Pigment Nephropathy: Novel Insights into Inflammasome-Mediated Pathogenesis
by
 , and
, and
Kurt T. K. Giuliani
1,2,3 ,
,
Andrew J. Kassianos
1,2,3,4,
Helen Healy
1,2,3 and
Pedro H. F. Gois
1,2,3,* 1
Kidney Health Service, Royal Brisbane and Women’s Hospital, Brisbane, QLD 4029, Australia
2
Conjoint Kidney Research Laboratory, Chemical Pathology—Pathology Queensland, Brisbane, QLD 4029, Australia
3
Faculty of Medicine, University of Queensland, Brisbane, QLD 4006, Australia
4
Institute of Health and Biomedical Innovation/School of Biomedical Sciences, Queensland University of Technology, Brisbane, QLD 4059, Australia
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(8), 1997; https://doi.org/10.3390/ijms20081997
Submission received: 29 March 2019
/
Revised: 17 April 2019
/
Accepted: 17 April 2019
/
Published: 23 April 2019
(This article belongs to the Special Issue Kidney Inflammation, Injury and Regeneration)
Abstract
:Pigment nephropathy is an acute decline in renal function following the deposition of endogenous haem-containing proteins in the kidneys. Haem pigments such as myoglobin and haemoglobin are filtered by glomeruli and absorbed by the proximal tubules. They cause renal vasoconstriction, tubular obstruction, increased oxidative stress and inflammation. Haem is associated with inflammation in sterile and infectious conditions, contributing to the pathogenesis of many disorders such as rhabdomyolysis and haemolytic diseases. In fact, haem appears to be a signalling molecule that is able to activate the inflammasome pathway. Recent studies highlight a pathogenic function for haem in triggering inflammatory responses through the activation of the nucleotide-binding domain-like receptor protein 3 (NLRP3) inflammasome. Among the inflammasome multiprotein complexes, the NLRP3 inflammasome has been the most widely characterized as a trigger of inflammatory caspases and the maturation of interleukin-18 and -1β. In the present review, we discuss the latest evidence on the importance of inflammasome-mediated inflammation in pigment nephropathy. Finally, we highlight the potential role of inflammasome inhibitors in the prophylaxis and treatment of pigment nephropathy.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Haem complexes consist of an Fe atom which is coordinated within the centre of a heterocyclic ring known as a protoporphyrin [1]. Haem-containing proteins are a large class of metalloproteins that play a pivotal role in maintaining basic biological functions [2]. Their broad activities range from mitochondrial electron transfer, oxygen transport and storage to signal transduction and control of gene expression [2].
Among the different haem group variants, haem a, b and c are the main biological types [3,4]. Of the haem variants, haem b is the most abundant form and is present biologically within myoglobin and haemoglobin, whilst haem a and c are present in cytochromes. Haem function as a prosthetic group in haemoproteins and are essential for reversible oxygen binding and transport [5,6]. However, under pathological conditions, an excess of circulating free haem may be highly cytotoxic and result in tissue damage, including within the kidney [3,6].
Pigment nephropathy (PN) is an acute decline in kidney function following the breakdown and deposition of endogenous haem pigment-containing proteins (myoglobin, haemoglobin) within renal tissue [7]. Both myoglobin and haemoglobin are freely filtered by glomeruli and when oxidised, release their haem moiety into the urinary space [8,9]. However, within the nephron, excess haem pigments may cause renal vasoconstriction, tubular obstruction, increased oxidative stress and inflammation [10,11,12,13].
Inflammation is an essential response of the innate immune system to harmful stimuli [14]. Haem is associated with inflammation in sterile and infectious conditions, contributing to the pathogenesis of many disorders such as rhabdomyolysis and haemolytic diseases [15]. There is an increasing body of evidence that haem trigger the inflammasome signalling cascade and ultimately, the innate immune response [16,17].
In the present review, we discuss the potential role of inflammasome activation as a driver of inflammation in PN. We explore the rationale of translating small molecule inhibitors of inflammasome activation already in clinical use, for diseases outside the kidney, in the prevention and treatment of PN.
2. The Nucleotide-Binding domain-Like Receptor Protein 3 (NLRP3) Inflammasome
The inflammasomes are a family of cytosolic signalling complexes with a central role in the activation of innate immune responses via the maturation and secretion of pro-inflammatory cytokines (interleukin (IL)-1β and IL-18) [18]. In particular, the nucleotide-binding domain-like receptor protein 3 (NLRP3) inflammasome, an extensively characterized inflammasome family member, is widely implicated in a variety of renal injuries, including acute and chronic kidney disease (CKD) [19,20,21]; oxalate and uric acid crystal nephropathy [22,23]; and diabetic nephropathies [24]. Inflammasomes respond to a diverse range of pathogen-associated molecular patterns (PAMPs) and endogenously derived damage-associated molecular patterns (DAMPs) via a suite of pattern recognition receptors (PRR). Of particular note, endogenous particulate matter, such as haem [16,17], monosodium urate (MSU) [25], oxalate [23,26] and cholesterol crystals [27,28] have all been identified as potent triggers of NLRP3 inflammasome activation and the subsequent release of pro-inflammatory cytokines [24].
Recently, Liston and Masters [29] proposed a mechanism of inflammasome activation in addition to the PAMP-DAMP axis. This mechanism responds to a loss of homeostasis via ‘homeostasis-altering molecular processes’ (HAMPs). They hypothesized that the PAMP-DAMP-HAMP axis was, collectively, likely to be sufficient for effective immunity and that deficiencies in this axis may cause the pathological inflammatory activation observed in sterile injury [29]. Examples of HAMPs which activate the inflammasome are perturbed membrane potential through K+ efflux and Ca2+ influx [30], extracellular adenosine triphosphate (ATP) [31,32,33], and mitochondrial damage through reactive oxygen species (ROS) [34], altered mitochondrial membrane potential (ΔΨm) [35] and oxidised mitochondrial DNA (mtDNA) [36]. While their activation triggers may be diverse, the signalling pathways of inflammasome activation can be categorized into either canonical or non-canonical activation.
2.1. Canonical Inflammasome Activation
Following the detection of PAMPs or DAMPs (Signal 1) by PRRs, the NLRP3 inflammasome is canonically activated in an orchestrated cascade of signals [37], see Figure 1. The transmembrane protein family of Toll-like receptors (TLRs) play an important role as PRRs, activating the downstream signalling cascade. This signalling cascade is known as the “priming” phase of inflammasome activation. Once primed, the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signalling complex translocates to the cell nucleus where it promotes the upregulation of NLRP3 and immature forms of IL-1β and IL-18 [38].
Following the priming phase, a second signal (Signal 2) is required to elicit the activation of the inflammasome, see Figure 1b. These signals can include interrupted phagocytosis [39], extracellular ATP [31,32,33], K+ and Ca2+ flux [39,40,41], endoplasmic reticulum stress [42], mitochondrial ROS [34], ΔΨm [35] and the release of oxidised mtDNA [36]. Particulate matter are also potent secondary signals which can activate the NLRP3 inflammasome via cell-surface contact [39]. The mechanism for detection of these PAMP/DAMP/HAMPs by NLRP3 remains poorly understood.
Once activated by these molecular signalling patterns, NLRP3 proteins self-oligomerize and nucleate the formation of the NLRP3 inflammasome complex. This inflammasome complex consists of the NLRP3 protein, the ASC (Apoptosis-associated Speck-like protein containing a Caspase-activation-and-recruitment domain) adaptor protein and pro-caspase-1. Boucher, et al. [43] recently showed that pro-caspase-1 proteins dimerize following their recruitment to the inflammasome complex, before self-cleaving into an active state. The transiently active caspase-1 dimer undergoes additional cleavage, forming a proteolytically active holoenzyme with the inflammasome, capable of processing the pro-inflammatory cytokines IL-1β and IL-18 into their active forms [18,43]. Caspase-1 also cleaves Gasdermin-D (GSDMD) into its active form. Active GSDMD translocates to the cell membrane and forms GSDMD pores in the plasma membrane, driving pyroptosis and the consequent rapid release of IL-1β and IL-18 into the surrounding extracellular micro-environment [44,45,46,47,48].
2.2. Non-Canonical Inflammasome Activation
Non-canonical activation of the inflammasome differs in that it is dependent on caspase-11 (murine) or caspase-4 (human) activity [49,50,51]. Gram-negative bacteria-derived PAMPs are established triggers of non-canonical activation, directly sensed by and activating caspase-11/-4 [51]. Active caspase-11/-4 proteolytically cleave pro-GSDMD into its active state, effecting cell death by pyroptosis [49,50]. Kayagaki, et al. [50] showed that murine caspase-11 also triggers an NLRP3-inflammasome response through an as-yet-to-be identified mechanism, resulting in the release of IL-1β and IL-18 [50]. In humans, caspase-4 is required for the maturation and release of IL-18 via a non-canonical inflammasome pathway [51]. However, the role of non-canonical inflammasome activation in kidney disease remains to be elucidated.
2.3. Inflammasomes in the Kidney
Inflammasome activation is a key driver of the pathobiology in a variety of murine models and human etiologies of acute kidney injury (AKI) and CKD. Several murine studies investigating NLRP3 function, using small-molecule inflammasome-specific inhibitors or gene knockout models, have provided strong evidence for inflammasome activity in renal tissue injury. Specifically, Nlrp3-/-, Asc-/- and Casp1-/- knock-out models have less kidney tissue damage and disease phenotype in unilateral ureteral obstruction (UUO) [52,53], diabetic kidney disease (DKD) [54] and crystal nephropathy [23,26]. However, the PAMPs/DAMPs/HAMPs that trigger inflammasome activation in these models are under active investigation.
Elevated soluble uric acid levels have been reported in the obstructed kidney of UUO mice [53]. Uric acid is an established activator of the inflammasome [55]. Furthermore, ROS derived from the activity of xanthine oxidase (XO), an enzyme which produces uric acid via purine catabolism, has also been reported to elicit an inflammasome response [56]. Allopurinol is a widely prescribed pharmaceutical used in the treatment of gout and directly inhibits XO activity. Notably, UUO mice treated with allopurinol exhibit less NLRP3 and IL-1β expression within the UUO kidney compared to untreated UUO controls [53]. These studies suggest a dual protective role for allopurinol by inhibiting both uric acid production and XO activity, thus preventing inflammasome activation.
Shahzad, et al. [54] reported NLRP3 activation in podocytes, an important cell type in the glomerular filtration barrier, in a murine DKD model [54]. Interestingly, this study demonstrated increased IL-1β and IL-18 expression within plasma and renal cortical extracts of diabetic animals, correlating with the functional kidney biomarker urine albumin/creatinine ratio [54].
IL-1β and IL-18 are produced by infiltrating hematopoietic cells, such as dendritic cells (DC) and macrophages, in mouse kidneys [57]. Supporting this concept, DC depletion in a crystal-induced model of murine renal fibrosis, resulted in reduced fibrosis and improved kidney function [20]. Furthermore, a similar outcome was achieved by treatment with a specific small molecule NLRP3 inflammasome inhibitor (MCC950; detailed below in Section 6.1) that blocked NLRP3 activation in kidney DC, reduced IL-1β and IL-18 production and inhibited the progression of renal fibrosis [20].
In contrast to these murine studies, the examination of inflammasome-mediated renal pathology in humans is less extensive. Whilst human proximal tubular epithelial cells (PTEC) appear to have the necessary inflammasome-related machinery, there is a paucity of evidence for its activation, particularly, whether these cells secrete IL-1β and IL-18 [58]. Intriguingly Kim, et al. [58] recently described an inflammasome-independent role for NLRP3 in human PTEC. In this study, hypoxic injury to PTEC increased NLRP3 expression independent of ASC, caspase-1, and IL-1β. Instead, the NLRP3 protein bound to the mitochondrial antiviral signal (MAVS), resulting in mitochondrial dysfunction (increased mitochondrial ROS) and cell death [58]. There is also emerging evidence that human tubular cells in acute oxalate nephropathy undergo a form of regulated cell death termed necroptosis. Products of necroptosis include DAMPs with the capacity to activate the canonical inflammasome pathway in innate immune cells (DC, macrophages) within the tubulointerstitium [20]. Our group has indeed shown increased numbers of activated human DC within the tubulointerstitium of fibrotic kidney biopsies, accumulating adjacent to injured PTEC [59].
The kidneys play a major role in maintaining homeostasis and regulating blood pressure. Renal inflammation and fibrosis are well-known contributing factors in the pathogenesis of hypertension [60]. In a murine model of salt-induced hypertension, NLRP3 inhibition by treatment with MCC950 reduced hypertension and heart rate, in addition to reduced inflammasome priming, inflammatory cytokines, kidney immune cell infiltration and kidney fibrosis [60]. Nevertheless, the specific mechanisms by which the inflammasome contributes to systemic hypertension are still unclear. Furthermore, the inflammasome-dependent interactions between specialized renal parenchymal and innate immune cells, in particular, the role of NLRP3 signalling in driving the pathobiology of human PN, remains to be elucidated.
3. Haem Catabolism and Role in Immune-Mediated Pathology
Excess haem pigments are highly cytotoxic in the kidney, leading to oxidative stress and inflammation under injurious conditions [61,62]. Our understanding of immune-mediated pathological conditions is that oxidative stress and inflammation are interdependent processes rather than discrete pathways of injury [63].
Free haem catalyses the formation of highly toxic free radicals—hydroxyl radicals (OH∙)—from hydrogen peroxide (H2O2) via the Fenton reaction. Under homeostatic conditions, excess free cellular haem is catabolized by haem oxygenases (HO)—stress-responsive HO-1 and constitutive HO-2, as summarized in Figure 2. Catabolism of free haem by HO leads to the production of: (1) carbon monoxide (CO); (2) biliverdin (BV), that is converted by biliverdin reductase (BVR) to the antioxidant bilirubin; and (3) the release of labile Fe, which is promptly bound to ferritin (FtH), collectively preventing cellular oxidative stress [64,65,66]. However, under pathological conditions, the accumulation of intracellular free haem can exceed the rate of haem degradation by the HO-1 isoenzyme. Furthermore, levels of cellular Fe can be greater than the scavenging capacity of FtH. When this occurs, free haem and/or labile Fe accumulate in cells and drive oxidative stress in the micro-environment. The uncontrolled generation of free radicals and the subsequent imbalance between reactive metabolites and endogenous anti-oxidants constitutes the stress response and ultimately lead to cellular damage and inflammation.
Haem directly regulates inflammatory leukocyte migration and retention in vitro and in vivo [68]. In rodent models, intraperitoneal and intrapleural injection of haem results in dose-dependent neutrophil migration into the respective body compartments [68,69]. Haem inhibits neutrophil apoptosis, resulting in the accumulation of neutrophils at sites of haem deposition, and drives expression of proinflammatory cytokines [69,70,71]. Haem has also been reported to induce surface expression of adhesion molecules—i.e., intercellular adhesion molecule-1 (ICAM-l), vascular adhesion molecule-1 (VCAM-l) and endothelial leukocyte adhesion molecule (E-selectin)—in human endothelial cells, thereby driving the adhesion/retention of leukocytes [72].
Recent evidence suggests haem can trigger activation of innate immune cells via the NLRP3 inflammasome. Dutra et al. showed that haem activation of the NLRP3 inflammasome in bone marrow macrophages was dependent on NADPH oxidases, K+ efflux and generation of mitochondrial ROS [8]. Notably, NLRP3 activation was independent of haem internalization, lysosomal damage and cell death [8]. Inflammasome activity within immortalized human endothelial cells in response to haem has also been reported in vitro, where haem was sufficient to induce significantly increased IL-1β mRNA transcripts and cytokine release [16]. Intriguingly, HO-1 activity appears to attenuate NLRP3 activity. However, this may be an indirect consequence of haem catabolism by HO-1, rather than direct interactions between HO-1 and NLRP3 [73]. Although recent studies suggest haem is an important trigger of the canonical inflammasome pathway [8,73,74], its functioning via non-canonical NLRP3 inflammasome activation in renal cells has not been explored.
4. Myoglobin-Mediated Pigment Nephropathy
Rhabdomyolysis is a clinical syndrome following physical, thermal, toxic, metabolic, ischaemic, infective and inflammatory insults to muscles [13]. The final step of the skeletal muscle breakdown is the release of toxic intracellular components, such as the hemoprotein myoglobin, into the circulation [10,75].
Myoglobinuric AKI is the most severe complication of rhabdomyolysis [76]. Myoglobin is one of the pathogenic drivers of renal injury following rhabdomyolysis. Myoglobin is cytotoxic, activating both pro-oxidant and inflammatory pathways. Cytotoxicity is augmented in the presence of volume depletion and aciduria, common features of AKI [77,78]. Renal vasoconstriction, tubular obstruction and apoptosis are additional pathological processes in myoglobin toxicity, see Figure 3 [10,12,79].
There is a large volume of published studies describing oxidative stress in myoglobinuric AKI [10,12,13,80]. As for other hemoproteins, myoglobin possesses a haem centre that can catalyse the production of ROS within the kidneys. The haem group within myoglobin is capable of cycling between various oxidative states (ferrous = Fe2+; ferric = Fe3+; and ferryl = Fe4+) that may lead to lipid peroxidation independently of the Fenton reaction and iron release, see Figure 3 [12,13,80].
Most studies of the inflammatory pathogenic processes in myoglobinuric AKI are derived from experimental animal models and transformed cell lines. In a rat model of glycerol-induced rhabdomyolysis, macrophage infiltration was evident in the renal cortex as early as six hours following glycerol injection [79]. In vitro evidence suggests myoglobin polarizes macrophages toward both M1 (pro-inflammatory) and M2 (anti-inflammatory/pro-fibrotic) phenotypes, whilst in vivo research indicates that a reduction in oxidative stress may facilitate kidney tissue repair via a skewing of macrophages toward an M2 subtype [10,81].
Indeed, inflammation is involved in the pathogenesis of rhabdomyolysis-induced AKI, with emerging evidence of a functional role for the NLRP3 inflammasome in this disease process. Komada, et al. [17] reported greater expression of inflammasome-related molecules (NLRP3, ASC, caspase-1 and IL-1β) in the renal parenchyma following glycerol-induced myoglobinuric AKI [17]. Furthermore, activation of the inflammasome pathway correlated with leukocyte infiltration, tubular injury and dysfunction in the diseased kidney. Notably, these endpoints were markedly attenuated in Nlrp3-/-, Asc-/- and Casp1-/- knockout mice [17].
At present, many questions regarding the potential triggers of the inflammasome cascade in myoglobinuric AKI remain unanswered. Komada, et al. [17] carried out in vitro experiments using renal tubular epithelial cells incubated with hemin (the oxidised form of haem), ferrous and ferric myoglobin, all potential stimuli of the NLRP3 inflammasome in myoglobinuric AKI. Although these experimental data were not published, the authors reported that these stimuli were insufficient to activate NLRP3 [17]. Although innate immune cells (DC, macrophages) have the required components for canonical inflammasome activation [74,82], the ability of tubular epithelial cells to secrete mature IL-1β via this two-step process remains uncertain [58,74,82]. Therefore, the absence of inflammatory cells in the in vitro experiments of Komada et al. may explain why they failed to demonstrate triggering of canonical inflammasome activation.
Finally, as the pathogenesis of rhabdomyolysis is multifactorial, the role of other concomitant factors, acting either as priming stimuli or directly activating the NLRP3 inflammasome, should not be ignored. For instance, data from several studies suggest that different types of crystals, such as calcium oxalate, monosodium urate and cholesterol, can function as DAMPs to trigger NLRP3 inflammasome activation [22,25,83]. Recently, we highlighted a potential role for urate crystals in generating oxidative stress and activating the NLRP3 inflammasome in an animal model of rhabdomyolysis-associated AKI [10]. Thus, additional research is required to validate this hypothesis as well as to further elucidate the mechanisms underlying inflammation in human myoglobinuric AKI.
5. Haemoglobin-Mediated Pigment Nephropathy
Haemolysis is defined as the rupture of red blood cells (RBC) as a result of intrinsic or extrinsic stresses, leading to the release of their intracellular contents, including hemoprotein haemoglobin [84]. Massive intravascular haemolysis is uncommon but occurs in life-threatening conditions such as poisoning, snake and insect envenomation, idiosyncratic drug reactions, haemolytic uraemic syndrome, paroxysmal nocturnal hemoglobinuria, malaria, haemorrhagic fevers, leptospirosis and septic shock [85,86,87,88,89,90,91].
In the event of haemolysis, plasma proteins such as haemoglobin-binding haptoglobin and haem-binding hemopexin effectively remove intravascular-produced haemoglobin/haem, thus mitigating haem-mediated deleterious effects [69]. However, under pathological conditions, the binding capacity of these plasma proteins is saturated, resulting in excess free haemoglobin in circulating blood [69]. Haemoglobulin and haem are filtered by the glomerulus, and free haemoglobin in the resultant ultrafiltrate is reabsorbed by the proximal tubules in an endocytic process involving the megalin-cubilin receptor system [65]. However, this absorption transport pathway is also concentration-dependent and large quantities of haemoglobin in the proximal tubules will saturate it, with free haemoglobulin/haem retained in the proximal tubules, leading to nephrotoxicity.
Many diseases featuring massive or recurrent haemolysis are complicated by AKI [9,64,84]. Prior to modern transfusion practices, ABO incompatibility was the most common cause of hemolysis-associated AKI [9]. With the exception of ABO-incompatible blood transfusions, haemolysis is now considered a contributing, rather than sole, trigger in the pathogenesis of haemoglobinuria-related AKI [9]. In fact, some conditions such as poisoning, envenomation and leptospirosis, may present with both haemolysis and rhabdomyolysis [85,92,93,94,95]. Furthermore, in malaria-associated AKI, other mechanisms play a greater pathogenic role than haemolysis, including mechanic obstruction by parasitized RBCs, the pro-inflammatory cytokine storm and immune-complex deposition [9,96].
The pathogenesis of haemoglobinuric AKI is multi-factorial, with aciduria, dehydration and renal ischaemia being the established contributing factors in the pathobiological processes [61,62]. These concomitant conditions are thought to enhance haem toxicity by favouring iron release and thus, pro-oxidant cytotoxic conditions [61].
As in myoglobin-derived PN, haemoglobin-derived free haem can drive oxidative stress, increased expression of adhesion molecules and elevated leukocyte infiltration into the diseased kidney [66,72]. Haemolysis also generates DAMP activity that triggers sterile inflammatory responses via the NLRP3 inflammasome [97]. In addition to haem, ruptured RBCs release heat shock proteins, ATP, IL-33 and mtDNA that are recognized triggers of the inflammasome cascade [97]. A correlate is found in humans with the disease of sickle cell, where patients commonly present with a state of chronic low-grade inflammation [69,98].
Intravascular hemolysis may also lead to haemoglobin in different oxidative states, i.e., hemoglobin (Fe2+), methemoglobin (Fe3+), and ferryl haemoglobin (Fe4+) [99]. Nyakundi, et al. [99] demonstrated both haem and ferryl haemoglobin stimulated LPS-primed macrophages to upregulate IL-1β mRNA and induce active IL-1β secretion. Further experiments conducted by Dutra et al. showed that the iron present within the haem molecule, not free iron, was the most important stimulus triggering the NLRP3 inflammasome and IL-1β secretion in macrophages and ultimately contributed to hemolysis-associated lethality [8]. Understanding these molecular pathways triggered by distinct haem motifs may prove useful in identifying novel therapeutic targets for haemoglobin/myoglobin-mediated pigment nephropathies.
6. Inflammasome Inhibition as a Potential Therapeutic Target
The significant pathological role of inflammasome activation in several chronic inflammatory diseases has made it an attractive target for therapeutic intervention. There are two approaches in current strategies inhibiting the inflammasome: (1) Targeting inflammasome activation directly and/or (2) targeting down-stream effects of IL-1β. Here, we review several compounds that could be repurposed, in combination with existing therapies, to ameliorate inflammatory immune responses in PN.
6.1. NLRP3 Inflammasome Inhibitors
Several compounds have been identified and developed for therapeutic inhibition of NLRP3 inflammasome activation. These established inflammasome-inhibiting compounds have been extensively reviewed by Lopez-Castejon and Pelegrin [100] and, more recently, by Baldwin, et al. [101]. Several preclinical studies have already investigated the use of these inflammasome inhibitors in AKI and CKD nephropathies, but their therapeutic efficacy has not been tested in PN.
The second-generation sulfonylurea drug, glyburide (also glibenclamide), is an established compound for the treatment of human type II diabetes mellitus [100,101]. Glyburide blocks KATP channels, depolarizing the cell membrane, triggering the release of insulin from pancreatic β-cells [100,101]. Glyburide’s actions were originally thought to be mediated via its role as a KATP channel blocker, but emerging evidence suggests that it, in fact, prevents the formation of ASC specks [101]. However, the specific mechanism of the interactions of glyburide and NLRP3 remain poorly understood.
Glyburide has been used in an adenine-rich diet rat model of CKD. In this study, glyburide treatment attenuated NLRP3 expression, improved renal function and ameliorated the CKD histopathology [102]. Unfortunately, glyburide is generally not a recommended treatment in CKD patients due to the increased risk of hypoglycemia [103]. In addition, patients with glucose-6-phosphate dehydrogenase deficiency are susceptible to developing haemolytic anemia following glyburide treatment [104,105].
A novel subclass of sulfonylurea containing compounds, derived from glyburide, was identified by Perregaux, et al. [106]. These compounds inhibited post-translational processing of IL-1β, resulting in little-to-no maturation or extracellular release of the cytokine. One of these compounds, MCC950 (also CP-456,773), was reported by Coll, et al. [107] as a potent, specific inhibitor of the NLRP3 inflammasome. Whilst the mechanism of MCC950-mediated NLRP3 inhibition is still poorly understood, MCC950 has been studied in several disease models, including colitis [108], Parkinson’s disease [109], diabetic encephalopathy [110] and non-alcoholic steatohepatitis [111]. Recent studies also evaluated MCC950 in pre-clinical models of AKI and CKD. MCC950 treatment attenuated kidney fibrosis in a murine model of diet-induced oxalate crystal-nephropathy [20]. Furthermore, MCC950 treatment abrogated kidney damage and ameliorated systemic blood pressure in a murine model of hypertension, induced by both surgery (uninephrectomy) and treatment with deoxycorticosterone [60]. MCC950’s relatively short half-life and its specificity for the NLRP3 inflammasome [107] make it, and its derivatives, ideal candidates for further investigations in PN.
6.2. Anti-IL-1β and IL-1 Receptor Antagonists
Inhibition of the down-stream IL-1β-signalling pathways has been widely adopted in rheumatology for treatment of auto-inflammatory diseases [112]. Strategies for these therapies involve: (1) Reducing the amount of IL-1β available for activating the endogenous IL-1 receptor (IL-1R) or (2) inhibiting the endogenous receptor directly.
Canakinumab is a potent monoclonal antibody specific for IL-1β [113,114] and an established therapeutic in the treatment of rheumatoid arthritis [112]. Canakinumab has been evaluated in patients with CKD, reducing the risk of major adverse cardiovascular event rates among high-risk atherosclerosis patients [115]. However, no differences in kidney function (as measured by the estimated Glomerulus Filtration Rate) were reported between placebo and Canakinumab-treated CKD patients [115]. A common CKD co-morbidity is gout, which arises as a consequence of increased uric acid. Inflammasome activation is imputed to be the prime mechanism of this auto-inflammatory condition [25]. A clinical trial using Canakinumab showed significantly reduced rates of gout attacks in patients, although no changes in serum uric acid concentrations were observed [116]. Studies such as these provide important foundational evidence for further pre-clinical studies of Canakinumab for the treatment of PN.
Therapeutic strategies targeting the IL-1 receptor (IL-1R) are also used in current clinical practice. Anakinra is a recombinant human IL-1R antagonist, competing with IL-1β for binding with the IL-1R [117]. Anakinra is another established therapeutic in the treatment of auto-inflammatory diseases in rheumatology. Notably, it has been successfully used in patients with Familial Mediterranean Fever (FMF) [118,119], an auto-inflammatory disease associated with mutations in the inflammasome component pyrin that results in triggering inflammasome activation [120,121]. The therapeutic use of anakinra for treating acute gout attacks in CKD patients is currently in clinical trials (ASGARD study), with the results yet to be published [117]. Interestingly, anakinra is being investigated as a third-line therapy in this ASGARD study, following non-response to second-line therapy, where the development of rhabdomyolysis was a reported side-effect [117,122].
Although these IL-1β- and IL-1R-targeting drugs are proving to be effective inflammasome inhibitors, pre-clinical studies investigating their efficacy for the treatment of PN are yet to be performed. These studies need to include in vivo and in vitro models of PN to not only establish therapeutic efficacy but also any unforeseen off-target effects.
7. Concluding Remarks
The release of haem by myoglobin and haemoglobin catabolism is pivotal in the pathogenesis of PN. Whilst haem toxicity is clinically recognized as important, the role of haem in the mechanism of the associated kidney inflammation may be overlooked. Irrespective of its source, haem triggers NLRP3 inflammasome activation, but this mechanistic pathway of disease in PN is still poorly understood. Contemporary studies have shifted to the role of haem driving kidney inflammation via NLRP3 inflammasome activation. The research is focused on the canonical activation of the inflammasome within immune cell populations by haem. The non-canonical activation of the inflammasome in immune cell populations by haem has not been investigated. Furthermore, neither canonical nor non-canonical mechanisms of inflammasome activation within kidney parenchymal cells are fully understood. Well-designed studies are required to address both, focusing on haemolytic driven AKI for which there is currently a lack of information.
The aim of future PN research is to provide evidence to move to pre-clinical studies of potential treatments for both myoglobinuric and haemolytic AKI. Non-renal studies with IL-1R antagonists and direct NLRP3 inflammasome inhibitors are advanced, with small molecules in clinical use for auto-immune rheumatological diseases. Several pre-clinical studies have investigated their therapeutic role in different patterns of kidney disease, but not PN. These studies provide the rationale for translation into clinical trials for the prevention and treatment of PN.
Author Contributions
P.H.F.G, A.J.K, H.H., and K.G. conceptualized the review article. P.H.F.G, K.T.K.G, A.J.K and H.H. contributed in writing, review and editing of the original draft. K.T.K.G. compiled the manuscript and P.H.F.G. supervised the study.
Funding
This research was supported by funds from Pathology Queensland, a Royal Brisbane and Women’s Hospital Research Grant, the Kidney Research Foundation, a National Health and Medical Research Council Project Grant (GNT1099222). K.T.K.G. is supported by an Australian Government Research Training Program (RTP) Scholarship.
Acknowledgments
The authors wish to thank Madeleine Kersting-Flynn for her advice and assistance with the illustrations presented in this document. The authors also would like to express our profound thanks for the continued support from the staff and patients at the Royal Brisbane and Women’s Hospital and Pathology Queensland.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AKI | Acute Kidney Injury |
| ASC | Apoptosis-associated speck-like protein containing a CARD domain |
| ATP | Adenosine Triphosphate |
| CARD | Caspase activation and recruitment domain |
| CD | Cluster of differentiation |
| CKD | Chronic Kidney Disease |
| DAMPs | Damage-associated molecular patterns |
| DC | Dendritic cells |
| DKD | Diabetic kidney disease |
| ESCRT | Endosomal sorting complexes required for transport |
| FMF | Familial Mediterranean Fever |
| GSDMD | Gasdermin-D |
| HAMPs | Homeostasis-altering molecular processes |
| HO | Haem Oxygenase |
| HO-1 | Haem Oxygenase-1 |
| ICAM-l | Intercellular Adhesion Molecule-1 |
| IL | Interleukin |
| IL-1R | IL-1 receptor |
| LPS | Lipopolysaccharide |
| MAVS | Mitochondrial antiviral signal |
| mtDNA | Mitochondrial DNA |
| NADPH | Dihydronicotinamide-adenine dinucleotide phosphate |
| NLRP3 | Nucleotide-binding domain-like receptor protein 3 |
| PAMPs | Pathogen-associated molecular patterns |
| PN | Pigment Nephropathy |
| PRRs | Pattern recognition receptors |
| PTEC | Proximal Tubule Epithelial Cells |
| RBC | Red Blood Cells |
| ROS | Reactive Oxygen Species |
| TLRs | Toll-like receptors |
| UUO | Unilateral ureteral obstruction |
| VCAM-l | Vascular Adhesion Molecule-1 |
| XO | Xanthine Oxidase |
References
- Sikorski, Z.E. Chemical and Functional Properties of Food Components; CRC Press: Boc Raton, FL, USA, 2007. [Google Scholar]
- Paoli, M.; Marles-Wright, J.; Smith, A. Structure–Function Relationships in Heme-Proteins. DNA Cell Biol. 2002, 21, 271–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, R.; Gouveia, Z.; Soares, M.P.; Gozzelino, R.; Kapitulnik, J.; Hebrew, T.; Ryter, S.W.; Immenschuh, S. Heme cytotoxicity and the pathogenesis of immune-mediated inflammatory diseases. Front. Pharmacol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.J.; Kahraman, A.; Thornton, J.M. Heme proteins-Diversity in structural characteristics, function, and folding. Proteins Struct. Function Bioinform. 2010, 78, 2349–2368. [Google Scholar] [CrossRef] [PubMed]
- Mense, S.M.; Zhang, L. Heme: A versatile signaling molecule controlling the activities of diverse regulators ranging from transcription factors to MAP kinases. Cell Res. 2006, 16, 681–692. [Google Scholar] [CrossRef] [PubMed]
- Immenschuh, S.; Vijayan, V.; Janciauskiene, S.; Gueler, F. Heme as a Target for Therapeutic Interventions. Front Pharmacol. 2017, 8, 146. [Google Scholar] [CrossRef]
- Nangaku, M. Hypoxia and Tubulointerstitial Injury: A Final Common Pathway to End-Stage Renal Failure. Nephron Exp. Nephrol. 2004, 98, e8–e12. [Google Scholar] [CrossRef] [PubMed]
- Dutra, F.F.; Alves, L.S.; Rodrigues, D.; Fernandez, P.L.; de Oliveira, R.B.; Golenbock, D.T.; Zamboni, D.S.; Bozza, M.T. Hemolysis-induced lethality involves inflammasome activation by heme. Proc. Natl. Acad. Sci. USA 2014. [Google Scholar] [CrossRef]
- Perazella, M.A.; Rosner, M.H. Clinical features and diagnosis of heme pigment-induced acute kidney injury. Available online: https://www.uptodate.com/contents/clinical-features-and-diagnosis-of-heme-pigment-induced-acute-kidney-injury (accessed on 7 February 2019).
- Gois, P.H.F.; Canale, D.; Volpini, R.A.; Ferreira, D.; Veras, M.M.; Andrade-Oliveira, V.; Câmara, N.O.S.; Shimizu, M.H.M.; Seguro, A.C. Allopurinol attenuates rhabdomyolysis-associated acute kidney injury: Renal and muscular protection. Free Radic. Biol. Med. 2016, 101, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Heyman, S.N.; Rosen, S.; Fuchs, S.; Epstein, F.H.; Brezis, M. Myoglobinuric acute renal failure in the rat: a role for medullary hypoperfusion, hypoxia, and tubular obstruction. J. Am. Soc. Nephrol. 1996, 7, 1066–1074. [Google Scholar]
- Moore, K.P.; Holt, S.G.; Patel, R.P.; Svistunenko, D.A.; Zackert, W.; Goodier, D.; Reeder, B.J.; Clozel, M.; Anand, R.; Cooper, C.E.; et al. A causative role for redox cycling of myoglobin and its inhibition by alkalinization in the pathogenesis and treatment of rhabdomyolysis-induced renal failure. J. Biol. Chem. 1998, 273, 31731–31737. [Google Scholar] [CrossRef] [PubMed]
- Zager, R.A.; Burkhart, K. Myoglobin toxicity in proximal human kidney cells: roles of Fe, Ca2+, H2O2, and terminal mitochondrial electron transport. Kidney Int. 1997, 51, 728–738. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Callaway, J.B.; Ting, J.P.Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Dutra, F.F.; Bozza, M.T. Heme on innate immunity and inflammation. Front. Pharmacol. 2014, 5, 115. [Google Scholar] [CrossRef] [Green Version]
- Erdei, J.; Tóth, A.; Balogh, E.; Nyakundi, B.B.; Bányai, E.; Ryffel, B.; Paragh, G.; Cordero, M.D.; Jeney, V. Induction of NLRP3 Inflammasome Activation by Heme in Human Endothelial Cells. Oxid. Med. Cell. Longev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Komada, T.; Usui, F.; Kawashima, A.; Kimura, H.; Karasawa, T.; Inoue, Y.; Kobayashi, M.; Mizushina, Y.; Kasahara, T.; Taniguchi, S.I.; et al. Role of NLRP3 inflammasomes for rhabdomyolysis-induced acute kidney injury. Sci. Rep. 2015. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Brähler, S.; Zinselmeyer, B.H.; Raju, S.; Nitschke, M.; Suleiman, H.; Saunders, B.T.; Johnson, M.W.; Böhner, A.M.C.; Viehmann, S.F.; Theisen, D.J.; et al. Opposing Roles of Dendritic Cell Subsets in Experimental GN. J. Am. Soc. Nephrol. 2017. [Google Scholar] [CrossRef]
- Ludwig-Portugall, I.; Bartok, E.; Dhana, E.; Evers, B.D.G.; Primiano, M.J.; Hall, J.P.; Franklin, B.S.; Knolle, P.A.; Hornung, V.; Hartmann, G.; et al. An NLRP3-specific inflammasome inhibitor attenuates crystal-induced kidney fibrosis in mice. Kidney Int. 2016, 90, 525–539. [Google Scholar] [CrossRef]
- Anders, H.J.; Muruve, D.A. The Inflammasomes in Kidney Disease. J. Am. Soc. Nephrol. 2011, 22, 1007–1018. [Google Scholar] [CrossRef] [Green Version]
- Wilson, G.J.; Gois, P.H.F.; Zhang, A.; Wang, X.; Law, B.M.P.; Kassianos, A.J.; Healy, H.G. The Role of Oxidative Stress and Inflammation in Acute Oxalate Nephropathy Associated with Ethylene Glycol Intoxication. Kidney Int. Rep. 2018, 3, 1217–1221. [Google Scholar] [CrossRef]
- Knauf, F.; Asplin, J.R.; Granja, I.; Schmidt, I.M.; Moeckel, G.W.; David, R.J.; Flavell, R.A.; Aronson, P.S. NALP3-mediated inflammation is a principal cause of progressive renal failure in oxalate nephropathy. Kidney Int. 2013, 84, 895–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaribeygi, H.; Katsiki, N.; Butler, A.E.; Sahebkar, A. Effects of antidiabetic drugs on NLRP3 inflammasome activity, with a focus on diabetic kidneys. Drug Discov. Today 2018. [Google Scholar] [CrossRef]
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237. [Google Scholar] [CrossRef] [PubMed]
- Mulay, S.R.; Kulkarni, O.P.; Rupanagudi, K.V.; Migliorini, A.; Darisipudi, M.N.; Vilaysane, A.; Muruve, D.; Shi, Y.; Munro, F.; Liapis, H.; et al. Calcium oxalate crystals induce renal inflammation by NLRP3-mediated IL-1β secretion. J. Clin. Investig. 2013, 123, 236–246. [Google Scholar] [CrossRef]
- Rajamäki, K.; Lappalainen, J.; Öörni, K.; Välimäki, E.; Matikainen, S.; Kovanen, P.T.; Eklund, K.K. Cholesterol Crystals Activate the NLRP3 Inflammasome in Human Macrophages: A Novel Link between Cholesterol Metabolism and Inflammation. PLoS ONE 2010, 5, e11765. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357. [Google Scholar] [CrossRef] [PubMed]
- Liston, A.; Masters, S.L. Homeostasis-altering molecular processes as mechanisms of inflammasome activation. Nat. Rev. Immunol. 2017, 17, 208. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zeng, M.Y.; Yang, D.; Motro, B.; Núñez, G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016, 530, 354–357. [Google Scholar] [CrossRef]
- Amores-Iniesta, J.; Barberà-Cremades, M.; Martínez, C.M.; Pons, J.A.; Revilla-Nuin, B.; Martínez-Alarcón, L.; Di Virgilio, F.; Parrilla, P.; Baroja-Mazo, A.; Pelegrín, P. Extracellular ATP Activates the NLRP3 Inflammasome and Is an Early Danger Signal of Skin Allograft Rejection. Cell Rep. 2017, 21, 3414–3426. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Zhang, J.; Zhang, W.; Zhang, J.; Yang, J.; Li, K.; He, Y. ATP-P2X4 signaling mediates NLRP3 inflammasome activation: A novel pathway of diabetic nephropathy. Int. J. Biochem. Cell Biol. 2013, 45, 932–943. [Google Scholar] [CrossRef]
- Sadatomi, D.; Nakashioya, K.; Mamiya, S.; Honda, S.; Tanimura, S.; Yamamura, Y.; Kameyama, Y.; Takeda, K. Mitochondrial function is required for extracellular ATP-induced NLRP3 inflammasome activation. J. Biochem. 2017, 161, 503–512. [Google Scholar] [CrossRef]
- Elliott, E.I.; Miller, A.N.; Banoth, B.; Iyer, S.S.; Stotland, A.; Weiss, J.P.; Gottlieb, R.A.; Sutterwala, F.S.; Cassel, S.L. Cutting Edge: Mitochondrial Assembly of the NLRP3 Inflammasome Complex Is Initiated at Priming. J. Immunol. 2018, 200, 3047. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.j.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.-M.; Hu, W.; Troutman, T.D.; Jennings, M.; Brewer, T.; Li, X.; Nanda, S.; Cohen, P.; Thomas, J.A.; Pasare, C. IRAK-1 bypasses priming and directly links TLRs to rapid NLRP3 inflammasome activation. Proc. Natl. Acad. Sci. USA 2014, 111, 775–780. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Hari, A.; Zhang, Y.; Tu, Z.; Detampel, P.; Stenner, M.; Ganguly, A.; Shi, Y. Activation of NLRP3 inflammasome by crystalline structures via cell surface contact. Sci. Rep. 2014, 4, 7281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847. [Google Scholar] [CrossRef] [PubMed]
- Bronner, D.N.; Abuaita, B.H.; Chen, X.; Fitzgerald, K.A.; Nuñez, G.; He, Y.; Yin, X.-M.; O’Riordan, M.X.D. Endoplasmic Reticulum Stress Activates the Inflammasome via NLRP3- and Caspase-2-Driven Mitochondrial Damage. Immunity 2015, 43, 451–462. [Google Scholar] [CrossRef]
- Boucher, D.; Monteleone, M.; Coll, R.C.; Chen, K.W.; Ross, C.M.; Teo, J.L.; Gomez, G.A.; Holley, C.L.; Bierschenk, D.; Stacey, K.J.; et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J. Exp. Med. 2018. [Google Scholar] [CrossRef]
- Monteleone, M.; Stanley, A.C.; Chen, K.W.; Brown, D.L.; Bezbradica, J.S.; von Pein, J.B.; Holley, C.L.; Boucher, D.; Shakespear, M.R.; Kapetanovic, R.; et al. Interleukin-1β Maturation Triggers Its Relocation to the Plasma Membrane for Gasdermin-D-Dependent and -Independent Secretion. Cell Rep. 2018, 24, 1425–1433. [Google Scholar] [CrossRef] [PubMed]
- Sborgi, L.; Rühl, S.; Mulvihill, E.; Pipercevic, J.; Heilig, R.; Stahlberg, H.; Farady, C.J.; Müller, D.J.; Broz, P.; Hiller, S. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016, 35, 1766–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Rühl, S.; Shkarina, K.; Demarco, B.; Heilig, R.; Santos, J.C.; Broz, P. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 2018, 362, 956–960. [Google Scholar] [CrossRef]
- Sun, G.; Guzman, E.; Balasanyan, V.; Conner, C.M.; Wong, K.; Zhou, H.R.; Kosik, K.S.; Montell, D.J. A molecular signature for anastasis, recovery from the brink of apoptotic cell death. J. Cell Biol. 2017, 216, 3355–3368. [Google Scholar] [CrossRef] [Green Version]
- Pellegrini, C.; Antonioli, L.; Lopez-Castejon, G.; Blandizzi, C.; Fornai, M. Canonical and non-canonical activation of NLRP3 inflammasome at the crossroad between immune tolerance and intestinal inflammation. Front. Immunol. 2017, 8, 36. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Knodler, L.A.; Crowley, S.M.; Sham, H.P.; Yang, H.; Wrande, M.; Ma, C.; Ernst, R.K.; Steele-Mortimer, O.; Celli, J.; Vallance, B.A. Noncanonical Inflammasome Activation of Caspase-4/Caspase-11 Mediates Epithelial Defenses against Enteric Bacterial Pathogens. Cell Host Microbe 2014, 16, 249–256. [Google Scholar] [CrossRef]
- Vilaysane, A.; Chun, J.; Seamone, M.E.; Wang, W.; Chin, R.; Hirota, S.; Li, Y.; Clark, S.A.; Tschopp, J.; Trpkov, K.; et al. The NLRP3 Inflammasome Promotes Renal Inflammation and Contributes to CKD. J. Am. Soc. Nephrol. 2010, 21, 1732–1744. [Google Scholar] [CrossRef] [Green Version]
- Braga, T.T.; Forni, M.F.; Correa-Costa, M.; Ramos, R.N.; Barbuto, J.A.; Branco, P.; Castoldi, A.; Hiyane, M.I.; Davanso, M.R.; Latz, E.; et al. Soluble Uric Acid Activates the NLRP3 Inflammasome. Sci. Rep. 2017, 7, 39884. [Google Scholar] [CrossRef] [Green Version]
- Shahzad, K.; Bock, F.; Dong, W.; Wang, H.; Kopf, S.; Kohli, S.; Al-Dabet, M.D.M.; Ranjan, S.; Wolter, J.; Wacker, C.; et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int. 2015, 87, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Gersch, M.S.; Johnson, R.J. Uric acid and the immune response. Nephrol. Dial. Transplant. 2006, 21, 3046–3047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ives, A.; Nomura, J.; Martinon, F.; Roger, T.; LeRoy, D.; Miner, J.N.; Simon, G.; Busso, N.; So, A. Xanthine oxidoreductase regulates macrophage IL1β secretion upon NLRP3 inflammasome activation. Nat. Commun. 2015, 6, 6555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The Interleukin-1 Family: Back to the Future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef]
- Kim, S.M.; Kim, Y.G.; Kim, D.J.; Park, S.H.; Jeong, K.H.; Lee, Y.H.; Lim, S.J.; Lee, S.H.; Moon, J.Y. Inflammasome-Independent Role of NLRP3 Mediates Mitochondrial Regulation in Renal Injury. Front. Immunol. 2018, 9, 2563. [Google Scholar] [CrossRef] [PubMed]
- Kassianos, A.J.; Wang, X.; Sampangi, S.; Muczynski, K.; Healy, H.; Wilkinson, R. Increased tubulointerstitial recruitment of human CD141hi CLEC9A+ and CD1c+ myeloid dendritic cell subsets in renal fibrosis and chronic kidney disease. AJP Renal Physiol. 2013, 305, F1391–F1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, S.M.; Ling, Y.H.; Huuskes, B.M.; Ferens, D.M.; Saini, N.; Chan, C.T.; Diep, H.; Kett, M.M.; Samuel, C.S.; Kemp-Harper, B.K.; et al. Pharmacological inhibition of the NLRP3 inflammasome reduces blood pressure, renal damage, and dysfunction in salt-sensitive hypertension. Cardiovasc. Res. 2019, 115, 776–787. [Google Scholar] [CrossRef]
- Paller, M.S. Hemoglobin- and myoglobin-induced acute renal failure in rats: Role of iron in nephrotoxicity. Am. J. Physiol. 1988, 255, F539–F544. [Google Scholar] [CrossRef]
- Zager, R.A.; Gamelin, L.M. Pathogenetic mechanisms in experimental hemoglobinuric acute renal failure. Am. J. Physiol. 1989, 256, F446–F455. [Google Scholar] [CrossRef]
- Biswas, S.K. Does the Interdependence between Oxidative Stress and Inflammation Explain the Antioxidant Paradox? Oxid. Med. Cell. Longev. 2016. [Google Scholar] [CrossRef]
- Qian, Q.; Nath, K.A.; Wu, Y.; Daoud, T.M.; Sethi, S. Hemolysis and acute kidney failure. Am. J. Kidney Dis. 2010. [Google Scholar] [CrossRef] [PubMed]
- Tracz, M.J.; Alam, J.; Nath, K.A. Physiology and Pathophysiology of Heme: Implications for Kidney Disease. J. Am. Soc. Nephrol. 2007. [Google Scholar] [CrossRef]
- Wagener, F.A.D.T.G.; Eggert, A.; Boerman, O.C.; Oyen, W.J.G.; Verhofstad, A.; Abraham, N.G.; Adema, G.; Van Kooyk, Y.; De Witte, T.; Figdor, C.G. Heme is a potent inducer of inflammation in mice and is counteracted by heme oxygenase. Blood 2001. [Google Scholar] [CrossRef]
- Rose, A.S.; Bradley, A.R.; Valasatava, Y.; Duarte, J.M.; Prlić, A.; Rose, P.W. NGL viewer: Web-based molecular graphics for large complexes. Bioinformatics 2018, 34, 3755–3758. [Google Scholar] [CrossRef] [PubMed]
- Porto, B.N.; Alves, L.S.; Fernández, P.L.; Dutra, T.P.; Figueiredo, R.T.; Graça-Souza, A.V.; Bozza, M.T. Heme induces neutrophil migration and reactive oxygen species generation through signaling pathways characteristic of chemotactic receptors. J. Biol. Chem. 2007. [Google Scholar] [CrossRef]
- Graça-Souza, A.V.; Arruda, M.A.B.; De Freitas, M.S.; Barja-Fidalgo, C.; Oliveira, P.L. Neutrophil activation by heme: Implications for inflammatory processes. Blood 2002. [Google Scholar] [CrossRef]
- Arruda, M.A.; Rossi, A.G.; de Freitas, M.S.; Barja-Fidalgo, C.; Graça-Souza, A.V. Heme inhibits human neutrophil apoptosis: Involvement of phosphoinositide 3-kinase, MAPK, and NF-κB. J. Immunol. 2004, 173, 2023–2030. [Google Scholar] [CrossRef]
- Figueiredo, R.T.; Fernandez, P.L.; Mourao-Sa, D.S.; Porto, B.N.; Dutra, F.F.; Alves, L.S.; Oliveira, M.F.; Oliveira, P.L.; Graça-Souza, A.V.; Bozza, M.T. Characterization of heme as activator of toll-like receptor 4. J. Biolog. Chem. 2007. [Google Scholar] [CrossRef]
- Wagener, F.A.; Feldman, E.; de Witte, T.; Abraham, N.G. Heme induces the expression of adhesion molecules ICAM-1, VCAM-1, and E selectin in vascular endothelial cells. Proc. Soc. Exp. Biol. Med. Soc. Exp. Biol. Med. 1997, 216, 456–463. [Google Scholar] [CrossRef]
- Lv, J.; Su, W.; Yu, Q.; Zhang, M.; Di, C.; Lin, X.; Wu, M.; Xia, Z. Heme oxygenase-1 protects airway epithelium against apoptosis by targeting the proinflammatory NLRP3–RXR axis in asthma. J. Biol. Chem. 2018, 293, 18454–18465. [Google Scholar] [CrossRef]
- Lorenz, G.; Darisipudi, M.N.; Anders, H.J. Canonical and non-canonical effects of the NLRP3 inflammasome in kidney inflammation and fibrosis. Nephrol. Dial. Transplant. 2014, 29, 41–48. [Google Scholar] [CrossRef]
- Bosch, X.; Poch, E.; Grau, J.M. Rhabdomyolysis and Acute Kidney Injury. N. Engl. J. Med. 2009, 361, 62–72. [Google Scholar] [CrossRef]
- Korrapati, M.C.; Shaner, B.E.; Schnellmann, R.G. Recovery from Glycerol-Induced Acute Kidney Injury Is Accelerated by Suramin. J. Pharmacol. Exp. Ther. 2012. [Google Scholar] [CrossRef]
- Desforges, J.F.; Better, O.S.; Stein, J.H. Early Management of Shock and Prophylaxis of Acute Renal Failure in Traumatic Rhabdomyolysis. N. Engl. J. Med. 1990, 322, 825–829. [Google Scholar] [CrossRef]
- Zager, R.A. Rhabdomyolysis and myohemoglobinuric acute renal failure. Kidney Int 1996, 49, 314–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homsi, E.; Janino, P.; De Faria, J.B.L. Role of caspases on cell death, inflammation, and cell cycle in glycerol-induced acute renal failure. Kidney Int. 2006, 69, 1385–1392. [Google Scholar] [CrossRef] [Green Version]
- Zager, R.A.; Foerder, C.A. Effects of inorganic iron and myoglobin on in vitro proximal tubular lipid peroxidation and cytotoxicity. J. Clin. Investig. 1992, 89, 989–995. [Google Scholar] [CrossRef] [PubMed]
- Belliere, J.; Casemayou, A.; Ducasse, L.; Zakaroff-Girard, A.; Martins, F.; Iacovoni, J.S.; Guilbeau-Frugier, C.; Buffin-Meyer, B.; Pipy, B.; Chauveau, D.; et al. Specific macrophage subtypes influence the progression of rhabdomyolysis-induced kidney injury. J. Am. Soc. Nephrol. 2015, 26, 1363–1377. [Google Scholar] [CrossRef]
- Lichtnekert, J.; Kulkarni, O.P.; Mulay, S.R.; Rupanagudi, K.V.; Ryu, M.; Allam, R.; Vielhauer, V.; Muruve, D.; Lindenmeyer, M.T.; Cohen, C.D.; et al. Anti-GBM Glomerulonephritis Involves IL-1 but Is Independent of NLRP3/ASC Inflammasome-Mediated Activation of Caspase-1. PLoS ONE 2011, 6, e26778. [Google Scholar] [CrossRef] [PubMed]
- Karasawa, T.; Takahashi, M. The crystal-induced activation of NLRP3 inflammasomes in atherosclerosis. Inflamm. Regen. 2017, 37, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nath, K.A.; Murali, N.S. Myoglobinuric and Hemoglobinuric Acute Kidney Injury, 5th ed.; Saunders Elsevier: Philadelphia, PA, USA, 2009; pp. 298–304. [Google Scholar]
- Anuradha, S.; Arora, S.; Mehrotra, S.; Arora, A.; Kar, P. Acute renal failure following para-phenylenediamine (PPD) poisoning: A case report and review. Renal Fail. 2004, 26, 329–332. [Google Scholar] [CrossRef]
- Fernandez, P.L.; Dutra, F.F.; Alves, L.; Figueiredo, R.T.; Mourão-Sa, D.; Fortes, G.B.; Bergstrand, S.; Lönn, D.; Cevallos, R.R.; Pereira, R.M.S.; et al. Heme Amplifies the Innate Immune Response to Microbial Molecules through Spleen Tyrosine Kinase (Syk)-dependent Reactive Oxygen Species Generation. J. Biol. Chem. 2010, 285, 32844–32851. [Google Scholar] [CrossRef]
- Gois, P.H.F.; Martines, M.S.; Ferreira, D.; Volpini, R.; Canale, D.; Malaque, C.; Crajoinas, R.; Girardi, A.C.C.; Massola Shimizu, M.H.; Seguro, A.C. Allopurinol attenuates acute kidney injury following Bothrops jararaca envenomation. PLoS Negl. Trop. Dis. 2017, 11, e0006024. [Google Scholar] [CrossRef]
- Mate-Kole, M.O.; Yeboah, E.D.; Affram, R.K.; Adu, D. Blackwater fever and acute renal failure in expatriates in Africa. Renal Fail. 1996, 18, 525–531. [Google Scholar] [CrossRef]
- Schrier, S.L. Diagnosis of Hemolytic Anemia in the Adult. Available online: https://www.uptodate.com/contents/diagnosis-of-hemolytic-anemia-in-the-adult (accessed on 12 March 2019).
- Viraraghavan, R.; Chakravarty, A.G.; Soreth, J. Cefotetan-induced haemolytic anaemia: A review of 85 cases. Adverse Drug React. Toxicol. Rev. 2002, 21, 101–107. [Google Scholar] [CrossRef]
- Chapman, A.B.; Rahbari-Oskoui, F.F.; Bennett, W.M. Acquired cystic disease of the kidney in adults. Available online: https://www.uptodate.com/contents/acquired-cystic-disease-of-the-kidney-in-adults (accessed on 11 March 2019).
- Abreu, P.A.E.; Seguro, A.C.; Canale, D.; Silva, A.M.G.d.; Matos, L.d.R.B.; Gotti, T.B.; Monaris, D.; Jesus, D.A.d.; Vasconcellos, S.A.; de Brito, T.; et al. Lp25 membrane protein from pathogenic Leptospira spp. is associated with rhabdomyolysis and oliguric acute kidney injury in a guinea pig model of leptospirosis. PLoS Negl. Trop. Dis. 2017, 11, e0005615. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, P.L.; Jacinto, C.N.; Silva Junior, G.B.; Lima, J.B.; Veras, M.d.S.B.; Daher, E.F.; Daher, E.F. Acute kidney injury caused by Crotalus and Bothrops snake venom: A review of epidemiology, clinical manifestations and treatment. Rev. Inst. Med. Trop. Sao Paulo 2013, 55, 295–301. [Google Scholar] [CrossRef]
- De Bragança, A.C.; Moreau, R.L.M.; De Brito, T.; Shimizu, M.H.M.; Canale, D.; De Jesus, D.A.; Silva, A.M.G.; Gois, P.H.; Seguro, A.C.; Magaldi, A.J. Ecstasy induces reactive oxygen species, kidney water absorption and rhabdomyolysis in normal rats. Effect of N-acetylcysteine and Allopurinol in oxidative stress and muscle fiber damage. PLoS ONE 2017, 12, e0179199. [Google Scholar] [CrossRef]
- Trowbridge, A.A.; Green, J.B.; Bonnett, J.D.; Shohet, S.B.; Ponnappa, B.D.; McCombs, W.B. Hemolytic anemia associated with leptospirosis. Morphologic and lipid studies. Am. J. Clin. Pathol. 1981, 76, 493–498. [Google Scholar] [CrossRef]
- Da Silva Junior, G.B.; Pinto, J.R.; Barros, E.J.G.; Farias, G.M.N.; Daher, E.D.F. Kidney involvement in malaria: An update. Rev. Inst. Med. Trop. Sao Paulo 2017. [Google Scholar] [CrossRef]
- Mendonça, R.; Silveira, A.A.A.; Conran, N. Red cell DAMPs and inflammation. Inflamm. Res. 2016. [Google Scholar] [CrossRef]
- Wagener, F.A.; Abraham, N.G.; Van Kooyk, Y.; De Witte, T.; Figdor, C.G. Heme-induced cell adhesion in the pathogenesis of sickle-cell disease and inflammation. Trends Pharmacol. Sci. 2001. [Google Scholar] [CrossRef]
- Nyakundi, B.B.; Tóth, A.; Balogh, E.; Nagy, B.; Erdei, J.; Ryffel, B.; Paragh, G.; Cordero, M.D.; Jeney, V. Oxidized hemoglobin forms contribute to NLRP3 inflammasome-driven IL-1β production upon intravascular hemolysis. Biochim. Biophys. Acta Mol. Basis Dis. 2019. [Google Scholar] [CrossRef]
- López-Castejón, G.; Pelegrín, P. Current status of inflammasome blockers as anti-inflammatory drugs. Expert Opin. Investig. Drugs 2012, 21, 995–1007. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.G.; Brough, D.; Freeman, S. Inhibiting the Inflammasome: A Chemical Perspective. J. Med. Chem. 2016, 59, 1691–1710. [Google Scholar] [CrossRef] [PubMed]
- Diwan, V.; Gobe, G.; Brown, L. Glibenclamide improves kidney and heart structure and function in the adenine-diet model of chronic kidney disease. Pharmacol. Res. 2014, 79, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Berns, J.S.; Glickman, J.D. Management of Hyperglycemia in Patients with Type 2 Diabetes and Pre-Dialysis Chronic Kidney Disease or End-Stage Renal Disease. Available online: https://www.uptodate.com/contents/management-of-hyperglycemia-in-patients-with-type-2-diabetes-and-pre-dialysis-chronic-kidney-disease-or-end-stage-renal-disease (accessed on 15 March 2019).
- Meloni, G.; Meloni, T. Glyburide-induced acute haemolysis in a G6PD-deficient patient with NIDDM. Br. J. Haematol. 1996, 92, 159–160. [Google Scholar] [CrossRef] [PubMed]
- Vinzio, S.; Andrès, E.; Perrin, A.-E.; Schlienger, J.-L.; Goichot, B. Glibenclamide-induced acute haemolytic anaemia revealing a G6PD-deficiency. Diabetes Res. Clin. Pract. 2004, 64, 181–183. [Google Scholar] [CrossRef]
- Perregaux, D.G.; McNiff, P.; Laliberte, R.; Hawryluk, N.; Peurano, H.; Stam, E.; Eggler, J.; Griffiths, R.; Dombroski, M.A.; Gabel, C.A. Identification and characterization of a novel class of interleukin-1 post-translational processing inhibitors. J. Pharmacol. Exp. Ther. 2001, 299, 187–197. [Google Scholar] [PubMed]
- Coll, R.C.; Robertson, A.A.B.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perera, A.P.; Fernando, R.; Shinde, T.; Gundamaraju, R.; Southam, B.; Sohal, S.S.; Robertson, A.A.B.; Schroder, K.; Kunde, D.; Eri, R. MCC950, a specific small molecule inhibitor of NLRP3 inflammasome attenuates colonic inflammation in spontaneous colitis mice. Sci. Rep. 2018, 8, 8618. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.B.; Butler, M.S.; Rowe, D.B.; O’Neill, L.A.; et al. Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 2018, 10, eaah4066. [Google Scholar] [CrossRef]
- Zhai, Y.; Meng, X.; Ye, T.; Xie, W.; Sun, G.; Sun, X. Inhibiting the NLRP3 Inflammasome Activation with MCC950 Ameliorates Diabetic Encephalopathy in db/db Mice. Molecules 2018, 23, 1–14. [Google Scholar] [CrossRef]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.H.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef] [Green Version]
- Vitale, A.; Insalaco, A.; Sfriso, P.; Lopalco, G.; Emmi, G.; Cattalini, M.; Manna, R.; Cimaz, R.; Priori, R.; Talarico, R.; et al. A Snapshot on the On-Label and Off-Label Use of the Interleukin-1 Inhibitors in Italy among Rheumatologists and Pediatric Rheumatologists: A Nationwide Multi-Center Retrospective Observational Study. Front. Pharmacol. 2016, 7, 380. [Google Scholar] [CrossRef]
- Alten, R.; Gram, H.; Joosten, L.A.; Berg, W.B.v.d.; Sieper, J.; Wassenberg, S.; Burmester, G.; van Riel, P.; Diaz-Lorente, M.; Bruin, G.J.M.; et al. The human anti-IL-1β monoclonal antibody ACZ885 is effective in joint inflammation models in mice and in a proof-of-concept study in patients with rheumatoid arthritis. Arthritis Res. Ther. 2008, 10, R67. [Google Scholar] [CrossRef]
- Rondeau, J.-M.; Ramage, P.; Zurini, M.; Gram, H. The molecular mode of action and species specificity of canakinumab, a human monoclonal antibody neutralizing IL-1β. mAbs 2015, 7, 1151–1160. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; MacFadyen, J.G.; Glynn, R.J.; Koenig, W.; Libby, P.; Everett, B.M.; Lefkowitz, M.; Thuren, T.; Cornel, J.H. Inhibition of Interleukin-1beta by Canakinumab and Cardiovascular Outcomes in Patients With Chronic Kidney Disease. J. Am. Coll. Cardiol. 2018, 71, 2405–2414. [Google Scholar] [CrossRef] [PubMed]
- Solomon, D.H.; Glynn, R.J.; MacFadyen, J.G.; Libby, P.; Thuren, T.; Everett, B.M.; Ridker, P.M. Relationship of Interleukin-1β Blockade With Incident Gout and Serum Uric Acid Levels: Exploratory Analysis of a Randomized Controlled TrialInterleukin-1β Blockade, Incident Gout, and Serum Uric Acid Levels. Ann. Intern. Med. 2018, 169, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, G.; Parker, T.; Turner, D.; Parker, M.; Scales, J.; Harnett, P.; Harrison, M.; Ahmed, K.; Bhagat, S.; Marianayagam, T.; et al. Feasibility randomised multicentre, double-blind, double-dummy controlled trial of anakinra, an interleukin-1 receptor antagonist versus intramuscular methylprednisolone for acute gout attacks in patients with chronic kidney disease (ASGARD): Protocol study. BMJ Open 2017, 7, e017121. [Google Scholar] [PubMed]
- Ben-Zvi, I.; Kukuy, O.; Giat, E.; Pras, E.; Feld, O.; Kivity, S.; Perski, O.; Bornstein, G.; Grossman, C.; Harari, G.; et al. Anakinra for Colchicine-Resistant Familial Mediterranean Fever: A Randomized, Double-Blind, Placebo-Controlled Trial. Arthritis Rheumatol. 2017, 69, 854–862. [Google Scholar] [CrossRef] [PubMed]
- Ugurlu, S.; Ergezen, B.; Ozdogan, H. Anakinra treatment in patients with Familial Mediterranean Fever: A single-center experience. Pediatr. Rheumatol. 2015, 13, P123. [Google Scholar] [CrossRef]
- Moghaddas, F.; Llamas, R.; De Nardo, D.; Martinez-Banaclocha, H.; Martinez-Garcia, J.J.; Mesa-del-Castillo, P.; Baker, P.J.; Gargallo, V.; Mensa-Vilaro, A.; Canna, S.; et al. A novel Pyrin-Associated Autoinflammation with Neutrophilic Dermatosis mutation further defines 14-3-3 binding of pyrin and distinction to Familial Mediterranean Fever. Ann. Rheum. Dis. 2017, 76, 2085–2094. [Google Scholar] [CrossRef] [PubMed]
- Masters, S.L.; Lagou, V.; Jéru, I.; Baker, P.J.; Van Eyck, L.; Parry, D.A.; Lawless, D.; De Nardo, D.; Garcia-Perez, J.E.; Dagley, L.F.; et al. Familial autoinflammation with neutrophilic dermatosis reveals a regulatory mechanism of pyrin activation. Sci. Transl. Med. 2016, 8, 332–345. [Google Scholar] [CrossRef] [PubMed]
- Curiel, R.V.; Guzman, N.J. Challenges Associated with the Management of Gouty Arthritis in Patients with Chronic Kidney Disease: A Systematic Review. Semin. Arthritis Rheum. 2012, 42, 166–178. [Google Scholar] [CrossRef] [PubMed]
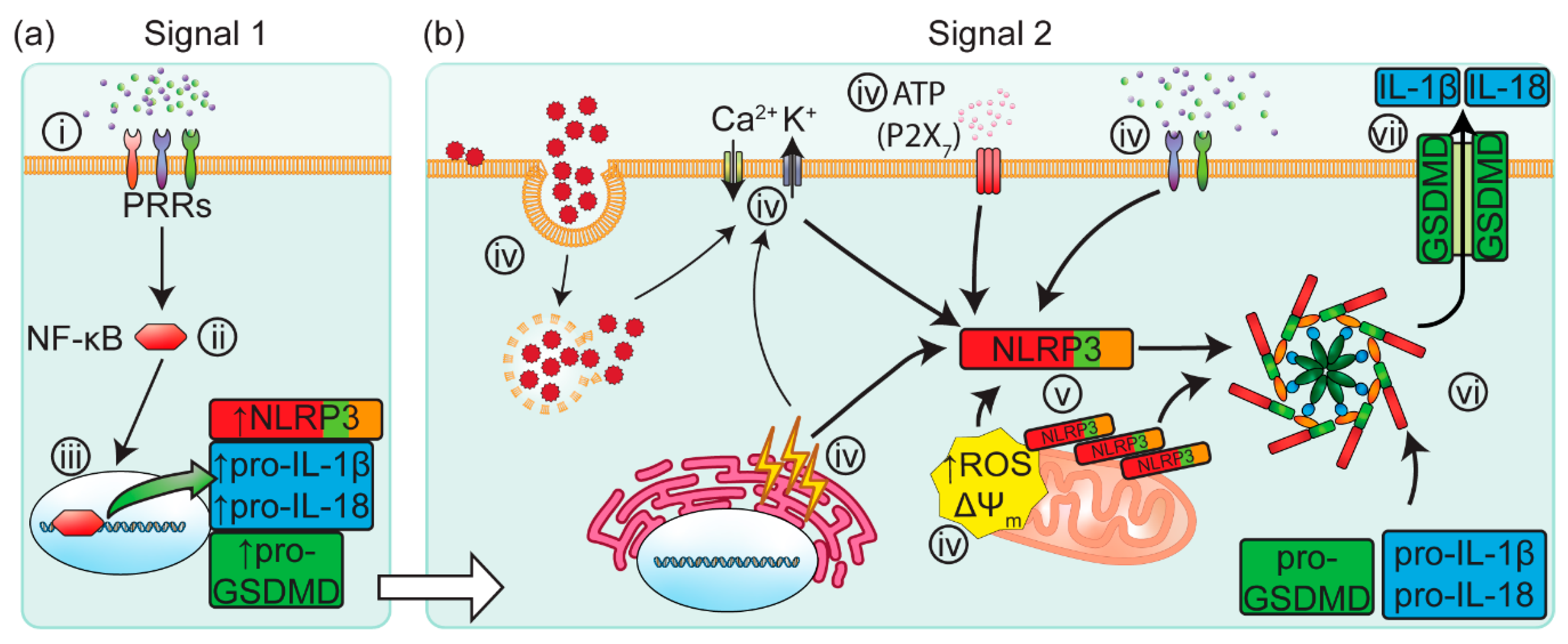
Figure 1.
The canonical inflammasome activation signalling cascade is initiated by signal 1 PAMPs and DAMPs. (a) Signal 1 elicits the activation of PRRs on the cell surface (i). The activation of PRRs results in a downstream signalling cascade, triggering the translocation of NF-κB into the nucleus (ii), where NF-κB upregulates the expression of NLRP3, pro-GSDMD, pro-IL-1β and pro-IL-18 (iii). (b) Signal 2 is provided by an array of PAMPs, DAMPs and HAMPs (iv), including arrested phagocytosis, perturbed membrane potential (ΔΨm), endoplasmic reticulum stress, extracellular ATP, and mitochondrial dysfunction. NLPR3 proteins which have co-localized to the mitochondria (v) are ideally located to rapidly respond to these markers of cellular stress. NLRP3 then oligomerizes with ASC and pro-Caspase-1, forming the NLRP3 inflammasome complex (vi). Caspase-1 undergoes self-cleavage whilst bound to the inflammasome complex (vi), driving the post-translational processing of IL-1β, IL-18 and GSDMD. Once cleaved, GSDMD proteins self-oligomerize to form pores in the cell membrane (vii), allowing for the rapid release of IL-1β and IL-18. In addition, these GSDMD pores may also drive cell-death via pyroptosis. ASC: Apoptosis-associated Speck-like protein containing a Caspase-activation-and-recruitment domain; PRR: pattern recognition receptor; PAMP: pathogen-associated molecular pattern; DAMP: damage-associated molecular pattern; nuclear factor kappa-light-chain-enhancer of activated B cells: NF-κB; NLRP3: nucleotide-binding domain-like receptor protein 3; IL: interleukin; GSDMD: Gasdermin D; ROS: reactive oxygen species; ΔΨm: mitochondrial membrane potential.
Figure 1.
The canonical inflammasome activation signalling cascade is initiated by signal 1 PAMPs and DAMPs. (a) Signal 1 elicits the activation of PRRs on the cell surface (i). The activation of PRRs results in a downstream signalling cascade, triggering the translocation of NF-κB into the nucleus (ii), where NF-κB upregulates the expression of NLRP3, pro-GSDMD, pro-IL-1β and pro-IL-18 (iii). (b) Signal 2 is provided by an array of PAMPs, DAMPs and HAMPs (iv), including arrested phagocytosis, perturbed membrane potential (ΔΨm), endoplasmic reticulum stress, extracellular ATP, and mitochondrial dysfunction. NLPR3 proteins which have co-localized to the mitochondria (v) are ideally located to rapidly respond to these markers of cellular stress. NLRP3 then oligomerizes with ASC and pro-Caspase-1, forming the NLRP3 inflammasome complex (vi). Caspase-1 undergoes self-cleavage whilst bound to the inflammasome complex (vi), driving the post-translational processing of IL-1β, IL-18 and GSDMD. Once cleaved, GSDMD proteins self-oligomerize to form pores in the cell membrane (vii), allowing for the rapid release of IL-1β and IL-18. In addition, these GSDMD pores may also drive cell-death via pyroptosis. ASC: Apoptosis-associated Speck-like protein containing a Caspase-activation-and-recruitment domain; PRR: pattern recognition receptor; PAMP: pathogen-associated molecular pattern; DAMP: damage-associated molecular pattern; nuclear factor kappa-light-chain-enhancer of activated B cells: NF-κB; NLRP3: nucleotide-binding domain-like receptor protein 3; IL: interleukin; GSDMD: Gasdermin D; ROS: reactive oxygen species; ΔΨm: mitochondrial membrane potential.

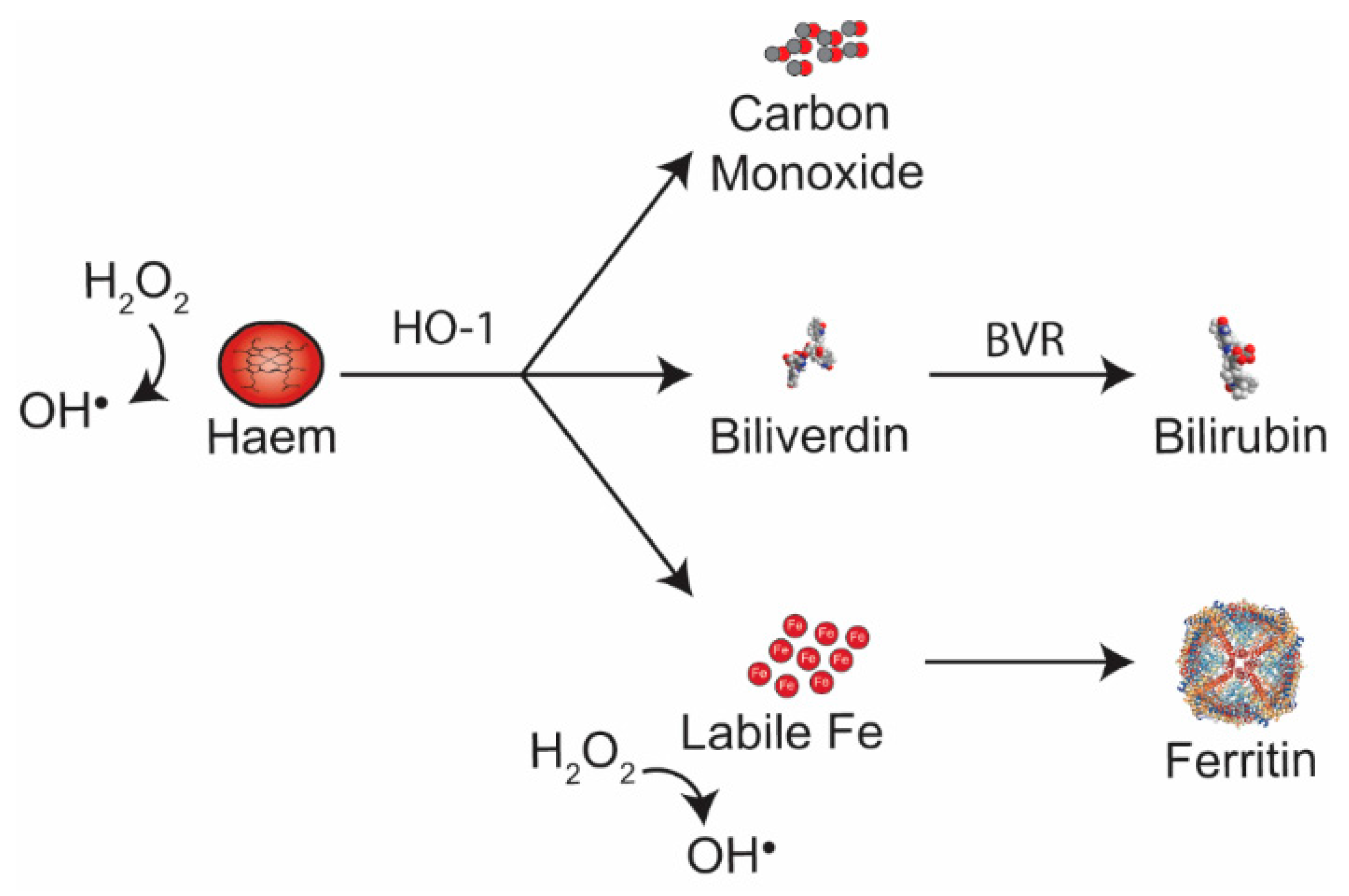
Figure 2.
Haem catabolism by HO-1 produces equimolar amounts of carbon monoxide, Biliverdin and labile iron. Biliverdin is converted to bilirubin by biliverdin reductase. Labile Fe can produce ROS, but is rapidly bound to ferritin. Ferritin (PBD ID: 5Z8U) image generated using the RCSB PDB NGL viewer [67]. BVR: Bilirubin reductase; HO-1: Haem oxygenase-1.
Figure 2.
Haem catabolism by HO-1 produces equimolar amounts of carbon monoxide, Biliverdin and labile iron. Biliverdin is converted to bilirubin by biliverdin reductase. Labile Fe can produce ROS, but is rapidly bound to ferritin. Ferritin (PBD ID: 5Z8U) image generated using the RCSB PDB NGL viewer [67]. BVR: Bilirubin reductase; HO-1: Haem oxygenase-1.

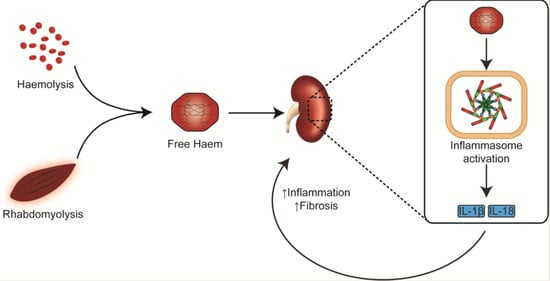
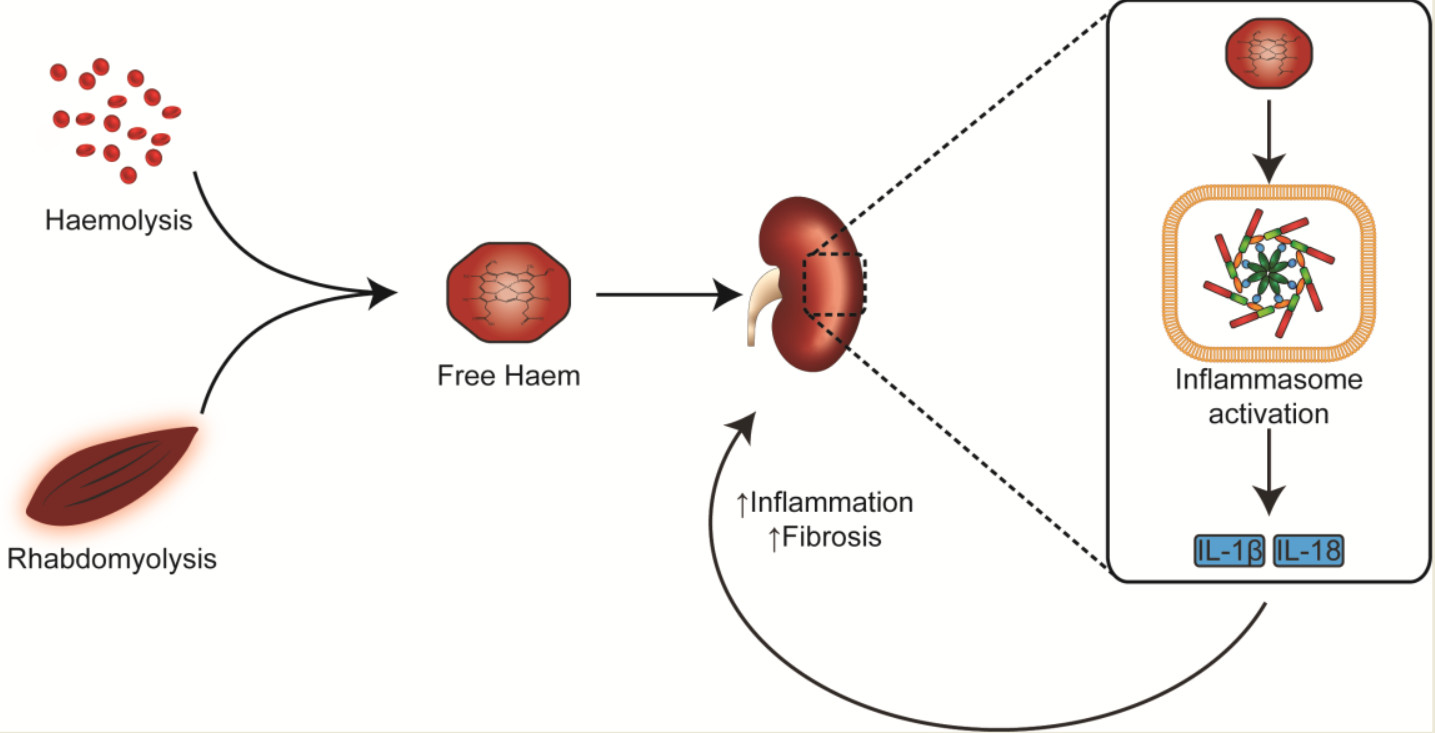
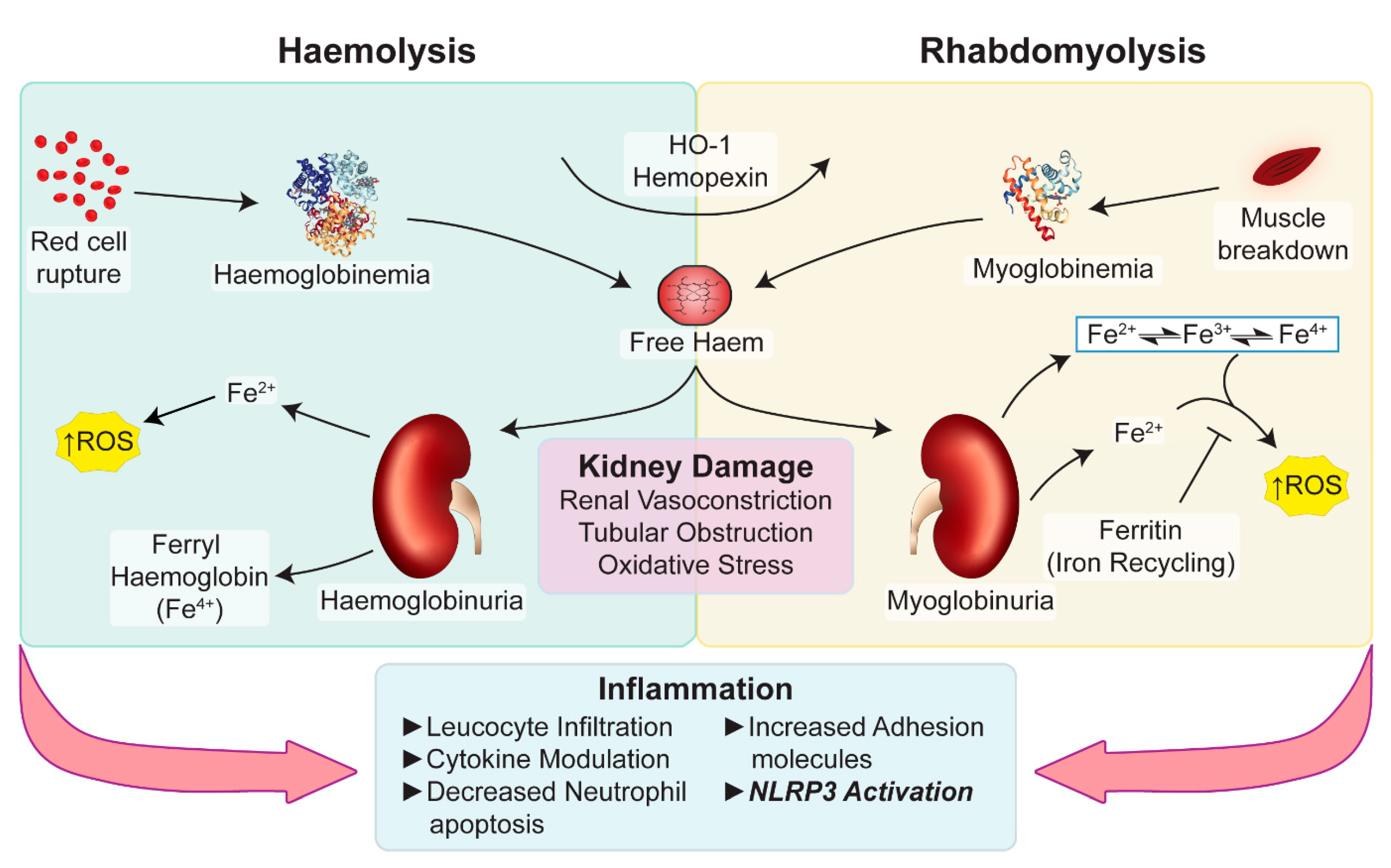
Figure 3.
Potential pathways underlying haem-associated kidney injury. Free haem generated by rhabdomyolysis and haemolysis are effectively removed by HO-1 and hemopexin. The binding capacity of these proteins is saturated in pathological conditions and free haem continues to be present. Haemoglobin, myoglobin and plasma free-haem are freely filtered by the glomerulus and can be deposited within the tubules. Oxidative stress, renal vasoconstriction, tubular obstruction by casts, iron-mediated tubular toxicity and inflammation play an important role in acute pigment nephropathy. Myoglobin (PBD ID: 1MBN) and haemoglobin (PBD ID: 1BIJ) structures generated using the RCSB PDB NGL viewer [67]. NLRP3: nucleotide-binding domain-like receptor protein 3; HO-1: Haem Oxygenase-1; ROS: Reactive Oxygen Species.
Figure 3.
Potential pathways underlying haem-associated kidney injury. Free haem generated by rhabdomyolysis and haemolysis are effectively removed by HO-1 and hemopexin. The binding capacity of these proteins is saturated in pathological conditions and free haem continues to be present. Haemoglobin, myoglobin and plasma free-haem are freely filtered by the glomerulus and can be deposited within the tubules. Oxidative stress, renal vasoconstriction, tubular obstruction by casts, iron-mediated tubular toxicity and inflammation play an important role in acute pigment nephropathy. Myoglobin (PBD ID: 1MBN) and haemoglobin (PBD ID: 1BIJ) structures generated using the RCSB PDB NGL viewer [67]. NLRP3: nucleotide-binding domain-like receptor protein 3; HO-1: Haem Oxygenase-1; ROS: Reactive Oxygen Species.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Giuliani, K.T.K.; Kassianos, A.J.; Healy, H.; Gois, P.H.F. Pigment Nephropathy: Novel Insights into Inflammasome-Mediated Pathogenesis. Int. J. Mol. Sci. 2019, 20, 1997. https://doi.org/10.3390/ijms20081997
AMA Style
Giuliani KTK, Kassianos AJ, Healy H, Gois PHF. Pigment Nephropathy: Novel Insights into Inflammasome-Mediated Pathogenesis. International Journal of Molecular Sciences. 2019; 20(8):1997. https://doi.org/10.3390/ijms20081997
Chicago/Turabian StyleGiuliani, Kurt T. K., Andrew J. Kassianos, Helen Healy, and Pedro H. F. Gois. 2019. "Pigment Nephropathy: Novel Insights into Inflammasome-Mediated Pathogenesis" International Journal of Molecular Sciences 20, no. 8: 1997. https://doi.org/10.3390/ijms20081997
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.