Green Version]
- Lindqvist, L.; Heinlein, M.; Huang, D.; Vaux, D. Prosurvival Bcl-2 family members affect autophagy only indirectly, by inhibiting Bax and Bak. Proc. Natl. Acad. Sci. USA 2014, 111, 8512–8517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crighton, D.; Wilkinson, S.; Ryan, K. DRAM links autophagy to p53 and programmed cell death. Autophagy 2007, 3, 72–74. [Google Scholar] [CrossRef]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Lorin, S.; Borges, A.; Ribeiro Dos Santos, L.; Souquère, S.; Pierron, G.; Ryan, K.; Codogno, P.; Djavaheri-Mergny, M. c-Jun NH2-terminal kinase activation is essential for DRAM-dependent induction of autophagy and apoptosis in 2-methoxyestradiol-treated Ewing sarcoma cells. Cancer Res. 2009, 69, 6924–6931. [Google Scholar] [CrossRef] [PubMed]
- Yousefi, S.; Perozzo, R.; Schmid, I.; Ziemiecki, A.; Schaffner, T.; Scapozza, L.; Brunner, T.; Simon, H.-U. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell Biol. 2006, 8, 1124–1132. [Google Scholar] [CrossRef]
- Francisco, R.; Pérez-Perarnau, A.; Cortés, C.; Gil, J.; Tauler, A.; Ambrosio, S. Histone deacetylase inhibition induces apoptosis and autophagy in human neuroblastoma cells. Cancer Lett. 2012, 318, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Ahn, M.Y.; Kim, T.H.; Yoon, S.; Kang, K.W.; Lee, J.; Moon, H.R.; Jung, J.H.; Chung, H.Y.; Kim, H.S. A new synthetic HDAC inhibitor, MHY218, induces apoptosis or autophagy-related cell death in tamoxifen-resistant MCF-7 breast cancer cells. Investig. New Drugs 2012, 30, 1887–1898. [Google Scholar] [CrossRef]
- Martin, A.; Park, M.; Mitchell, C.; Walker, T.; Rahmani, M.; Thorburn, A.; Häussinger, D.; Reinehr, R.; Grant, S.; Dent, P. BCL-2 family inhibitors enhance histone deacetylase inhibitor and sorafenib lethality via autophagy and overcome blockade of the extrinsic pathway to facilitate killing. Mol. Pharmacol. 2009, 76, 327–341. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, Y.; Zhou, L.; Leng, Y.; Lin, H.; Kmieciak, M.; Pei, X.-Y.; Jones, R.; Orlowski, R.Z.; Dai, Y.; et al. A Bim-targeting strategy overcomes adaptive bortezomib resistance in myeloma through a novel link between autophagy and apoptosis. Blood 2014, 124, 2687–2697. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Goldstein, L.A.; Hou, W.; Gastman, B.R.; Rabinowich, H. Regulation of Mitochondrial Apoptotic Events by p53-mediated Disruption of Complexes between Antiapoptotic Bcl-2 Members and Bim. J. Biol. Chem. 2010, 285, 22473–22483. [Google Scholar] [CrossRef]
- Zhan, Y.; Gong, K.; Chen, C.; Wang, H.; Li, W. P38 MAP kinase functions as a switch in MS-275-induced reactive oxygen species-dependent autophagy and apoptosis in Human colon cancer cells. Free Radic. Biol. Med. 2012, 53, 532–543. [Google Scholar] [CrossRef] [PubMed]
- Torgersen, M.L.; Engedal, N.; Bøe, S.; Hokland, P.; Simonsen, A. Targeting autophagy potentiates the apoptotic effect of histone deacetylase inhibitors in t ( 8; 21 ) AML cells. Blood 2018, 122, 2467–2477. [Google Scholar] [CrossRef] [PubMed]
- Butler, L.M.; Webb, Y.; Agus, D.B.; Higgins, B.; Tolentino, T.R.; Kutko, M.C.; LaQuaglia, M.P.; Drobnjak, M.; Cordon-Cardo, C.; Scher, H.I.; et al. Inhibition of transformed cell growth and induction of cellular differentiation by pyroxamide, an inhibitor of histone deacetylase. Clin. Cancer Res. 2001, 7, 962–970. [Google Scholar] [PubMed]
Figure 1.
Known mechanisms of histone deacetylase inhibitor-induced (HDACi) re-activation of apoptosis. These activate the intrinsic (mitochondrial) the extrinsic (death-receptor) or both pathways of apoptosis. Also non-coding RNAs, of which microRNAs prevail, have been described to mediate HDACi-stimulated apoptosis.
Figure 1.
Known mechanisms of histone deacetylase inhibitor-induced (HDACi) re-activation of apoptosis. These activate the intrinsic (mitochondrial) the extrinsic (death-receptor) or both pathways of apoptosis. Also non-coding RNAs, of which microRNAs prevail, have been described to mediate HDACi-stimulated apoptosis.

Figure 2.
Known mechanisms and signaling pathways involved in histone deacetylase inhibitor-activated induction or suppression of autophagy. In most cases mTOR inhibition representing the canonical pathway, ROS accumulation, NF-κB hyperacetylation, p21 upregulation, or the involvement of p53 signaling is observed.
Figure 2.
Known mechanisms and signaling pathways involved in histone deacetylase inhibitor-activated induction or suppression of autophagy. In most cases mTOR inhibition representing the canonical pathway, ROS accumulation, NF-κB hyperacetylation, p21 upregulation, or the involvement of p53 signaling is observed.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mrakovcic, M.; Kleinheinz, J.; Fröhlich, L.F. p53 at the Crossroads between Different Types of HDAC Inhibitor-Mediated Cancer Cell Death. Int. J. Mol. Sci. 2019, 20, 2415. https://doi.org/10.3390/ijms20102415
AMA Style
Mrakovcic M, Kleinheinz J, Fröhlich LF. p53 at the Crossroads between Different Types of HDAC Inhibitor-Mediated Cancer Cell Death. International Journal of Molecular Sciences. 2019; 20(10):2415. https://doi.org/10.3390/ijms20102415
Chicago/Turabian StyleMrakovcic, Maria, Johannes Kleinheinz, and Leopold F. Fröhlich. 2019. "p53 at the Crossroads between Different Types of HDAC Inhibitor-Mediated Cancer Cell Death" International Journal of Molecular Sciences 20, no. 10: 2415. https://doi.org/10.3390/ijms20102415
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.